

Abstract
Intracellular transport after endosomal escape presents one of the major barriers for efficient non-viral gene delivery because plasmid DNA and synthetic nanoparticulate carriers suffer from significantly restricted diffusion in the cytoplasm. We postulate that forces generated by actin polymerization, a mechanism used by several bacterial pathogens such as Listeria monocytogenes, can be harnessed to propel nanoparticles within the cytoplasm and thereby overcome diffusional limitations associated with gene transport in the cell cytoplasm. In this work, we synthesized and characterized plasmid DNA-containing nanoparticles modified with ActA protein, the single protein in L. monocytogenes responsible for activating actin polymerization and initiating actin comet-tail propulsion. The motility of the ActA-modified nanoparticles was assessed in Xenopus laevis cytoplasmic extract supplemented with fluorescently labeled actin. Nanoparticle motility was monitored using multi-color, time-lapse fluorescence microscopy for the formation of actin comet tails attached to the fluorescently labeled vehicle. We observed particle motility with velocities ∼0.06 μm/s with anionic-charged plasmid carriers formed from either poly(lactic-co-glycolic acid) (PLGA) or 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes, but interestingly not with cationic particles assembled by encapsulation of plasmid with either polyethylenimine (PEI) or 1,2-dioleoyl-3-trimethylammonium-propane/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOTAP/DOPE) lipids. Control particles coated with albumin instead of ActA also showed no motility. Taken together, we have demonstrated the feasibility of translating the comet-tail propulsion mechanism to synthetic drug carriers as a potential approach to overcome intracellular transport barriers, and also have identified appropriate gene delivery systems that can be employed for this mechanism.
Keywords: Gene therapy, Bio-mimetic material, Nanoparticle, Gene transfer
1. Introduction
Non-viral gene delivery using polymeric and lipid materials offers an attractive alternative to viral gene delivery in terms of greater control of material composition, the ability to tune vehicle surface properties and relatively lower immunogenicity [1]. However, synthetic vehicles are generally less efficient than viral counterparts. For efficient delivery, synthetic vehicles must be engineered with the ability to overcome the numerous barriers encountered between the site of administration and localization to the cell nucleus [2]. This series of barriers includes (1) the physical and chemical stability of DNA and its delivery vehicle in the extracellular space, (2) cellular uptake by endocytosis, (3) escape from the endosomal compartments prior to trafficking to lysosomes, (4) cytosolic transport, and (5) nuclear localization of the plasmid for transcription.
Cytosolic transport is one of the least addressed barriers in gene delivery. Upon endosomal escape, the gene carrier or DNA must traverse the cytosol to access the nucleus. Passive diffusion of gene carriers such as polyethylenimine (PEI) polyplexes may take hours to reach the perinuclear region [3] while diffusion of microinjected DNA in the cytoplasm (to mimic the released DNA from cationic lipid complexes after endosomal escape) has been found to be substantially less than that observed in water [4,5]. For DNA larger than 2000 base pairs in length microinjected into the cytoplasm of HeLa cells, Lukacs and co-researchers reported little or no diffusion, suggesting that the cytoplasm is a substantial diffusional barrier [5]. Furthermore, DNA is enzymatically degraded during the diffusional transport, resulting in a half-life of 50–90 min in the cytoplasm [6,7]. There is conflicting evidence for active transport of DNA in the cytoplasm [1], although Suh et al. have previously reported that up to 17% of endocytosed PEI polyplexes can undergo microtubule-mediated active transport [3].
Besides passive diffusion and active microtubule-mediated transport, another mode of intracellular transport is the one utilized by Listeria monocytogenes, a bacterial pathogen. Listeria is able to move rapidly through host cells and cytoplasmic extracts (Fig. 1a), propelled by polymerization of hijacked actin from the host cell [8–10]; this associated actin-rich structure resembles a comet tail [11]. Previously, a single L. monocytogenes gene product, ActA, was identified as being responsible for coordinating actin-based motility in the host cell. ActA, a 65 kD protein, is expressed on the bacterial surface in a polar fashion, and the pole with the higher ActA density is associated with the comet tail [12]. ActA-deficient mutant strains cannot associate with actin or move intracellularly [13–15]. ActA initiates actin polymerization via interactions with host cell proteins by activating actin filament nucleation through the Arp2/3 complex and recruitment of vasodilator-stimulated phosphoprotein (VASP), which is an F-actin associated protein. Host cell profilin then interacts with VASP to enhance actin filament elongation while capping protein binds to the barbed end of actin filaments to prevent elongation of older filaments away from the bacterial surface. Host cell α-actinin crosslinks filaments to stabilize the actin structure, and ADF/cofilin disassembles old filaments. The physical mechanism by which actin polymerization generates force for propulsion remains unclear [16] although several models such as tethered ratchet model [17], actoclampin filament end-tracking motor model [18–20], and the elastic propulsion model [21–23] have been proposed.
Fig. 1.

(a) Listeria (labeled with DAPI, top) exhibit actin comet-tail motility (arrows) in Xenopus cytosolic extracts (with Alexa Fluo®-488 fluorescently labeled actin added, bottom); (b) proposed translation of actin motility to gene carrier to overcome diffusional restrictions in the cytoplasm.
Previously, Cameron et al. successfully translated this actin-based motility to polystyrene beads in Xenopus laevis egg extracts [24]. The ActA-coated polystyrene beads were demonstrated to first form actin clouds, break symmetry and undergo actin polymerization-based movement in the cytoplasmic extract [24,25]. Biophysical parameters such as particle size, protein surface density and quality of the cytoplasmic extracts were found to influence the speed, trajectory, and initiation of the actin-based motility [25]. The goal of this study is to investigate if this actin comet-tail motility is translatable to synthetic gene delivery systems as a means of overcoming cytoplasmic diffusional restrictions and enhancing intracellular transport of these systems (Fig. 1b). To reach this goal, several polymer and lipid-based formulations were functionalized with ActA and evaluated for actin-based cytoplasmic motility. Standard gene carriers including polyethylenimine-based poly-plexes (PEI/DNA complexes), poly(lactic-co-glycolic acid) (PLGA) particles and lipoplexes (lipid/DNA complexes) were used to investigate the effects of particle composition, size and surface charge on the ability of these modified materials to form comet-tail propulsion.
