

Abstract
AIM: To investigate alternative or subordinate pathways involved in colorectal tumorigenesis and tumor growth, possibly determining at-risk populations and predicting responses to treatment.
METHODS: Using microarray gene-expression analysis, we analyzed patterns of gene expression relative to canonical molecular changes and clinicopathological features in 84 sporadic colorectal cancer patients, standardized by tumor location. Subsets of differentially expressed genes were confirmed by real-time reverse-transcript polymerase chain reaction (RT-PCR).
RESULTS: The largest number of genes identified as being differentially expressed was by tumor location, and the next largest number by lymphovascular or neural invasion of tumor cells and by mismatch repair (MMR) defects. Amongst biological processes, the immune response was significantly implicated in entire molecular changes observed during colorectal tumorigenesis (P < 0.001). Amongst 47 differentially expressed genes, seven (PISD, NIBP, BAI2, STOML1, MRPL21, MRPL16, and MKKS) were newly found to correlate with tumorigenesis and tumor growth. Most location-associated molecular changes had distinct effects on gene expression, but the effects of the latter were sometimes contradictory.
CONCLUSION: We show that several differentially expressed genes were associated with canonical molecular changes in sporadic colorectal cancers, possibly constituting alternative or subordinate pathways of tumorigenesis. As tumor location was the dominant factor influencing differential gene expression, location-specific analysis may identify location-associated pathways and enhance the accuracy of class prediction.
Keywords: Colorectal adenocarcinomas, Sporadic, Gene expression, Profiling, Tumorigenesis
INTRODUCTION
Analysis of genetic alterations in hereditary colorectal cancers have identified several molecular changes, including those involving APC-Wnt signaling, mismatch repair (MMR) defects, RAF cascades, and p53 alterations[1-3]. The pattern of molecular changes observed in hereditary colon cancers suggested a stepwise model for colorectal tumorigenesis. About 80% of colorectal cancers, however, are sporadic, and the pattern of genetic alterations observed in hereditary tumors has been consistently observed in only a small number of sporadic tumors[1]. These findings suggest the existence of alternative or subordinate and crossover pathways of colorectal tumorigenesis.
The APC protein is thought to contribute to all processes governing tumor tissues, including proliferation, migration, apoptosis, and differentiation[4]. Loss of APC function leads to intracellular β-catenin stabilization, the key component of canonical Wnt signaling, and constitutive signaling of β-catenin within the nucleus[5,6]. The current model of colon tumorigenesis suggests that MMR defects cause tumors primarily through two mechanisms, mutations in tumor suppressor gene pathways and inappropriate apoptosis[7]. Sporadic colorectal cancers with MMR defects, including almost all those with BRAF mutations, are thought to arise through the CpG island methylator phenotype (CIMP) associated with methylation of MLH1[3]. These alterations initiate cellular processes directed towards either proliferation or differentiation, depending on signal intensity and duration[8]. Alternatively, RAS mutations may be early events in the adenoma-carcinoma sequence, and RAF alterations may be related to the progression and development of de novo colorectal cancer[9].
The p53 pathway is ubiquitously lost in human cancers, either by p53 mutations, observed in 60% of tumors, or by loss of cell signaling upstream and downstream of p53 in the 40% of cancers expressing wild-type p53[10]. Following disruption of p21WAF1, p53 expression is enhanced because of p53 stabilization, which correlates with the increased expression of the tumor suppressor p14ARF, an inhibitor of the ubiquitin ligase activity of MDM2[11]. Apart from these molecular changes, however, little is known about crossover pathways between APC-Wnt signaling and MMR or RAF alterations. APC and RAS mutations have been shown to be synergistic in promoting β-catenin nuclear translocation, thus enhancing canonical Wnt signal transduction[12]. Moreover, APC was shown to regulate cellular proliferation and transformation induced by the activation of both RAS and β-catenin signaling[13].
To identify alternative or subordinate pathways involved in colorectal tumorigenesis and tumor growth, we assessed gene expression patterns, relative to canonical molecular changes and clinicopathological features in patients with colorectal tumors. Individual steps and pathways were sorted into various biological processes. We also performed location-specific analysis to determine whether this exercise might improve the accuracy of class prediction. Our results may also be used to determine at-risk populations and to predict responses to treatment.
MATERIALS AND METHODS
Patients and tissue samples
We prospectively enrolled 84 consecutive patients with sporadic colorectal cancer scheduled to undergo curative resection between 2006 and 2007 at the Asan Medical Center (Seoul, Korea) (Table 1). Tumors were standardized by location, and samples of tumor and normal colonic mucosa, taken at least 5 cm from the tumor borders, were obtained at the time of surgery. The tissue samples were snap-frozen in liquid nitrogen. Total RNA was extracted using RNeasy RNA extraction kits (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions, and DNA was extracted from lymphocytes and tumors using standard methods. Cancer staging was determined by imaging studies and operative findings with histological diagnosis according to the American Joint Committee on Cancer (6th ed., 2001). Our sample size was determined for competent cluster analysis using an efficient annealing algorithm with error rates of < 10%. All patients provided written informed consent, and the study protocol was approved by the Institutional Review Board for Human Genetic and Genomic Research, in accordance with the Declaration of Helsinki.
Table 1.
Clinicopathological features relative to location of sporadic colorectal cancers
| Clinicopathologic features |
Tumor location1 (No. of patients) |
P | ||
| R (n = 27) | L (n = 29) | P (n = 28) | ||
| Male/Female | 18/9 | 15/14 | 20/8 | 0.273 |
| Age | 62 ± 7 | 60 ± 12 | 62 ± 10 | 0.646 |
| AJCC stage2, I/II/III/IV | 4/13/6/4 | 4/15/6/4 | 4/10/9/5 | 0.926 |
| Tumor differentiation, WD | 22/5 | 29/0 | 24/4 | 0.021 (R vs L) |
| + MD/PD + muc | 0.052 (L vs P) | |||
| Synchronous adenoma, -/+ | 18/9 | 23/6 | 14/14 | 0.052 (L vs P) |
| LVN invasion, -/+ | 15/12 | 22/7 | 20/8 | 0.236 |
R: Cecum-splenic flexure of transverse colon; L: Splenic flexure of transverse colon-sigmoid colon; P: Rectum.
According to the American Joint Committee on Cancer (6th ed., 2001). WD, MD, PD, and muc, well-, moderately-, poorly-differentiated, and mucinous. LVN: Lymphovascular or neural invasion of tumor cells.
