

Abstract
Uronate isomerase (URI) catalyzes the reversible isomerization of D-glucuronate to D-fructuronate and of D-galacturonate to D-tagaturonate. URI is a member of the amidohydrolase superfamily (AHS), a highly divergent group of enzymes that catalyzes primarily hydrolytic reactions. The chemical mechanism and active site structure of URI was investigated in an attempt to obtain a greater understanding of how an active site template that apparently evolved to catalyze hydrolytic reactions has been re-forged to catalyze an isomerization reaction. The pH-rate profiles for kcat and kcat/Km for URI from Escherichia coli are bell-shaped and indicate that one group must be unprotonated and another residue must be protonated for catalytic activity. Primary isotope effects on the kinetic constants with [2-2H]-D-glucuronate and the effects of changes in solvent viscosity are consistent with product release as the rate limiting step. The X-ray structure of Bh0493, a URI from Bacillus halodurans, was determined in the presence of the substrate D-glucuronate. The bound complex showed that the mononuclear metal center in the active site is ligated to the C-6 carboxylate and the C-5 hydroxyl group of the substrate. This hydroxyl group is also hydrogen bonded to Asp-355 in the same orientation as the hydroxide/water is bound in those members of the AHS that catalyze hydrolytic reactions. In addition, the C-2 and C-3 hydroxyl groups of the substrate are hydrogen bonded to Arg-357 and the carbonyl group at C-1 is hydrogen bonded to Tyr-50. A chemical mechanism is proposed that utilizes a proton transfer from C-2 of D-glucuronate to C-1 that is initiated by the combined actions of Asp-355 from the end of β-strand 8 and the C-5 hydroxyl of the substrate that is bound to the metal ion. The formation of the proposed cis-enediol intermediate is further facilitated by the shuttling of the proton between the C-2 and C-1 oxygens by the conserved Tyr-50 and/or Arg-355.
Uronate isomerase (URI1) catalyzes the first step in the pathway for the metabolism of D-glucuronate and D-galacturonate. In this transformation, D-glucuronate and D-galacturonate are initially isomerized into their corresponding keto products, D-fructuronate and D-tagaturonate, respectively (1). D-Fructuronate and D-tagaturonate are then reduced to D-mannonate and D-altronate, respectively, by mannonate and altronate dehydrogenase in the presence of NADH (2). The pathways converge through a dehydration reaction where mannonate dehydrase and altronate dehydrase convert mannonate and altronate to 2-keto-3-deoxy-D-gluconic acid (KDG). This product is then phosphorylated by the enzyme ketodeoxygluconic acid kinase with ATP to form 2-keto-3-deoxy-6-phosphogluconic acid (KDG-6-P). In the final step of this pathway, 2-keto-3-deoxy-6-phosphogluconic acid is cleaved by an aldolase to yield pyruvate and D-glyceraldehyde-3-phosphate, which enter the citric acid cycle and glycolysis. The entire pathway is summarized in Scheme 1 (1,2).
Scheme 1.

We have demonstrated that uronate isomerase is a member of the amidohydrolase superfamily of enzymes based on sequence alignments and three dimensional structural comparisons (3). The majority of the functionally characterized members of the amidohydrolase superfamily catalyze the hydrolysis of amide or ester bonds to carbon or phosphorus centers (4,5). Well characterized examples include dihydroorotase (6), urease (7) and phosphotriesterase (8). Members of this superfamily also catalyze the deamination of many nucleotides including adenosine (9), cytosine (10), and guanine (11). The active sites of these enzymes generally contain a mononuclear or binuclear metal center that is perched at the C-terminal end of the β-barrel within a (β/α)8 structural fold. The most highly conserved residues in the AHS include two histidines from β-strand 1, histidines after the ends of β-stands 5 and 6, and an aspartic acid from β-strand 8. Since URI catalyzes an isomerization of an aldose sugar to the corresponding ketose product, this enzyme is one of the most divergent members of the amidohydrolase superfamily. The mechanistic details of this transformation are therefore of significant interest for a greater understanding of how an active site that originally evolved to catalyze hydrolytic reactions has been re-forged to undergo an isomerization reaction.
We have previously demonstrated that the hydrogen originally at C-2 of D-glucuronate is ultimately found at the pro-R position at C-1 of D-fructuronate and that this hydrogen slowly exchanges with solvent (12). These results are consistent with a proton transfer mechanism with a cis-enediol intermediate. The general mechanism, shown in Scheme 2, indicates a requirement for at least two residues that participate in the transformation of D-glucuronate into D-fructuronate. A general base (:B1) abstracts the proton from C-2 of D-glucuronate and a general acid (H:B2) facilitates the transfer of a proton to the carbonyl oxygen at C-1 to produce the cis-enediol intermediate. In the subsequent step, the ketose product is generated by a proton transfer from the hydroxyl group at C-2 of the proposed intermediate and protonation of C-1 by H:B1. For compounds such as D-glucuronate the enzymatic transformation is made more complicated by the fact that in solution the substrate exists almost entirely as a mixture of two anomeric cyclic hemiacetals.
Scheme 2.

This paper focuses on a determination of the chemical mechanism for the isomerization reaction catalyzed by URI from E. coli. The rate limiting steps have been interrogated by measuring the primary kinetic isotope effects with [2-2H]-D-glucuronate and solvent isotope effects with D2O for the wild type and mutant enzymes. The rate limitation imposed by product release has been examined using solvent viscosity effects. The identity of the residues involved in the proton transfer events has been probed by pH-rate profiles and characterization of the kinetic constants for mutant enzymes. These approaches have been augmented by the determination of the X-ray structure of a uronate isomerase from Bacillus halodurans (Bh0493) in the presence of D-glucuronate, D-fructuronate, and two mimics of the cis-enediol intermediate.
Materials and Methods
Materials
D-Glucuronic acid (I), NADH, buffers, and all other chemicals were purchased from Sigma-Aldrich or Acros, unless otherwise stated. D-arabinaric acid (III) and the monohydroxamate derivative of this compound (II) were synthesized as previously described (12). 2,6-Anhydro-L-gulonic acid (IV) was synthesized starting from L-xylose (13, 14). The structures of these compounds are presented in Scheme 3. Oligonucleotide syntheses and DNA sequencing were performed by the Gene Technologies Lab of Texas A&M University. Metal analyses were done using inductively coupled plasma mass spectrometry (ICP-MS) as previously described (12).
Scheme 3.

Site-Directed Mutagenesis
Site-directed mutagenesis of URI was performed using the QuikChange mutagenesis kit from Stratagene. The following mutants were obtained by this method: H33N, H33A, H35N, H35A, H59N, H59A, Y60F, Y60A, R186K, R186M, D238N, H297N, R302K, R302M, H297A, W381F, W381A, D412N, D412A, R414K, and R414M. The mutations were confirmed by DNA sequencing of the modified plasmids.
Protein Expression and Purification
The uxaC gene encoding uronate isomerase in E. coli was cloned into the expression vector pET28. The protein was expressed in the E. coli strain BL21(DE3) and purified as previously described (12). The enzymes contained up to 1 equivalent of zinc (depending on the mutant) as measured by ICP-MS.