2. Materials and methods
2.1. Particle formulation
2.1.1. PEI complexes
Branched PEI with an average molecular weight of 25 k was obtained from Sigma–Aldrich (St Louis, MO). Polyplexes were formulated at a molar ratio of PEI nitrogen to DNA phosphate (N/P ratio) of 3 by mixing the polycation solution (in ultra-pure mQ-H2O) with an equal volume of DNA (0.1 mg/ml in the same buffer) previously labeled with POPO3 nucleic dye (Invitrogen), followed by incubating at room temperature for at least 15 min.
2.1.2. PLGA/DNA particles
PLGA nanospheres encapsulating DNA were prepared using a double emulsion-solvent evaporation method, as described previously [26]. Briefly, 0.2 ml of fluorescently labeled salmon sperm DNA (0.1 mg/ml) in ultra-pure H2O and 0.02 ml ActA (5 mg/ml) were emulsified in 6 ml of 5% w/v PLGA (lactide:glycolide 50:50, MW 40,000–70,000, Sigma–Aldrich) in methylene chloride using homogenizer (T-18 basic, IKA, Wilmington, DE) for 1 min at 13,500 rpm for micro-sized particles and at 25,000 rpm for nano-sized particles. The water-in-oil emulsion was further emulsified in 50 ml of a 2% (w/v) aqueous solution of polyvinyl alcohol (PVA, MW 146,000–186,000, Sigma–Aldrich) using homogenizer for 4 min to form a water-in-oil-in-water multiple emulsion. The emulsion was stirred for 24 h to remove the methylene chloride by evaporation. The PLGA nanospheres were recovered by ultracentrifugation (15,000 rpm for 60 min at 4 °C) and washed five times to remove PVA. Preparation of PLGA-PEI particles was performed similarly except PEI 25 k was added to the 5% w/v PLGA at w/w ratio of 1:10 and 1:2 prior to the first emulsification [27,28].
2.1.3. Lipoplexes and liposomes
Lipoplexes and liposomes were formulated based on manufacturer's instructions (Avanti Polar Lipids) and the protocol of Upadhyaya [29]. Briefly, DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids), DOGS-Ni-NTA nickel-chelating lipids (1,2-dioleoyl-sn-glycero-3-{[N-(5-amino-1-carboxypentyl)iminodiacetic acid] succinyl}, Avanti Polar Lipids) and Marina Blue® DHPE (Marina Blue® 1,2-dihex-adecanoyl-sn-glycero-3-phosphoethanolamine, Invitrogen), each dissolved in chloroform, were mixed at a weight ratio of 85:15:0.5 and evaporated on the inner surface of a round-bottom glass flask. The flask with the dried lipid layer was placed in a vacuum desiccator for several hours. Lipoplex formation was initiated by rehydration with 1 ml of fluorescently labeled DNA (0.1 mg/ml) at the lipid: DNA w/w ratio of 10:1 with gentle agitation for 1 h. For liposomes, only buffer of the same volume was used for rehydration. To prepare the DOPE/DOTAP lipid particles, DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, Avanti Polar Lipids) and DOTAP (1,2-dioleoyl-3-trimethylammonium-propane, Avanti Polar Lipids) at 1:1 w/w ratio were used in place of the phosphatidylcholine lipids. Extrusion of lipoplexes and liposomes if required was performed using the Mini-Extruder (Avanti Polar Lipids) according to manufacturer's instructions.
2.2. ActA protein isolation
ActA-His was purified with minor modifications as described previously [24,29] from strain DP-L2723 of L. monocytogenes expressing a truncated ActA gene encoding amino acids 1–613 [30]. Cysteine-terminated ActA-His-cys was purified from strain DP-L4363 of L. monocytogenes (both strains a gift from Dr. Julie Theriot, Stanford University) expressing a truncated ActA gene encoding amino acids 1–613 with a COOH-terminal six-histidine tag replacing the transmembrane domain and containing an additional cysteine amino acid [30]. A flask containing 50 ml of brain heart infusion media with 30 μg/ml chloramphenicol was inoculated with one colony and was grown overnight (12 h) at 37 °C. The bacterial culture was further inoculated in 1.8 L of LB media with 10 μg/ml chloramphenicol and was grown for 12-18 h at 37 °C in the dark. After bacteria were pelleted by centrifugation, protein in the supernatant was precipitated with ammonium sulfate and after centrifugation, resuspended in a Ni Load Buffer (20 mm Tris pH 7.9, 250 mm NaCl and 20 mm imidazole) and mixed with nickel nitrilotriacetic acid (Ni-NTA) superflow (Qiagen, Valencia, CA) for 1 h at room temperature before loading into a 5-ml polypropylene column. ActA was eluted with gradient of Elute Buffer (20 mm Tris pH 7.9, 500 mm NaCl and 1 m imidazole). Isolated protein was verified by running SDS-PAGE gel electrophoresis and for further purification was dialyzed into buffer QA (20 mm bis-Tris pH 6.5, 0.5 mm EDTA) overnight at 4 °C and mixed with Q Sepharose Fast Flow (GE Healthcare, Piscataway, NJ) before loading into a column made from glass Pasteur pipette for elution. The protein was eluted with gradients of buffer QB (20 mm bis-Tris pH 6.5, 0.5 mm EDTA, 1 M NaCl), dialyzed with ultra-pure mQ-H2O and concentrated to 5 mg/ml for flash-freezing in liquid nitrogen and storage at −80 °C.