Clinicopathological features and molecular changes in colorectal tumorigenesis
Methods of representative molecular changes in tumor tissues, including APC mutations, Wnt-activated alterations, MMR defects, RAF-mediated changes, and p53 alterations have been described using different samples[14]. Briefly, APC mutations were assessed throughout all exons and introns, whereas Wnt-activated alterations were assessed by immune staining for β-catenin, Axin2, GSK3β, and E-cadherin. The search for MMR alterations included microsatellite instability (MSI) assays using the Bethesda panel, assays of methylation status at the 5'-promoter site and the 3'-small site of hMLH1, and immune staining for hMLH1 and hMSH2. We assessed RAF-mediated alterations by determining BRAF codon 600 mutations, mutations in KRAS exons 12 and 13, and immune staining for MEK. Alterations in p53 were assessed by immune staining for altered p53. Crossover was defined when a tumor carried both APC/Wnt-activated changes and MMR defects or RAF-mediated alterations.
cDNA microarray and data analyses
The 21k cDNA microarray chips were prepared using Korean Unigene Information (KUGI) cDNA clones (http://kugi.kribb.re.kr/) and Incyte Human 10k cDNA clones. The PCR products of each clone were spotted on type-7 glass slides using an Array Spotter Generation III (Amersham Pharmacia, Piscataway, NJ, USA). Aliquots of tumor and non-tumor RNAs (20 mg respectively) were used as templates for the synthesis of cDNA, labeled with Cy5 or Cy3, respectively, using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) for 2 h at 42°C. The two labeled cDNAs were mixed, filtered through Microcon YM-30 filters (Millipore, Bedford, MA, USA) to exclude unincorporated dNTPs, and hybridized to the microarray slides at 50°C overnight using a 3DNA Array 50 kit (Genisphere Inc., Hatfield, PA, USA). After hybridization, each microarray was washed twice with 2 × SSC with 0.2% (w/v) SDS at room temperature for 5 min, and finally with 95% (v/v) ethanol at room temperature for 1 min. The slides were scanned using a ScanArray 5000 Scanner (Axon Instruments, Union City, CA, USA), and scanned images were analyzed using the GenePix Pro 4.0 program (Axon Instruments). The raw data were normalized using the print-tip Lowess method available in the OLIN package of the Bioconductor project (http://www.bioconductor.org)[15]. Missing values were imputed using the k-nearest neighbor method (available at the GEPAS web service: http://gepas.bioinfo.cipf.es/cgi-bin/preprocess/). The raw data have been deposited in the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/projects/geo/) under the accession number GSE10982.
Quantitative reverse-transcript polymerase chain reaction (RT-PCR)
Total cellular RNA (5 μg) was reverse transcribed into cDNA using SuperScript II (Invitrogen). Real-time (RT)-PCR was performed using the Exicycler Quantitative Thermal Block (Bioneer, Daejeon, Korea). The RT-PCR reaction product (100 ng) was amplified in a 15 μL reaction volume with 2 × SYBR Premix EX Taq (Takara, Shiga, Japan). Primers were designed using the Primer3 program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Following an initial denaturation at 95°C for 1 min, the amplification protocol consisted of 45 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s, followed by a final extension step of 72°C for 10 min. The β-actin protein was used as an internal control. Relative quantification of each mRNA was analyzed by the comparative threshold cycle (TC) method.
Parametric analysis of gene set enrichment (PAGE)
We applied the PAGE method to identify significant changes in expression of gene sets[16]. Diverse categories of gene sets included molecular changes associated with colorectal tumorigenesis, namely cell cycle and apoptosis pathways, receptor protein tyrosine kinase signaling, Wnt/cadherin signaling, DNA MMR, and TGF-β signaling pathway. They were prepared from Affymetrix annotation files (http://www.affymetrix.com/netaffy) and annotation files were downloaded from the Source web service (http://genome-www5.stanford.edu/cgi-bin/source/sourceBatchSearch). Gene sets identified by gene ontology (GO) protocols included those involved in various biological processes, genes responsible for cellular components and molecular functions, genes defined by chromosomal locations, genes related by InterPro domains, and genes involved in distinct metabolic pathways. Pathway information was obtained from the BioCarta (http://www.biocarta.com) and KEGG (http://www.genome.ad.jp/kegg/) databases. Publications on differentially expressed genes were accessed to understand gene effects on biological functions and tumorigenesis, using PubMed (http://www.ncbi.nlm.nih.gov/sites/entrez).
Statistical analysis
The associations of molecular changes and clinico-pathologic features with tumor location were examined by cross-table analysis using Fisher’s exact or Pearson’s χ2 tests with their significance level at 5%. The statistical significance of between-group comparisons was analyzed using Student’s t-test and Q-values were calculated from corresponding P-values to control the false discovery rate (FDR) that may occur when testing multiple hypotheses[17]. Differential gene expression between tumors and normal epithelia were deemed to be significant at P < 0.01 for initial screening and P < 0.001 for individual gene candidates. Class prediction was examined using the BRB-Array Tools package (version 3.6) available at http://linus.nci.nih.gov/BRB-ArrayTools.html. All computations were performed using R statistical programming language (http://cran.r-project.org/) and the Bioconductor packages.
RESULTS
Differentially expressed genes relative to molecular changes and clinicopathological features
Assays for genes differentially expressed relative to molecular changes and clinicopathological features (Tables 2 and 3) showed that tumor location was associated with the highest numbers of differentially expressed genes. When we compared the right colon with the left colon and rectum taken together, we found that 1628 genes were differentially expressed, and when we compared the right colon with the left colon and rectum considered separately, we found that 1263 genes were differentially expressed. The next greatest extent of differential gene expression was seen when lymphovascular or neural invasion (LVI) of tumor cells occurred, and an analysis by defects in MMR yielded the next largest differentially expressed gene set. The differentially expressed genes significantly associated with canonical tumorigenesis and tumor progression are collectively shown in Table 3. The differential expression of several candidate and novel genes was confirmed by real time RT-PCR (Figure 1).
Table 2.