Enzyme Assays
The conversion of D-glucuronate to D-fructuronate by URI was coupled to the reduction of D-fructuronate with NADH by mannonate dehydrogenase (MDH) as previously described (12). The assays were monitored spectrophotometrically by following the decrease in absorbance at 340 nm. The standard assay conditions contained 50 mM HEPES (pH 8.0), varying concentrations of D-glucuronate, 0.2 mM NADH, excess MDH, and URI in a final volume of 250 μL. The pH-dependence of the kinetic parameters, kcat and kcat/Km, were measured over the pH range of 5.3–10.3 at 0.20 pH intervals. The buffers used for the pH-rate profiles were MES, PIPES, HEPES, CHES, and CAPS. The pH values were recorded after the completion of the assays. The effects of solvent viscosity on the kinetic constants were determined at pH 8.0 using sucrose as the micro-viscogen at 25 °C. The concentrations of sucrose were 0%, 10%, 14%, 20%, 24%, and 32% (w/w), and the corresponding relative viscosities were 1, 1.3, 1.5, 1.9, 2.2, and 3.2 (15, 16). The solvent isotope effects on the kinetic parameters for URI and two mutant enzymes (D412N and R414M) were measured in 99% D2O at a pD of 8.4. The primary deuterium kinetic isotope effects were obtained by direct comparison of the kinetic constants at pH 8.0 for [2-1H]-D-glucuronate and [2-2H]-D-glucuronate.
Preparation of [2-2H]-Glucuronic Acid
[2-2H]-D-glucuronate was prepared from [2-2H]-D-glucose in three steps. The [2-2H]-D-glucose was refluxed in methanol in the presence of dry Dowex-50(H+) for 12 h to form a mixture of α- and β-methyl [2-2H]-D-glucopyranoside (17). The solvent was removed under reduced pressure to yield crystals of the pure α-anomer. Methyl [2-2H]-α-D-glucopyranoside was quantitatively oxidized at C-6 using 2,2,6,6-tetramethyl-1-piperidinyloxy/sodium bromide/sodium hypochlorite at pH 10, to form methyl [2-2H]-α-D-glucuronopyranoside (18, 19). The product was washed with methanol and purified using a column of DEAE Sephadex with a gradient of sodium bicarbonate. The fractions containing the desired product were evaporated to dryness. The methyl [2-2H]-D-glucuronopyranoside was demethylated with concentrated HCl at 4 °C and the pH adjusted to ~10 with sodium hydroxide. The concentration of [2-2H]-D-glucuronate was determined enzymatically and was produced in an overall yield of 46%. The products from each step in the synthesis were characterized by 1H and 13C NMR and mass spectrometry.
Data Analysis
The kinetic parameters, kcat and kcat/Km, for uronate isomerase with D-glucuronate as the substrate were determined by fitting the initial velocity data to equation 1 where ν is the initial velocity, Et is the total enzyme concentration, kcat is the turnover number, [A] is the substrate concentration, and Km is the Michaelis constant. The profiles for the variation of kcat or kcat/Km with pH were fit to equation 2, where c is the pH-independent value of y, Ka and Kb are the dissociation constants of the ionizable groups and H is the proton concentration. The competitive inhibition patterns were fit to equation 3, where Kis is the slope inhibition constant and I is the concentration of the inhibitor.
| (1) |
| (2) |
| (3) |
Crystallization and Data Collection
Five different crystalline complexes (Table 1) were grown by the hanging drop method at room temperature for Bh0493 from B. halodurans: (a) complex with D-arabinarate, crystal form 1; (b) complex with D-arabinarate, crystal form 2; (c) complex with arabinohydroxamate; (d) complex with D-glucuronate; and (e) complex with D-fructuronate. The initial protein solution for all 5 crystallizations contained Bh0493 (16 mg/mL) in 10 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM methionine, 10% glycerol, 1.0 mM DTT, 0.2 mM ZnCl2, and 40 mM of the corresponding substrate or inhibitor. The crystallization conditions utilized the following conditions: For the Bh0493·D-arabinarate complex (form 1), the protein solution contained 40 mM D-arabinarate and the precipitant contained 25% PEG 3350, 0.1 M Tris (pH 8.5), and 0.2 M NaCl. Crystals appeared in 4–5 days and exhibited diffraction consistent with the space group R32, with two copies of the complex per asymmetric unit. For the Bh0493·D-arabinarate complex (form 2), the protein solution contained 40 mM D-arabinarate and the precipitant contained 20% PEG 3350, and 0.2 M sodium citrate (pH 6.0). Crystals appeared in 4 days and exhibited a diffraction pattern consistent with space group C2, with 12 copies of the complex per asymmetric unit. For the Bh0493·arabinohydroxamate complex, the protein solution contained 40 mM arabinohydroxamate and the precipitant contained 25% PEG 3350, 0.1 M Tris (pH 8.5), and 0.2 M NaCl. Crystals appeared in 2 days and exhibited a diffraction pattern consistent with space group R32 with two copies of the complex per asymmetric unit. For the Bh0493·D-glucuronate complex, the protein solution contained 40 mM D-glucuronate and the precipitant contained 20% PEG 3350, and 0.2 M ammonium citrate (pH 6.0). For this complex the protein solution was incubated on ice with D-glucuronate for two hours before crystallization. The crystals appeared in 9 days and exhibited diffraction consistent with the space group C2 with 12 copies of the complex per asymmetric unit. For the Bh0493·D-fructuronate complex, the protein solution was incubated on ice for ~ two months with 40 mM D-glucuronate before crystallization. The precipitant contained 25% PEG 3350, 0.1 M Tris (pH 8.5), and 0.2 M NaCl. Crystals appeared in two weeks and exhibited diffraction consistent with the space group P4122 with three copies of the complex per asymmetric unit.
Table 1.
Data collection and refinement statistics for crystals of the complexes of uronate isomerase from Bacillus Halodurans with various ligands.
| URI·D-Arabinarate | URI·D-Arabinarate | URI·Arabinohydroxamate | URI·D-Glucuronate | URI·D-Fructuronate | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | R32 | C2 | R32 | C2 | P41 22 |
| No. of mol. in asym. unit | 2 | 12 | 2 | 12 | 3 |
| Cell dimensions | |||||
| a, b, c (Å) | 149.35,149.35,254.16 | 274.82,156.52,185.96 | 149.30, 149.30, 254.02 | 274.09,156.88,185.21 | 84.35,99.14,126.31 |
| β(°) | 116.20 | 115.78 | |||
| Resolution (Å) | 2.2 | 2.2 | 2.2 | 2.1 | 1.9 |
| No. of unique reflections | 55294 | 352288 | 55174 | 404122 | 128635 |
| Rmerge | 0.058 | 0.065 | 0.071 | 0.074 | 0.086 |
| I/σI | 27.1 | 19.3 | 20.6 | 15.2 | 17.1 |
| Completeness (%) | 99.7 | 98.8 | 99.8 | 98.7 | 93.9 |
| Refinement | |||||
| Resolution (Å) | 25.0-2.2 | 25.0-2.2 | 25.0-2.2 | 25.0-2.1 | 25.0-1.9 |
| Rcryst | 0.218 | 0.213 | 0.204 | 0.227 | 0.213 |
| Rfree | 0.238 | 0.245 | 0.226 | 0.258 | 0.241 |
| Protein atoms | 6766 | 40860 | 6744 | 40860 | 10164 |
| Waters | 259 | 1880 | 312 | 1824 | 572 |
| Rmsd, bond lengths (Å) | 0.007 | 0.006 | 0.007 | 0.006 | 0.006 |
| Rmsd, bond angles (°) | 1.3 | 1.3 | 1.3 | 1.3 | 1.3 |
| Bound inhibitor | D-Arabinarate | D-Arabinarate | Arabinohydroxamate | D-Glucuronate | D-Fructuronate |
| Inhibitor atoms | 24 | 144 | 26 | 156 | 39 |
| Bound ions | 4Zn2+, 2CO3 2−, 2Cl− | 12Zn2+,12CO32,4Na+1,4Cl− | 4Zn2+, 2CO32−, 2Cl− | 12Zn2+, 12CO32−,4Cl− | 4Zn2+, 3CO32−, 1Cl − |
| PDB entry | 3HK5 | 3HK7 | 3HK8 | 3HK9 | 3HKA |
Prior to data collection, the crystals of all Bh0493 complexes (Table 1) were transferred to cryoprotectant solutions composed of their mother liquids and 20% glycerol and flash-cooled in a nitrogen stream. All data sets were collected at the NSLS X4A beamline (Brookhaven National Laboratory) on an ADSC CCD detector (Table 1). Diffraction intensities were integrated and scaled with programs DENZO and SCALEPACK (20). The data collection statistics are given in Table 1.
