

Abstract
Studies on rodents and humans demonstrate an inherited predisposition to hepatocellular carcinoma (HCC). Analysis of the molecular alterations involved in the acquisition of a phenotype resistant or susceptible to hepatocarcinogenesis showed a deregulation of G1 and S phases in HCC of genetically susceptible F344 rats and a G1-S block in lesions of resistant Brown norway (BN) rats. Unrestrained extracellular signal-regulated kinase (ERK) activity linked to proteasomal degradation of dual-specificity phosphatase 1 (DUSP1), a specific ERK inhibitor, by the CKS1-SKP2 ubiquitin ligase complex occurs in more aggressive HCC of F344 rats and humans. This mechanism is less active in HCC of BN rats and human HCC with better prognosis. Upregulation of iNos cross-talk with IKK/NF-κB and RAS/ERK pathways occurs in rodent liver lesions at higher levels in the most aggressive models represented by HCC of F344 rats and c-Myc-TGF-α transgenic mice. iNOS, IKK/NF-κB, and RAS/ERK upregulation is highest in human HCC with a poorer prognosis and positively correlates with tumor proliferation, genomic instability and microvascularization, and negatively with apoptosis. Thus, cell cycle regulation and the activity of signal transduction pathways seem to be modulated by HCC modifier genes, and differences in their efficiency influence the susceptibility to hepatocarcinogenesis and probably the prognosis of human HCC.
Keywords: Hepatocarcinogenesis, Genetic predisposition, Polygenic disease, Redifferentiation, Signal transduction pathways, Cell cycle, Cell proliferation, Apoptosis, Proteasomal degradation
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most frequent human cancers, with 1 million of newly diagnosed cases each year. The highest frequencies are found in sub-Saharan Africa and far eastern Asia, where hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are endemic, and in regions where food contaminated with Aflatoxin B1 is consumed[1-3]. Other risk factors associated with the development of HCC include: long term use of oral contraceptive (female), high dose of androgen steroids, type 2 diabetes, genetic disorders such as hemochromatosis, hereditary tyrosinemia, glycogen storage disease (types 1 and 2), α1-antitrypsin deficiency, Wilson’s disease; porphyria cutanea tarda, galactosemia, orotic aciduria, congenital cholestatic syndrome, and environmental agents (Thorotrast, Aflatoxins, Cycasin, Pyrrolizidine alkaloids, Vinyl chloride, tobacco smoke, N-nitrosylated compounds). HCC incidence appears to be rising, even in countries with relatively low incidence[4]. HCC is prevalently male associated with M:F ratios ranging from 1.3 to 12.9 according to the geographic area[1]. It is a rapidly fatal disease, with a life expectancy of about 6 mo from the time of diagnosis. Partial liver resection or liver transplantation are potentially curative, but only a minority of the cases is amenable to these treatments.
The frequency of HCC, similarly to that of other tumor types, shows great differences within a human population as a response to environmental risk agents[5]. This suggests that additional environmental and/or genetic factors may be involved in the pathogenesis of the disease. Genetic polymorphisms of Cytochromes P450 2E1 and 2D6, Aldehyde dehydrogenase, Arylamine N-acetyltransferase 2, Epoxide hydrolase and L-MYC, and mutation of the Glutathione S-transferase gene have been associated with increased risk of HCC[6-10]. The genetic susceptibility has been shown to be one of the factors involved in familial aggregations of HCCs, even in HBV endemic areas where perinatal transmission of HBV is mainly responsible of familial HBV clustering[11]. The absence of an obviously inherited predisposition to the majority of liver cancers and of familial aggregations of HCC, independent of environmental agents such as HBV and HCV infections indicates that high-penetrance mutations are rare for this tumor, and suggests instead the involvement of low-penetrance genetic variants. Indeed, studies of families at risk seem to confirm the implication of a polygenic control of cancer incidence[12,13], and are in keeping with a polygenic model of autosomal recessive inheritance with a major gene involved in the genetic predisposition of HCC onset at an earlier age[14].
The development of cancers in mammals depends on accumulation within somatic cells of a number of genetic alterations including activation of proto-oncogenes and inactivation of oncosuppressor genes. Genetic instability plays an important role in the accumulation of these alterations[1]. Although various interindividual and interspecies differences have been documented, several morphological, biochemical and biological commonalties have been found in human and rodent preneoplastic and neoplastic liver lesions[3,15-17]. This suggests that the basic mechanisms of HCC development, in different species, are closely similar. Furthermore, studies with experimental models of complex diseases, different from cancer, indicate that the genetic variants controlling susceptibility map to orthologous regions of the mouse and human genomes, and various diseases are caused by polymorphism of equivalent mouse and human genes[18]. On the basis of these observations and considerations; in recent years, mouse and rat hepatocarcinogenesis models have been used to map susceptibility genes, define the genetic model responsible for an increased risk of HCC, and examine the effector mechanisms of tumor susceptibility genes.
RESISTANT PHENOTYPE
Liver carcinogenesis is a multistage process[1,15-17,19]. The clonal expansion of carcinogen-initiated cells (promotion) leads to the development of foci of altered hepatocytes (FAHs) that, in the rat, can be identified by immunohistochemical staining of glutathione S-transferase 7-7 (GST 7-7). Most liver preneoplastic lesions re-differentiate or no further evolve to cancer, whereas a subset of lesions acquires the capacity of autonomous growth and progress to neoplastic nodules (dysplastic nodules, adenomas) and HCCs[19,20]. Genomic instability, up-regulation of oncogenes and downregulation of oncosuppressor genes, and in late stages, alteration of cell adhesion mechanisms drive the process evolution[15,21].
A striking behavior of rodents’ genetic resistance to hepatocarcinogenesis is the fact that resistance genes apparently do not affect the initiation of the process, but only the capacity of initiated cells to grow autonomously[22-25]. In recent years the Brown norway (BN)[26] and Copenhagen (Cop)[27,28] rat strains have been shown to be strongly resistant to hepatocarcinogenesis. Both strains, crossed with F344 rats, susceptible to hepatocarcinogenesis, dominantly transmit their resistance to (BN × F344) F1 (BFF1) and (Cop × F344) F1 (CFF1) rats. Treatment of rats with DENA/AAF/partial hepatectomy, according to the “resistant hepatocyte” model of hepatocarcinogenesis[19], results in the development in the liver of resistant strains of an elevated number of fast-growing early preneoplastic lesions, positive for GST 7-7 expression. After exhaustion of the promoting stimulus, GST 7-7 positive lesions undergo a progressive decrease in growth capacity and intensive phenotypic reversion (remodeling)[29], thus showing inability to grow autonomously. In carcinogen-treated Cop rat strain, the block of progression of preneoplastic lesions exclusively depends on re-differentiation, whereas DNA synthesis in these lesions proceeds at the same rate as in the lesions of susceptible strains[27,28,30]. The analysis of BN and Cop rats excluded apoptosis in the preneoplastic lesions as a mechanism of resistance to hepatocarcinogenesis and attributed a preeminent role to redifferentiation[26-28]. However, apoptosis occurs in HCC from the resistant BN[31] and DHB[32] rats. These observations indicate that in the presence of dominant resistance alleles the subset of autonomously growing preneoplastic lesions, selected during promoting treatments, is very small or absent.
MAPPING OF THE LOCI CONTROLLING THE SUSCEPTIBILITY TO HEPATOCARCINOGENESIS
Linkage analysis experiments, made in various models of rodent hepatocarcinogenesis, led to the identification of different hepatocarcinogenesis susceptibility (Hcs) and resistance (Hcr) loci[25]. Mouse Hcs1, Hcs2, and Hcs3 loci were identified on chromosomes 7, 8, and 12, respectively, in urethane-treated F2 male mice generated by crossing the susceptible C3H/HeJ strain with the resistant A/J strain[33]. Interspecific testcrosses between the phylogenetically distant C3H/HeJ and Mus spretus mice, followed by the cross of the resulting F1 with the resistant C57BL/6J (B6) strain, to increase interstrain polymorphism[23], led to the identification of 3 additional Hcs loci (numbered from 4 to 6), mapping to chromosomes 2, 5, and 19, respectively. More recently a seventh Hcs locus (Hcs7) was mapped to distal chromosome 1 by analysis of the backcrosses and intercrosses between the susceptible C3H/HeJ or CBA/J strains and the resistant B6 strain[35]. Congenic B6.C3H (D1Mit5-D1Mit17) and B6.BR (D1Mit5-D1Mit17) were generated in which a approximate 70 cm segment (between D1Mit5 and D1Mit17) from C3H or C57BR/cdj (Br) susceptible strains, was introgressed onto a B6 background. These recombinant congenic strains (RCSs) develop more liver tumors than B6 mice, indicating that distal chromosome 1 carries potent modifier gene(s) sufficient to confer susceptibility to liver cancer.
Two loci involved in the susceptibility to HCC have been identified in crosses between BR and B6 mice[35]. BR females are extremely sensitive to HCC induction, since they are genetically insensitive to the inhibition of hepatocarcinogenesis exerted by ovarian hormones. This property is dominantly transmitted to B6BRF1 and BRB6F1 mice. BR alleles at two loci, on chromosomes 17 and 1, identified in backcrosses and F2 progeny, are associated with increased susceptibility in both sexes. They were denominated Hcf1 and Hcf2 (hepatocarcinogenesis in females) loci. Hcf1 and at a lower extent Hcf2 accounted for the higher sensitivity of BR mice to hepatocarcinogenesis.
In addition to susceptibility loci, two resistance loci, with negative phenotypic effects have been discovered in mouse genome. Hcr1 and Hcr2 loci map on chromosomes 4 and 10, respectively[36]. Further work[37] has shown that a resistant F1 mouse may be generated by crossing the resistant BXD-15 recombinant inbred mouse, presumably carrying Hcr genes contributed by the parental strain DBA/2J, to susceptible recombinant BXD-11 mice, which should carry DBA/2J Hcs genes. This strongly suggests that Hcr genes may modify the activity of several sensitivity loci.
The genome of the BALB/c mouse strain provides alleles that semi dominantly inhibit hepatocellular tumor development in F1 crosses with the highly hepatocarcinogenesis-susceptible C3H/He strain[39]. Recent genome-wide linkage analysis in a F2 population produced by intercrossing the BALB/c to the C3H/He mouse strain revealed a hepatocarcinogen resistance 3 (Hpcr3) locus with a major role in the resistance to urethane-induced hepatocarcinogenesis[40]. This locus, mapping to central Chromosome 15, accounts for 40% of the phenotypic variance. A gene expression profile of normal adult mouse liver showed a significant association with susceptibility of BALB/c, C3H/He, and F1 mice to hepatocarcinogenesis, and identified the genes expressed in the Hpcr3 locus region. This analysis implicated the E2F1 pathway in the modulation of the phenotype susceptibility to hepatocarcinogenesis.
The first locus regulating the susceptibility of rats to chemical hepatocarcinogenesis, denominated rcc locus, has been identified in the telomeric end of chromosome 20 of MHC-recombinant rat strains, congenic for the MHC genes and its linked region grc (growth reproduction complex)[41,42]. The rcc+ locus has many properties in common with tumor-suppressor genes: it is recessive, its deletion causes phenotypic susceptibility to various carcinogens, and it inhibits tumor development in many organs and tissues, including liver, skin, kidney and mesenchyma[41].