Number of differentially expressed genes in terms of molecular changes and clinicopathological features
| Parameters | No. of patients (missing) | No. of differentiallyexpressed genes (P < 0.01), total (up/down) |
| Molecular changes1, -/+ | ||
| APC mutations | 55/27 (2) | 83 (41/42) |
| Wnt-activated | 45/38 (1) | 82 (37/45) |
| MMR defects | 70/14 | 238 (122/116) |
| RAF-mediated | 58/26 | 108 (59/49) |
| Altered p53 expression | 24/59 (1) | 125 (57/68) |
| Crossover | 64/19 (1) | 92 (44/48) |
| Clinicopathologic features | ||
| Tumor location2, R/L + P | 27/57 | 1628 (936/692) |
| R/L/P | 27/29/28 | 1263 |
| AJCC stage3, I + II/III + IV | 50/34 | 195 (103/92) |
| Tumor differentiation, WD + MD/PD + muc | 75/9 | 151 (69/82) |
| Synchronous adenoma, -/+ | 55/29 | 152 (92/60) |
| LVN invasion, -/+ | 57/27 | 279 (147/132) |
Wnt-activated: explored by β-catenin assay, AXIN2 measurement, and GSK-3β immune staining; MMR defect: analyzed by MSI assay, MLH1 5'-promoter measurement or 3'-methylation, and MLH1 or MSH2 immune staining; RAF-mediated alterations: assayed by detection of mutations in BRAF V600E and KRAS exons 12 and 13, and MEK immune staining; Crossover: When a tumor carried both APC/Wnt-mediated alterations and MMR defects or RAF-mediated alterations.
R: Caecum-splenic flexure of transverse colon; L: Splenic flexure of transverse colon-sigmoid colon; P: Rectum.
According to the American Joint Committee on Cancer (6th ed., 2001). WD, MD, PD, and muc, well-, moderately-, poorly-differentiated, and mucinous; MMR: Mismatch repair.
Table 3.
Differential gene expression associated with molecular changes and clinicopathological features1
| Parameters | Symbol | Name | Log2 fold changes | Unadjusted P |
| APC mutations | CDH7 | Cadherin 7, type 2 | 0.419885 | 0.00033 |
| DYRK1A | DS tyr-(Y)-phosphorylation regulated kinase 1A | 0.326912 | 0.000343 | |
| SLC19A2 | Solute carrier family 19 member 2 | -0.48574 | 0.000427 | |
| PISD | Phosphatidylserine decarboxylase | 0.272127 | 0.000545 | |
| NDUFC1 | NADH dehydrogenase 1, subcomplex unknown | 0.539581 | 0.000572 | |
| Wnt-activated alterations | PRAF2 | PRA1 domain family, member 2 | 0.620619 | 0.000205 |
| FOXF1 | Forkhead box F1 | -0.93359 | 0.000524 | |
| CD99L2 | CD99 molecule-like 2 | 0.753489 | 0.000772 | |
| MMR defects | HMGB1 | High-mobility group box 1 | -0.38141 | 3.74E-06 |
| MT1X | Metallothionein 1X | 0.965429 | 0.000252 | |
| MT1A | Metallothionein 1A | 1.17894 | 0.000351 | |
| SUGT1 | SGT1, G2 allele of SKP1 (S. cerevisiae) | -0.34799 | 0.00039 | |
| VTI1B | Vesicle transport with t-SNAREs homolog 1B (yeast) | -0.28513 | 0.000435 | |
| SST | Somatostatin | 1.24407 | 0.000564 | |
| TDG | Thymine-DNA glycosylase | 0.479829 | 0.000946 | |
| RAF-mediated alterations | RAB22A | RAB22A, member RAS oncogene family | -0.36017 | 0.000289 |
| PPP1R13L | Protein phosphatase 1, regulatory subunit 13 like | 0.823379 | 0.000614 | |
| CAST | Calpastatin | 0.285773 | 0.000872 | |
| Altered p53 expression | HLA-F | Major histocompatibility complex, class I, F | -0.55645 | 0.000429 |
| XRCC3 | XRCC in Chinese hamster cells 3 | -0.26678 | 0.000588 | |
| CCDC24 | Coiled-coil domain containing 24 | -0.28995 | 0.000996 | |
| Crossover2 | NID2 | Nidogen 2 (osteonidogen) | 0.938223 | 0.000214 |
| EGLN3 | egl nine homolog 3 (C. elegans) | 0.740933 | 0.000375 | |
| ITIH1 | Inter-alpha (globulin) inhibitor H1 | -0.34777 | 0.0004 | |
| CFH | Complement factor H | -0.33826 | 0.000542 | |
| ABI3BP | ABI gene family, member 3 binding protein | -0.82558 | 0.000637 | |
| NIBP | NIK and IKK binding protein | 0.649949 | 0.000688 | |
| SPRR3 | Small praline-rich protein 3 | 0.99738 | 0.000946 | |
| AJCC stage3 | PNPT1 | Polyribonucleotide nucleotidyltransferase 1 | 0.716381 | 4.94E-06 |
| BAI2 | Brain-specific angiogenesis inhibitor 2 | 0.545369 | 1.57E-05 | |
| ADCY1 | Adenylate cyclase 1 (brain) | -0.43437 | 1.97E-05 | |
| VEGFC | Vascular endothelial growth factor C | 0.329341 | 9.11E-05 | |
| ATAD3B | ATPase family, AAA domain containing 3B | -0.40978 | 0.000108 | |
| CAP1 | CAP, adenylate cyclase-associated protein 1 (yeast) | -0.66905 | 0.000405 | |
| RPS6KA6 | Ribosomal protein S6 kinase, 90kDa, polypeptide 6 | 0.301665 | 0.000586 | |
| FGF5 | Fibroblast growth factor 5 | 0.464162 | 0.000701 | |
| LVN invasion | MMP12 | Matrix metallopeptidase 12 (macrophage elastase) | -0.69015 | 7.76E-05 |
| RAP1GDS1 | RAP1, GTP-GDP dissociation stimulator 1 | -0.60949 | 9.63E-05 | |
| STOML1 | Stomatin (EPB72)-like 1 | -0.39407 | 0.000255 | |
| CCL16 | Chemokine (C-C motif) ligand 16 | 0.475838 | 0.000542 | |
| NOTCH3 | Notch homolog 3 (Drosophila) | 0.592567 | 0.000679 | |
| DHPS | Deoxyhypusine synthase | -0.33131 | 0.000965 | |
| Synchronous adenoma | PARP2 | Poly (ADP-ribose) polymerase family, member 2 | 0.573461 | 6.00E-05 |
| MRPL21 | Mitochondrial ribosomal protein L21 | 0.243204 | 0.000149 | |
| MRPL16 | Mitochondrial ribosomal protein L16 | 0.267004 | 0.000432 | |
| MKKS | McKusick-Kaufman syndrome | 0.439584 | 0.000447 | |
| LHX2 | LIM homeobox 2 | 0.663108 | 0.000782 |
Differential gene expression in tumor tissues compared to normal epithelia was examined in terms of molecular changes and clinicopathological features, as was considered to be significant when P < 0.001;
When a tumor carried both APC/Wnt-mediated alterations and MMR defects or RAF-mediated alterations;
According to the American Joint Committee on Cancer (6th ed., 2001).