Structure Determination and Model Refinement
All five URI structures (Table 1) were determined by molecular replacement with the fully automated molecular replacement pipeline BALBES (21), using only input diffraction and sequence data. The native uronate isomerase from B. halodurans (PDB ID: 2Q08) was used by BALBES as a template in all five structure determinations. Partially refined structures of all URI crystal forms (Table 1) were output from BALBES without manual intervention. Several iterative cycles of refinement were performed for each crystal form including: manual model rebuilding with TOM (22), refinement with CNS (23), automatic model rebuilding with ARP (24), and solvent building with the CCP4 suite (25).
The rhombohedral crystal form of the complex Bh0493·D-arabinarate contains 2 copies of the complex in the asymmetric unit of the cell; the monoclinic crystal form of the same complex contains 12 copies of the complex in the asymmetric unit packed as four trimers. The first residue and last 13 residues of every molecule are disordered in the first crystal form of this complex. The four N-terminal residues and 14 last residues are disordered in every molecule of the second crystal form of the Bh0493·D-arabinarate complex. The disordered residues are not included in the final models. The asymmetric unit of the crystalline complex Bh0493·arabinohydroxamate contains two molecules of the complex where the first residue and the last 14 residues of every molecule are disordered. The asymmetric unit of the crystalline complex Bh0493·D-glucuronate contains 12 copies of the complex packed as four trimers. The first residue and the last 13 residues are disordered in every molecule of this complex. The asymmetric unit of the crystalline complex Bh0493·D-fructuronate contains one trimer of the complex. The first two residues and last 14 residues are disordered in every molecule and are not included in the final models. This complex was produced by a long incubation and subsequent co-crystallization of Bh0493 with D-glucuronate but the electron density of the bound inhibitor can be interpreted only as the product, D-fructuronate. The Zn2+ ions bound in the active sites were clearly visible in every molecule of every URI complexes shown in Table 1. Additional ions (Na+, Zn2+, and Cl−) located on local 3-fold axis of every Bh0493 trimer also exhibited good density in all five URI crystalline complexes. Final crystallographic refinement statistics for all of the URI complexes are provided in the Table 1.
Results
Requirement for Divalent Cation
The importance of a metal ion for the catalytic activity of uronate isomerase was reinvestigated. The apo-enzyme was prepared and subsequently tested for enzymatic activity using D-glucuronate as the substrate. The wild-type URI from E. coli was found to contain 0.9 equivalents of zinc after purification. This protein (3 mL) at a concentration of 3.0 mg/mL was dialyzed against 1 L of dialysis buffer containing 20 mM dipicolinate in 50 mM MES, pH 6.0. The buffer was changed three times over the course of 48 h, after which the catalytic activity and metal content of the enzyme were determined. The chelator affectively removed more than 98% of the bound zinc as indicated by ICP-MS. The activity of the enzyme was assayed with mannonate dehydrogenase and NADH to detect the formation of D-fructuronate in the presence of 10 μM dipicolinate. The apo-enzyme exhibited less than 1% activity of the native enzyme with bound zinc. This result is at variance with our previous report which had concluded that the activity of uronate isomerase is independent of the presence or absence of divalent cations within the active site (12). The reason for this difference in results has not been determined but was perhaps due to contaminating divalent cations in the assay solution.
Inhibition by 2,6-Anhydro-L-Gulonic Acid (IV)
Compound IV was synthesized as a cyclic analogue mimic of the pyranose form of D-glucuronate. The inhibitory properties of IV were determined with the wild-type uronate isomerase from E. coli and B. halodurans (Bh0493). This compound was found to be a competitive inhibitor for both enzymes. The data were fit to equation 3 and the values of Kis were determined to be 45 ± 4 and 24 ± 2 μM for the URI from E. coli and Bh0493, respectively.
pH-Rate Profiles
The kinetic constants for the conversion of D-glucuronate to D-fructuronate were obtained as a function of pH. The pH-rate profiles for the effects of pH on kcat and kcat/Km are presented in Figures 1A and 1B, respectively. The pH profiles are bell-shaped and are consistent with a single functional group that must be unprotonated for activity and another functional group that must be protonated for catalytic activity. From a fit of the data to equation 2 the kinetic pKa values from the kcat/Km plot are 5.5 ± 0.1 and 9.5 ± 0.1, respectively. From the plot of kcat vs. pH, kinetic pKa values of 5.8 ± 0.1 and 10.2 ± 0.1 were obtained.
Figure 1.

pH-rate profile for the wild type uronate isomerase from E. coli containing 1 equivalent of zinc. The data were fit to equation 2. (A) Plot of log kcat vs. pH. (B) Plot of log kcat/Km vs. pH.
Site-Directed Mutants
Site-directed mutagenesis was utilized to identify the involvement of specific amino acids in metal binding, substrate recognition, and the catalytic mechanism of uronate isomerase. Conserved residues were chosen based on the location within the active site of Bh0493, a uronate isomerase found in B. halodurans. His-33 and His-35 were mutated to investigate the importance of metal binding and the potential role of the divalent cation on catalytic activity. Mutations at either of these two resides resulted in the dramatic loss of affinity for the divalent cation and a significant reduction in the catalytic activity. The diminution of catalytic activity for the mutants is more severe when these histidine residues are changed to alanine than to asparagine. The highly conserved histidine at the end of β-strand 5 (His-297) and the invariant aspartate at the end of β-strand 8 (Asp-412) were mutated to asparagine and alanine. For the mutations at His-297 there were substantial increases in the Michaelis constant. In contrast, with the mutation of Asp-412 the reduction in catalytic activity was more pronounced on kcat. These four residues are broadly conserved in all members of the amidohydrolase superfamily.
Additional residues that are conserved among all of the known uronate isomerases were mutated as a probe of functional participation in binding and catalysis. These residues include Trp-381 from the conserved WWF motif after β-strand 7, His-59 and Tyr-60 after β-strand 1, and three conserved arginines (Arg-186, Arg-302, and Arg-414). Mutation of residues Trp-381 and and Arg-302 resulted in increases in the value of Km and small changes in kcat, indicating that these residues most likely take part in substrate recognition and binding. For the methionine mutation at Arg-186 there were significant changes in both kcat and kcat/Km. For His-59 and Arg-414 there were relatively small changes in Km but drastic reductions in the value of kcat. The kinetic constants for the mutants constructed for this investigation are presented in Table 2.
Table 2.