Numerous other loci have been identified by linkage analysis of male backcrosses and intercrosses of resistant BN and/or Cop rats to susceptible F344 rats. Seven susceptibility loci have been identified: rat Hcs1 and Hcs2 loci on chromosomes 7 and 1 respectively, in BN × BFF1 backcross progeny[43], Hcs3 and Hcs4 loci in BFF2 rats[44], and Hcs3, Hcs5, Hcs6 and Hcs7 in CFF2 intercrosses[45]. Hcr loci numbered 1 to 3 have been mapped to chromosomes 10, 4, and 8, respectively, in BN × BFF1 backcrosses[43]. Four additional Hcr loci, numbered from 9 to 12, (Rat genome database, www.rgd.mcw.edu/; previously numbered from 4 to 7) were identified on chromosomes 4, 6, and 8 of BFF2 rats[44]. Hcr13 and Hcr14 (RGD; previously numbered 8 and 9) were mapped to chromosomes 4 and 18 of CFF2 rats[45].
The results of genomic scanning of crosses of BN and Cop rats with F344 rats are consistent with some observations on a resistant mutant of Donryu rats strain, the DRH rats[46,47], indicating the presence of two clusters of genes on chromosomes 1 and 4 of (DRH × F344) F2 rats, designated collectively as Drh1 and Drh2, and considered to be resistance genes. These genes are transmitted dominantly from DHR rats to the F2 progeny. The Drh1 locus affects the development of FAH induced by 3’-Me-DAB[46,47], whereas Drh2 seems to control the progression of FAH to carcinoma. On the basis of the chromosomal localization, Dhr2 seems to correspond to Hcr2 on chromosome 4, while Drh1 corresponds to Hcs3 and Hcs5.
The phenotypic effect of Hcs3 locus in BFF2 rats, consisting in a marked increase in the volume of neoplastic nodules, accounts for 49% of the total phenotypic traits[44]. In CFF2 rats, Hcs3 and Hcs5 loci apparently account for only 14.6% and 8.4% of the phenotypic trait, respectively, consisting in about a 100% rise in number of non-remodeling nodules[45]. These nodules represent less than 20% of the total lesions in these rats. Thus, a diluting effect of the large number of remodeling lesions may be responsible for the apparently low penetrance of the C allele at the Hcs3 locus. In DRH rats, the presence of F alleles at the Drh1 locus has a dominant positive effect on the number of FAH (about 100% increases)[46]. These phenotypic effects of Hcs3/Drh1 have been recently confirmed in a congenic DRH.F344-Drh1 strain in which an about 43 cm segment of Drh1 from F344 rats has been introgressed onto a DRH background[32]. Above observations are consistent with a major role of Hcs3 in the predisposition to liver cancer. It should be noted that the analysis of phenotypic effects of susceptibility/resistance loci in rats showed the presence of susceptibility loci in resistant BN rats. Given that BFF1 rats, heterozygous at all loci, are resistant to hepatocarcinogenesis[26], the behavior of backcross rats suggests the existence of inhibitory mechanisms of susceptibility genes in these animals.
Interestingly, the Hpcr3 locus in the middle region of mouse chromosome 15 is synthetic to the rat Hcs1 locus which has been linked with dominant resistance to hepatocarcinogenesis in a backcross between the genetically resistant BN and the susceptible F344 rat strains[43]. Thus, it is possible that the mouse Hpcr3 and the rat Hcs1 locus modify hepatocarcinogenesis susceptibility by functional alleles of the same gene in both species. Moreover, both the mouse Hpcr3 and the rat Hcs1 regions are homologous to the human chromosome 8q region which undergoes frequent structural alterations in HCCs, including copy number gains that have been associated with tumor growth[48-50]. Consistent with these findings, the gain of chromosome 15 occurs in primary HCCs of B6C3F1 mice[51]. These findings suggest that Hpcr3 represents a major modifier locus for hepatocarcinogenesis.
The progressive disappearance of molecular markers of preneoplastic lesions, followed by the disappearance of histological evidence of these lesions (phenotypic reversion, remodeling), during rat liver carcinogenesis, has been interpreted, at least in some instances, as re-differentiation[19,20,26-28]. The regulation of marker expression may involve the activity of various genes. We have identified in BFF2 rats’ two loci, denominated liver neoplastic nodule remodeling, Lnnr1 and Lnnr2 whose phenotypic effect was the reduction in the percentage of remodeling lesions[52]. Due to an intensive remodeling of preneoplastic lesions in the Cop strain, more detailed information was obtained by genomic scanning of CFF2 rats, in which four Lnnr loci were discovered: Lnnr3 downregulated the number of remodeling neoplastic nodule, whereas Lnnr4 and Lnnr5 had the opposite effect. An additional locus, on chromosome 6, Lnnr6, had a negative phenotypic effect on the volume of remodeling nodules[45]. These observations are consistent with a model of hepatocarcinogenesis in which only a relatively small subset of early preneoplastic lesions is genetically programmed to evolve to HCC, whereas the remainder undergoes phenotypic reversion. The importance of this phenomenon is underlined by the observation of hepatocarcinogenesis prevention by compounds inducing remodeling[20], as well as by its implication in some cases of spontaneous regression of liver nodules and carcinomas in humans[53].
The recent construction of the F344.BN-Hcs4 RCS, by introgressing a 4.41 cm portion of Hcs4 from BN strain in an isogenic F344 background, allowed the important identification of a high penetrance gene(s), activated by estrogens and inhibited/unaffected by testosterone, conferring resistance to females towards liver cancer[54]. The volume and positivity for Proliferating Cell Nuclear Antigen (PCNA) were much higher in chemically induced preneoplastic lesions of F344 than BN rats, of both sexes. These parameters were lower in females than males. It was found that lesion volume and PCNA values of male RCS were similar to those of F344 rats, but corresponded in females to those of BN females. Carcinomatous nodules and HCC developed, at 32 and 60 wk, respectively, in male F344 and congenics and, rarely, in F344 females. Gonadectomy of congenic males, followed by β-estradiol administration, caused decrease in Ar (Androgen receptor) gene expression, increase in Er-α (Estrogen receptor-α) expression, and development of preneoplastic lesions comparable to those from BN females. Administration of testosterone to gonadectomized females leads to Ar increase and development of preneoplastic lesions as in F344 males. This indicates a role of homozygous B alleles at Hcs4 in determination of phenotypic patterns of female RCS.
Research on rodent models of liver cancer have clearly shown a model-based on the polygenic inheritance of low penetrance genes with a few predominant susceptibility/resistance loci. Furthermore, the study of epistatic interactions between microsatellite loci, inducing phenotypic effects not predictable on the basis of the sum of their separate effect, resulted in the identification of several novel tumor modifier loci in rats, indicating that gene-gene interactions have a major role in hepatocarcinogenesis[25]. We can envisage the existence of different subsets of low-penetrance genes at play in different subsets of population.
Overall, these findings prove the existence of a great complexity of the inherited predisposition to liver cancer. A complex combination and interplay of susceptibility or resistance alleles determines the individual risk. Some individuals may inherit a predominance of susceptibility alleles and/or a major allele and may be highly cancer prone. However, since human individuals are generally casually assorted, independently of genetic factors, a situation of high or low genetic risk should be rare, and most humans should be at average risk. A corollary of this situation is that the effect of polygenic inheritance can be masked by a predominant presence of environmental high risk factors. Nevertheless, taking into account the effect of susceptibility/resistance genes on the proliferative activity and re-differentiation of initiated cells, it may be expected that the genetic substrate largely influences the prognosis.
PUTATIVE TARGETS OF SUSCEPTIBILITY/RESISTANCE GENES
Cell cycle deregulation
The deregulation of signal transduction pathways, cell cycle control, and genes involved in cell death signals characterizes initiated cells and influences their evolution to malignancy[1-3,15]. Several genes implicated in these activities, in mouse and rats, can be targeted by the susceptibility/resistance genes. This mechanism could be responsible for the acquisition of a phenotype susceptible or resistant to HCC development.
According to recent evidence HCCs developing in c-Myc transgenic mice undergo sustained regression, associated with re-differentiation of tumor cells, following inactivation of c-Myc transgene expression[55]. The re-differentiation is not terminal, and tumor growth starts again after restoration of c-Myc expression. These results, linking re-differentiation of tumor cells to the expression of c-Myc, a gene located at Lnnr1[52], are in keeping with the observation that inhibition of c-Myc expression by antisense strategy contributes to the deregulation of E2f1 expression, and blocks in vitro growth of human and rat HCC cells[56]. In addition, in vivo studies have shown c-Myc hypomethylation in preneoplastic and neoplastic rat liver lesions[57]. The decrease in c-Myc expression, induced by prolonged administration of the methyl donor S-adenosylmethionine to rats, is associated with growth restraint and re-differentiation of liver lesions[20,57,58]. No differences in c-Myc expression occur in the liver of normal F344 and BN rats[56,58,59]. However, the expression of this gene is much higher in preneoplastic and neoplastic lesions of F344 rats than in the lesions of BN rats[56,58,59]. c-Myc is frequently amplified in preneoplastic and neoplastic lesions of the susceptible strain, but not in the lesions of the resistant BN and Wistar strains[60]. Taken together, these observations suggest the existence of some connections between c-Myc and the susceptibility genes that regulate re-differentiation.
Since c-Myc regulates the pRb-E2F pathway, we evaluated cell cycle gene expression in neoplastic nodules and HCCs, induced by initiation/selection protocols, 40 and 70 wk after diethylnitrosamine treatment, in susceptible F344 rats, and resistant Wistar and BN rats[59]. No interstrain differences in gene expression were observed in normal livers. Overexpression of c-Myc, Cyclins D1, E, and A, and E2f1 genes, at mRNA and protein levels, rise in Cyclin D1-Cyclin-dependent kinase 4 (CDK4), Cyclin E-CDK2 and E2f1-DP1 complexes, and pRb hyperphosphorylation occurred in nodules and HCCs of F344 rats. In nodules and/or HCCs of Wistar and BN rats, low or no increases in c-Myc, Cyclins D1, E, and A, and E2f1 expression, and Cyclin-CDKs complexes formation were associated with pRb hypophosphorylation. These results are consistent with a deregulation of the G1 and S phases in liver lesions of susceptible rats, and a block of G1-S transition in lesions of resistant strains, which explains their low progression capacity.
Autonomously growing preneoplastic liver nodules develop in susceptible rat strains, initiated by diethylnitrosamine and subjected to the initiation/selection treatments of the “resistant hepatocyte” protocol, after the cessation (about 6 wk after initiation) of the promotion stimulus represented by partial hepatectomy[19,20,61]. In contrast, only very few preneoplastic lesions of resistant BN rats grow autonomously after the end of the promoting stage; the majority of lesions remodel or no further evolve to neoplasia[61]. To evaluate the molecular mechanisms underlying the appearance of the resistant phenotype in BN rats[61], the behavior of the p16INK4A and some genes regulating cell cycle inhibition by p16INK4A have been analyzed. Preneoplastic liver (7 wk after initiation), neoplastic nodules (32 wk) and HCC (57 wk) showed high p16INK4A expression at mRNA and protein levels, in both F344 and BN rat strains. This was associated with increase in the expression of Heat shock protein 90 (Hsp90) and Cell division cycle 37 (Cdc37) protein, and Cdc37-Cdk4 complex. The HSP90-CDC37 complex protects several kinases, including Cdk4 and Cdk6 from the formation of inhibitory complexes with p16INK4A[62-64] (Figure 1). Consequently, the increase in Cdc37-Cdk4 complex resulted in a decrease in p16INK4A-Cdk4 complex in the lesions of F344 rats, whereas lower/no changes occurred in BN rats[61].
Figure 1.