Figure 1.

Quantitative RT-PCR of selected genes associated with molecular changes and clinicopathological features from microarray gene expression data. These were NUDFC1 and SCL19A2 (with APC mutations), MT1X and MT1A (with MMR defects), SPRR3 (with crossover), CCL16 (with lymphovascular or neural invasion), and MRPL16 and MKKS (with synchronous adenoma). Genes differentially expressed between two groups were selected and their expression patterns measured using RT-PCR. WT: Without molecular or clinicopathological changes; MT: Molecular or clinicopathological changes. P-values from unpaired t-tests are shown.
Gene sets associated with APC and Wnt pathways
APC mutations are related to expression of constituents of the extracellular matrix (ECM) and to formation of the axonemal dynein complex, whereas Wnt-associated alterations are associated with the immune response, ECM formation, and filopodium expression. In addition, changes in pyruvate and arginine/proline metabolism have been associated with APC mutations; whereas alterations in G-protein receptor binding, the activities of various chemokines, phosphatase binding efficiency, and glycolysis/gluconeogenesis rates are associated with mutations in Wnt. We found that upregulation of three genes (CDH7, DYRK1A, PISD) and downregulation of one (SLC19A2) were associated with APC mutations, whereas upregulation of two genes (PRAF2, CD99L2) and downregulation of one (FOXF1) were associated with Wnt-activated changes (P < 0.001).
Gene set alterations associated with the MMR and RAF pathways
Biologically, MMR defects affect the immune response (including antigen processing), chromosome functions, and cytoskeleton structure, whereas RAF-mediated alterations are related to thyroid hormone generation and cytoplasmic effects. Cadmium and copper ion binding, MHC class II receptor activity, and fatty acid metabolism have been associated with MMR defects, and protein dimerization activity with RAF-mediated alterations. We found that four upregulated genes (MT1X, MT1A, SST, TDG) and three downregulated genes (HMGB1, SUGT1, VTI1B) were associated with MMR defects, and that two were upregulated (PPP1R13L, CAST) and one was downregulated (RAB22A) in association with RAF-mediated alterations (P < 0.001).
Gene set alterations associated with p53 and crossover pathways
Alterations in p53 have been associated with the immune response (including antigen processing), ECM structure, and sensory perception, whereas crossover was related to cell cycle stage and protein localization. MHC class I receptor activity, oxidoreductase activity, and glycolysis were associated with p53 alterations, and protein kinase binding and renyltransferase activity were associated with crossover. No upregulated but three downregulated genes (HLA-F, XRCC3, CCDC24) were associated with p53 alterations, whereas four upregulated genes (NID2, EGLN3, NIBP, SPRR3) and three downregulated genes (ITIH1, CFH, ABI3BP), were associated with crossover (P < 0.001).
Tumor location-specific analysis shows distinct patterns of gene expression
As colon tumor location had a very marked effect on differential gene expression, genes differentially expressed as a result of particular molecular changes and clinicopathological features may be concealed by tumor location. Interestingly, the number of genes differentially expressed as a result of tumor location increased slightly in association with several clinicopathological variables, although the sample size was much smaller (about one-third, data not shown) than those of other tumor sets. More importantly, most molecular changes had distinct location-associated effects on gene expression, but these were sometimes accompanied by contradictory effects. For example, p53 alterations inhibited the expression of antigen presentation-related genes only in right colon cancers, but had no effect in left colon or rectal cancers (Figure 2). After separation into classes showing various molecular and clinicopathological characteristics, the accuracy of binary outcome prediction was estimated using seven different machine learning algorithms available at BRBArrayTools. As expected, the best prediction accuracy (85%-94%) was achieved by tumor location (Table 4), followed by MMR defects (76%-83%). Most of the other molecular and clinicopathological variables had prediction accuracies < 75%. The prediction accuracies for each variable were also examined after restricting the analysis by the three tumor locations mentioned above. As expected, location-specific analysis generally increased the prediction accuracy significantly (Figure 3), especially in the case of variables associated with synchronous adenoma, tumor stage, RAF-mediated changes, and Wnt-activated alterations. Accuracy, however, decreased when variables associated with APC mutations, p53 alterations, and crossover, were analyzed.
Figure 2.

Pattern of expression of genes in the “antigen presentation, endogenous antigen” gene set as distinguished by tumor location and p53 status. Protein p53 alterations in the ascending colon coordinately decreased the expression of genes in the gene set whereas p53 alterations in the descending colon or rectum had no effect on gene expression. R: Cecum-splenic flexure of transverse colon; L: Splenic flexure of transverse colon-sigmoid colon; P: Rectum.
Table 4.
Class prediction accuracies (%) relative to molecular changes and clinicopathological features
| Parameters | Genes1 | CCP | DLDA | 1-NN | 3-NN | NC | SVM | BCCP |
| Tumor location | 620 | 85 | 86 | 92 | 90 | 86 | 94 | 94 |
| Synchronous adenoma | 12 | 58 | 60 | 64 | 63 | 57 | 67 | 65 |
| Tumor stage | 22 | 62 | 58 | 58 | 60 | 61 | 54 | 60 |
| APC mutations | 12 | 72 | 72 | 57 | 72 | 67 | 73 | 67 |
| LVN invasion | 27 | 70 | 69 | 57 | 60 | 68 | 64 | 68 |
| MMR defects | 44 | 76 | 77 | 82 | 82 | 76 | 81 | 83 |
| p53 alterations | 8 | 65 | 70 | 61 | 73 | 66 | 72 | 79 |
| RAF-mediated alterations | 3 | 36 | 35 | 43 | 51 | 36 | 42 | 69 |
| Wnt-activated alterations | 9 | 54 | 54 | 61 | 61 | 58 | 49 | 54 |
Number of classifier genes. CCP: Compound covariate predictor; DLDA: Diagonal linear discriminant analysis; 1-NN: One nearest-neighbor; 3-NN: Three nearest-neighbor; NC: Nearest centroid; SVM: Support vector machine; BCCP: Bayesian compound covariate predictor; LVN: Lymphovascular or neural.
Figure 3.