2.3. ActA modification of particles
Carboxylated polystyrene beads (Bangs Laboratories, Inc, Fishers, IN) with an average diameter of 210 nm were incubated in ActA-His solution of 2 mg/ml. 2 μl of the beads (1% solids) were added to 10 μl of protein solution. For all ActA-modified nanoparticles, the amount of ActA protein added to the various gene carrier formulations was determined to be in excess quantities in order to achieve particle surface saturation. Beads were incubated in the protein solution for 1 h at room temperature, then pelleted and washed in PBS pH 7.4 before resuspension in 30 μl of PBS pH 7.4. Protein-coated beads were stored on ice or at 4 °C and were used within 1 week. BSA-coated beads were prepared by substituting the ActA protein solution with 2 mg/ml BSA protein solution.
PEI/DNA complexes were modified in a manner similar to carboxylated polystyrene beads via adsorption. The complexes were first concentrated using Nano-sep® centrifugal devices (Pall Life Sciences) to a final volume of ∼30 to 40 μl. Then concentrated complexes were incubated with 10 μl of the protein solution for 1 h at room temperature. The complexes were subsequently washed in PBS pH 7.4 using Nanosep® centrifugal devices before resuspending in ∼30 μl of PBS pH 7.4.
PLGA/DNA particles were also modified similarly. 100 μl of the formulated particles were pelleted before 10 μl of protein solution at 2 mg/ml was added for 1 h incubation. The particles were then pelleted and washed in PBS pH 7.4, and were resuspended in 30 μl of PBS pH 7.4.
For lipoplexes and liposomes, equal volumes of lipoplex or liposome suspension (45 μl) and ActA stock (diluted down to 0.5 mg/ml) were incubated for 1 h at room temperature while shaking and then transferred to ice [29]. For each experiment, new lipids were rehydrated and immobilized with ActA before use.
ActA-conjugated PEGylated polyplexes were prepared by first complexing the PEI and DNA at N/P ratio of 3 as described above. The complex was then PEGylated with maleimide-PEG5000-NHS (Nektar Therapeutics, Huntsville, AL) and reacted with the ActA-His-cys protein. ActA-conjugated PEGylated lipoplexes were prepared according to the hydration method as described above except maleimide-derivatized PEG2000-DSPE lipids (Avanti Polar Lipids) were used in place of DOGS-Ni-NTA lipids. ActA-His-cys protein was then conjugated to the lipoplexes via thiol reaction.
Successful immobilization of the ActA protein on the various formulations was indirectly supported by observations that the particles formed actin clouds in the cytoplasmic extracts as described next whereas control bovine serum albumin (BSA)-coated counterparts did not.
2.4. Motility assays
To assay for particle motility, ActA-modified particles were mixed with X. laevis egg cytoplasmic extract (10 μl) prepared as previously described [24] supplemented with Alexa Fluor® 488-conjugated actin (Invitrogen, Carlsbad, CA) and ATP regenerating mix consisting of 7.5 mm creatine phosphate, 1 mm ATP and 1 mm MgCl2 [31]. The assay mix was allowed to incubate on ice for 1 h. Then, a 2 μl sample was removed and was squashed between a microscope slide and 22-mm-square glass coverslip was sealed with vaseline:lanolin:paraffin (at 1:1:1), and was incubated at room temperature for 1 h in the dark before observing on the microscope. All observations were performed on a Nikon TE2000-U inverted epifluorescence microscope. Time-lapse dual-fluorescence channel microscopy at 5 s interval was captured at 100–150× magnification with an intensified charge-coupled device camera using METAMORPH (Universal Imaging, Media, PA) software for 3–5 min at 2–6 h after preparation.
2.5. Sizing and zeta potential
For quantification, sizing and zeta potential measurements were performed using a ZetaPALS dynamic light scattering detector (Brookhaven Instruments Corporation, Holtsville, NY) or for larger particles of >1 μm, the size of particles was determined from frames of acquired time series by image analysis using NIH ImageJ software (NIH, Bethesda, MD) measure tool. For fraction of beads with clouds and tails, the number of particles with actin clouds and tails per time series was manually counted before dividing over the total number of particles, also manually counted to determine the fractions. Rates of movement were determined by averaging the distance moved in 3 min using NIH ImageJ software with Manual Particle Tracking Plugin developed by Fabrice Cordelires (Institut Curie, Orsay, France).
3. Results and discussion
Several ActA-modified nanoparticles were prepared and evaluated for actin-mediated motility. Except for studies involving polystyrene beads and liposomes, all formulations, including PEI, PLGA, PLGA-PEI, DOPC and DOPE/DOTAP, were formulated to carry DNA. Prior to evaluating the ActA-modified particles, the activity of the ActA protein and integrity of the cytoplasmic extract were first verified with ActA-coated polystyrene beads to replicate the motility observations previously reported by Cameron and coworkers [24,25]. Carboxylated polystyrene beads coated with ActA were observed to form comet clouds and also propel in the cytoplasmic extracts by actin polymerization, whereas control BSA-coated polystyrene beads did not form actin clouds or move by actin comet tails (Supplementary Fig. 1). The observed fraction of ActA-coated polystyrene particles with clouds and tails was 0.85 ± 0.22 and 0.21 ± 0.31 respectively (Supplementary Fig. 1).