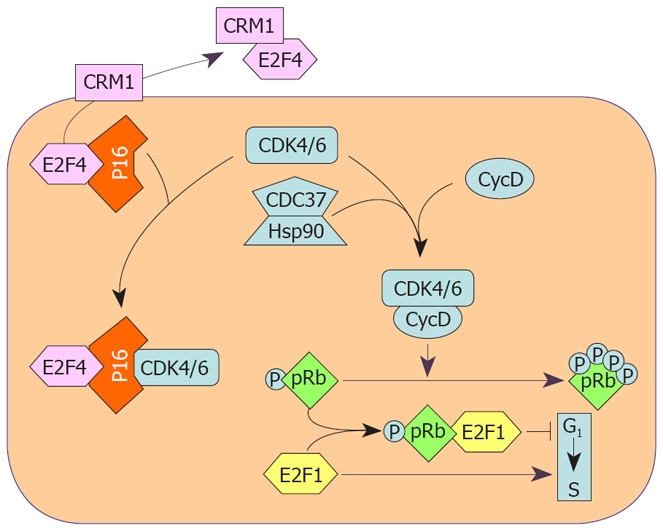
Cell cycle protection from inhibition by P16INK4A through the CDC37-HSP90 complex and CRM1 transporter protein. P16INK4A forms complexes with CDK4 and CDK6 which, as a consequence, cannot by activated by Cyclin D1 and cannot phosphorylate pRb. The chaperons CDC37 and HSP90 form complexes with CDKs protecting them from inactivation by P16INK4A. CRM1 forms a complex with E2F4, a P16INK4A effector, transporting it outside of the nucleus, thus inactivating P16INK4A.
The transcription factor E2f4, is a p16INK4A effector, acting as a growth repressor[65], equally expressed in the lesions of both F344 and BN rats. Required for chromosome region maintenance 1 protein (Crm1) is a receptor for various proteins containing a specific nuclear export sequence, including E2f4[66]. Crm1 and the cytoplasmic E2f4-Crm1 complex are highest in preneoplastic and neoplastic lesions of F344 rats. This indicates more elevated nuclear E2f4 efflux in the susceptible rats leading to a decrease in the interaction of p16INK4A with G1 kinases (Figure 1). Furthermore, lower P16INK4A level and highest upregulation of the HSP90/CDC37, and E2F4/CRM1 systems occur in human HCCs with a poorer prognosis (HCCP), based on survival rate, compared to HCCs with a better prognosis (HCCB)[61]. Accordingly, the P16INK4A-CDK4 complex is higher in HCCB than in HCCP, whereas the complexes of CDK4 with HSP90 and CDC37 are higher in HCCP than in HCCB. Consistent with a protective role of CDC37 against growth inhibition is the observation that a decrease in its expression, induced by specific siRNAs, leads to inhibition of DNA synthesis in HepG2 cells, without modifying P16INK4A expression[61]. This suggests that CDC37 could be a target for HCC chemoprevention and therapy. These findings underline the role of the Hsp90/Cdc37 and E2f4/Crm1 systems in the acquisition of a susceptible or resistant phenotype in rats and suggest that the protection by CDC37 and CRM1 against cell cycle inhibition by P16INK4A may influence the prognosis of human HCC.
Overall, these observations indicate that susceptible F344 rats develop various adaptive mechanisms for protection against stress-responsive tumor suppressors, such as p16INK4A, that confer to their liver cells the ability to proliferate under stressful conditions, such as hypoxia, oxidative stress, DNA damage, abnormal conditions of growth and differentiation, inappropriate extracellular matrix, and improper cell-to-cell contracts. In the resistant BN strain, a shut off of these mechanisms, associated with cell cycle deregulation and growth inhibition, occurs in coincidence with the exhaustion of promoting stimuli.
Mitogen activated protein kinase pathway
The best characterized RAS effector promoting cell cycle progression is the mitogen activated protein kinase (MAPK) pathway[67] (Figure 2). Active RAS (RAS-GTP) drives the RAF1-MAPK kinase kinase 1/2 (MEK1/2)-extracellular signal-regulated kinase 1/2 (ERK1/2) cascade which mediates proliferative and survival signals and, through the binding to RAS association domain family 1A (RASSF1A) and the related protein Novel RAS effector 1A (NORE1A), may induce apoptosis[68,69]. RASSF1A and NORE1A are members of the RASSF family of RAS inhibitors and form homo- and hetero-dimers, which activate the mammalian sterile twenty kinase 1 (MST1 kinase), an upstream effector of the p38MAPK and JNK pathways[67-72]. Following activation, phosphorylated MST1 induces apoptosis via caspase-dependent and -independent mechanisms[60-72]. Furthermore, RASSF1A may inhibit the ERK pathway by its association with the plasma membrane calcium pump (PMCA) 4b protein[73]. Finally, p38MAPK may be also activated by Disabled homolog 2 (DAB2)-interacting protein (DAB2IP), a RAS-GTP inhibitor, via activation of Apoptosis signal-regulating kinase 1 (ASK1), which phosphorylates p38MAPK[74].
Figure 2.

Schematic representations of the activated RAS-MAPK, RASSF1A/NORE1A, and Dab2IP/ASK1 pathways, iNOS signalling, and FOXM1-related pathways involved in the dysregulation of cell growth and apoptosis in HCC. Active Ha-RAS (GTP-RAS) triggers the MAPK pathway leading to activation of ERK1/2. Active ERK can down-regulate DUSP1 by phosphorylation at ser296 allowing the formation of the SKP2/CSK1/DUSP1 complex, which facilitates DUSP1 ubiquitination and proteasomal degradation. In addition, ERK may further contribute to DUSP1 proteolysis via induction of its target FOXM1, leading to transcriptional activation of SKP2 and CKS1. These mechanisms result in decreased inhibition of ERK by DUSP1 (blunt arrow). Moreover, FOXM1 favors the growth of neoplastic cells by targeting genes involved in G2 ← M transition, genomic instability, angiogenesis, NF-κB activation, and anti-apoptosis. iNOS activates IKK that allows proteasomal degradation of the NF-κB inhibitor, IkB-α. This results in NF-κB activation. iNOS also activates Ha-RAS, thus triggering the MAPK pathway leading to activation of ERK1/2. NF-κB activation by ERK may occur through AURORA-A (AURKA) which inhibits IkB-α. NF-κB activates various antiapoptotic genes (XIAP, cIAP1, BCL-xL) and inhibits the proapoptotic gene JNK. The inhibition of RAS activation by DAB2IP leads to the activation of ASK1, whereas active RAS favors the formation of the RASSF1-NORE1A complex. Both the ASK1 and RASSF1-NORE1A complexes trigger pro-apoptotic pathways. ASK1: Apoptosis signal-regulating kinase 1; BCL-Xl: BCL2-related protein, long isoform; CDC37: Cell division cycle 37; CKS1: CDC28 protein kinase 1b; cIAP: Iinhibitor-of-apoptosis protein 1; DAB2: Disabled homolog 2; DAB2IP: DAB2-interacting protein; DUSP1: Dual-specificity phosphatase 1; EPO: Erythropoietin; ERK: Extracellular signal-regulated kinase; GLI1: Glioblastoma associated oncogene 1; HIF-1α: Hypoxia-inducible factor 1 α; HSP90: Heat shock protein 90; HXK II: Hexokinase II; IKK: Inhibitor of kB kinase; iNOS: Inducible nitric oxide synthase; MST1: Mammalian sterile twenty kinase 1; NEK2: NIMA-related kinase 2; NF-κB: Nuclear factor-κB; NORE1A: Novel RAS effector 1A; RASSF1A: RAS association domain family 1A; RKIP: RAF kinase inhibitory protein; SKP2: S-phase kinase-associated protein 2; VEGF-α: Vascular endothelial growth factor α; XIAP: Inhibitor of apoptosis, X-linked. Pointed and blunt arrows indicate activation and inhibition, respectively.
Studies on the role of the inhibitors of RAS/ERK pathway in the acquisition of a phenotype resistant or susceptible to hepatocarcinogenesis, showed moderate activation of Ras, Raf1, and Mek1/2 proteins, paralleled by strong induction of Dab2 and Raf kinase inhibitory protein (Rkip) inhibitors, in neoplastic nodules and HCC of both F344 and BN rats, induced by the “resistant hepatocyte” protocol[31]. This is compatible with the limitation of Ras-GTP-mediated activation of Raf1 due to the upregulation of the Raf1 inhibitor, Dab2[31,75] (Figure 2). Indeed, in the lesions developed in the resistant BN strain, lower Dab2 expression is associated with relatively low Ras-GTP and Raf1 expression and, consequently, low pRaf1 level[31]. Presumably, similar mechanisms may be envisaged for the inhibitory effect of Rkip1 on Mek activation[76] and determine the absence of interstrain differences in pMek1/2 expression[31]. However, the possibility that other phosphatases interfere with Raf and Mek activation, during rat liver carcinogenesis, cannot be excluded and should be the object of further research.
High levels of Dual-specificity phosphatase 1 (Dusp1), a specific Erk inhibitor (Figure 2), may be found only in neoplastic nodules and HCC of the resistant BN rat lesions, leading to modest Erk activation, whereas a progressive Dusp1 decline occurs in corresponding lesions from susceptible F344 rats and is accompanied by elevated Erk activation[31]. Interestingly, Dusp1 is slightly upregulated in preneoplastic liver of both F344 and BN strain, but its level progressively decreases in early liver nodules (12 wk after initiation), as well as in neoplastic nodules and HCC of F344 rats[31], i.e. in coincidence with the development of autonomously growing lesions in susceptible rats[25,61]. In contrast, Dusp1 further increases in the lesions of BN rats after the 12th week. This suggests that even in the presence of a limited increase in the levels of upstream activators (Raf1, Mek1/2) of Erk1/2, a failure of Dusp1 induction might sustain Erk1/2 activation and contributes to the development of autonomously growing liver lesions in F344 rats. This conclusion is further substantiated by the observed overexpression of pErk1/2 target genes, such as Hif1-α (hypoxia-inducible factor 1α) and Vegf-α (Vascular endothelial growth factor α), regulating angiogenesis, and Hxk II[31] (Hexokinase II). The latter gene is a key glycolytic enzyme whose expression is correlated with Hif1-α mRNA, and may promote HCC development by different mechanisms, including enhanced energy production, overproduction of antiapoptotic enzymes and metabolic precursors for cell growth[77]. In BN rats, progressive increases in Dusp1 expression in preneoplastic and neoplastic lesions is associated with low expression of pErk1/2 and its target genes. This could contribute to the low propensity of BN rat lesions to progress.
The mechanisms underlying the effect of susceptibility genes on the deregulation of the inhibitors of Ras/Erk cascade in rat hepatocarcinogenesis are not yet completely known. The progressive rise in expression of the inhibitors of Ras/Erk pathway, including Dab2 and Rkip, in preneoplastic and neoplastic liver lesions of F344 rats, suggests a compensatory mechanism controlling (at least in part) Ras-GTP, pRaf1, and pMek1/2 upregulation in this rat model. Sustained Erk activation in F344 nodules and HCC promotes the phosphorylation of Dusp1 at the ser296 residue. This is followed by proteasomal degradation of Dusp1[78,79] (see below) and enhancement of Erk-driven HCC growth. The presence of Dusp1 overexpression in late lesions of BN rats speaks in favor of a possible low proteasomal disruption of Dusp1 protein, thus resulting in a further inhibition of pErk1/2. These findings support the hypothesis that Dusp1 is at least one of the genes involved in the acquisition of a resistant phenotype. Interestingly, Dusp1 co-localizes with the resistance locus Hcr1 on chromosome 10, in correspondence of the LOD score peak, a region frequently affected by loss of heterozygosis (LOH) during rat hepatocarcinogenesis[80]. Nevertheless, the existence of localization and functional plausibility does not prove per se that Dusp1 is a hepatocarcinogenesis “modifier” gene[25]. Dusp1 could be indeed controlled by modifiers of hepatocarcinogenesis. In accordance with the latter statement, the absence of functional polymorphisms at the Dusp1 in F344 and BN rat liver tissues as detected by DNA sequencing (Feo et al, unpublished data) speaks against a role of Dusp1 as a tumor modifier gene. Further work is necessary to clarify this point.