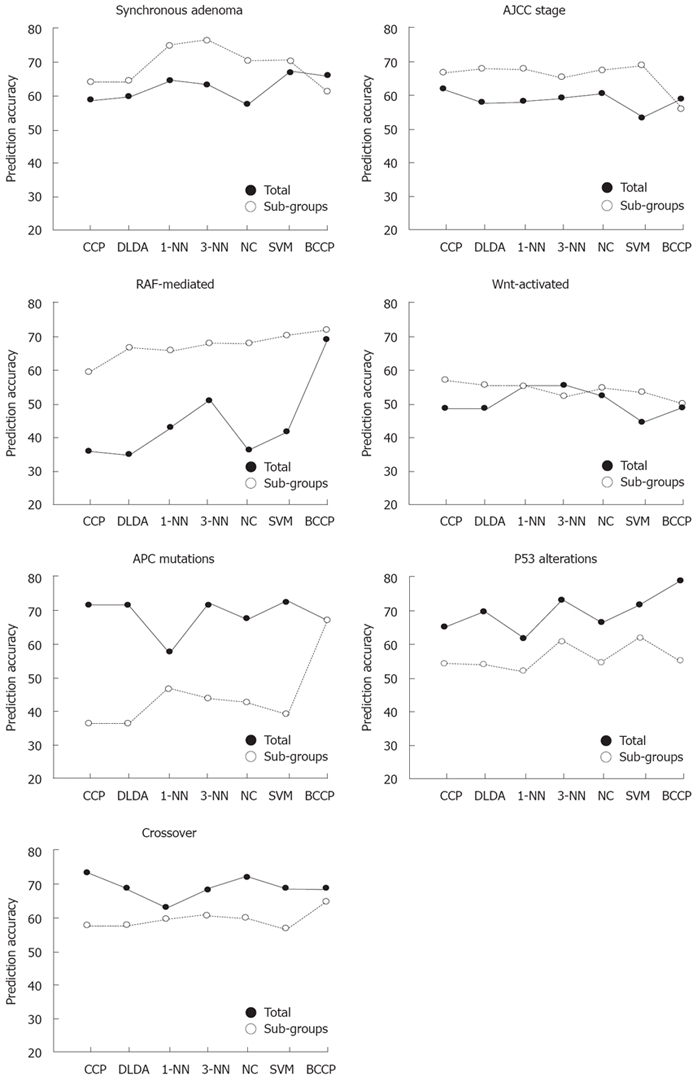
Accuracy of class prediction increases with tumor location-specific analysis. Samples were divided into three subgroups corresponding to three tumor locations (right colon, left colon, and rectum). Class prediction was performed using either all samples or samples within each subgroup. For tumor location-specific analysis, the results of class prediction (true or false) from each of the three locations were combined to calculate the overall prediction accuracy. Ten genetic or clinicopathological parameters were analyzed.
Clinicopathological features correlated with genomic alterations
Biologically, we found that tumor stage was related to antigen presentation, cell adhesion and migration, bone mineralization, and epithelial cell differentiation, and that lymphovascular or neural invasion was related to cell adhesion, immune response, and sensory perception. We also found that serine-type enzyme activities, high-density lipoprotein binding, pancreatic RNase activity, and glycolysis/gluconeogenesis were related to tumor stage, and that various structural molecules, hormones, serine-type enzyme activities, phosphate transport, the metabolism of the ECM and related molecules, and high density lipoprotein binding were related to lymphovascular or neural invasion. Synchronous adenoma was related to protein biosynthesis, ribosomal proteins, and MHC class I receptor activity. We found that five genes (PNPT1, BAI2, VEGFC, RPS6K6A, FGF5) were upregulated and three (ADCY1, ATAD3B, CAP1) were downregulated in association with tumor stages; that two genes (CCL16, NOTCH3) and four genes (MMP12, RAP1GDS1, STOML1, DHPS) were up- or down-regulated, respectively, in association with lymphovascular or neural invasion, and that five genes (PARP2, MRPL21, MRPL16, MKKS, LHX2) were upregulated but no gene was downregulated in association with synchronous adenoma (P < 0.001).
DISCUSSION
Distinctive molecular changes, such as APC mutations and MMR defects, are respectively associated with two types of hereditary colorectal cancers, familial adenomatous polyposis and hereditary non-polyposis colorectal cancer. Although these hereditary tumors constitute fewer than 5%-8% of all colorectal cancers, the molecular changes identified in hereditary tumors are important in sporadic colorectal cancers[14,18,19]. The tumor suppressor APC is the major regulator of canonical Wnt signaling; these two proteins form a multi-protein complex encompassing kinases such as GSK-3β, CK1, and Axins, to prevent colorectal tumorigenesis[5]. Mutations in the oncogenes RAF and RAS are closely associated with MMR defects, and may act as alternative tumor-initiating steps that synergize with DNA methylation and occur within the context of serrated polyps[19,20]. The p53 protein, which normally induces G1 cell cycle arrest to facilitate DNA repair during replication, cannot induce cell cycle arrest when mutated in later stages of the adenoma-carcinoma sequence, thus leading to cell proliferation[21].
In our study, ECM interactions and the immune response were down- and up-regulated in tumors with APC mutations and Wnt-activated alterations, respectively. Gene set analysis earlier showed that the structural motif of osteopontin mediated critical cell-matrix and cell-cell signaling whose transcriptional regulation involves multiple pathways including Wnt/β-catenin/APC/GSK-3β/Tcf-4[22]. Expression of the E-cadherin β-catenin was observed in dendritic cells and loss of E-cadherin adhesion triggered a functionally distinct pathway of maturation linked more closely to the maintenance of tolerance than to the initiation of immunity[23]. In ApcMin/+ mice, in which APC mutations are upregulated, dietary arginine increased colon tumorigenesis[24]. Amongst the eight genes we identified that were associated with APC mutations and Wnt activation, we found that one, PISD, was a novel gene upregulated in tumor cells with these alterations. Phosphatidylserine decarboxylation may provide a functionally important source of phosphatidylethanolamine in mitochondria[25].
We found that MMR defects correlated positively with an enhanced immune response and metal ion binding, whereas RAF alterations correlated with activation of cellular processes and thyroid hormone generation. Many tumor-infiltrating lymphocytes are present in MSI+ tumors, along with activated CD8+ cytotoxic T cells[26,27]. Furthermore, tumor-specific peptides generated by MSI may be involved in anti-tumor immune responses and may be useful in the diagnosis and treatment of patients with MSI+ colorectal cancers[27]. The deleterious effects of Cd2+ reported to date include generation of reactive oxygen species, inhibition of DNA repair, depletion of glutathione, and alteration of apoptosis[28]. In contrast, RAS/RAF/MEK/ERK-transduced signals can initiate cellular processes directed towards either proliferation or differentiation, depending on signal intensity and duration[29]. RAF mutations are associated with advanced clinical stages and early recurrence in patients with papillary thyroid cancer[30]. Amongst the genes up-regulated by MMR defects are the metallothionein genes, including MT1X and MT1A, which are expressed differentially in various tissues, during several developmental stages, and in response to metals, steroids, and stress[30]. Several of these genes, including MT1X, were overexpressed in MSI+ colorectal and gastric cancers[31].