Kinetic parameters and metal content of mutants of URI from E. colia
| Enzyme | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | URI/Zn |
|---|---|---|---|---|
| WT | 196 ± 6 | 0.50 ± 0.05 | (4.0 ± 0.4) × 105 | 0.90 |
| H33N (H26)b | 2.1 ± 0.1 | (5.0 ± 0.4) × 10−2 | (4.7 ± 0.4) × 104 | 0.07 |
| H33A | 0.60 ± 0.01 | 0.20 ± 0.01 | (3.0 ± 0.2) × 103 | 0.20 |
| H35N (H28) | 4.0 ± 0.2 | 9.4 ± 1.1 | (4.3 ± 0.5) × 102 | <0.05 |
| H35A | 0.70 ± 0.04 | 39 ± 5 | 18 ± 2 | <0.05 |
| H59N (H49) | 15 ± 1 | 0.70 ± 0.04 | (2.1 ± 0.1) × 104 | 0.96 |
| H59A | 0.60 ± 0.01 | 0.70 ± 0.07 | (8.3 ± 0.1 × 102 | 0.95 |
| Y60F (Y50) | 21.7 ± 0.1 | 0.16 ± 0.01 | (1.4 ± 0.1) × 105 | 0.8 |
| Y60A | 13.9 ± 0.1 | 0.21 ± 0.01 | (6.6 ± 0.3) × 104 | 0.8 |
| R186K (R170) | 54 ± 2 | 2.6 ± 0.2 | (2.1 ± 0.2) × 104 | 0.94 |
| R186M | 4.7 ± 0.1 | 38 ± 3 | (1.3 ± 0.1) × 102 | 0.91 |
| D238N | 60 ± 1 | 1.3 ± 0.1 | (4.6 ±0.1) × 104 | 0.70 |
| H297N (M258) | 30 ± 2 | 56 ± 5 | (5.0 ± 0.5) × 102 | 1.00 |
| H297A | 10 ± 1 | (2.2 ± 0.3) × 102 | 43 ± 7 | 0.41 |
| R302K (K303) | 160 ± 4 | 2.5 ± 0.2 | (6.3 ± 0.5) × 104 | 0.90 |
| R302M | 180 ± 9 | (2.0 ± 0.3) × 102 | (8.8 ± 1.3) × 102 | 0.99 |
| W381F (W325) | 16 ± 1 | 1.7 ± 0.1 | (9.5 ± 0.4) × 103 | 0.90 |
| W381A | 250 ± 6 | 21± 2 | (1.2 ± 0.1) × 104 | 0.69 |
| D412N (D355) | 0.60 ± 0.01 | 1.00 ± 0.04 | (6.0 ± 0.3) × 102 | 0.36 |
| D412A | (9.0 ± 0.3) × 10−3 | 0.40 ± 0.05 | 21 ± 3 | 0.12 |
| R414K (R357) | 5.8 ± 0.1 | 0.82 ± 0.02 | (7.1 ± 0.2) × 103 | 0.92 |
| R414M | 0.70 ± 0.01 | 1.4 ± 0.1 | (5.4 ± 0.2) × 102 | 0.91 |
These data were obtained at 30 °C, pH 8.0, with D-glucuronate as the substrate.
The corresponding residue numbers for Bh0493.
Kinetic Isotope Effects
Primary deuterium kinetic isotope effects on the isomerization of D-glucuronate were measured as a probe of the rate limiting steps in the overall reaction mechanism. For the solvent deuterium isotope effects, the kinetic parameters were obtained for the wild type enzyme in H2O and D2O with D-glucuronate as the substrate. The double reciprocal plots are presented in Figure 2. The solvent isotope effects for D2Okcat and D2O(kcat/Km) are 1.22 ± 0.02 and 1.10 ± 0.09, respectively. Solvent isotope effects were also determined for four of the site directed mutants, H59N, Y60F, D412N and R414M. The solvent isotope effects for the H59N mutant were 1.7 ± 0.1 for D2Okcat and 1.4 ± 0.1 for D2O(kcat/Km), while the effects for the Y60F mutant were 2.1 ± 0.1 and 1.2 ± 0.1 for D2Okcat and D2O(kcat/Km), respectively. For the D412N mutant, the solvent isotope effects were determined to be 1.3 ± 0.1 and 1.5 ± 0.2 for D2Okcat and D2O(kcat/Km), respectively. For the R414M mutant, the solvent isotope effects were 1.8 ± 0.1 for D2Okcat and 2.0 ± 0.3 for D2O(kcat/Km).
Figure 2.

Double-reciprocal plots for the solvent isotope effect (H2O vs D2O) where 1/v (s) is plotted against 1/[D-glucuronate] (mM−1). The data sets in H2O are represented by circles and the squares correspond to the values obtained in D2O. (A) represents the plot for the wild type enzyme, (B) for the R414M mutant, and (C) is the plot for the D412N mutant.
The primary deuterium isotope effects for abstraction of the proton from C-2 of D-glucuronate were determined for the wild type enzyme and two mutants, D412N and R414M. The double-reciprocal plots are shown in Figure 3. For the wild-type enzyme the primary deuterium isotope effects on kcat and kcat/Km were determined to be 1.4 ± 0.1 and 1.2 ± 0.1, respectively. For the D412N mutant, the primary deuterium isotope effects were determined to be 2.0 ± 0.2 for Dkcat and 1.9 ± 0.3 for D(kcat/Km). For the R414M mutant, the isotope effect on kcat was 3.2 ± 0.1 and the effect on kcat/Km was 3.5 ± 0.4.
Figure 3.

Primary isotope effects using protonated and deuterated D-glucuronate at the C-2 position are presented as double-reciprocal plots where (A) is the plot for the wild-type enzyme, (B) R414M mutant, and (C) D412N mutant. The data for the protonated substrate are represented as circles, and the values for the deuterated substrate are denoted as squares.
Solvent Viscosity Effects
Alterations in solvent viscosity were utilized to probe the degree of rate limitation by the binding and dissociation of products and substrates on the kinetic constants of uronate isomerase (15). The effects of changes in solvent viscosity on kcat and kcat/Km were made by the addition of sucrose (26). A plot of okcat/ηkcat versus the relative solvent viscosity for the wild-type enzyme exhibits a slope of 0.72 ± 0.08. The slope for the effect of solvent viscosity on kcat/Km is 0.64 ± 0.05 for the wild-type enzyme. For the two mutant enzymes, D412N and R414M, the slopes for the effect of solvent viscosity on kcat were found to be 0.07 ± 0.05 and 0.05 ± 0.01, respectively. For the effect on kcat/Km the slopes were 0.03 ± 0.05 and −0.17 ± 0.02, respectively, with the D412N and R414M mutant enzymes. The kinetic data are presented in Figures 4A and 4B.
Figure 4.

The effect of viscosity on the relative values of kcat (A) and kcat/Km (B) using sucrose as the microviscogen. The circles indicate the wild type enzyme from E. coli, the squares represent the data set for the D412N mutant, and the triangles signify the values for the R414M mutant.
Structures of Inhibitor Complexes
The crystal structure of Bh0493 from B. halodurans was previously determined in the absence of bound substrates or inhibitors (PDB code: 2Q08, 3). In that structure zinc is bound in the active site and coordinated to two histidines from β-strand 1 (H26 and H28) and the invariant aspartate found at the end of β-strand 8 (D355). The structure of Bh0493 was determined in the presence of the substrates D-glucuronate (I), D-fructuronate, and two inhibitors that mimic the proposed cis-enediol(ate) intermediate, D-arabinarate (III) and its hydroxamate derivative (II). Portions of the electron density maps that show the D-glucuronate and D-fructuronate in the active site of Bh0493 are presented in Figures 5a and 5b. These compounds occupy the active site in a similar manner and interact with the same set of amino acid residues. Stereoscopic images of the active site complexes for compounds I, II, and III are presented in Figures 6A–C and the distances between specific amino acid residues and the bound ligands are provided in Figures 7A–C.
Figure 5.