Following confirmation of ActA activity using polystyrene beads, ActA-immobilized particles (formulated using PEI, DOPC, DOPE/DOTAP, or PLGA) containing DNA cargo were assayed in the cytosolic extracts. PEI/DNA complexes at N/P=3 (of size comparable to the polystyrene beads) were able to form actin clouds but were unable to break symmetry and form comet tails (Fig. 2). This charge ratio was selected because it is the minimum charge ratio for consistent, complete plasmid DNA condensation. In contrast, synthetic nano- and micro-sized PLGA carriers were found to polymerize actin and exhibit comet-tail motility (Fig. 2 and Supplementary Movies 1 and 2). ActA-modified DOPC lipoplexes also exhibited comet-tail motility (Fig. 2 and Supplementary Movie 3). ActA-modified DOPE/DOTAP/DNA particles were able to form actin clouds, but without subsequent comet-tail motility. To our knowledge, this is the first demonstration of ActA-mediated motility applied to synthetic gene carriers. This is also the first time that a material other than polystyrene beads and natural phospholipid vesicles consisting of egg phosphatidylcholine or phosphatidylethanolamine has been shown to exhibit comet-tail motility when modified with the ActA protein.
Fig. 2.
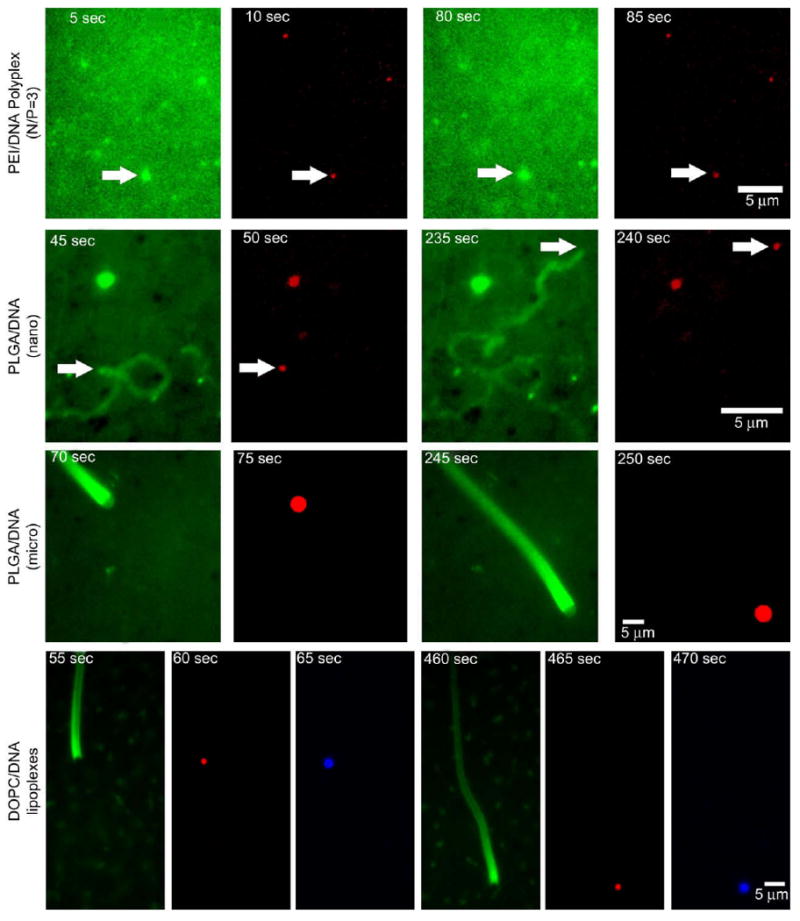
Image excerpts of representative dual or tri-fluorescence channels time series of PEI, PLGA and DOPC gene carriers with red fluorescence-labeled DNA in Xenopus cytoplasmic extract supplemented with green fluorescence-conjugated actin. PEI polyplexes formed actin clouds but did not display comet-tail motility while both small and large PLGA/DNA particles (0.25 μm and 2.5 μm), and DOPC lipoplexes (with Marina Blue-labeled lipid) were observed to polymerize actin and exhibit actin comet-tail motility.
Interestingly, delivery formulations that were able to polymerize actin and display comet-tail propulsion were found to possess anionic charge from zeta potential measurements (Fig. 3a). In contrast, the formulations that did not successfully form actin comet tails such as PEI polyplexes and DOPE/DOTAP lipoplexes were of cationic nature. The effect of surface charge on actin motility has not been previously investigated with the polystyrene beads but it may not be surprising that it has a role since the ActA-modified particles need to avoid non-specific “sticking” in the cytoplasm while recruiting specific host-cell proteins such as Arp2/3 complexes and actin to initiate actin polymerization. When the surface charge of ActA-coated polystyrene bead controls was measured, it was also negative (zeta potential of −23.58 mV), which was similar to ActA-modified PLGA particles. To further investigate the effect of surface charge, cationic PLGA–PEI particles were prepared using the same double emulsion method used for PLGA particles, except PEI was included during formulation [27,28]. When these were modified with ActA and assayed in cytoplasmic extract, no comet tails were observed. In addition, there was also a lower fraction of particles with clouds in comparison to the anionic PLGA particles. This result was similar to the other cationic PEI polyplexes and DOPE/DOTAP lipoplexes assayed (Fig. 3). Thus, the observations here suggest that surface charge might be a strong determinant of actin motility. Future investigations varying the mixing ratio of the biomaterial and DNA (N/P ratio) of the various gene carriers should be performed in order to understand this charge effect dependency more thoroughly. The actin comet tails associated with ActA-modified particles were observed to revert back to actin clouds at the end of their motility (Supplementary movie 4).
Fig. 3.