We found that alterations in p53 downregulated immune responses and ascorbic acid binding. Anti-p53 IgG has been detected in the sera of subjects with various types of cancer, indicating induction of anti-p53 CD4+ Th cells[32]. Ascorbic acid can block the effects of TNF-α on endothelial cell proliferation and apoptosis by inhibiting TNF-α-induced p53 expression and Rb hypophosphorylation, as well as by promoting collagen IV production[33]. We also observed that the crossover pathway between APC/Wnt-activated and MMR defects or RAF-mediated alterations, which has rarely been observed in human colorectal cancers, was associated with cell cycle and protein localization. Recently, mice carrying compound Apc and Ras mutations were characterized as having a striking increase in intestinal tumor multiplicity and progression, compared with Apc-only mutant animals[12]. Amongst the seven genes we identified as associated with the crossover pathway, one, NIBP, was a novel gene upregulated in tumor cells with these alterations. NIBP has been reported to enhance the cytokine-induced NF-κB signaling pathway by interacting with NIK and IKKβ[34], which may activate the TNF-induced invasive activity of tumor cells.
Embryologically, the right and left colon has different origins, the midgut and hindgut, respectively, and is supplied by different circulation and innervation[35]. We found that tumor location was the dominant factor for differential gene expression in colorectal cancers. Thus, location-specific analysis may more precisely discriminate between alterations in gene expression caused by canonical molecular changes. The dependence of gene expression differences on tumor location has been reported previously[36-39]. The dominant expression pattern has been shown to be consistent with different embryonic origins and a second pattern reveals a gradual change from the caecum to the rectum[39]. We found that the prediction accuracy by tumor location-specific analysis was increased using analyses by synchronous adenoma, tumor stage, and RAF-mediated and Wnt-activated alterations, but decreased by analyses using APC mutations and p53 alterations. These findings suggest that APC mutations and p53 alterations may affect tumorigenesis as initiators and terminators, respectively, along the entire colon. In the absence of APC mutations and p53 alterations, however, synchronous adenoma, tumor stage, RAF-mediated changes, and Wnt-activated alterations may determine tumorigenesis at different locations. In addition, we found that several biological processes were affected differently by tumor location, often in opposite senses. One of the most significantly altered biological processes was the immune response. We observed that genes involved in the immune response were coordinately downregulated in left colon cancers with p53 alterations but not in right colon or rectal cancers. The same trend was observed with APC mutations, but the opposite trend was observed with MMR defects. Location-specific analysis also allowed the prediction of gene class by expression profiling in 6 of 10 parameters in our analysis, and in agreement with previous findings[38]. Gene expression profiling has been used to predict metastasis or recurrence in patients with stage II colon cancer, thus enhancing the selection of chemosensitive patients for adjuvant chemotherapy[40,41]. Our finding that distinct molecular pathways of tumorigenesis occur in right and left colon cancers suggests that prediction of responsiveness to adjuvant therapy will benefit from location stratification.
In our study, both tumor stage and lymphovascular or neural invasion were associated with antigen presentation, ECM metabolism, and cellular and extracellular processes that determine tumor initiation and progression. Amongst the 14 differentially expressed genes associated with these biological functions, one encodes CCL16, a chemoattractant for monocytes and lymphocytes that can increase tumor rejection, antigen presentation by macrophages, T cell cytotoxicity, and the angiogenic activity of vascular endothelial cells[42]. BAI2 and STOML1 are novel genes, upregulated in advanced cancers and downregulated in lymphovascular and neural tumor invasion, respectively. Human BAI2, probably a G-protein-coupled receptor in the brain, participates in the early stages of neovascularization of the cerebral cortex after ischemia[43]. The stomatin homolog (UNC-24) of C. elegans, a protein similar to the human stomatin homolog STOML1 (SLP-1), is required for normal locomotor response to volatile anesthetics and contains a region of sequence homologous to the nonspecific lipid transfer protein[44].
As the traditional adenoma-carcinoma sequence, which is instigated in adenomas (or aberrant crypt foci) by the APC-Wnt signaling pathway, accounts for more than two-thirds of all colorectal cancers[3], we examined the molecular association of tumorigenesis in patients with synchronous adenoma. We found that three novel genes, MRPL21, MRPL16, and MKKS, were upregulated in tumors with synchronous adenoma. A mitochondrial ribosomal protein, MRPL21, arrests the cell cycle by increasing p21WAF1/CIP1 and p27Kip1 levels under growth inhibitory conditions[45]. The MRPL16 gene originated via duplication of a pre-existing mitochondrial ribosomal protein gene as well as by recruitment of some DNA sequence from outside of the mitoribosomal genome[46]. McKusick-Kaufman syndrome (MKKS) is a human developmental anomaly syndrome featuring hydrometrocolpos, postaxial polydactyly, and congenital heart disease[47]. In protein biosynthesis, MKKS is similar in function to type II chaperonins, which are responsible for folding a wide range of proteins[48].
In conclusion, we found that the differential expression of 47 genes was associated with canonical molecular changes and clinicopathological characteristics of sporadic colorectal cancers, possibly constituting alternative or subordinate pathways of tumorigenesis and tumor growth. Currently, the seven novel genes of our study that correlate with tumorigenesis and tumor growth, are functionally assessed to be possible candidates as diagnostic or therapeutic targets for colorectal cancers. Amongst these biological processes, the immune response was uniformly involved in all molecular changes, that is, APC/Wnt-activated alterations, changes arising from MMR defects, RAF-mediated changes, and p53-caused alterations. As tumor location was the dominant factor for differential gene expression in colorectal cancers, location-specific analysis may precisely discriminate particular gene expression profiles and enhance the accuracy of tumor class prediction.
COMMENTS
Background
Although various molecular changes have been identified in colorectal cancers, a clear pattern is detected in only 6.6% of these tumors, indicating the need to identify alternative or subordinate pathways involved in colorectal tumorigenesis and tumor growth.
Research frontiers
To identify alternative or subordinate pathways involved in colorectal tumorigenesis and tumor growth, this study assessed gene expression patterns, relative to canonical molecular changes and clinicopathological features, in patients with colorectal tumors. Individual steps and pathways were sorted into various biological processes.