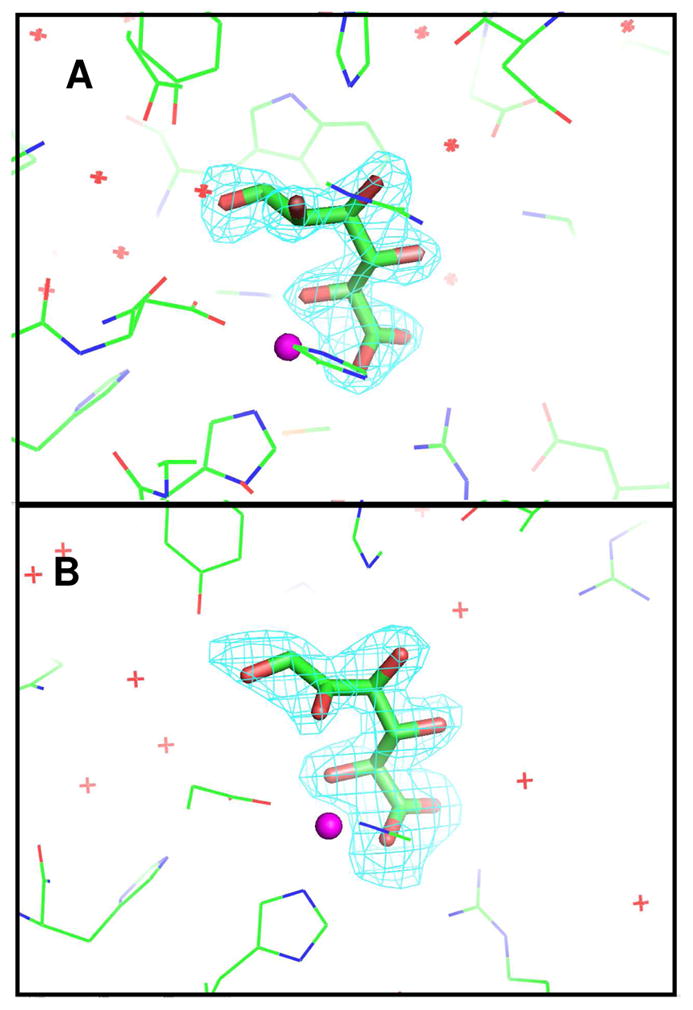
View of the active site structures of Bh0493 with bound ligands: (A) D-glucuronate; (B) D-fructuronate. Omit electron density maps (Fo−Fc) are contoured at 3.5σ. The corresponding ligands were omitted from the model and the remainder of the unit cell was subjected to a cycle of simulated annealing with CNS at 3000 °C.
Figure 6.

Stereoview images of Bh0493 in the presence of bound (A) D-glucuronate (I), (B) hydroxamate of arabinarate (II), and (C) arabinarate (III). This figure was created with PyMOL v0.99.
Figure 7.
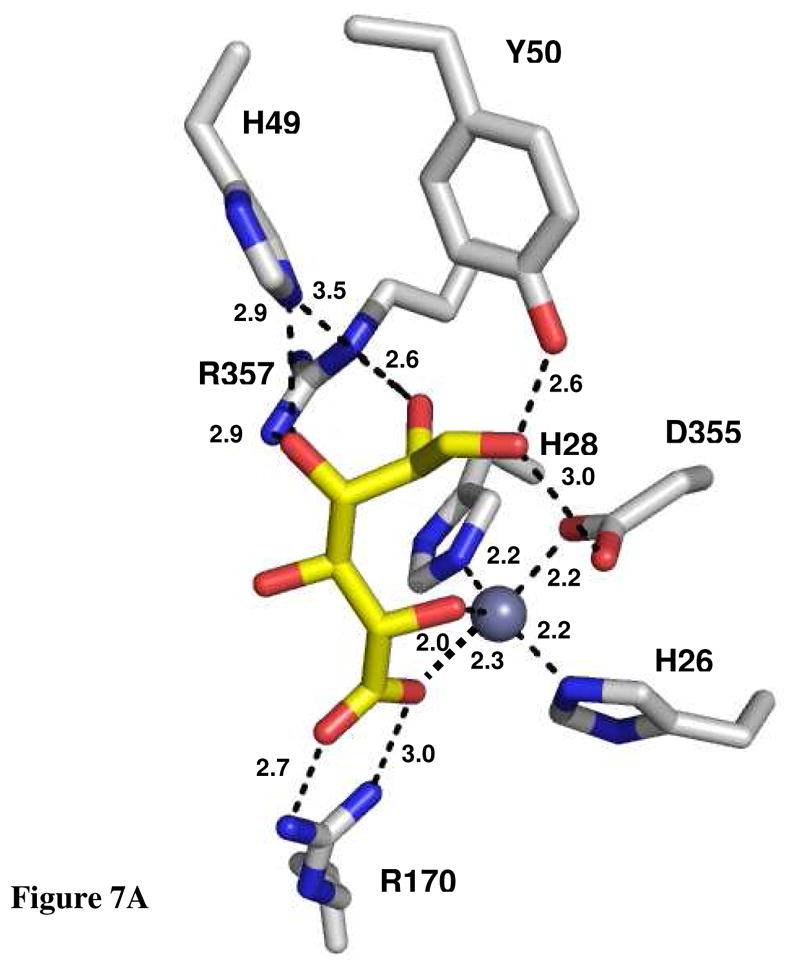
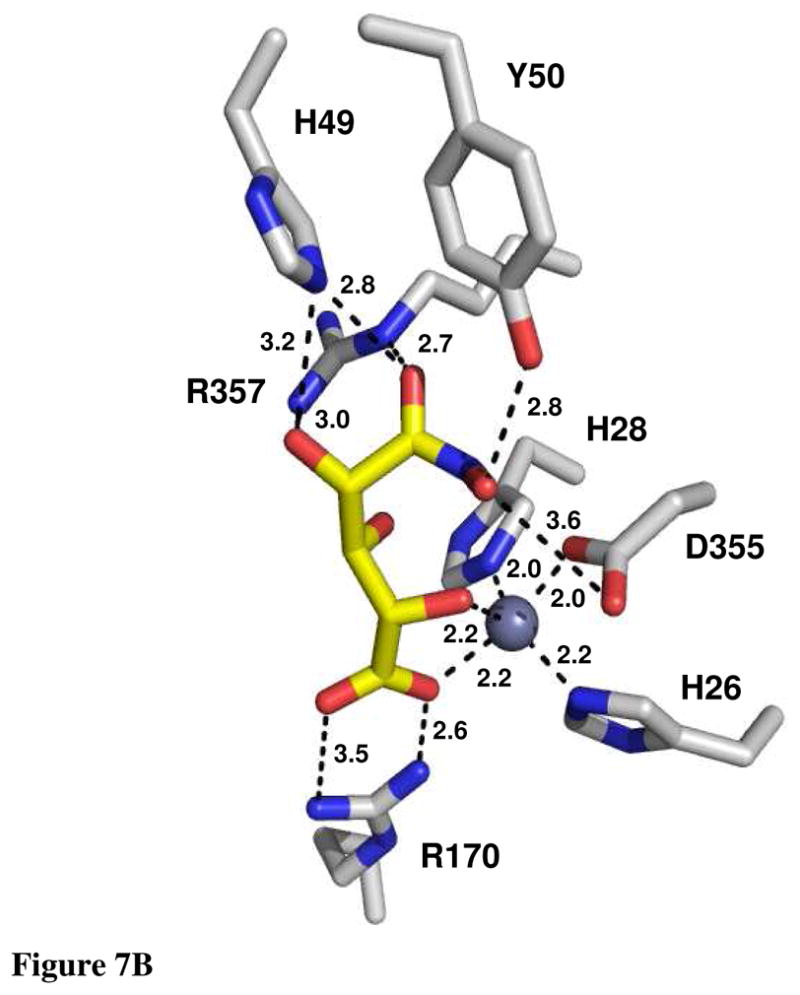
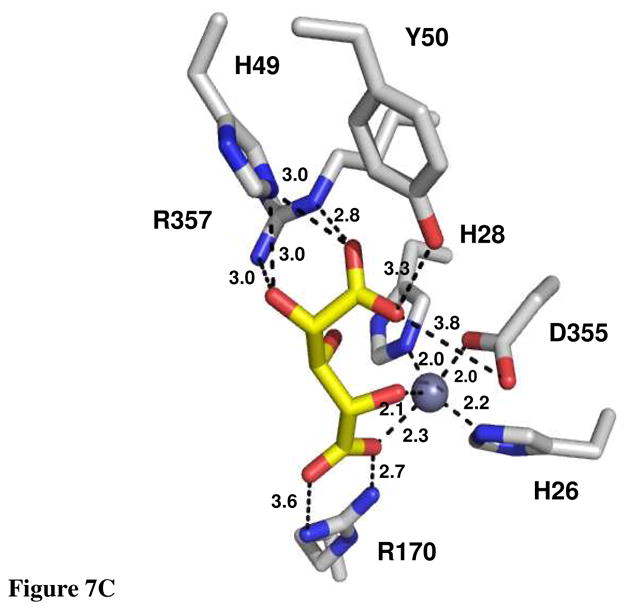
Active site of Bh0493 with (A) D-glucuronate, (B) hydroxamate of D-arabinarate, and (C) D-arabinarate. Interactions between the enzyme and ligand with the distances are shown.