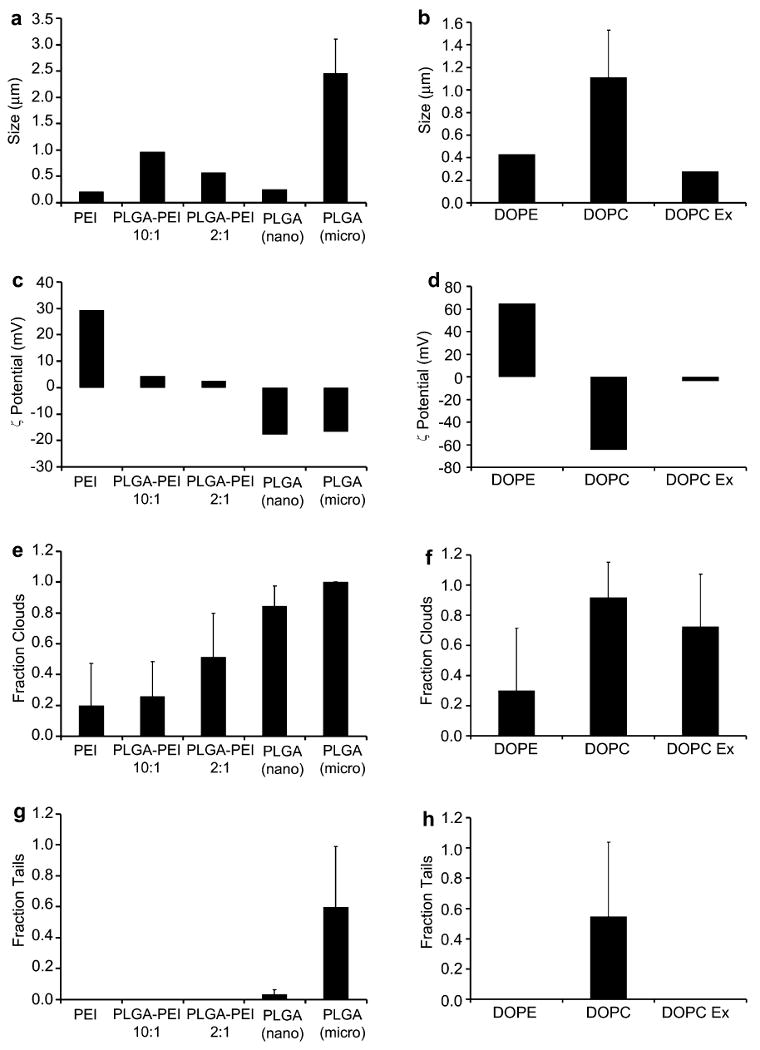
Characterization of particle properties (a–d) and actin-based motility (e–h) for various ActA-modified gene carriers. Polymer-based (left graphs) and lipid-based (right graphs) formulations were assessed for particle size, zeta potential, and the ability to initiate formation of actin clouds and actin comet tails.
To examine the effect of particle size on actin-based motility, nano-sized (0.25 μm) and micro-sized (2.5 μm), ActA-modified PLGA particles were prepared. A significantly higher fraction of particles with comet tails was observed for the larger particles compared to the smaller particles (0.60 ± 0.39 vs. 0.04 ± 0.03 respectively) (Fig. 3). This trend was also observed when comparing comet-tail formation between non-extruded DOPC lipoplexes (1.1 μm diameter) and smaller 200 nm extruded lipoplexes (0.55 ± 0.49 vs. 0.00 ± 0.00 respectively). The correlation of comet-tail formation efficiency with particle size was also observed with 200 nm and 400 nm extruded liposomes (0.004 ± 0.009 vs. 0.02 ± 0.03 respectively) that did not contain DNA (Supplementary Fig. 2). This is in contrast to the observations of Cameron et al. using polystyrene beads. In their studies, smaller particles have a higher fraction of particles with tails than the large particles. In addition, particles larger than 0.75 μm in diameter were not observed to form comet tails in cytoplasmic extracts [24]. However, Upadhyaya et al. reported that ActA-chelated vesicles of size 1–6 μm in diameter were able to display actin motility in a bovine brain extract assay [29] while Berheim-Groswasser et al. reported that polystyrene beads of up to 10 μm diameter coated with another Arp2/3 complex activator protein (Wiskott–Aldrich syndrome protein (WASP) subdomain VCA) exhibit actin comet-tail propulsion in an in vitro purified protein system [32]. The results suggest that material properties in addition to size may be a determinant of actin motility since the material used may have certain characteristics such as density, flexibility or surface properties that affect the optimal configuration of the attached ActA protein and its ability to initiate actin polymerization.
Next, the speeds of the particles displaying actin motility were analyzed using NIH ImageJ software and compared with speeds of control ActA-modified polystyrene beads and Listeria in the cytoplasmic extract (Fig. 4). Overall, the particles with actin polymerization were able to move more than 10 mm within minutes. In contrast, passive diffusion of PEI/DNA particles (∼150 nm) requires about 9 h to traverse 10 μm (diffusional speed of 0.00032 μm/s) [3]. The speeds of the ActA-modified PLGA particles (nano: 0.08 ± 0.04 μm/s; micro: 0.19 ± 0.10 μm/s) and ActA-modified DOPC lipoplexes (0.06 ± 0.02 μm/s) in extracts (Fig. 4a) were comparable to the polystyrene controls (0.06 ± 0.02 μm/s). These speeds were about 2-fold faster than the Listeria control (0.04 ± 0.01 μm/s) in the cytosolic extracts (Fig. 4b). The values here were also similar in order of magnitude to those previously reported by Cameron et al. and Upadhyaya et al. [24,25,29]. Compared to the passive diffusion value for nanoparticles reported by Suh et al., actin polymerization motility was about 100-fold faster than thermal-driven motion. It should be mentioned that these gene carriers can also undergo active transport via microtubules; the average velocities reported for this movement range from about 0.2 μm/s [3,33] to up to 2 μm/s [34]. This is about 1–2 orders of magnitude faster than actin polymerization motility.
Fig. 4.