A gradual increase of Rassf1A/Nore1A/Mst1-driven apoptosis has been detected in HCC of both F344 and BN strains, with highest levels in BN HCC, whereas loss of Dab2IP (Figure 2) occurs only in F344 rat HCC[31]. These changes are associated with significantly higher apoptosis in BN than F344 HCC. Taken together, these results indicate a control of the Ras/Erk pathway, as well as of the pro-apoptotic Rassf1A/Nore1A and Dab2IP/Ask1 pathways by HCC susceptibility genes. Dusp1 possesses a prominent role in the acquisition of the phenotype resistant to HCC by BN rats, whereas late activation of RassF1A/Nore1A and Dab2IP/Ask1 axes is implicated in the highest apoptosis characteristic of BN HCC[31].
Sustained ERK activity is associated with various types of human tumors, including lung, breast, colon, pancreas, and kidney[81-83]. This frequently depends on upregulation of the RAS/MEK cascade. However, constitutive ERK overexpression may also occur independently of the RAS/MEK signaling[84,85]. Recent studies[86] indicate that prolonged activation of ERK promotes phosphorylation at the Ser296 residue of DUSP1. Phosphorylation of this specific residue renders the DUSP1 protein susceptible to ubiquitination and proteasomal degradation by the Skp1/Cul1/F-box protein SKP2 (SCFSKP2)[79]. On the other hand, constitutive ERK expression may induce SKP2/CSK1 ubiquitin ligase which can phosphorylate DUSP1 and determine its ubiquitination (Figure 2). This mechanism, which contributes to maintain elevated ERK expression, could be at least partially attributed to induction by ERK of the FOXM1 (Forkhead box M1B)[86,87] (Figure 2) which, in turn, upregulates the SKP2/CSK1 ligase[88]. It should be noted, however, that transient activation of ERK induces the catalytic activation of DUSP1, followed by inactivation of ERK[87,88]. This body of evidence indicates that DUSP1 feedback inhibits its activation by ERK and that DUSP1 might be a crucial regulator of ERK activity in the cell. DUSP1 inactivation is frequent in prostate and urothelial tumors[89,90], and recent observations indicate that immunohistochemical positivity for DUSP1 in human HCC is associated with longer patients’ survival[91].
The interactions of DUSP1 with CKS1-SKP2 ubiquitin ligase have been recently evaluated in human HCC subtypes with different survival times, in the attempt to correlate the effects of DUSP1 molecular interactions with tumor growth and patients’ survival, and explore DUSP1 prognostic role[92]. It was found that the levels of DUSP1 are significantly higher in HCCB when compared with both normal and non-tumorous surrounding livers, whereas DUSP1 protein expression sharply declines in HCCP[92]. In the latter subtype, DUSP1 inactivation is due to either ERK/CKS1/SKP2-dependent ubiquitination or promoter hypermethylation associated with loss of heterozygosity at the DUSP1 locus. Notably, expression levels of DUSP1 inversely correlate with those of activated ERK as well as with HCC proliferation index and microvessel density, and directly correlate with HCC apoptosis and patients’ survival rate. Functional studies revealed that DUSP1 reactivation leads to suppression of ERK, CKS1 and SKP2 activities, inhibition of proliferation and induction of apoptosis in human hepatoma cell lines. Taken together, these data indicate that ERK achieves unrestrained activity during HCC progression by triggering ubiquitin-mediated proteolysis of its specific inhibitor DUSP1. Thus, DUSP1 may represent a valuable prognostic marker and ERK, CKS1 or SKP2 potential therapeutic targets for human HCC.
FOXM1: A pleiotropic regulator of hepatocarcinogenesis
FOXM1 transcription factor is a major downstream effector of ERK whose overexpression occurs in various experimental and human tumors[93,94]. FOXM1 promotes cell proliferation through its ability to influence various cell cycle phases. Indeed, FOXM1 triggers the activation of SKP2/CKS1 ubiquitin ligase, which targets P21WAF1, P27KIP1 and P57KIP2 proteins for degradation during the G1-S transition[93-98] (Figure 2). Furthermore, FOXM1 induces transcription of genes promoting cell cycle progression (AURKA, CDC2, CYCLIN B1, NEK2, and CDC25B), genomic instability generators (NEK2, CDC25B), suppressors of cell cycle inhibitors (SKP2, CKS1), and apoptosis inhibitors (SURVIVIN)[93-97]. In the mouse liver, Foxm1 depletion results in block of proliferation and resistance to hepatocarcinogenesis[98-102]. Recently, the role of FOXM1 during hepatocarcinogenesis has been studied in both, the susceptible/resistant comparative rat model and the human HCC (Feo et al, unpublished results). Activation of Foxm1 and its targets (AurkA, Cdc2, Cyclin B1, Nek2) occurs earlier and is most pronounced in liver lesions from F344 than BN rats, leading to highest Cdc2-Cyclin B1 complexes (implying highest G2-M transition) in F344 rats. In humans, FOXM1 is ubiquitously and progressively induced from surrounding non-tumorous liver to HCC, reaching highest levels in tumors with poorer prognosis and its expression levels directly correlate with proliferation index, genomic instability rate, and microvessel density, and inversely correlate with apoptosis. Interestingly, the strong correlation between FOXM1 levels and both genomic instability rate and adverse outcome in HCC agrees with the existence of a molecular signature, including FOXM1 overexpression, which is significantly associated with degree of genomic instability and accurately predicts patients survival in multiple tumors[87,103,104].
Some reports showed FOXM1 upregulation following either ERK or Glioblastoma associated oncogene 1 (GLI1) induction[105-107] (Figure 2). FOXM1 is a direct transcriptional target of GLI1[105,106]. GLI family proteins, including GLI1, 2, and 3, are the terminal effectors of the Hedgehog signaling[107,108]. The interaction of Sonic hedgehog (SHH) with its plasmamembrane receptor PTCH1, releases PTCH-induced inhibition of the membrane protein Smoothened (SMO). This results in GLI proteins activation and nuclear translocation, where they activate target gene transcription[109]. According to recent observations GLI2 is overexpressed in some HCC cell lines, and its inhibition by antisense oligonucleotides inhibits cell proliferation[110]. GLI1 overexpression occurs in a lower number of HCC cell lines and its inhibition causes lower decrease in growth rate[110].
High levels of phosphorylated ERK1/2 (pERK1/2) and Gli1 proteins occur in neoplastic nodules and HCC induced by the resistant hepatocyte protocol in F344 rats and, at a lower extent, in BN rats (Feo et al, unpublished results). Furthermore, pERK1/2 and GLI1 expression are higher in human HCC than normal and non-neoplastic surrounding livers, and most pronounced in HCCP. Silencing of either ERK2 or GLI1 via siRNA in human HCC cell lines, leads to strong decreases in FOXM1 levels, whereas forced ERK2 or GLI1 overexpression results in a remarkable elevated rise in FOXM1 level[107]. These findings suggest a reciprocal activation of ERK2 and GLI1, but the mechanisms underlying this phenomenon and its role in the activation of Hedgehog signaling are unclear and require further investigation.
Interestingly, a recent report implies the combined overexpression of HSP90 and CDC37 in sustaining elevated Fused Homolog expression[111]. Accordingly, a previous report from our laboratory showed a strong induction of HSP90 and CDC37 in F344 rat liver lesions and human HCCP[61]. Thus, it might be hypothesized a role of HSP90 and CDC37 combined activity in the highest activation of GLI1 observed in F344 neoplastic lesions and human HCCP.
The observation that FOXM1 induces SKP2 and CKS1 expression, involved in DUSP1 degradation, underlines the role of FOXM1 in the active proliferation of HCC cells, though its implication in a positive feedback loop reinforcing ERK cascade, by its ability to inhibit DUSP1[78] (Figure 2).
Inducible nitric oxide synthase signaling
Inducible nitric oxide synthase (iNOS) produces sustained nitric oxide (NO) concentrations in response to pro-inflammatory agents. NO is a major mediator of chronic inflammation and may modulate tumorigenesis by regulating cell proliferation, survival, and migration, angiogenesis, drug resistance, and DNA repair[112,113]. In particular, iNOS might promote unrestrained cell growth via its ability to inactivate the retinoblastoma (pRb) pathway[114]. Some observations envisage a cross talk between iNOS and inhibitor of kB kinase (IKK)/nuclear factor-κB (NF-κB) and RAS/ERK pathways. The Ikk and NF-κB activities are strongly reduced in iNos knockout mice[115]. NO activates Ha-RAS/ERK pathway in T lymphocytes[116]. Phosphorylated ERK activates iNos in melanoma[117] and NF-κB in HeLa cells[118].
iNOS, NF-κB, RAS, and ERK are upregulated in preneoplastic rat liver lesions[119], dysplastic and neoplastic liver from c-Myc/TGF-α transgenic mice[120], and human HCCs[121,122], and elevated NO plasma levels are present in patients with cirrhosis and HCC[123]. A recent analysis of iNos function and interactions with NF-κB and Ha-RAS/ERK signalling was performed during hepatocarcinogenesis in F344 and BN rats, possessing different genetic predisposition to HCC, and TGF-α and c-Myc-TGF-α transgenic mice, characterized by different susceptibility to HCC[124]. iNos upregulation was found always at higher levels in the most aggressive preneoplastic and neoplastic liver lesions of F344 rats and c-Myc-TGF-α transgenic mice. Moreover, the determination of iNOS expression in human HCC shows highest values in HCC with poorer prognosis[124]. The suppression of iNOS signaling by Aminoguanidine[125] in c-Myc/Tgf-α mice and human HCC cell lines results in decreased HCC growth and NF-κB and RAS/ERK expression, and increased apoptosis[124]. In contrast, NO production by Glyco-S-nitroso-N-acetyl penicillamine 2 (Glyco-Snap-2) inhibits apoptosis of in vitro growing human HCC cells. Conversely, the block of NF-κB signalling by Sulfasalazine[126] or siRNA, or ERK signaling by the MEK inhibitor UO126[127] causes iNOS downregulation in HCC cell lines. In transgenic mice and human HCC cell lines, iNOS anti-apoptotic effect seems to be mediated by the NF-κB cascade. The latter induces various antiapoptotic proteins, such as BCL2-related protein, long isoform (BCL-xL), Inhibitor of apoptosis, X-linked (XIAP), and Inhibitor-of-apoptosis protein 1 (cIAP1), and inhibits the proapoptotic pJNK[124]. Accordingly, iNOS suppression by Aminoguanidine triggers downregulation of NF-κB and antiapoptotic proteins, and upregulation of pJNK, in c-Myc/TGF-α and HCC cell lines. However, these findings cannot exclude the contribution of other mechanisms to the antiapoptotic action of iNOS.
Above results assign a role to iNOS upregulation in the control of the proliferative phenotype of preneoplastic and neoplastic liver cells through the activation of the IKK/NF-κB axis (Figure 2). They also imply a cross-talk between iNOS and Ha-RAS/ERK. The mechanism of NF-κB regulation by pERK1/2 is not fully understood. Recent observations show that pERK1/2 activates AURORA-A (AURKA), which in turn may activate NF-κB through Inhibitor of kB (IKB-α) inhibition[128]. The possibility that pERK1/2 contributes to NF-κB upregulation via direct activation of iNOS may also be taken into account, but requires experimental support.
