

Abstract
Co-assembly of KCNQ1 with different accessory, or beta, subunits that are members of the KCNE family results in potassium (K+) channels that conduct functionally distinct currents. The alpha subunit KCNQ1 conducts a slowly activated delayed rectifier K+ current (IKs), a major contributor to cardiac repolarization, when co-assembled with KCNE1 and channels that favor the open state when co-assembled with either KCNE2 or KCNE3. In the heart, stimulation of the sympathetic nervous system enhances IKs. A macromolecular signaling complex of the IKs channel including the targeting protein Yotiao coordinates up or downregulation of channel activity by protein kinase A (PKA) phosphorylation and dephosphorylation of molecules in the complex. β-adrenergic receptor mediated IKs upregulation, a functional consequence of PKA phosphorylation of the KCNQ1 amino terminus (N-T), requires co-expression of KCNQ1/Yotiao with KCNE1. Here, we report that co-expression of KCNE2, like KCNE1, confers a functional channel response to KCNQ1 phosphorylation, but co-expression of KCNE3 does not. Amino acid sequence comparison among the KCNE peptides, and KCNE1 truncation experiments, reveal a segment of the predicted intracellular KCNE1 carboxyl terminus (C-T) that is necessary for functional transduction of PKA phosphorylated KCNQ1. Moreover, chimera analysis reveals a region of KCNE1 sufficient to confer cAMP-dependent functional regulation upon the KCNQ1_ KCNE3_Yotiao channel. The property of specific beta subunits to transduce post-translational regulation of alpha subunits of ion channels adds another dimension to our understanding molecular mechanisms underlying the diversity of regulation of native K+ channels.
Keywords: ancillary subunit, delayed rectifier, sympathetic nervous system, phosphorylation, functional diversity, carboxyl-terminal domain
Introduction
Heteromultimeric assembly of KV channels and accessory subunits provides a multiplicity of native K+ currents that are important in maintaining the electrical activity in most cells.1 KCNQ1, a KV channel found in a variety of tissues including the heart, brain, small intestine, colon and stomach, conducts functionally distinct K+ currents.2 The tissue specificity of currents is largely due to assembly of the pore forming KCNQ1 alpha subunit with different KCNE accessory (beta) subunits.3 When co-assembled with the beta subunit KCNE1 (minK), KCNQ1 conducts a slowly-activating delayed rectifier K+ current (IKs) that contributes to cardiac repolarization.4–6 KCNQ1 also forms K+ channels that heavily favor the open state when co-assembled with the beta subunits KCNE2 (MiRP1) or KCNE3 (MiRP2).7,8 The functional consequences of expression with distinct beta subunit variants may contribute to various physiological responses not only in cardiac cells but also in epithelial cells in which KCNQ1 is expressed.7–9 In the heart, contributions from diverse heteromultimeric KCNQ1-KCNE channels to cardiac repolarization is important because inherited mutations in KCNQ1, KCNE1 and KCNE2 have been associated with variants of the heritable long-QT syndrome (LQT-1, LQT-5 and LQT-6) as well as congenital atrial fibrillation;10,11 and overexpression of KCNE3 has been shown to alter cardiac repolarization in an animal model.12
Co-assembly of KCNQ1 with different members of the KCNE family may also affect physiologically critical posttranslational modification of expressed channel activity. In turn, this may contribute to or underlie distinct regulation of K+ channels by a variety of cellular modulators of the multitude of native K+ currents.1 This may be particularly important in the response of these channels to modulation by stimulation of the sympathetic nervous system (SNS). Control of the cardiac electrical system by the SNS in response to exercise or emotional stress, a fundamental property of the cardiovascular system, is mediated by the activation of β-adrenergic receptors that regulate the activity of select ion channels by phosphorylation via the cAMP-dependent protein kinase (PKA) pathway. SNS stimulation increases the intracellular second messenger cAMP, enhances IKs, and consequently, shortens the cardiac ventricular action potential duration (APD) to ensure adequate time for inter beat diastolic filling. SNS mediated APD shortening underlies SNS control of QT intervals in humans.13 The cAMP-mediated regulation of the IKs channel is particularly important because carriers of LQT-1 mutations in the IKs alpha subunit are at greatest risk of lethal arrhythmias in the face of SNS stimulation.14 The regulation is mediated by a macromolecular signaling complex consisting of KCNQ1, KCNE1 and Yotiao that regulates channel activity by phosphorylation and de-phosphorylation of Ser27 in the N-T of KCNQ1,15 and of Ser43 in the N-T of Yotiao (AKAP9).16,17 KCNE1, which is not a substrate of PKA-phosphorylation, is required for the transduction of the functional consequences of the negative charge at Ser27 in KCNQ1.18 Although it has been shown that KCNE1 is not required for PKA phosphorylation of KCNQ1,18 neither the mechanism of the transduction by KCNE1 nor roles of other KCNE variants in the cAMP-mediated regulation via the macromolecular complex have been explored.