Innovations and breakthroughs
The largest number of genes identified as differentially expressed was by tumor location, and the next largest number by lymphovascular or neural invasion of tumor cells and by mismatch repair (MMR) defects. Amongst biological processes, the immune response was significantly implicated in entire molecular changes observed during colorectal tumorigenesis (P < 0.001). Amongst 47 differentially expressed genes, seven (PISD, NIBP, BAI2, STOML1, MRPL21, MRPL16, and MKKS) were newly found to correlate with tumorigenesis and tumor growth. Most location-associated molecular changes had distinct effects on gene expression, but the effects of the latter were sometimes contradictory.
Applications
This study found that the differential expression of 47 genes was associated with canonical molecular changes and clinicopathological characteristics of sporadic colorectal cancers, possibly constituting alternative or subordinate pathways of tumorigenesis and tumor growth. The seven novel genes of this study correlate with tumorigenesis and tumor growth and can functionally be assessed as possible candidates for diagnostic or therapeutic targets of colorectal cancers.
Terminology
The cDNA microarray becomes a fundamental tool to gain direct molecular insight into tumorigenesis. Additionally, as phenotypic diversities of cancer occur from genetic alterations, genomic expression profiling might have been recognized as the first step to find useful therapeutic targets.
Peer review
This paper describes alternative or subordinate pathways involved in colorectal tumorigenesis and tumor growth, constituting an individual geno-pathogenesis map for colorectal cancer. As the study strengthened tumor location as a dominant factor for differential gene expression in colorectal cancers, location-specific analysis precisely discriminate particular gene expression profiles, possibly providing individual responses to respective regimen. It’s an interesting paper.
Footnotes
Supported by The Basic Research Program of the Korea Science & Engineering Foundation, No. R01-2006-000-10021-0; and the Korea Health 21 R&D Project, Ministry of Health & Welfare No. A062254
Peer reviewer: Finlay A Macrae, MD, Professor, Royal Melbourne Hospital, Po Box 2010, Victoria 3050, Australia
S- Editor Li DL L- Editor Kremer M E- Editor Zheng XM
References
- 1.Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, Garner RC, Steele RJ, Wolf CR. Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci USA. 2002;99:9433–9438. doi: 10.1073/pnas.122612899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–818. [PMC free article] [PubMed] [Google Scholar]
- 3.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 4.Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Thorstensen L, Lind GE, Løvig T, Diep CB, Meling GI, Rognum TO, Lothe RA. Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability. Neoplasia. 2005;7:99–108. doi: 10.1593/neo.04448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chao EC, Lipkin SM. Molecular models for the tissue specificity of DNA mismatch repair-deficient carcinogenesis. Nucleic Acids Res. 2006;34:840–852. doi: 10.1093/nar/gkj489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6:322–327. doi: 10.1016/S1470-2045(05)70168-6. [DOI] [PubMed] [Google Scholar]
- 9.Ikehara N, Semba S, Sakashita M, Aoyama N, Kasuga M, Yokozaki H. BRAF mutation associated with dysregulation of apoptosis in human colorectal neoplasms. Int J Cancer. 2005;115:943–950. doi: 10.1002/ijc.20957. [DOI] [PubMed] [Google Scholar]
- 10.Bourdon JC. p53 and its isoforms in cancer. Br J Cancer. 2007;97:277–282. doi: 10.1038/sj.bjc.6603886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Javelaud D, Besancon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio. J Biol Chem. 2002;277:37949–37954. doi: 10.1074/jbc.M204497200. [DOI] [PubMed] [Google Scholar]
- 12.Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, Rosty C, Abal M, El Marjou F, Smits R, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096–1109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 13.Park KS, Jeon SH, Kim SE, Bahk YY, Holmen SL, Williams BO, Chung KC, Surh YJ, Choi KY. APC inhibits ERK pathway activation and cellular proliferation induced by RAS. J Cell Sci. 2006;119:819–827. doi: 10.1242/jcs.02779. [DOI] [PubMed] [Google Scholar]
- 14.Kim JC, Cho YK, Roh SA, Yu CS, Gong G, Jang SJ, Kim SY, Kim YS. Individual tumorigenesis pathways of sporadic colorectal adenocarcinomas are associated with the biological behavior of tumors. Cancer Sci. 2008;99:1348–1354. doi: 10.1111/j.1349-7006.2008.00819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Futschik ME, Crompton T. OLIN: optimized normalization, visualization and quality testing of two-channel microarray data. Bioinformatics. 2005;21:1724–1726. doi: 10.1093/bioinformatics/bti199. [DOI] [PubMed] [Google Scholar]
- 16.Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Early DS, Fontana L, Davidson NO. Translational approaches to addressing complex genetic pathways in colorectal cancer. Transl Res. 2008;151:10–16. doi: 10.1016/j.trsl.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jass JR. Colorectal cancer: a multipathway disease. Crit Rev Oncog. 2006;12:273–287. doi: 10.1615/critrevoncog.v12.i3-4.50. [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara T, Stolker JM, Watanabe T, Rashid A, Longo P, Eshleman JR, Booker S, Lynch HT, Jass JR, Green JS, et al. Accumulated clonal genetic alterations in familial and sporadic colorectal carcinomas with widespread instability in microsatellite sequences. Am J Pathol. 1998;153:1063–1078. doi: 10.1016/S0002-9440(10)65651-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan KM, Vousden KH. Cancer: pinning a change on p53. Nature. 2002;419:795, 797. doi: 10.1038/419795a. [DOI] [PubMed] [Google Scholar]
- 22.Wai PY, Kuo PC. Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev. 2008;27:103–118. doi: 10.1007/s10555-007-9104-9. [DOI] [PubMed] [Google Scholar]
- 23.Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, Whitney JA, Connolly J, Banchereau J, Mellman I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity. 2007;27:610–624. doi: 10.1016/j.immuni.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yerushalmi HF, Besselsen DG, Ignatenko NA, Blohm-Mangone KA, Padilla-Torres JL, Stringer DE, Guillen JM, Holubec H, Payne CM, Gerner EW. Role of polyamines in arginine-dependent colon carcinogenesis in Apc(Min) (/+) mice. Mol Carcinog. 2006;45:764–773. doi: 10.1002/mc.20246. [DOI] [PubMed] [Google Scholar]