The observation that the expression of iNOS and its downstream targets is highest in HCCs prone to progression both in rodents and humans, and that iNOS levels are directly correlated with genomic instability, proliferation rate and microvessel density of HCC, and inversely correlated with apoptosis and patients’ survival[124], suggests that iNOS upregulation and changes in iNOS/NF-κB and iNOS/Ha-RAS/ERK cross-talks are prognostic markers for HCC. These results agree with the observation of a significant association of iNOS and Metalloproteinase-9 expression with HCC recurrence[121], and iNOS overexpression with poor prognosis for gastric cancer[129], adenoid cystic carcinoma of salivary glands[130], fibrous histiocytoma[131], colorectal cancer[132]. These iNOS effects seem to be mediated by its angiogenic properties and intensified by Cycloxygenase 2 (COX2) upregulation[133]. However, no correlation of iNOS overexpression with prognosis has been reported for pancreatic and ovarian tumors[134,135]. Furthermore, iNos ablation did not prevent hepatocarcinogenesis induced by a choline-deficient, L-aminoacid-deficient diet in mice[136], suggesting a relatively minor role of iNOS signaling. In this model of hepatocarcinogenesis, high production of lipid peroxides in hepatocyte nuclei[137] may cause DNA damage and contribute to HCC development via generation of genomic instability in an iNOS-independent manner.
In conclusion, iNOS overexpression contributes to growth deregulation in preneoplastic and neoplastic liver cells through a cross-talk with Ha-RAS/ERK and IKK-NF-κB axis. This does not exclude per se the activation of iNOS signaling by other mechanisms, such as inflammatory cytokines or the Wnt/β-catenin signaling[138]. However, the role of Wnt/β-catenin signaling in iNOS upregulation seems to be unlikely due to the observation of equal β-catenin activation (nuclear localization) in HCCs from both F344 and BN rats (M. Frau, unpublished data) expressing sharply different iNos mRNA levels. β-Catenin activation also occurs in a lower percentage of HCC cells from c-Myc/TGF-α than TGF-α transgenics (12% vs 30%)[139]. The highest iNos expression occurs in HCC from double transgenic mice.
The association of the block of iNOS signaling by a specific inhibitor such as Aminoguanidine with a consistent decrease in HCC growth and increase in apoptosis in vitro indicates that the key component of this pathway could represent therapeutic targets that may contribute to create networked biological therapies[140]. Thus, determination of iNOS immunoreactivity status can be proposed as a promising candidate for the identification of high risk patients who may benefit from new anticancer drugs targeting iNOS and its interplay with IKK/NF-κB and Ha-RAS/ERK signalling.
CONCLUSION
Studies on mouse and rat models of hepatocarcino-genesis have shown the existence of a great complexity of the inherited predisposition to liver cancer, and support a model based on the polygenic inheritance of low penetrance genes, with several gene-gene interactions and a main susceptibility locus (i.e. Hcs7, Hpcr3 for mice, and Hcs3/Hcr5 for rats)[25]. A complex combination and interplay of susceptibility or resistance alleles determines the individual risk. It has been proposed that the polymorphic variants of these “cancer modifier” genes can foster phenotypic expression of previously unexpressed alleles with consequent positive or negative influences on cell growth and differentiation[25]. Thus, the type of influence of modifiers on the carcinogenesis process may largely depend on interindividual differences in gene polymorphism, gene-gene interactions and gene-environment interactions. Epidemiologic and segregation studies strongly suggest a similar genetic model for the inherited predisposition to human HCC[11-14]. Taking into account the effect of susceptibility/resistance genes on the proliferative activity and re-differentiation of initiated cells, it may be expected that the genetic substrate largely influences the prognosis.
The mechanisms underlying the acquisition of the resistant phenotype have not been completely defined, as yet. A lower genomic instability of liver lesions developing in the resistant animals compared to the susceptible strains has been documented[80,141,142]. This may be tentatively attributed to interstrain differences in the activity of care taker and DNA repair genes, resulting in the prevention of the accumulation of DNA damage by initiated cells of the resistant rat strains. This hypothesis, however, needs experimental support. Nevertheless, accumulating evidence indicates that the predominance of susceptibility or resistance genes in individuals can largely influence the molecular control of cell proliferation and cell death. The susceptible rats develop various adaptive mechanisms for protection against stress-responsive tumor suppressors, such as p16INK4A, that confer to their liver cells the ability to proliferate under stressful conditions. In the resistant BN strain, a shut off of these mechanisms, associated with strong cell cycle deregulation and growth inhibition, occurs in coincidence with the exhaustion of promoting stimuli.
Among the effectors promoting cell cycle progression, ERK achieves unrestrained activity during HCC progression by triggering ubiquitin-mediated proteolysis of its specific inhibitor DUSP1. This mechanism is much more active in neoplastic nodules and HCC of susceptible rats than in the lesions of the resistant rats. The observation that FOXM1 induces the expression of CKS1-SKP2 ligase, involved in DUSP1 degradation, underlines the role of FOXM1 in the active proliferation of HCC cells, though its implication in a positive feedback loop reinforcing ERK cascade, by its ability to inhibit DUSP1[77]. These data indicate that FOXM1 upregulation is associated with the acquisition of a susceptible phenotype in rats and may influence human HCC development and prognosis. A role in the control of the proliferative phenotype of preneoplastic and neoplastic liver cells has been also assigned to higher iNos upregulation, inducing higher activation of the IKK/NF-κB and Ha-RAS/ERK signaling, in neoplastic nodules and HCCs of susceptible than resistant rats[124]. In the latter rats, HCC growth is also contrasted by relatively high cell death by apoptosis, which, at least in part depends on the activation of pro-apoptotic Rassf1A/Nore1A and Dab2IP/Ask1 pathways. Thus, Dusp1 possesses a prominent role in the acquisition by BN rats of a phenotype resistant to HCC. Late activation of RassF1A/Nore1A and Dab2IP/Ask1 axes is implicated in the highest apoptosis of BN HCC.
Importantly, most of the alterations responsible for the acquisition of a resistant or susceptible phenotype by rats have also been found in human HCC with better of poorer prognosis. A link between fast growth and signaling deregulation characterizes human HCC with poor prognosis, whereas the behaviour of HCC with better prognosis is more similar to that of the lesions of resistant rats. This does not necessarily imply a genetic regulation of signaling pathways in humans like that found in rodents. Further studies are needed to clarify the influence of susceptibility genes on signaling pathways supporting tumor growth and progression in humans.
The study of signal transduction pathways in rats differently predisposed to HCC development and prone to HCC progression, allowed the identification of numerous potential prognostic markers of hepatocarcinogenesis, such as the cell cycle protective mechanisms against p16INK4A, represented by CDC37-HSP90 complex and CRM1 protein, the ERK inhibitor DUSP1, iNOS, FOXM1. The prognostic role of these proteins was confirmed by analyzing the correlation of their expression with clinicopathological parameters of human HCC[91,124]. They therefore represent promising candidates for the identification of high risk patients who may benefit from new anticancer drugs against key components of signaling pathways.
Future work should focus on HCC prevention obtained by blocking key compounds of signal transduction network. Early blockage of signaling pathways may result in more efficient prevention, and rodent models may be useful to identify progression markers and therapeutic targets in early stages of the process, and in a large number of HCC subtypes. The study of experimental models recapitulating early preneoplastic alterations of human liver carcinogenesis may lead to the discovery of biomarkers of the risk of cirrhosis evolution to full malignancy, as well as of new key genes and signal transduction pathways involved in hepatocarcinogenesis. The existence of numerous interspecies commonalties in the biological behaviour and molecular changes of preneoplastic and neoplastic liver lesions[1-3,15,25] underlines the usefulness of this approach. The complexity of molecular changes of HCC predicts the impossibility to cure HCC development by interfering with only one signaling pathway. To overcome this difficulty and the occurrence of resistance to therapy, networked biologic therapies have been proposed[140,143,144] in which a combination of non-cytotoxic interventions must be performed to interrupt the damage. These interventions may be directed to interfere with different cell survival pathways, enhance apoptosis, block angiogenesis and extrahepatic fibrosis, induce the lysis of tumor cells, stimulate antitumor immunity, decrease HBV and HCV replication, etc. Furthermore, the combination of gene therapy with conventional therapeutic approaches with cytotoxic drugs may improve the treatment and reduce the doses of toxic compounds.
Footnotes
Supported by Grants from the “Associazione Italiana Ricerche sul Cancro”
Peer reviewer: Carlos J Pirola, PhD, Department of Molecular Genetics and Biology of Complex Dis, Institute of Medical Research A. Lanari, University of Buenos Aires-CONICET, Combatiente de Malvinas 3150, Buenos Aires 1427, Argentina
S- Editor Li DL L- Editor Alpini GD E- Editor Lin YP
References
- 1.Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–219. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 2.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 3.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka Y, Hanada K, Mizokami M, Yeo AE, Shih JW, Gojobori T, Alter HJ. Inaugural Article: A comparison of the molecular clock of hepatitis C virus in the United States and Japan predicts that hepatocellular carcinoma incidence in the United States will increase over the next two decades. Proc Natl Acad Sci USA. 2002;99:15584–15589. doi: 10.1073/pnas.242608099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Indulski JA, Lutz W. Metabolic genotype in relation to individual susceptibility to environmental carcinogens. Int Arch Occup Environ Health. 2000;73:71–85. doi: 10.1007/pl00007942. [DOI] [PubMed] [Google Scholar]