In the present study, we investigated the roles of two other auxiliary subunits of KCNQ1 in cAMP-dependent regulation of heteromultimeric KCNQ1-KCNE channels in the presence of Yotiao. We find that expression of KCNQ1 with KCNE2, but not KCNE3, also results in assembled channels that respond functionally to KCNQ1 phosphorylation. In addition, we show that the KCNE1 C-T domain is necessary for the functional response of KCNQ1_ KCNE1 channels to KCNQ1 phosphorylation and is sufficient to transfer functional responses to KCNQ1_KCNE3 channels when added to the KCNE3 C-T domain in a chimerical construct.
Results
We previously have shown that the KCNQ1 protein can be PKA phosphorylated in response to cAMP independent of co-assembly with KCNE1, but that the functional response of IKs channels to cAMP requires co-assembly of KCNE1 and KCNQ 1.18 To begin this investigation, we first tested whether or not PKA-dependent phosphorylation, was affected by co-expression of KCNQ1 with either KCNE2 or KCNE3. Because we have previously shown that the response to cAMP of the KCNE1_KCNQ1 channel is phosphorylation of Ser27 in the KCNQ1 N-T, we used a previously described antibody that specifically detects KCNQ1 pSer27 to assay KCNQ1 phosphorylation in response to cAMP and okadaic acid (OA) application (Methods). We repeated this measurement when KCNQ1 was expressed with KCNE1, KCNE2 or KCNE3. The results of these experiments, summarized in Figure 1, indicate that KCNQ1 is phosphorylated in response to cAMP independent of co-expression of any of these beta subunits.
Figure 1.
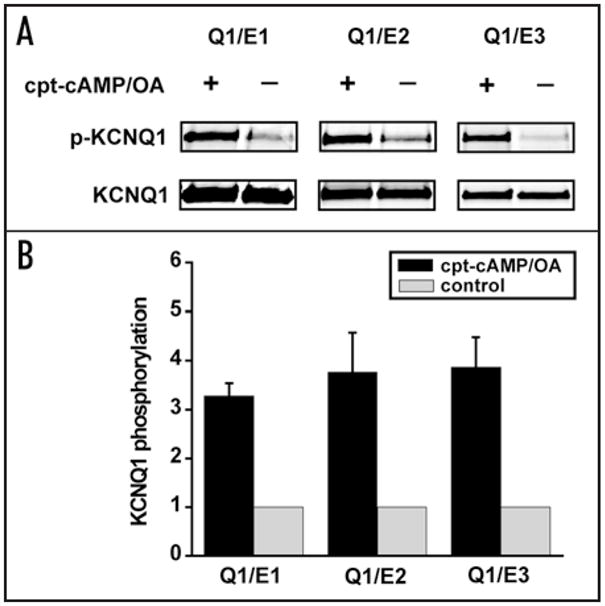
KCNQ1 (Q1) phosphorylation is independent of co-expressed beta subunit variant. (A) Cells expressing KCNQ1_KCNE1, KCNQ1_KCNE2 or KCNQ1_KCNQE3 were treated with cpt-cAMP/OA (+) or none (−, control). Representative western blots for phosphorylated KCNQ1 (p-KCNQ1) are shown in the upper panels and blots for total KCNQ1 are shown in the lower panels. (B) Summary western blots, quantified as described in Methods, are plotted as bar graphs. Signals of the phosphorylated KCNQ1 and total KCNQ1 from the dual western blot on the same gel were detected and analyzed by an Odyssey machine. Loading differences between cpt-cAMP/OA treated cells and control cells were first corrected based on the values of total KCNQ1. Phosphorylation signals of the treated cells were then measured and normalized to that of the control cells. Relative phosphorylation values are expressed as mean ± standard error and plotted. ANOVA test shows no significant difference in phosphorylation among KCNQ1_KCNE1, KCNQ1_KCNE2 and KCNQ1_KCNE3 (n = 4 for each group).