In the complex with D-glucuronate, the substrate is bound in the open chain configuration. The terminal carboxylate is ion-paired with the guanidino group of Arg-170 and there is a monodentate coordination with the bound zinc. The zinc is also ligated with the hydroxyl group attached to C-5 of the substrate. The hydroxyl group at C-4 does not make any specific interactions with the protein, which is consistent with the observation that both D-glucuronate and D-galacturonate are substrates for this enzyme (1). The hydroxyl at C-3 interacts with both Arg-357 and His-49. The nearest residue to the hydroxyl at C-2 of D-glucuronate is Arg-357. The hydroxyl group from Tyr-50 hydrogen bonds with the carbonyl group at C-1 of the substrate. The closest residue to the hydrogen that is abstracted from C-2 of D-glucuronate is the side chain carboxylate of Asp-355. Similar interactions are found in the complexes with D-fructuronate, D-arabinarate and the corresponding hydroxamate derivative. The D-fructuronate complex was identified in a crystal that was grown after a long incubation and subsequent co-crystallization of Bh0493 with D-glucuronate.
Discussion
The uronate isomerase from E. coli has previously been shown to catalyze the isomerization of D-glucuronate and D-galacturonate to D-fructuronate and D-tagaturonate, respectively (1, 2). With D-glucuronate the hydrogen at C-2 was shown to be transferred to the pro-R position at C-1 of the product (12). This hydrogen exchanges with solvent at a rate that is 4 orders of magnitude slower than the net interconversion of substrate and product. These results were interpreted to be consistent with a reaction mechanism that was initiated by proton abstraction at C-2, formation of a cis-enediol(ate) intermediate and subsequent reprotonation at C-1 (12). This minimal reaction mechanism thus requires at least two different residues within the active for these proton transfers. A general base (B1:) is required for the removal of the proton at C-2 and delivery to C-1 and a general acid (B2:H) is needed to shuttle a proton between the oxygens attached to C-1 and C-2 of the substrate/product pair. This transformation has been summarized in Scheme 2. A requirement for a minimum of two amino acids that must be in a specific state of protonation is experimentally supported by the measurement of the effects of pH on the magnitude of kcat and kcat/Km for the conversion of D-glucuronate to D-fructuronate. The pH-rate profiles for both kcat and kcat/Km are bell-shaped and indicate that one residue must be unprotonated and another protonated for catalytic activity. In these profiles the general base has a kinetic pKa between 5.5 and 5.8 whereas the general acid has a kinetic pKa between 9.5 and 10.2. Candidates for these residues were identified through the elucidation of the three-dimensional crystal structure of a uronate isomerase from B. halodurans (Bh0493) in the presence of D-glucuronate, D-fructuronate, and two mimics of the cis-enediol(ate) intermediate.
The crystal structure of Bh0493 determined with D-glucuronate has identified those residues in the active site that interact directly with the substrate. In this structure the zinc is ligated by three amino acids from the protein: the two conserved histidine residues from β-strand 1 and the aspartate from β-strand 8. The C-6 carboxylate group of the substrate is ligated to the zinc and also ion-paired with Arg-170. The hydroxyl group from C-5 is ligated to the zinc in the active site and hydrogen bonded to the aspartate from β-strand 8. At the other end of the substrate, Arg-357 hydrogen bonds with the two hydroxyls from C-3 and C-2. The phenolic group of Tyr-50 forms a hydrogen bond with the carbonyl group at C1. The closest residue from the protein to the hydrogen at C-2 that must be abstracted during the chemical transformation is Asp-355 at 3.15 Å. In addition, His-49 is within hydrogen bonding distance to the hydroxyl at C-2 in the hydroxamate inhibitor (II) but not in the complex with the bound D-glucuronate (I) Thus, the most likely residues that are required for the isomerization reaction (in Bh0493) are His-49, Tyr-50, Asp-355 and Arg-357. In the E. coli enzyme these residues are equivalent to His-59, Tyr-60, Asp-412, and Arg-414, respectively. These residues, in addition to His-33 and His-35 (ligands to the zinc) Arg-186 (equivalent to R170 in Bh0493), His-297 (a conserved histidine at the end of β-stand 5 in most members of the amidohydrolase superfamily), Arg-302 and Trp-381, were mutated as probes of functional significance.
The mutation of specific residues within the active site of URI results in significant perturbations to the magnitude of the kinetic constants for substrate turnover. Changes to either of the two histidine residues that originate from the end of β-strand 1 weakens the binding of zinc to the active site and this results in a diminution of catalytic activity. This observation is consistent with the proposed role of zinc in the direct ligation of the substrate through the C-6 carboxylate and the hydroxyl from C-5. A drastic reduction in the affinity of the substrate occurs with the mutation of Arg-186 to methionine. In this case the Km for the substrate increases by nearly two orders of magnitude and the value of kcat/Km is reduced by more than three orders of magnitude. This result is consistent with an ion-pair interaction between the C-6 carboxylate and the guanidino group of Arg-186 (R170 in Bh0493) that is observed in the X-ray structure of Bh0493. There is also a substantial increase in the Km for D-glucuronate when His-297 is mutated to alanine or asparagine. This residue originates from the end of β-strand 5 and is highly conserved in nearly all members of the amidohydrolase superfamily but this residue is not conserved in Bh0493 and thus it is not easy to discern the effect on the structure of URI from E. coli.
The most dramatic reductions in kcat occur with the mutation of Asp-412 to alanine or Arg-414 to methionine. Arg-414 is equivalent to Arg-357 from Bh0493, which is hydrogen bonded to the C-2 and C-3 hydroxyls of the bound D-glucuronate. Therefore, this residue is a suitable candidate for assisting in the movement of the proton from the hydroxyl at C-2 during the transformation to D-fructuronate. The other candidate for this process is the phenolic side chain from Tyr-60. This group is hydrogen bonded to the carbonyl oxygen at C-1 of D-glucuronate in the X-ray crystal structure. Mutation of Tyr-60 to phenylalanine results in reductions in kcat and kcat/Km of about an order of magnitude. The only residue from the protein that is suitably positioned to function as the general base for the abstraction of the proton from C-2 and delivery to C-1 is Asp-412. Many members of the amidohydrolase superfamily that catalyze hydrolytic reactions have been shown to use this residue from the end of β-strand 8 to abstract a proton from the hydrolytic hydroxide or water (6, 8, 27, 28).