- 25.Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005;280:40032–40040. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michael-Robinson JM, Biemer-Hüttmann A, Purdie DM, Walsh MD, Simms LA, Biden KG, Young JP, Leggett BA, Jass JR, Radford-Smith GL. Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status. Gut. 2001;48:360–366. doi: 10.1136/gut.48.3.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houston AM, Michael-Robinson JM, Walsh MD, Cummings MC, Ryan AE, Lincoln D, Pandeya N, Jass JR, Radford-Smith GL, O'Connell J. The "Fas counterattack" is not an active mode of tumor immune evasion in colorectal cancer with high-level microsatellite instability. Hum Pathol. 2008;39:243–250. doi: 10.1016/j.humpath.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 28.Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28:742–762. doi: 10.1210/er.2007-0007. [DOI] [PubMed] [Google Scholar]
- 30.Mayo KE, Warren R, Palmiter RD. The mouse metallothionein-I gene is transcriptionally regulated by cadmium following transfection into human or mouse cells. Cell. 1982;29:99–108. doi: 10.1016/0092-8674(82)90094-0. [DOI] [PubMed] [Google Scholar]
- 31.Giacomini CP, Leung SY, Chen X, Yuen ST, Kim YH, Bair E, Pollack JR. A gene expression signature of genetic instability in colon cancer. Cancer Res. 2005;65:9200–9205. doi: 10.1158/0008-5472.CAN-04-4163. [DOI] [PubMed] [Google Scholar]
- 32.Ito D, Albers A, Zhao YX, Visus C, Appella E, Whiteside TL, DeLeo AB. The wild-type sequence (wt) p53(25-35) peptide induces HLA-DR7 and HLA-DR11-restricted CD4+ Th cells capable of enhancing the ex vivo expansion and function of anti-wt p53(264-272) peptide CD8+ T cells. J Immunol. 2006;177:6795–6803. doi: 10.4049/jimmunol.177.10.6795. [DOI] [PubMed] [Google Scholar]
- 33.Saeed RW, Peng T, Metz CN. Ascorbic acid blocks the growth inhibitory effect of tumor necrosis factor-alpha on endothelial cells. Exp Biol Med (Maywood) 2003;228:855–865. doi: 10.1177/15353702-0322807-12. [DOI] [PubMed] [Google Scholar]
- 34.Hu WH, Pendergast JS, Mo XM, Brambilla R, Bracchi-Ricard V, Li F, Walters WM, Blits B, He L, Schaal SM, et al. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J Biol Chem. 2005;280:29233–29241. doi: 10.1074/jbc.M501670200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wexner SD, Jorge JM. Colon and rectal surgery. 5th ed. Corman ML, editor. Philadelphia: Lippincott Williams & Wilkins; 2005. pp. 1–29. [Google Scholar]
- 36.Glebov OK, Rodriguez LM, Nakahara K, Jenkins J, Cliatt J, Humbyrd CJ, DeNobile J, Soballe P, Simon R, Wright G, et al. Distinguishing right from left colon by the pattern of gene expression. Cancer Epidemiol Biomarkers Prev. 2003;12:755–762. [PubMed] [Google Scholar]
- 37.Birkenkamp-Demtroder K, Olesen SH, Sørensen FB, Laurberg S, Laiho P, Aaltonen LA, Orntoft TF. Differential gene expression in colon cancer of the caecum versus the sigmoid and rectosigmoid. Gut. 2005;54:374–384. doi: 10.1136/gut.2003.036848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komuro K, Tada M, Tamoto E, Kawakami A, Matsunaga A, Teramoto K, Shindoh G, Takada M, Murakawa K, Kanai M, et al. Right- and left-sided colorectal cancers display distinct expression profiles and the anatomical stratification allows a high accuracy prediction of lymph node metastasis. J Surg Res. 2005;124:216–224. doi: 10.1016/j.jss.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 39.LaPointe LC, Dunne R, Brown GS, Worthley DL, Molloy PL, Wattchow D, Young GP. Map of differential transcript expression in the normal human large intestine. Physiol Genomics. 2008;33:50–64. doi: 10.1152/physiolgenomics.00185.2006. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Jatkoe T, Zhang Y, Mutch MG, Talantov D, Jiang J, McLeod HL, Atkins D. Gene expression profiles and molecular markers to predict recurrence of Dukes' B colon cancer. J Clin Oncol. 2004;22:1564–1571. doi: 10.1200/JCO.2004.08.186. [DOI] [PubMed] [Google Scholar]
- 41.Barrier A, Boelle PY, Roser F, Gregg J, Tse C, Brault D, Lacaine F, Houry S, Huguier M, Franc B, et al. Stage II colon cancer prognosis prediction by tumor gene expression profiling. J Clin Oncol. 2006;24:4685–4691. doi: 10.1200/JCO.2005.05.0229. [DOI] [PubMed] [Google Scholar]
- 42.Strasly M, Doronzo G, Cappello P, Valdembri D, Arese M, Mitola S, Moore P, Alessandri G, Giovarelli M, Bussolino F. CCL16 activates an angiogenic program in vascular endothelial cells. Blood. 2004;103:40–49. doi: 10.1182/blood-2003-05-1387. [DOI] [PubMed] [Google Scholar]
- 43.Kee HJ, Koh JT, Kim MY, Ahn KY, Kim JK, Bae CS, Park SS, Kim KK. Expression of brain-specific angiogenesis inhibitor 2 (BAI2) in normal and ischemic brain: involvement of BAI2 in the ischemia-induced brain angiogenesis. J Cereb Blood Flow Metab. 2002;22:1054–1067. doi: 10.1097/00004647-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 44.Barnes TM, Jin Y, Horvitz HR, Ruvkun G, Hekimi S. The Caenorhabditis elegans behavioral gene unc-24 encodes a novel bipartite protein similar to both erythrocyte band 7.2 (stomatin) and nonspecific lipid transfer protein. J Neurochem. 1996;67:46–57. doi: 10.1046/j.1471-4159.1996.67010046.x. [DOI] [PubMed] [Google Scholar]
- 45.Kim MJ, Yoo YA, Kim HJ, Kang S, Kim YG, Kim JS, Yoo YD. Mitochondrial ribosomal protein L41 mediates serum starvation-induced cell-cycle arrest through an increase of p21(WAF1/CIP1) Biochem Biophys Res Commun. 2005;338:1179–1184. doi: 10.1016/j.bbrc.2005.10.064. [DOI] [PubMed] [Google Scholar]
- 46.Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, Ettema TJ. Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res. 2007;35:4686–4703. doi: 10.1093/nar/gkm441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone DL, Slavotinek A, Bouffard GG, Banerjee-Basu S, Baxevanis AD, Barr M, Biesecker LG. Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat Genet. 2000;25:79–82. doi: 10.1038/75637. [DOI] [PubMed] [Google Scholar]
- 48.Agashe VR, Hartl FU. Roles of molecular chaperones in cytoplasmic protein folding. Semin Cell Dev Biol. 2000;11:15–25. doi: 10.1006/scdb.1999.0347. [DOI] [PubMed] [Google Scholar]