To investigate whether KCNE2 or KCNE3, like KCNE1, transduces the phosphorylation-induced negative charge at Ser27 KCNQ1 into a functional change in expressed channel activity, we measured perforated patch whole cell currents in CHO cells expressing Yotiao, the appropriate beta-subunits and a construct that mimics phosphorylated KCNQ1. Co-expression of KCNE2 or KCNE3 with wild type KCNQ1 results in channels that that dramatically favor the open state (Fig. 2) consistent other studies.7,8 To mimic PKA phosphorylation of KCNQ1, we replaced KCNQ1 Ser27 by Asp (S27D) to change the charge at this residue and refer to the construct as pseudo phosphorylated KCNQ1 as we have previously reported.16,18 Expression of S27D KCNQ1 with KCNE2, but not KCNE3, resulted in an increase current amplitudes (Fig. 2A and B). Much like KCNQ1 regulation in the presence of KCNE1,16 a negative charge at Ser27 is required to cause this increase in channel current. Ala substitution at Ser27 (S27A) in KCNQ1 did not change the current amplitudes (Fig. 2A) when co-expressed with KCNE2.
Figure 2.

Co-expression of KCNE2, but not KCNE3, alters function of pseudo phosphorylated KCNQ1 channels. Whole-cell currents were measured with perforated patch-clamp configuration from CHO cells transfected with S27D_KCNQ1 and Yotiao plus either KCNE2 (A) or KCNE3 (B). The S27D mutation was used to mimic the PKA phosphorylation of the IKs channels at Ser-27 in KCNQ1 in the absence of cAMP16 (see text). Membrane currents were elicited by 2-s test pulses from −120 mV to 100 mV (20-mV increments) following 2-s pulses to −40 mV. Pulse frequency was 0.067 Hz. The membranes were held at −65 mV for KCNE2 and −80 mV for KCNE3. I–V curves (wild type KCNQ1 (WT): open squares, S27D KCNQ1 (SD): blue squares, S27A KCNQ1 (SA): red squares) were plotted with amplitudes at the first test pulses, which are pointed by arrows in the representative traces. The numbers of experiments were as follows: KCNE2 WT, n = 15, SD, n = 10, and SA, n = 7; KCNE3 WT, n = 6, and SD, n = 7. *p < 0.05 vs. WT KCNQ1, ANOVA for KCNE2 and Student’s t-test for KCNE3.
Because Figure 2 suggests that co-expression of KCNE2 and KCNQ1 should result in functionally responsive channels to cAMP challenges, we next tested whether an external application of a membrane permeable cAMP (cpt-cAMP) enhances KCNE2/KCNQ1 currents (note that in all experiments Yotiao was also expressed). In comparison with the time control without cAMP application, external application of cpt-cAMP for longer than 3 min significantly increased KCNE2/wild type KCNQ1 currents. The cAMP-induced enhancement was then reversed by washout of cAMP (Fig. 3A). Addition of CPT-cAMP increased the KCNE2/KCNQ1 currents elicited by test pulses to the range between −60 mV and +40 mV (Fig. 3B). Mutations at KCNQ1 residue 27 ablated the cAMP-induced enhancement of KCNE2/KCNQ1 currents regardless of substituted amino acid residue (Fig. 4), suggesting that the cAMP-dependent functional regulation is a consequence of PKA-mediated phosphorylation at Ser27 of KCNQ1.