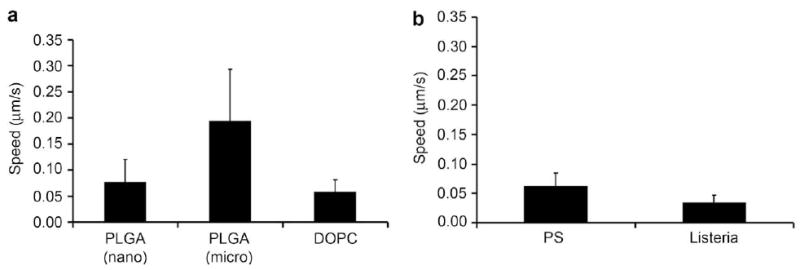
Speed of (a) ActA-modified gene carriers demonstrating actin comet-tail motility in the Xenopus cytoplasmic extract in comparison to (b) 200 nm ActA-modified polystyrene beads and Listeria.
In comparing the effect of size on the particle speed for ActA-modified PLGA formulations, the larger particles were faster than the smaller ones (0.19 ± 0.10 μm/s vs. 0.08 ± 0.04 μm/s respectively). One possible explanation is that the larger particles were able to form more extensive tails and generate more force for propulsion than smaller particles [17]. Indeed, we observed that the actin polymerization for larger particles was more intense than smaller particles as indicated by the higher signal to noise ratio in the images (Fig. 2). Furthermore, previous electron micrographs of ActA-coated polystyrene beads have shown a very dense array of twisted actin filaments in the tail for 0.5 μm beads compared to a single actin filament for 0.05 μm [35]. However, the trend reported here was in contrast to the observations of Cameron et al. where speed as a function of size peaked with 0.5 μm polystyrene beads and of Bernheim-Groswasser et al. where a reduced rate of motility with increasing diameter was observed [32]. In contrast, Wiesner and co-workers found that the speed of full length N-WASP-coated beads was independent of their size in purified protein systems [36]. Furthermore, Schwartz and co-workers studied the role of substrate curvature using polystyrene disks and reported results similar to the observations here [37]. These contradictory observations could be due to variations such as the quality of the assay and the protein isolated, but may also suggest that factors other than particle curvature or size may influence particle speed. It may be possible that composition of the materials used for particle formation, which influences particle density and surface characteristics, might also be a determinant of actin motility behavior. Lastly, Cameron and co-workers found that speed may also be reduced by extract dilution and by addition of excess skeletal muscle actin; enhanced speed was observed when non-muscle (platelet) actin was used in place of skeletal muscle actin [25].
Interestingly, the paths of larger particles were observed to be less tortuous than that of smaller particles (Fig. 2). This effect of size on path curvature has also similarly been observed previously with ActA-coated polystyrene beads by Cameron et al. [25] and VCA-coated beads moving in a mixture of purified proteins [32]. In addition, Cameron and co-workers noted that the trajectory that a particle follows may also be strongly influenced by the surrounding environment, with less concentrated extract resulting in greater path curvature. The physical basis of trajectory curvature remains to be elucidated, but it may be due to local variations in actin gel assembly rate from the outside to the inside of the curve [38–40].
Unlike active transport which is directed to the perinuclear region of the cell, it should be noted that the direction of comet-tail motility transport is rather random. However, the fact that these particles can overcome restricted diffusion suggests that there is a higher probability that some of these particles will end up in the perinuclear region rather than getting degraded in the process of diffusional transport. Furthermore, one of the intriguing aspects of actin motility is that the pathogen Listeria also uses this mechanism for cell-to-cell spread [11,41]. Thus, such ActA-modified particles may be capable of cell-to-cell transmission. This might be an intriguing approach to overcome extracellular barriers in tissue, especially solid tumors.
In addition to the above formulations, ActA-conjugated PEGylated polyplexes and PEGylated DOPC lipoplexes prepared using bifunctional PEGs (maleimide–PEG–NHS and DSPE–PEG–maleimide lipids respectively) were assayed for motility (Supplementary Fig. 3). PEGylation of cargo carriers is of great interest in the gene and drug delivery field as it have been shown previously to provide a stealth-like shield around therapeutic agents, reducing non-specific interactions and protein immunogenicity, reducing degradation by metabolic enzymes, thereby prolonging the half-life of the complexes in the blood [42,43]. In addition, Mishra and co-workers have shown in vitro that PEGylation can significantly affect cellular uptake and intracellular trafficking of non-viral gene delivery vehicles by preventing aggregation of non-viral gene delivery vehicles both outside and within cells [44]. Despite the benefits that could come from formulating PEGylated particles, no comet-tail motility was observed in motility assays with PEGylated formulations, although the particles did form actin clouds. It is possible that PEG chains might interfere with the interactions of the ActA protein with the host cell proteins or that the flexibility of the PEG-linker may affect comet-tail formation.
4. Conclusion
We report here the demonstration of the concept that the comet-tail motility utilized by the bacterial pathogen L. monocytogenes can be translated to synthetic drug delivery vehicles to overcome diffusional barriers. ActA-modified gene carriers were demonstrated to undergo directional actin polymerization-based propulsion similar to that of Listeria. Based on the preliminary studies, we have also successfully identified suitable gene carriers for future studies and research. Surface charge was identified as a possible strong determinant of motility, while particle size and composition (Table 1) also have influences. Anionic particles such as DOPC lipoplexes and PLGA/DNA particles immobilized with ActA protein were able to polymerize actin and undergo directional comet-tail propulsion while cationic particles did not. In addition, larger anionic ActA-modified particles have better motility than smaller particles in terms of the fraction of particles with tails and the speed of particles. PLGA may be the more suitable candidate since it is more stable than lipid formulations which may dissociate in the cytosol during or upon endosomal release. Furthermore, the extruded lipoplexes and liposomes did not exhibit comet-tail motility in comparison to their less applicable non-extruded counterparts.
Table 1.