- 6.Kato S, Tajiri T, Matsukura N, Matsuda N, Taniai N, Mamada H, Yoshida H, Kiyam T, Naito Z. Genetic polymorphisms of aldehyde dehydrogenase 2, cytochrome p450 2E1 for liver cancer risk in HCV antibody-positive japanese patients and the variations of CYP2E1 mRNA expression levels in the liver due to its polymorphism. Scand J Gastroenterol. 2003;38:886–893. doi: 10.1080/00365520310004489. [DOI] [PubMed] [Google Scholar]
- 7.Agundez JA, Olivera M, Ladero JM, Rodriguez-Lescure A, Ledesma MC, Diaz-Rubio M, Meyer UA, Benitez J. Increased risk for hepatocellular carcinoma in NAT2-slow acetylators and CYP2D6-rapid metabolizers. Pharmacogenetics. 1996;6:501–512. doi: 10.1097/00008571-199612000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh LL, Huang RC, Yu MW, Chen CJ, Liaw YF. L-myc, GST M1 genetic polymorphism and hepatocellular carcinoma risk among chronic hepatitis B carriers. Cancer Lett. 1996;103:171–176. doi: 10.1016/0304-3835(96)04209-7. [DOI] [PubMed] [Google Scholar]
- 9.Taylor JA, Bell DA, Nagorney D. L-myc proto-oncogene alleles and susceptibility to hepatocellular carcinoma. Int J Cancer. 1993;54:927–930. doi: 10.1002/ijc.2910540610. [DOI] [PubMed] [Google Scholar]
- 10.Sonzogni L, Silvestri L, De Silvestri A, Gritti C, Foti L, Zavaglia C, Bottelli R, Mondelli MU, Civardi E, Silini EM. Polymorphisms of microsomal epoxide hydrolase gene and severity of HCV-related liver disease. Hepatology. 2002;36:195–201. doi: 10.1053/jhep.2002.33898. [DOI] [PubMed] [Google Scholar]
- 11.Yu MW, Chang HC, Liaw YF, Lin SM, Lee SD, Liu CJ, Chen PJ, Hsiao TJ, Lee PH, Chen CJ. Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. J Natl Cancer Inst. 2000;92:1159–1164. doi: 10.1093/jnci/92.14.1159. [DOI] [PubMed] [Google Scholar]
- 12.Cai RL, Meng W, Lu HY, Lin WY, Jiang F, Shen FM. Segregation analysis of hepatocellular carcinoma in a moderately high-incidence area of East China. World J Gastroenterol. 2003;9:2428–2432. doi: 10.3748/wjg.v9.i11.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez E, La Vecchia C, D'Avanzo B, Negri E, Franceschi S. Family history and the risk of liver, gallbladder, and pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 1994;3:209–212. [PubMed] [Google Scholar]
- 14.Hemminki K, Li X. Familial risks of cancer as a guide to gene identification and mode of inheritance. Int J Cancer. 2004;110:291–294. doi: 10.1002/ijc.20107. [DOI] [PubMed] [Google Scholar]
- 15.Feo F, Pascale RM, Simile MM, De Miglio MR, Muroni MR, Calvisi D. Genetic alterations in liver carcinogenesis: implications for new preventive and therapeutic strategies. Crit Rev Oncog. 2000;11:19–62. [PubMed] [Google Scholar]
- 16.Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- 17.Rebouissou S, Bioulac-Sage P, Zucman-Rossi J. Molecular pathogenesis of focal nodular hyperplasia and hepatocellular adenoma. J Hepatol. 2008;48:163–170. doi: 10.1016/j.jhep.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Korstanje R, Paigen B. From QTL to gene: the harvest begins. Nat Genet. 2002;31:235–236. doi: 10.1038/ng0702-235. [DOI] [PubMed] [Google Scholar]
- 19.Farber E, Sarma DS. Hepatocarcinogenesis: a dynamic cellular perspective. Lab Invest. 1987;56:4–22. [PubMed] [Google Scholar]
- 20.Garcea R, Daino L, Pascale R, Simile MM, Puddu M, Frassetto S, Cozzolino P, Seddaiu MA, Gaspa L, Feo F. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: role of remodeling and apoptosis. Cancer Res. 1989;49:1850–1856. [PubMed] [Google Scholar]
- 21.Zhou XD. Recurrence and metastasis of hepatocellular carcinoma: progress and prospects. Hepatobiliary Pancreat Dis Int. 2002;1:35–41. [PubMed] [Google Scholar]
- 22.Dragani TA, Manenti G, Della Porta G. Genetic susceptibility to murine hepatocarcinogenesis is associated with high growth rate of NDEA-initiated hepatocytes. J Cancer Res Clin Oncol. 1987;113:223–229. doi: 10.1007/BF00396377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manenti G, Binelli G, Gariboldi M, Canzian F, De Gregorio L, Falvella FS, Dragani TA, Pierotti MA. Multiple loci affect genetic predisposition to hepatocarcinogenesis in mice. Genomics. 1994;23:118–124. doi: 10.1006/geno.1994.1466. [DOI] [PubMed] [Google Scholar]
- 24.Denda A, Kitayama W, Konishi Y, Yan Y, Fukamachi Y, Miura M, Gotoh S, Ikemura K, Abe T, Higashi T, et al. Genetic properties for the suppression of development of putative preneoplastic glutathione S-transferase placental form-positive foci in the liver of carcinogen-resistant DRH strain rats. Cancer Lett. 1999;140:59–67. doi: 10.1016/s0304-3835(99)00051-8. [DOI] [PubMed] [Google Scholar]
- 25.Feo F, De Miglio MR, Simile MM, Muroni MR, Calvisi DF, Frau M, Pascale RM. Hepatocellular carcinoma as a complex polygenic disease. Interpretive analysis of recent developments on genetic predisposition. Biochim Biophys Acta. 2006;1765:126–147. doi: 10.1016/j.bbcan.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Pascale RM, Simile MM, DeMiglio MR, Muroni MR, Gaspa L, Dragani TA, Feo F. The BN rat strain carries dominant hepatocarcinogen resistance loci. Carcinogenesis. 1996;17:1765–1768. doi: 10.1093/carcin/17.8.1765. [DOI] [PubMed] [Google Scholar]
- 27.Wood GA, Korkola JE, Lee VM, Sarma DS, Archer MC. Resistance of Copenhagen rats to chemical induction of glutathione S-transferase 7-7-positive liver foci. Carcinogenesis. 1997;18:1745–1750. doi: 10.1093/carcin/18.9.1745. [DOI] [PubMed] [Google Scholar]
- 28.Wood GA, Sarma DS, Archer MC. Resistance to the promotion of glutathione S-transferase 7-7-positive liver lesions in Copenhagen rats. Carcinogenesis. 1999;20:1169–1175. doi: 10.1093/carcin/20.7.1169. [DOI] [PubMed] [Google Scholar]
- 29.Enomoto K, Farber E. Kinetics of phenotypic maturation of remodeling of hyperplastic nodules during liver carcinogenesis. Cancer Res. 1982;42:2330–2335. [PubMed] [Google Scholar]
- 30.Wood GA, Sarma DS, Archer MC. Inheritance of resistance to promotion of preneoplastic liver lesions in Copenhagen rats. Exp Biol Med (Maywood) 2001;226:831–835. doi: 10.1177/153537020122600904. [DOI] [PubMed] [Google Scholar]
- 31.Calvisi DF, Pinna F, Pellegrino R, Sanna V, Sini M, Daino L, Simile MM, De Miglio MR, Frau M, Tomasi ML, et al. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int J Cancer. 2008;123:2057–2064. doi: 10.1002/ijc.23720. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Higashi K, Hiai H. Role of resistant Drh1 locus in chemical carcinogen-induced hepatocarcinogenesis in rats: analysis with a speed congenic strain. Cancer Sci. 2005;96:164–169. doi: 10.1111/j.1349-7006.2005.00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gariboldi M, Manenti G, Canzian F, Falvella FS, Pierotti MA, Della Porta G, Binelli G, Dragani TA. Chromosome mapping of murine susceptibility loci to liver carcinogenesis. Cancer Res. 1993;53:209–211. [PubMed] [Google Scholar]
- 34.G. Manenti, G. Binelli, M. Gariboldi, F. Canzian, L. De Gregorio, F.S. Falvella, T.A. Dragani, M.A. Pierotti. Multiple loci affect genetic predisposition to hepatocarcinogenesis in mice. Genomics. 1994;23:118–124. doi: 10.1006/geno.1994.1466. [DOI] [PubMed] [Google Scholar]
- 35.Bilger A, Bennett LM, Carabeo RA, Chiaverotti TA, Dvorak C, Liss KM, Schadewald SA, Pitot HC, Drinkwater NR. A potent modifier of liver cancer risk on distal mouse chromosome 1: linkage analysis and characterization of congenic lines. Genetics. 2004;167:859–866. doi: 10.1534/genetics.103.024521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poole TM, Drinkwater NR. Two genes abrogate the inhibition of murine hepatocarcinogenesis by ovarian hormones. Proc Natl Acad Sci USA. 1996;93:5848–5853. doi: 10.1073/pnas.93.12.5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee GH, Bennett LM, Carabeo RA, Drinkwater NR. Identification of hepatocarcinogen-resistance genes in DBA/2 mice. Genetics. 1995;139:387–395. doi: 10.1093/genetics/139.1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee GH, Drinkwater NR. The Hcr (hepatocarcinogen resistance) loci of DBA/2J mice partially suppress phenotypic expression of the Hcs (hepatocarcinogen sensitivity) loci of C3H/HeJ mice. Carcinogenesis. 1995;16:1993–1996. doi: 10.1093/carcin/16.8.1993. [DOI] [PubMed] [Google Scholar]
- 39.Dragani TA, Manenti G, Della Porta G. Quantitative analysis of genetic susceptibility to liver and lung carcinogenesis in mice. Cancer Res. 1991;51:6299–6303. [PubMed] [Google Scholar]
- 40.Manenti G, Galvan A, Falvella FS, Pascale RM, Spada E, Milani S, Gonzalez Neira A, Feo F, Dragani TA. Genetic control of resistance to hepatocarcinogenesis by the mouse Hpcr3 locus. Hepatology. 2008;48:617–623. doi: 10.1002/hep.22374. [DOI] [PubMed] [Google Scholar]
- 41.Melhem MF, Kunz HW, Gill TJ 3rd. A major histocompatibility complex-linked locus in the rat critically influences resistance to diethylnitrosamine carcinogenesis. Proc Natl Acad Sci USA. 1993;90:1967–1971. doi: 10.1073/pnas.90.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao KN, Shinozuka H, Kunz HW, Gill TJ 3rd. Enhanced susceptibility to a chemical carcinogen in rats carrying MHC-linked genes influencing development (GRC) Int J Cancer. 1984;34:113–120. doi: 10.1002/ijc.2910340120. [DOI] [PubMed] [Google Scholar]
- 43.De Miglio MR, Canzian F, Pascale RM, Simile MM, Muroni MR, Calvisi D, Romeo G, Feo F. Identification of genetic loci controlling hepatocarcinogenesis on rat chromosomes 7 and 10. Cancer Res. 1999;59:4651–4657. [PubMed] [Google Scholar]
- 44.De Miglio MR, Pascale RM, Simile MM, Muroni MR, Calvisi DF, Virdis P, Bosinco GM, Frau M, Seddaiu MA, Ladu S, et al. Chromosome mapping of multiple loci affecting the genetic predisposition to rat liver carcinogenesis. Cancer Res. 2002;62:4459–4463. [PubMed] [Google Scholar]
- 45.De Miglio MR, Pascale RM, Simile MM, Muroni MR, Virdis P, Kwong KM, Wong LK, Bosinco GM, Pulina FR, Calvisi DF, et al. Polygenic control of hepatocarcinogenesis in Copenhagen x F344 rats. Int J Cancer. 2004;111:9–16. doi: 10.1002/ijc.20225. [DOI] [PubMed] [Google Scholar]
- 46.Zeng ZZ, Higashi S, Kitayama W, Denda A, Yan Y, Matsuo K, Konishi Y, Hiai H, Higashi K. Genetic resistance to chemical carcinogen-induced preneoplastic hepatic lesions in DRH strain rats. Cancer Res. 2000;60:2876–2881. [PubMed] [Google Scholar]
- 47.Yan Y, Zeng ZZ, Higashi S, Denda A, Konishi Y, Onishi S, Ueno H, Higashi K, Hiai H. Resistance of DRH strain rats to chemical carcinogenesis of liver: genetic analysis of later progression stage. Carcinogenesis. 2002;23:189–196. doi: 10.1093/carcin/23.1.189. [DOI] [PubMed] [Google Scholar]
- 48.Fujiwara Y, Monden M, Mori T, Nakamura Y, Emi M. Frequent multiplication of the long arm of chromosome 8 in hepatocellular carcinoma. Cancer Res. 1993;53:857–860. [PubMed] [Google Scholar]