Figure 3.

Membrane permeable CPT-cAMP enhances KCNE2/KCNQ1/Yotiao channel currents. Whole-cell currents were measured with perforated patch-clamp configuration from CHO cells transfected with wild type KCNQ1, KCNE2 and Yotiao. (A) Time course of the enhancement of the KCNE2/KCNQ1/Yotiao currents by membrane permeable cpt-cAMP (0.5 mM). Mean ± SEM current amplitude (normalized to amplitude recorded at 5-min after start of current recording, i.e., before cAMP treatment) is plotted vs. time after start of current recording. Membrane currents were elicited by test pulses (0 mV, −40 mV return) at 0.067 Hz from a holding potential of −65 mV. The time course of the plots for the cpt-cAMP application (filled squares, n = 8) is superimposed with the time course of the plots for time control without cAMP application (open squares, n = 16). cpt-cAMP was applied from extracellular side of the CHO cells as indicated by the gray bar above the time course. *p < 0.05 vs. time control, Student’s t-test. Data for I–V in (B) were obtained during periods indicated by horizontal bars. In Right, representative current traces before (control) and 7-min after the cpt-cAMP application were superimposed with 5-min washout of cAMP. (B) Voltage-dependence of the cAMP-induced enhancement of the KCNE2/KCNQ1/Yotiao currents. Shown are plots of mean current amplitude ± SEM vs. activating pulse voltage (Left, n = 8) as well as representative current traces that were obtained by the same protocol as in Figure 2A. The cAMP-induced changes are described by superimposing I–V plots before (control, open squares) and after the cAMP application (cpt-cAMP, filled squares). *p < 0.05 vs. control, Paired Student’s t-test.
Figure 4.

KCNQ1 Ser-27 is required for functional response of KCNE2/KCNQ1/Yotiao channels to cAMP. Ser-27 in the KCNQ1 N terminus was mutated to Asp (S27D) and Ala (S27A). CHO cells were transfected with KCNE2, the indicated KCNQ1 construct, and Yotiao. (A) Substitutions of Ser-27 abolish PKA-dependent regulation of KCNE2/KCNQ1/Yotiao channels. Filled bars indicate mean ± SEM current amplitudes 7-min after cAMP application (normalized to amplitude before the application), and are compared with time control (open bars) without application of cpt-cAMP (0.5 mM). Currents were measured, and cpt-cAMP was applied as described in Figure 3A. Data for WT in Figure 3A is re-plotted as indicated above, and then is compared with data for S27D and S27A. The numbers of experiments were as follows: WT, time control, n = 8, and cAMP, n = 16; S27D, time control, n = 8, and cAMP, n = 7; S27A, time control, n = 7, and cAMP, n = 8. *p < 0.05 vs. time control, Student’s t-test. Representative current traces before the cAMP application (control, black) are superimposed with the traces after the cAMP application (red) for each KCNQ1 construct. (Scale bars: 10 pA/pF, 1 s). (B) S27D (SD) abolishes the PKA-dependent regulation over activating pulse voltages. Shown are plots of mean current amplitude ± SEM (Left, n = 7) as well as representative current traces as in Figure 3B for WT KCNQ1 (control; open squares, and cpt-cAMP; filled squares). (C) S27A (SA) abolishes the PKA-dependent regulation over activating pulse voltages. Shown are plots of mean current amplitude ± SEM (Left, n = 8) as well as representative current traces as in Figure 3B for WT KCNQ1 (control; open squares, and cpt-cAMP; filled squares).
Amino-acid sequence comparison among the KCNE family (Fig. 5) reveals potential clues for a molecular mechanism underlying the transduction of a functional response to the negative charge at KCNQ1 Ser27 of channels that include KCNE1 and KCNE2 but not KCNE3. The overall amino acid sequence homology among the KCNE family members is not very high: KCNE1 and KCNE2 have about 28% sequence identity and KCNE1 and KCNE3 show only about 15% identity. The C-T of these beta-subunits shows very little sequence homology except for in one potentially important region. The proximal portion of the C-T domains of KCNE1 and KCNE2 are highly conserved from Arg67 through Ala93 whereas there is considerable divergence in sequence in this region when comparing KCNE1 with KCNE3. Furthermore, this region of KCNE1 contains 9 known LQT-5 mutations, suggesting that this region could be very important for channel function.19–22
Figure 5.