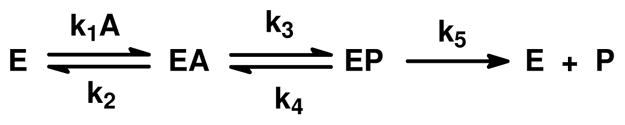
Changes in solvent viscosity and isotopic substitution were used to address the degree of rate limitation on the bond breaking steps and product release (15, 16). A simplified kinetic mechanism for the conversion of substrate to product for the uronate isomerization reaction is presented in Scheme 4, where EA represents the enzyme-glucuronate complex and EP represents the enzyme-fructuronate complex. In this mechanism the expression for kcat is given by (k3k5)/(k3 + k4 + k5). If one assumes that k5 is inversely proportional to the relative solvent viscosity then the value of k5, relative to the sum of k3 and k4 can be determined from a plot of the ratio of o(kcat)/η(kcat) as a function of the relative solvent viscosity, η. The slope of this plot is equal to (k3+ k4)/(k3 + k4 + k5). For the wild-type enzyme the slope was found to be ~0.7 and thus the sum of k3 and k4 is greater than the product release step, k5. This result is consistent with the release of the product as the rate limiting step for the wild type enzyme and the relatively small primary isotpe effect for [2-2H]-D-glucuronate. With the D412N and R412M mutants the slope of this plot, for changes in the relative value of kcat as a function of solvent viscosity, is reduced substantially. This result is consistent with a significant reduction in the rate constants for the interconversion of the substrate/product pair to the point where k5 is now greater than the sum of k3 and k4. This result is also consistent with the significant increase in the value of Dkcat. With the D412N and R414M mutants, the primary isotope effects are 2.0 and 3.2, respectively, and thus with these two mutants the interconversion of the substrate/product pair is substantially rate limiting.
Scheme 4.
Mechanism of Action
Based upon the X-ray crystal structure of Bh0493 in the presence of the bound substrate, the catalytic properties of selected site-directed mutants and the stereochemical constraints for the conversion of D-glucuronate to D-fructuronate, a minimal chemical mechanism can be written for uronate isomerase. In the proposed mechanism for URI from E. coli, D-glucuronate is bound in the active site through electrostatic interactions to five highly conserved amino acid residues and the divalent cation. The carboxylate group at C-6 is coordinated to the divalent cation and Arg-186. The hydroxyl group at C-5 is also coordinated to the zinc. The hydroxyl groups at C-3 and C-2 interact with the side chain guanidino group of Arg-414 and the hydroxyl at C-2 is also apparently able to hydrogen bond to His-59. The carbonyl group at C-1 is hydrogen bonded to the phenolic oxygen of Tyr-60. The pH-rate profiles for URI are consistent with two amino acid residues that must be in a specific state of protonation for catalytic activity. The general base, with a kinetic pKa of approximately 5.8, is consistent with Asp-412. This residue is conserved in all members of the amidohydrolase superfamily and for those enzymes that catalyze hydrolytic reactions it has been shown to initiate proton transfers from water/hydroxide to the leaving group (6, 8, 27, 28). The general acid, with a kinetic pKa of approximately 10.2 from the pH-rate profiles, may be due to Tyr-60 but it is difficult to exclude a role for Arg-414.
In the simplest mechanism D-glucuronate binds in the open chain conformation in the active site and then Asp-412 abstracts the proton from C-2 as the carbonyl group of C-1 is protonated by Tyr-60 to form a cis-enediol intermediate. In the subsequent step, Tyr-60 abstracts the proton from the hydroxyl at C-2 as Asp-414 delivers a proton to C-1 with proR stereochemistry. It should be noted that the mutation of Tyr-60 to alanine or phenylalanine diminishes kcat by only a factor of 10. This reduction in rate is perhaps smaller than what may be expected for this role in catalysis. However, the lack of a primary deuterium isotope effect for the wild-type enzyme indicates that the chemical step is not rate-limiting. In addition, a water molecule may substitute for the phenolic group in the mutant enzymes. Additional mechanisms can be written that utilize a combination of Tyr-60, Arg-414, and His-59 to facilitate the proton movements between the oxygens at C-2 and C-1 within the cis-enediol intermediate and in the opening of the hemiacetal. The proposed reaction mechanism for URI is different from that of metallo-enzyme xylose isomerase since that enzyme catalyzes a hydride transfer mechanism rather than a proton transfer mechanism (29).
An alternative mechanism can be proposed in which Asp-412 abstracts the proton from the hydroxyl at C-5, which then abstracts a proton from C-2 to initiate the formation of the cis-enediol intermediate. The hydroxyl at C-5 is additionally activated through direct ligation to the bound zinc. These two variations are presented graphically in Figure 8. This latter mechanism is particularly attractive since it retains elements that are common to nearly all members of the amidohydrolase superfamily that have been interrogated mechanistically (6, 8, 27, 28). The structural similarities in the active site of uronate isomerase with those members of the amidohydrolase superfamily nicely illustrates the evolutionary link between those members of the AHS that catalyze hydrolytic reactions and those that catalyze 1,2-proton transfers. A structural alignment of the active sites of Bh0493/D-glucuronate and dihydroorotase (DHO)/dihydroorotate (PDB entry code 1j79) supports this proposition as illustrated in Figure 9. In this structural alignment one of the C-6 carboxylate oxygens from D-glucuronate is positioned in nearly the same place as the carboxylate oxygen of the bridging carbamate functional group in DHO. These oxygen atoms interact directly with the alpha-metal (Mα) in their respective structures. Moreover, the C-5 oxygen of D-glucuronate is positioned in the same way as the nucleophilic hydroxide in DHO and is oriented to favor proton abstraction by Asp-412 at a distance of 2.9 Å. The C-5 hydroxyl is 2.0 Å away from the hydrogen at C-2 of the bound substrate.
Figure 8.

Proposed mechanisms for the isomerization of D-glucuronate by URI.
Figure 9.
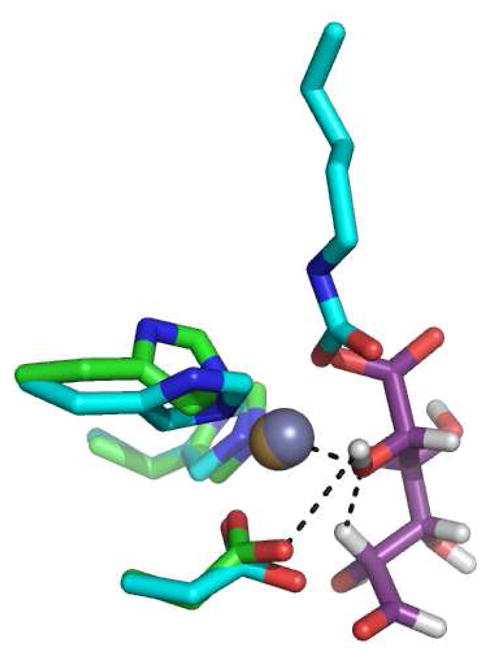
A structural alignment for portions of the active sites of Bh0493 (green) and DHO from E. coli (blue). The alpha metal (Mα) is shown as a yellow sphere for Bh0493 and in grey for DHO. The bound D-glucuronate for Bh0493, is shown in purple. The interactions between the C-5 oxygen of D-glucuronate and Mα, C-2 hydrogen and Asp-412 are indicated by dashed lines. The other ligands to the metal ion are shown as stick representations, including the HxH from β-strand 1 and aspartate from β-strand 8. The C-6 oxygen of D-glucuronate, aligns closely the the position occupied by one of the oxygens from the carboxyl group of the carboxylated lysine from strand 4 of DHO. The C-5 hydroxyl of D-glucuronate is orientated in the same position as the bridging hydroxide in the DHO structure.