Effects of surface charge, size and composition on the actin motility of ActA-modified particles.
| Motility | Chargea | Sizea,b | Composition | ||
|---|---|---|---|---|---|
| Fraction | Speed | ||||
| PEI/DNA NP 3 | − | − | ++ | Small | Polymer |
| PLGA–PEI | − | − | + | Large | Polymer |
| DOPE/DOTAP | − | − | +++ | Large | Lipid |
| DOPC (200 nm Ex) | − | − | − | Small | Lipid |
| PLGA (nano) | + | + | −− | Small | Polymer |
| DOPC (Non Ex) | ++ | + | −−− | Large | Lipid |
| PLGA (micro) | +++ | +++ | −− | Large | Polymer |
After ActA modification.
Small <350 nm; large >350 nm.
Future works may include characterizing the effect of material mixing ratio (such as N/P ratio, lipid:lipid w/w ratio) to more thoroughly evaluate their effect on actin motility, optimizing the protein density or conjugation ratio on the surface of the particles to increase the efficiency of particles with actin-based motility, and replicating this phenomenon in cultured cells. In addition, carriers identified here may also have wider implications in intracellular and intercellular cargo delivery. For example, the materials identified can be extended to encapsulate and transport other therapeutic cargo such as siRNA and drugs. Taken together, the findings in this study open the possibility to an alternative strategy to that of microtubule-mediated cytosolic transport for intracellular drug carriers to overcome restricted diffusional cytoplasmic barriers.
Supplementary Material
Acknowledgments
The authors thank Dr. Julie Theriot and Matthew Footer of Stanford University for generously donating the Listeria strains and their technical assistance and advice on ActA comet-tail motility, and Dr. David Kimelman and Hank Farr (University of Washington) for their technical assistance in X. laevis handling and egg extraction. We are grateful to Dr. James Nelson (Stanford University) for providing the MDCK cells expressing GFP-actin. Egg extraction from X. laevis was conducted under an IUCAC-approved laboratory-animal protocol. We also thank Ester Kwon for providing characterization data on PEI/DNA formulations. This work was supported by NIH/NCI grant 1R21CA114141, and the Alliance for Cancer Gene Therapy (SHP, Patricia Zoch Tate Young Investigator Award). TTG acknowledges a graduate fellowship provided by the National Science Foundation and IKP acknowledges the support from Korea Science and Engineering Foundation grant R13-2002-013-07001-0 to the Medical Research Center for Gene Regulation.
Appendix
Figures with essential colour discrimination. Fig. 2 of this article is difficult to interpret in black and white. The full colour image can be found in the on-line version, at doi:10.1016/j.biomaterials.2008. 10.059.
Footnotes
Appendix. Supplementary data: Supplementary figures associated with this article can be found, in the online version, at doi:10.1016/j.biomaterials.2008.10.059.
References
- 1.Wiethoff CM, Middaugh CR. Barriers to nonviral gene delivery. J Pharm Sci. 2003;92:203–17. doi: 10.1002/jps.10286. [DOI] [PubMed] [Google Scholar]
- 2.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4:581–93. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 3.Suh J, Wirtz D, Hanes J. Efficient active transport of gene nanocarriers to the cell nucleus. Proc Natl Acad Sci U S A. 2003;100:3878–82. doi: 10.1073/pnas.0636277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dauty E, Verkman AS. Actin cytoskeleton as the principal determinant of size-dependent DNA mobility in cytoplasm. J Biol Chem. 2005;280:7823–8. doi: 10.1074/jbc.M412374200. [DOI] [PubMed] [Google Scholar]
- 5.Lukacs GL, Haggie P, Seksek O, Lechardeur D, Freedman N, Verkman AS. Size-dependent DNA mobility in cytoplasm and nucleus. J Biol Chem. 2000;275:1625–9. doi: 10.1074/jbc.275.3.1625. [DOI] [PubMed] [Google Scholar]
- 6.Lechardeur D, Sohn KJ, Haardt M, Joshi PB, Monck M, Graham RW, et al. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Ther. 1999;6(4):482–97. doi: 10.1038/sj.gt.3300867. [DOI] [PubMed] [Google Scholar]
- 7.Pollard H, Toumaniantz G, Amos JL, Avet-Loiseau H, Guihard G, Behr JP, et al. Ca2+-sensitive cytosolic nucleases prevent efficient delivery to the nucleus of injected plasmids. J Gene Med. 2001;3:153–64. doi: 10.1002/jgm.160. [DOI] [PubMed] [Google Scholar]
- 8.Dabiri GA, Sanger JM, Portnoy DA, Southwick FS. Listeria-monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci U S A. 1990;87:6068–72. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Theriot JA, Rosenblatt J, Portnoy DA, Goldschmidtclermont PJ, Mitchison TJ. Involvement of profilin in the actin-based motility of L-monocytogenes in cells and in cell-free-extracts. Cell. 1994;76:505–17. doi: 10.1016/0092-8674(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 10.Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature. 1997;385:265–9. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- 11.Tilney LG, Portnoy DA. Actin-filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria-monocytogenes. J Cell Biol. 1989;109:1597–608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kocks C, Hellio R, Gounon P, Ohayon H, Cossart P. Polarized distribution of Listeria-monocytogenes surface protein ActA at the site of directional actin assembly. J Cell Sci. 1993;105:699–710. doi: 10.1242/jcs.105.3.699. [DOI] [PubMed] [Google Scholar]