Amino-acid sequence comparison of KCNE1 with KCNE2 and KCNE3. Gray background indicates amino acid residues that are identical in KCNE1 with KCNE2 (A) and KCNE3 (B). The bar indicates the predicted transmembrane region. Sites for KCNE1 truncation in Figure 5 are indicated as N79stop and Y94stop. Arrow indicates splice sites used in chimera construction in Figure 6.
We thus hypothesized that the C-T domains of KCNE peptides are critical for transduction of cAMP-dependent functional regulations of the KCNQ1 channel co-assembled with appropriate KCNE peptides. We first tested this hypothesis by employing previously reported KCNE1 deletion mutants lacking amino acid residues 94–129 (Y94stop) and amino acid residues 79–129 (N79stop)23 and examined the effects of these mutations on the cAMP-dependent regulation of the IKs channel (Fig. 6 and Table 1). As we have previously reported,18 dialysis of patch-clamped Chinese hamster ovary (CHO) cells co-expressing wild type KCNE1, KCNQ1, and Yotiao with cAMP (0.2 mM) and OA (0.2 μM) increases the activity of KCNE1/KCNQ1 channels that can be quantified by measuring the amplitude of deactivating current tails (after +100 mV test pulses: -cAMP/OA 72.0 ± 8.7 pA/pF, n = 10; cAMP/OA 133.7 ± 10.8 pA/pF, n = 10; p < 0.05) (Fig. 6A). We took advantage of this regulatory response to the cAMP stimulation to explore the effects of KCNE1 C-T domain truncations. In cells expressing Y94stop KCNE1, KCNQ1 and Yotiao (Fig. 6B), dialysis with cAMP/OA increased tail current amplitudes (after +100 mV test pulses: -cAMP/OA 28.6 ± 11.5 pA/pF, n = 6; cAMP/OA 68.4 ± 9.0 pA/pF, n = 6; p < 0.05). Although deletion of amino acids 94–129 in KCNE1 decreased overall tail current densities, dialysis of cAMP/OA increased total current density at every voltage in which the channel was open. However, in cells expressing N79stop KCNE1, KCNQ1 and Yotiao (Fig. 6B), there was no significant effect of intracellular cAMP and OA on KCNE1/KCNQ1 tail current amplitudes (after +100 mV test pulses: -cAMP/OA 13.8 ± 3.5 pA/pF, n = 6; cAMP/OA 21.2 ± 4.2 pA/pF, n = 6; p = N.S). Because the PKA phosphorylation at Ser27 KCNQ1 is independent of co-expression of KCNE1,18 it is very unlikely that the N79stop deletion affects the KCNQ1 phosphorylation. The characteristic IKs current seen in Figure 6C indicates that the truncated KCNE1 successfully co-assembled with KCNQ1, but the N79stop beta-subunit failed mediate the cAMP-depedent functional effect on the co-assembled channel
Figure 6.

Truncation of the KCNE1 carboxyl terminus impairs cAMP-dependent regulation of the KCNQ1/KCNE1/Yotiao channel. Whole-cell currents were recorded from patch-clamped CHO cells co-transfected with KCNE1/KCNQ1/Yotiao with or without cAMP (0.2 mM) and OA (0.2 μM) dialysis. Data were obtained 12-min after the membrane rupture in the absence (control) or the presence of cAMP/OA inside (cAMP/OA). Each KCNE1 construct is shown in Left. Shown are plots of mean tail current ± SEM vs. activating pulse voltage (Center) as well as mean currents (Right) in the absence (control, black) and presence of cAMP/OA (cAMP/OA, red). *p < 0.05 vs. control, Student’s t-test. Mean currents were obtained by +100-mV pulse (−40-mV return) indicated by arrows in the plots. (A) Full length of WT KCNE1 (Met1-Pro129). Control, n = 10; cAMP/OA, n = 10. (Scale, 100 pA/pF, 1 s). (B) Y94stop KCNE1 (Met1-Ala93). Control, n = 6; cAMP/OA, n = 6. (Scale, 100 pA/pF, 1 s). (C) N79stop KCNE1 (Met1-Phe78). Control, n = 6; cAMP/OA, n = 6. (Scale, 50 pA/pF, 1 s).