- 49.Wong N, Lai P, Lee SW, Fan S, Pang E, Liew CT, Sheng Z, Lau JW, Johnson PJ. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: relationship to disease stage, tumor size, and cirrhosis. Am J Pathol. 1999;154:37–43. doi: 10.1016/S0002-9440(10)65248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okamoto H, Yasui K, Zhao C, Arii S, Inazawa J. PTK2 and EIF3S3 genes may be amplification targets at 8q23-q24 and are associated with large hepatocellular carcinomas. Hepatology. 2003;38:1242–1249. doi: 10.1053/jhep.2003.50457. [DOI] [PubMed] [Google Scholar]
- 51.Ogawa K, Osanai M, Obata M, Ishizaki K, Kamiya K. Gain of chromosomes 15 and 19 is frequent in both mouse hepatocellular carcinoma cell lines and primary tumors, but loss of chromosomes 4 and 12 is detected only in the cell lines. Carcinogenesis. 1999;20:2083–2088. doi: 10.1093/carcin/20.11.2083. [DOI] [PubMed] [Google Scholar]
- 52.De Miglio MR, Simile MM, Muroni MR, Calvisi DF, Virdis P, Asara G, Frau M, Bosinco GM, Seddaiu MA, Daino L, et al. Phenotypic reversion of rat neoplastic liver nodules is under genetic control. Int J Cancer. 2003;105:70–75. doi: 10.1002/ijc.11044. [DOI] [PubMed] [Google Scholar]
- 53.Chang WY. Complete spontaneous regression of cancer: four case reports, review of literature, and discussion of possible mechanisms involved. Hawaii Med J. 2000;59:379–387. [PubMed] [Google Scholar]
- 54.De Miglio MR, Virdis P, Calvisi DF, Frau M, Muroni MR, Simile MM, Daino L, Careddu GM, Sanna-Passino E, Pascale RM, et al. Mapping a sex hormone-sensitive gene determining female resistance to liver carcinogenesis in a congenic F344.BN-Hcs4 rat. Cancer Res. 2006;66:10384–10390. doi: 10.1158/0008-5472.CAN-06-2881. [DOI] [PubMed] [Google Scholar]
- 55.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 56.Simile MM, De Miglio MR, Muroni MR, Frau M, Asara G, Serra S, Muntoni MD, Seddaiu MA, Daino L, Feo F, et al. Down-regulation of c-myc and Cyclin D1 genes by antisense oligodeoxy nucleotides inhibits the expression of E2F1 and in vitro growth of HepG2 and Morris 5123 liver cancer cells. Carcinogenesis. 2004;25:333–341. doi: 10.1093/carcin/bgh014. [DOI] [PubMed] [Google Scholar]
- 57.Garcea R, Daino L, Pascale R, Simile MM, Puddu M, Ruggiu ME, Seddaiu MA, Satta G, Sequenza MJ, Feo F. Protooncogene methylation and expression in regenerating liver and preneoplastic liver nodules induced in the rat by diethylnitrosamine: effect of variations of S-adenosylmethionine:S-adenosylhomocysteine ratio. Carcinogenesis. 1989;10:1183–1192. doi: 10.1093/carcin/10.7.1183. [DOI] [PubMed] [Google Scholar]
- 58.Pascale RM, Marras V, Simile MM, Daino L, Pinna G, Bennati S, Carta M, Seddaiu MA, Massarelli G, Feo F. Chemoprevention of rat liver carcinogenesis by S-adenosyl-L-methionine: a long-term study. Cancer Res. 1992;52:4979–4986. [PubMed] [Google Scholar]
- 59.Pascale RM, Simile MM, De Miglio MR, Muroni MR, Calvisi DF, Asara G, Casabona D, Frau M, Seddaiu MA, Feo F. Cell cycle deregulation in liver lesions of rats with and without genetic predisposition to hepatocarcinogenesis. Hepatology. 2002;35:1341–1350. doi: 10.1053/jhep.2002.33682. [DOI] [PubMed] [Google Scholar]
- 60.De Miglio MR, Simile MM, Muroni MR, Pusceddu S, Calvisi D, Carru A, Seddaiu MA, Daino L, Deiana L, Pascale RM, et al. Correlation of c-myc overexpression and amplification with progression of preneoplastic liver lesions to malignancy in the poorly susceptible Wistar rat strain. Mol Carcinog. 1999;25:21–29. [PubMed] [Google Scholar]
- 61.Pascale RM, Simile MM, Calvisi DF, Frau M, Muroni MR, Seddaiu MA, Daino L, Muntoni MD, De Miglio MR, Thorgeirsson SS, et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology. 2005;42:1310–1319. doi: 10.1002/hep.20962. [DOI] [PubMed] [Google Scholar]
- 62.Hunter T, Poon RY. Cdc37: a protein kinase chaperone? Trends Cell Biol. 1997;7:157–161. doi: 10.1016/S0962-8924(97)01027-1. [DOI] [PubMed] [Google Scholar]
- 63.Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996;10:1491–1502. doi: 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- 64.Dai K, Kobayashi R, Beach D. Physical interaction of mammalian CDC37 with CDK4. J Biol Chem. 1996;271:22030–22034. doi: 10.1074/jbc.271.36.22030. [DOI] [PubMed] [Google Scholar]
- 65.Crosby ME, Almasan A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther. 2004;3:1208–1211. doi: 10.4161/cbt.3.12.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohtani N, Brennan P, Gaubatz S, Sanij E, Hertzog P, Wolvetang E, Ghysdael J, Rowe M, Hara E. Epstein-Barr virus LMP1 blocks p16INK4a-RB pathway by promoting nuclear export of E2F4/5. J Cell Biol. 2003;162:173–183. doi: 10.1083/jcb.200302085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 68.Feig LA, Buchsbaum RJ. Cell signaling: life or death decisions of ras proteins. Curr Biol. 2002;12:R259–R261. doi: 10.1016/s0960-9822(02)00787-x. [DOI] [PubMed] [Google Scholar]
- 69.Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999–9006. doi: 10.1038/sj.onc.1207111. [DOI] [PubMed] [Google Scholar]
- 70.Johnston AM, Naselli G, Gonez LJ, Martin RM, Harrison LC, DeAizpurua HJ. SPAK, a STE20/SPS1-related kinase that activates the p38 pathway. Oncogene. 2000;19:4290–4297. doi: 10.1038/sj.onc.1203784. [DOI] [PubMed] [Google Scholar]
- 71.Ura S, Masuyama N, Graves JD, Gotoh Y. MST1-JNK promotes apoptosis via caspase-dependent and independent pathways. Genes Cells. 2001;6:519–530. doi: 10.1046/j.1365-2443.2001.00439.x. [DOI] [PubMed] [Google Scholar]
- 72.Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, Avruch J. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol. 2002;12:253–265. doi: 10.1016/s0960-9822(02)00683-8. [DOI] [PubMed] [Google Scholar]
- 73.Armesilla AL, Williams JC, Buch MH, Pickard A, Emerson M, Cartwright EJ, Oceandy D, Vos MD, Gillies S, Clark GJ, et al. Novel functional interaction between the plasma membrane Ca2+ pump 4b and the proapoptotic tumor suppressor Ras-associated factor 1 (RASSF1) J Biol Chem. 2004;279:31318–31328. doi: 10.1074/jbc.M307557200. [DOI] [PubMed] [Google Scholar]
- 74.Guicciardi ME, Gores GJ. AIP1: a new player in TNF signaling. J Clin Invest. 2003;111:1813–1815. doi: 10.1172/JCI18911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu XX, Yi T, Tang B, Lambeth JD. Disabled-2 (Dab2) is an SH3 domain-binding partner of Grb2. Oncogene. 1998;16:1561–1569. doi: 10.1038/sj.onc.1201678. [DOI] [PubMed] [Google Scholar]
- 76.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 77.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 78.Lin YW, Yang JL. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J Biol Chem. 2006;281:915–926. doi: 10.1074/jbc.M508720200. [DOI] [PubMed] [Google Scholar]
- 79.Wong J, Zhang J, Si X, Gao G, Luo H. Inhibition of the extracellular signal-regulated kinase signaling pathway is correlated with proteasome inhibitor suppression of coxsackievirus replication. Biochem Biophys Res Commun. 2007;358:903–907. doi: 10.1016/j.bbrc.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 80.De Miglio MR, Muroni MR, Simile MM, Virdis P, Asara G, Frau M, Calvisi DF, Seddaiu MA, Pascale RM, Feo F. Frequent loss of heterozygosity at the Hcr1 (hepatocarcinogenesis resistance) locus on chromosome 10 in primary hepatocellular carcinomas from LFF1 rat strain. Hepatology. 2001;33:1110–1117. doi: 10.1053/jhep.2001.23795. [DOI] [PubMed] [Google Scholar]
- 81.Wang HY, Cheng Z, Malbon CC. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003;191:229–237. doi: 10.1016/s0304-3835(02)00612-2. [DOI] [PubMed] [Google Scholar]
- 82.Bang YJ, Kwon JH, Kang SH, Kim JW, Yang YC. Increased MAPK activity and MKP-1 overexpression in human gastric adenocarcinoma. Biochem Biophys Res Commun. 1998;250:43–47. doi: 10.1006/bbrc.1998.9256. [DOI] [PubMed] [Google Scholar]
- 83.Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- 84.Grammer TC, Blenis J. Evidence for MEK-independent pathways regulating the prolonged activation of the ERK-MAP kinases. Oncogene. 1997;14:1635–1642. doi: 10.1038/sj.onc.1201000. [DOI] [PubMed] [Google Scholar]
- 85.Barry OP, Mullan B, Sheehan D, Kazanietz MG, Shanahan F, Collins JK, O'Sullivan GC. Constitutive ERK1/2 activation in esophagogastric rib bone marrow micrometastatic cells is MEK-independent. J Biol Chem. 2001;276:15537–15546. doi: 10.1074/jbc.M010847200. [DOI] [PubMed] [Google Scholar]
- 86.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol Cell Biol. 2004;24:2649–2661. doi: 10.1128/MCB.24.7.2649-2661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rauhala HE, Porkka KP, Tolonen TT, Martikainen PM, Tammela TL, Visakorpi T. Dual-specificity phosphatase 1 and serum/glucocorticoid-regulated kinase are downregulated in prostate cancer. Int J Cancer. 2005;117:738–745. doi: 10.1002/ijc.21270. [DOI] [PubMed] [Google Scholar]
- 90.Shimada K, Nakamura M, Ishida E, Higuchi T, Tanaka M, Ota I, Konishi N. c-Jun NH2 terminal kinase activation and decreased expression of mitogen-activated protein kinase phosphatase-1 play important roles in invasion and angiogenesis of urothelial carcinomas. Am J Pathol. 2007;171:1003–1012. doi: 10.2353/ajpath.2007.070010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsujita E, Taketomi A, Gion T, Kuroda Y, Endo K, Watanabe A, Nakashima H, Aishima S, Kohnoe S, Maehara Y. Suppressed MKP-1 is an independent predictor of outcome in patients with hepatocellular carcinoma. Oncology. 2005;69:342–347. doi: 10.1159/000089766. [DOI] [PubMed] [Google Scholar]
- 92.Calvisi DF, Pinna F, Meloni F, Ladu S, Pellegrino R, Sini M, Daino L, Simile MM, De Miglio MR, Virdis P, et al. Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 2008;68:4192–4200. doi: 10.1158/0008-5472.CAN-07-6157. [DOI] [PubMed] [Google Scholar]
- 93.Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV, Major ML, Gusarova GA, Yoder HM, Costa RH, et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–2161. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 94.Costa RH, Kalinichenko VV, Major ML, Raychaudhuri P. New and unexpected: forkhead meets ARF. Curr Opin Genet Dev. 2005;15:42–48. doi: 10.1016/j.gde.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 95.Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem. 2007;388:1257–1274. doi: 10.1515/BC.2007.159. [DOI] [PubMed] [Google Scholar]
- 96.Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007;132:1420–1431. doi: 10.1053/j.gastro.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 97.Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67:8293–8300. doi: 10.1158/0008-5472.CAN-07-1265. [DOI] [PubMed] [Google Scholar]
- 98.Zhao YY, Gao XP, Zhao YD, Mirza MK, Frey RS, Kalinichenko VV, Wang IC, Costa RH, Malik AB. Endothelial cell-restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J Clin Invest. 2006;116:2333–2343. doi: 10.1172/JCI27154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 100.Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci USA. 2002;99:16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Krupczak-Hollis K, Wang X, Dennewitz MB, Costa RH. Growth hormone stimulates proliferation of old-aged regenerating liver through forkhead box m1b. Hepatology. 2003;38:1552–1562. doi: 10.1016/j.hep.2003.08.052. [DOI] [PubMed] [Google Scholar]