- 13.Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria-monocytogenes ActA protein in mammalian-cells. Proc Natl Acad Sci U S A. 1993;90:11890–4. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Domann E, Wehland J, Rohde M, Pistor S, Hartl M, Goebel W, et al. A novel bacterial virulence gene in Listeria-monocytogenes required for host-cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J. 1992;11:1981–90. doi: 10.1002/j.1460-2075.1992.tb05252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. l-Monocytogenes-induced actin assembly requires the ActA gene-product, a surface protein. Cell. 1992;68:521–31. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- 16.Mogilner A. On the edge: modeling protrusion. Curr Opin Cell Biol. 2006;18:32–9. doi: 10.1016/j.ceb.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Mogilner A, Oster G. Force generation by actin polymerization II: the elastic ratchet and tethered filaments. Biophys J. 2003;84:1591–605. doi: 10.1016/S0006-3495(03)74969-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickinson RB, Purich DL. Clamped-filament elongation model for actin-based motors. Biophys J. 2002;82:605–17. doi: 10.1016/S0006-3495(02)75425-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickinson RB, Caro L, Purich DL. Force generation by cytoskeletal filament end-tracking proteins. Biophys J. 2004;87:2838–54. doi: 10.1529/biophysj.104.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dickinson RB, Purich DL. Diffusion rate limitations in actin-based propulsion of hard and deformable particles. Biophys J. 2006;91:1548–63. doi: 10.1529/biophysj.106.082362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernheim-Groswasser A, Prost J, Sykes C. Mechanism of actin-based motility: a dynamic state diagram. Biophys J. 2005;89:1411–9. doi: 10.1529/biophysj.104.055822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerbal F, Chaikin P, Rabin Y, Prost J. An elastic analysis of Listeria monocytogenes propulsion. Biophys J. 2000;79:2259–75. doi: 10.1016/S0006-3495(00)76473-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plastino J, Sykes C. The actin slingshot. Curr Opin Cell Biol. 2005;17:62–6. doi: 10.1016/j.ceb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Cameron LA, Footer MJ, van Oudenaarden A, Theriot JA. Motility of ActA protein-coated microspheres driven by actin polymerization. Proc Natl Acad Sci U S A. 1999;96:4908–13. doi: 10.1073/pnas.96.9.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameron LA, Robbins JR, Footer MJ, Theriot JA. Biophysical parameters influence actin-based movement, trajectory, and initiation in a cell-free system. Mol Biol Cell. 2004;15:2312–23. doi: 10.1091/mbc.E03-12-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang SW, Lim HW, Seo SW, Jeon O, Lee M, Kim BS. Nanosphere-mediated deliver of vascular endothelial growth factor gene for therapeutic angiogenesis in mouse ischemic limbs. Biomaterials. 2008;29:1109–17. doi: 10.1016/j.biomaterials.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Bivas-Benita M, Romeijn S, Junginger HE, Borchard G. PLGA-PEI nanoparticles for gene delivery to pulmonary epithelium. Eur J Pharm Biopharm. 2004;58:1–6. doi: 10.1016/j.ejpb.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang XQ, Intra J, Salem AK. Comparative study of poly (lactic-co-glycolic acid)-poly ethyleneimine-plasmid DNA microparticles prepared using double emulsion methods. J Microencapsul. 2008;25:1–12. doi: 10.1080/02652040701659347. [DOI] [PubMed] [Google Scholar]
- 29.Upadhyaya A, Chabot JR, Andreeva A, Samadani A, van Oudenaarden A. Probing polymerization forces by using actin-propelled lipid vesicles. Proc Natl Acad Sci U S A. 2003;100:4521–6. doi: 10.1073/pnas.0837027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science. 1998;281:105–8. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- 31.Murray AW. Cell cycle extracts. Methods Cell Biol. 1991;36:581–605. [PubMed] [Google Scholar]
- 32.Bernheim-Groswasser A, Wiesner S, Golsteyn RM, Carlier MF, Sykes C. The dynamics of actin-based motility depend on surface parameters. Nature. 2002;417:308–11. doi: 10.1038/417308a. [DOI] [PubMed] [Google Scholar]
- 33.Suh J, Dawson M, Hanes J. Real-time multiple-particle tracking: applications to drug and gene delivery. Adv Drug Deliv Rev. 2005;57:63–78. doi: 10.1016/j.addr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Bergen J, Pun S. Analysis of the intracellular barriers encountered by nonviral gene carriers in a model of spatially controlled delivery to neurons. J Gene Med. 2008;10:187–97. doi: 10.1002/jgm.1137. [DOI] [PubMed] [Google Scholar]
- 35.Cameron LA, Svitkina TM, Vignjevic D, Theriot JA, Borisy GG. Dendritic organization of actin comet tails. Curr Biol. 2001:130–5. doi: 10.1016/s0960-9822(01)00022-7. [DOI] [PubMed] [Google Scholar]
- 36.Wiesner S, Helfer E, Didry D, Ducouret G, Lafuma F, Carlier M, et al. A biomimetic motility assay provides insight into the mechanism of actin-based motility. J Cell Biol. 2003;160:387–98. doi: 10.1083/jcb.200207148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwartz IM, Ehrenberg M, Bindschadler M, McGrath JL. The role of substrate curvature in actin-based pushing forces. Curr Biol. 2004;14:1094–8. doi: 10.1016/j.cub.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 38.Carlsson AE. Growth of branched actin networks against obstacles. Biophys J. 2001;81:1907–23. doi: 10.1016/S0006-3495(01)75842-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlsson AE. Growth velocities of branched actin networks. Biophys J. 2003;84:2907–18. doi: 10.1016/S0006-3495(03)70018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutenberg AD, Grant M. Curved tails in polymerization-based bacterial motility. Phys Rev E. 2001;64:021904. doi: 10.1103/PhysRevE.64.021904. [DOI] [PubMed] [Google Scholar]
- 41.Robbins JR, Barth AI, Marquis H, de Hostos EL, Nelson WJ, Theriot JA. Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J Cell Biol. 1999;146:1333–49. doi: 10.1083/jcb.146.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E. PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999:595–605. doi: 10.1038/sj.gt.3300900. [DOI] [PubMed] [Google Scholar]
- 43.Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10:1451–8. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 44.Mishra S, Webster P, Davis ME. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur J Cell Biol. 2004;83:97–111. doi: 10.1078/0171-9335-00363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.