Footnotes
This work was supported in part by the NIH (GM71790) and the Robert A. Welch Foundation (A-840). The X-ray coordinates and structure factors for Bh0493 have been deposited in the Protein Data Bank (PDB accession codes: 3HK5, 3HK7, 3HK8, 3HK9, and 3HKA)
Abbreviations: URI, uronate isomerase; KDG, 2-keto-3-deoxy-D-gluconic acid; KDG-6-P, 2-keto-3-deoxy-6-phospho-D-gluconic acid; MDH, mannonate dehydrogenase, ICP-MS, inductively coupled plasma mass spectrometry.
References
- 1.Ashwell G, Wahba AJ, Hickman J. Uronic acid metabolism in bacteria. I. Purification and properties of uronic acid isomerases in Escherichia coli. J Biol Chem. 1960;235:1559–1565. [PubMed] [Google Scholar]
- 2.Wahba AJ, Hickman J, Ashwell G. Uronic acid metabolism in bacteria. III. Purification and properties of D-altronic acid and D-mannonic acid dehydrogenases in Escherichia coli. J Biol Chem. 1960;235:1566–1570. [PubMed] [Google Scholar]
- 3.Nguyen TT, Brown S, Fedorov AA, Fedorov EV, Babbitt PC, Almo SC, Raushel FM. At the periphery of the amidohydrolase superfamily: Bh0493 from Bacillus halodurans catalyzes the isomerization of D-galacturonate to D-tagaturonate. Biochemistry. 2008;47:1194–1206. doi: 10.1021/bi7017738. [DOI] [PubMed] [Google Scholar]
- 4.Siebert CM, Raushel FM. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 5.Holm L, Sander C. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins. 1997;28:72–82. [PubMed] [Google Scholar]
- 6.Porter TN, Li Y, Raushel FM. Mechanism for the dihydroorotase reaction. Biochemistry. 2004;43:16285–16292. doi: 10.1021/bi048308g. [DOI] [PubMed] [Google Scholar]
- 7.Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. A new proposal for urease mechanism based on the crystal structures of the native and inhibited enzyme from Bacillus pasteurii: Why urea hydrolysis costs two nickels. Struct Folding Des. 1999;7:205–216. doi: 10.1016/S0969-2126(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 8.Aubert SA, Li Y, Raushel FM. Mechanism for the hydrolysis of organophosphates by the bacterial phosphotriesterase. Biochemistry. 2004;43:5707–5715. doi: 10.1021/bi0497805. [DOI] [PubMed] [Google Scholar]
- 9.Wilson DK, Rudolph FB, Quiocho FA. Atomic structure of adenosine deaminase complexed with a transition state analog: Understanding catalysis and immunodeficiency mutation. Science. 1991;252:1278–1284. doi: 10.1126/science.1925539. [DOI] [PubMed] [Google Scholar]
- 10.Ireton GC, McDermott G, Black ME, Stoddard BL. The structure of Escherichia coli cytosine deaminase. J Mol Biol. 2002;315:687–697. doi: 10.1006/jmbi.2001.5277. [DOI] [PubMed] [Google Scholar]
- 11.Roy JE, Roy KL. The mechanism and specificity of guanine deaminase. Can J Biochem. 1967;45:1263–1269. doi: 10.1139/o67-147. [DOI] [PubMed] [Google Scholar]
- 12.Williams L, Nguyen T, Li Y, Porter TN, Raushel FM. Uronate isomerase: A nonhydrolytic member of the amidohydrolase superfamily with an ambivalent requirement for a divalent metal ion. Biochemistry. 2006;45:7453–7462. doi: 10.1021/bi060531l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sowden JC, Oftedahl ML. Anhydridization of 1-deoxy-1-nitrohexitols. J Org Chem. 1961;26:1974–1977. [Google Scholar]
- 14.Dromowicz M, Koll P. Synthesis of 2,6-anhydroaldonic acids from the corresponding anhydrodeoxynitroalditols (glycopyranosylnitromethanes) and their conversion into methyl esters, amides, and alditols. Carbohydrate Research. 1998;311:103–119. [Google Scholar]
- 15.Brouwer AC, Kirsch JF. Investigation of diffusion-limited rates of chymotrypsin reactions by viscosity variation. Biochemistry. 1982;21:1302–1307. doi: 10.1021/bi00535a030. [DOI] [PubMed] [Google Scholar]
- 16.Maurice M, Bearne SL. Kinetics and thermodynamics of mandalate racemase catalysis. Biochemistry. 2002;41:4048–4058. doi: 10.1021/bi016044h. [DOI] [PubMed] [Google Scholar]
- 17.Podlasek CA, Wu J, Atripe WA, Bondo PB, Serianni AS. [13C]-Enriched methyl aldopyranosides: Structural interpretations of 13C-1H spin-coupling constants and 1H chemical shifts. J Am Chem Soc. 1995;117:8635–8644. [Google Scholar]
- 18.de Nooy AEJ, Besemer AC, van Bekkum H. Highly selective nitroxyl radical-mediated oxidation of primary alcohol groups in water-soluble glucans. Carbohydrate Research. 1995;269:89–98. [Google Scholar]
- 19.Bragd PL, van Bekkum H, Besemer AC. TEMPO-mediated oxidation of polysaccharides: survey of methods and applications. Topics in Catalysis. 2004;27:49–66. [Google Scholar]
- 20.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CWJ, Sweet RM, Abelson JN, Simon MI, editors. Methods in Enzymology. Academic Pres; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 21.Long F, Vagin A, Young P, Murshudov GN. BALBES: a Molecular Replacement Pipeline. Acta Crystallogr D. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones AT. Interactive computer graphics: FRODO. Methods Enzymol. 1985;115:157–171. doi: 10.1016/0076-6879(85)15014-7. [DOI] [PubMed] [Google Scholar]
- 23.Brunger AT, Adams PD, Clore GM, DeLAno WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54 doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 24.Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Crystallogr. 1993;D49:129–147. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 25.Collaborative Computational Project No. 4. The CCP4 suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 26.Hardy LW, Kirsch JK. Diffusion-limited component of reactions catalyzed by Bacillus cereus β-lactamases I. Biochemistry. 1984;23:1275–1282. doi: 10.1021/bi00301a040. [DOI] [PubMed] [Google Scholar]
- 27.Hall RS, Xiang DF, Xu C, Raushel FM. N-acetyl-D-glucosamine-6-phosphate deacetylase: Substrate activate via a single divalent metal ion. Biochemistry. 2007;46:7942–7952. doi: 10.1021/bi700543x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marti-Arbona R, Fresquet V, Thoden JB, Davis ML, Holden HM, Raushel FM. Mechanism of the reaction catalyzed by isoaspartyl dipeptidase from Escherichia coli. Biochemistry. 2005;44:7115–7124. doi: 10.1021/bi050008r. [DOI] [PubMed] [Google Scholar]
- 29.Allen KN, Lavie A, Farber GK, Glasfeld A, Petsko GA, Ringe D. Isotopic exchange plus substrate and inhibition kinetics of D-xylose isomerase do not support a proton-transfer mechanism. Biochemistry. 1994;33:1481–1487. doi: 10.1021/bi00172a026. [DOI] [PubMed] [Google Scholar]