- 102.Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–850. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bektas N, Haaf A, Veeck J, Wild PJ, Luscher-Firzlaff J, Hartmann A, Knuchel R, Dahl E. Tight correlation between expression of the Forkhead transcription factor FOXM1 and HER2 in human breast cancer. BMC Cancer. 2008;8:42. doi: 10.1186/1471-2407-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 105.Katoh Y, Katoh M. Hedgehog signaling pathway and gastric cancer. Cancer Biol Ther. 2005;4:1050–1054. doi: 10.4161/cbt.4.10.2184. [DOI] [PubMed] [Google Scholar]
- 106.Teh MT, Wong ST, Neill GW, Ghali LR, Philpott MP, Quinn AG. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62:4773–4780. [PubMed] [Google Scholar]
- 107.Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3:903–911. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- 108.Kasper M, Regl G, Frischauf AM, Aberger F. GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer. 2006;42:437–445. doi: 10.1016/j.ejca.2005.08.039. [DOI] [PubMed] [Google Scholar]
- 109.Crompton T, Outram SV, Hager-Theodorides AL. Sonic hedgehog signalling in T-cell development and activation. Nat Rev Immunol. 2007;7:726–735. doi: 10.1038/nri2151. [DOI] [PubMed] [Google Scholar]
- 110.Kim Y, Yoon JW, Xiao X, Dean NM, Monia BP, Marcusson EG. Selective down-regulation of glioma-associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res. 2007;67:3583–3593. doi: 10.1158/0008-5472.CAN-06-3040. [DOI] [PubMed] [Google Scholar]
- 111.Kise Y, Takenaka K, Tezuka T, Yamamoto T, Miki H. Fused kinase is stabilized by Cdc37/Hsp90 and enhances Gli protein levels. Biochem Biophys Res Commun. 2006;351:78–84. doi: 10.1016/j.bbrc.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 112.Lasagna N, Fantappie O, Solazzo M, Morbidelli L, Marchetti S, Cipriani G, Ziche M, Mazzanti R. Hepatocyte growth factor and inducible nitric oxide synthase are involved in multidrug resistance-induced angiogenesis in hepatocellular carcinoma cell lines. Cancer Res. 2006;66:2673–2682. doi: 10.1158/0008-5472.CAN-05-2290. [DOI] [PubMed] [Google Scholar]
- 113.Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–2380. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 114.Ying L, Hofseth AB, Browning DD, Nagarkatti M, Nagarkatti PS, Hofseth LJ. Nitric oxide inactivates the retinoblastoma pathway in chronic inflammation. Cancer Res. 2007;67:9286–9293. doi: 10.1158/0008-5472.CAN-07-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zingarelli B, Hake PW, Yang Z, O'Connor M, Denenberg A, Wong HR. Absence of inducible nitric oxide synthase modulates early reperfusion-induced NF-kappaB and AP-1 activation and enhances myocardial damage. FASEB J. 2002;16:327–342. doi: 10.1096/fj.01-0533com. [DOI] [PubMed] [Google Scholar]
- 116.Deora AA, Hajjar DP, Lander HM. Recruitment and activation of Raf-1 kinase by nitric oxide-activated Ras. Biochemistry. 2000;39:9901–9908. doi: 10.1021/bi992954b. [DOI] [PubMed] [Google Scholar]
- 117.Ellerhorst JA, Ekmekcioglu S, Johnson MK, Cooke CP, Johnson MM, Grimm EA. Regulation of iNOS by the p44/42 mitogen-activated protein kinase pathway in human melanoma. Oncogene. 2006;25:3956–3962. doi: 10.1038/sj.onc.1209419. [DOI] [PubMed] [Google Scholar]
- 118.Zhao Q, Lee FS. Mitogen-activated protein kinase/ERK kinase kinases 2 and 3 activate nuclear factor-kappaB through IkappaB kinase-alpha and IkappaB kinase-beta. J Biol Chem. 1999;274:8355–8358. doi: 10.1074/jbc.274.13.8355. [DOI] [PubMed] [Google Scholar]
- 119.Simile MM, Pagnan G, Pastorino F, Brignole C, De Miglio MR, Muroni MR, Asara G, Frau M, Seddaiu MA, Calvisi DF, et al. Chemopreventive N-(4-hydroxyphenyl)retinamide (fenretinide) targets deregulated NF-{kappa}B and Mat1A genes in the early stages of rat liver carcinogenesis. Carcinogenesis. 2005;26:417–427. doi: 10.1093/carcin/bgh315. [DOI] [PubMed] [Google Scholar]
- 120.Calvisi DF, Ladu S, Hironaka K, Factor VM, Thorgeirsson SS. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer. J Hepatol. 2004;41:815–822. doi: 10.1016/j.jhep.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 121.Sun MH, Han XC, Jia MK, Jiang WD, Wang M, Zhang H, Han G, Jiang Y. Expressions of inducible nitric oxide synthase and matrix metalloproteinase-9 and their effects on angiogenesis and progression of hepatocellular carcinoma. World J Gastroenterol. 2005;11:5931–5937. doi: 10.3748/wjg.v11.i38.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ikeguchi M, Ueta T, Yamane Y, Hirooka Y, Kaibara N. Inducible nitric oxide synthase and survivin messenger RNA expression in hepatocellular carcinoma. Clin Cancer Res. 2002;8:3131–3136. [PubMed] [Google Scholar]
- 123.Moriyama A, Masumoto A, Nanri H, Tabaru A, Unoki H, Imoto I, Ikeda M, Otsuki M. High plasma concentrations of nitrite/nitrate in patients with hepatocellular carcinoma. Am J Gastroenterol. 1997;92:1520–1523. [PubMed] [Google Scholar]
- 124.Calvisi DF, Pinna F, Ladu S, Pellegrino R, Muroni MR, Simile MM, Frau M, Tomasi ML, De Miglio MR, Seddaiu MA, et al. Aberrant iNOS signaling is under genetic control in rodent liver cancer and potentially prognostic for the human disease. Carcinogenesis. 2008;29:1639–1647. doi: 10.1093/carcin/bgn155. [DOI] [PubMed] [Google Scholar]
- 125.Misko TP, Moore WM, Kasten TP, Nickols GA, Corbett JA, Tilton RG, McDaniel ML, Williamson JR, Currie MG. Selective inhibition of the inducible nitric oxide synthase by aminoguanidine. Eur J Pharmacol. 1993;233:119–125. doi: 10.1016/0014-2999(93)90357-n. [DOI] [PubMed] [Google Scholar]
- 126.Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206:193–199. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 127.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 128.Briassouli P, Chan F, Savage K, Reis-Filho JS, Linardopoulos S. Aurora-A regulation of nuclear factor-kappaB signaling by phosphorylation of IkappaBalpha. Cancer Res. 2007;67:1689–1695. doi: 10.1158/0008-5472.CAN-06-2272. [DOI] [PubMed] [Google Scholar]
- 129.Li LG, Xu HM. Inducible nitric oxide synthase, nitrotyrosine and apoptosis in gastric adenocarcinomas and their correlation with a poor survival. World J Gastroenterol. 2005;11:2539–2544. doi: 10.3748/wjg.v11.i17.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang J, Peng B, Chen X. Expressions of nuclear factor kappaB, inducible nitric oxide synthase, and vascular endothelial growth factor in adenoid cystic carcinoma of salivary glands: correlations with the angiogenesis and clinical outcome. Clin Cancer Res. 2005;11:7334–7343. doi: 10.1158/1078-0432.CCR-05-0241. [DOI] [PubMed] [Google Scholar]
- 131.Hoki Y, Hiraku Y, Ma N, Murata M, Matsumine A, Nagahama M, Shintani K, Uchida A, Kawanishi S. iNOS-dependent DNA damage in patients with malignant fibrous histiocytoma in relation to prognosis. Cancer Sci. 2007;98:163–168. doi: 10.1111/j.1349-7006.2006.00376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cianchi F, Cortesini C, Fantappie O, Messerini L, Sardi I, Lasagna N, Perna F, Fabbroni V, Di Felice A, Perigli G, et al. Cyclooxygenase-2 activation mediates the proangiogenic effect of nitric oxide in colorectal cancer. Clin Cancer Res. 2004;10:2694–2704. doi: 10.1158/1078-0432.ccr-03-0192. [DOI] [PubMed] [Google Scholar]
- 133.Rahman MA, Dhar DK, Yamaguchi E, Maruyama S, Sato T, Hayashi H, Ono T, Yamanoi A, Kohno H, Nagasue N. Coexpression of inducible nitric oxide synthase and COX-2 in hepatocellular carcinoma and surrounding liver: possible involvement of COX-2 in the angiogenesis of hepatitis C virus-positive cases. Clin Cancer Res. 2001;7:1325–1332. [PubMed] [Google Scholar]
- 134.Kong G, Kim EK, Kim WS, Lee KT, Lee YW, Lee JK, Paik SW, Rhee JC. Role of cyclooxygenase-2 and inducible nitric oxide synthase in pancreatic cancer. J Gastroenterol Hepatol. 2002;17:914–921. doi: 10.1046/j.1440-1746.2002.02829.x. [DOI] [PubMed] [Google Scholar]
- 135.Ozel E, Pestereli HE, Simsek T, Erdogan G, Karaveli FS. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in ovarian surface epithelial carcinomas: is there any correlation with angiogenesis or clinicopathologic parameters? Int J Gynecol Cancer. 2006;16:549–555. doi: 10.1111/j.1525-1438.2006.00567.x. [DOI] [PubMed] [Google Scholar]
- 136.Denda A, Kitayama W, Kishida H, Murata N, Tamura K, Kusuoka O, Tsutsumi M, Nishikawa F, Kita E, Nakae D, et al. Expression of inducible nitric oxide (NO) synthase but not prevention by its gene ablation of hepatocarcinogenesis with fibrosis caused by a choline-deficient, L-amino acid-defined diet in rats and mice. Nitric Oxide. 2007;16:164–176. doi: 10.1016/j.niox.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 137.Ghoshal AK. New insight into the biochemical pathology of liver in choline deficiency. Crit Rev Biochem Mol Biol. 1995;30:263–273. doi: 10.3109/10409239509083487. [DOI] [PubMed] [Google Scholar]
- 138.Du Q, Park KS, Guo Z, He P, Nagashima M, Shao L, Sahai R, Geller DA, Hussain SP. Regulation of human nitric oxide synthase 2 expression by Wnt beta-catenin signaling. Cancer Res. 2006;66:7024–7031. doi: 10.1158/0008-5472.CAN-05-4110. [DOI] [PubMed] [Google Scholar]
- 139.Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS. Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology. 2004;126:1374–1386. doi: 10.1053/j.gastro.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 140.Epstein RJ, Leung TW. Reversing hepatocellular carcinoma progression by using networked biological therapies. Clin Cancer Res. 2007;13:11–17. doi: 10.1158/1078-0432.CCR-06-1619. [DOI] [PubMed] [Google Scholar]
- 141.Gariboldi M, Pascale R, Manenti G, De Miglio MR, Calvisi D, Carru A, Dragani TA, Feo F. Analysis of loss of heterozygosity in neoplastic nodules induced by diethylnitrosamine in the resistant BFF1 rat strain. Carcinogenesis. 1999;20:1363–1368. doi: 10.1093/carcin/20.7.1363. [DOI] [PubMed] [Google Scholar]
- 142.Teeguarden JG, Newton MA, Dragan YP, Pitot HC. Genome-wide loss of heterozygosity analysis of chemically induced rat hepatocellular carcinomas reveals elevated frequency of allelic imbalances on chromosomes 1, 6, 8, 11, 15, 17, and 20. Mol Carcinog. 2000;28:51–61. doi: 10.1002/(sici)1098-2744(200005)28:1<51::aid-mc7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 143.Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25:3866–3884. doi: 10.1038/sj.onc.1209550. [DOI] [PubMed] [Google Scholar]
- 144.Calvisi DF, Pascale RM, Feo F. Dissection of signal transduction pathways as a tool for the development of targeted therapies of hepatocellular carcinoma. Rev Recent Clin Trials. 2007;2:217–236. doi: 10.2174/157488707781662715. [DOI] [PubMed] [Google Scholar]