Table 1.
Effects of cAMP/OA dialysis on voltage-dependence of activation and time constants of deactivation in KCNE1/KCNQ1/Yotiao channels
| Wild type KCNE1 | Y94stop KCNE1 | N79stop KCNE1 | |
|---|---|---|---|
| Activation | |||
| V1/2 (mV) | control; 27.7 ± 3.3 (10) | control; 83.6 ± 5.5# (6) | control; 93.2 ± 2.0# (6) |
| cAMP/OA; 14.8 ± 5.3* (10) | cAMP/OA; 60.1 ± 6.2*# (6) | cAMP/OA; 91.4 ± 2.4#§ (6) | |
| k | control; 18.5 ± 0.7 (10) | control; 21.1 ± 1.1 (6) | control; 16.2 ± 0.9§ (6) |
| cAMP/OA; 18.5 ± 0.5 (10) | cAMP/OA; 18.6 ± 1.2 (6) | cAMP/OA; 16.7 ± 0.4 (6) | |
| Deactivation | |||
| τ (ms) | control; 878 ± 69 (10) | control; 216 ± 18# (6) | control; 120 ± 11# (6) |
| cAMP/OA;1150 ± 105* (10) | cAMP/OA; 297 ± 29*# (6) | cAMP/OA; 145 ± 11# (6) | |
Data obtained in Figure 6 are analyzed here. Activation parameters (apparent V1/2 and slop factor, k) were obtained from data of individual isochronal activation curves. Deactivation time constants (τ) were obtained by fits to deactivating tail currents after test pulses to +100 mV. Numbers of the experiments are shown in parenthesis following data values.
p < 0.05 vs. control, Student’s t-test.
p < 0.05 vs. wild-type KCNE1, ANOVA.
p < 0.05 vs. Y94stop KCNE1, ANOVA.
In order to confirm the involvement of the KCNE1 C-T in the PKA-dependent functional regulation, we introduced chimeras of the N-T domain and transmembrane regions of KCNE3 (M1-Y92) spliced with the C-T domain of KCNE1 (N79-P129). We refer to this construct as KCNE3N/1C. We then tested for functional responses of KCNQ1_ KCNE3N/1C channels to cAMP. Figure 7A shows that the 5-min application of 0.5 mM cpt-cAMP, as done for KCNE2 in Figure 3, did not affect current amplitudes over the activating voltages in cells expressing KCNE3, KCNQ1 and Yotiao, which is consistent with the result using pseudo-phosphorylated KCNQ1 in Figure 2B. Without cAMP stimulation, current amplitudes recorded from cells expressing KCNQ1, Yotiao plus KCNE3N/1C were significantly smaller than those in the KCNQ1/KCNE3/Yotiao channel. In contrast to wild type KCNE3, the application of cpt-cAMP significantly increased the current amplitude of the KCNQ1_KCNE3N/1C_Yotiao channels (Fig. 7B), showing that the KCNE1 C-T domain is sufficient to transfer functional responsiveness to cAMP to the assembled channels. Taken together, our results indicate that C-T domains of the beta-subunits of the KCNE family are critical determinants of the PKA-mediated functional regulation of KCNE/KCNQ1/Yotiao channels.
Figure 7.

A KCNE1 carboxyl terminal fragment is sufficient to confer cAMP-dependent regulation of the KCNQ1/KCNE3/Yotiao channel. Whole-cell currents were measured with perforated patch-clamp configuration from CHO cells transfected with KCNQ1 and Yotiao plus either wild type KCNE3 (A) or a chimera of the N terminus KCNE3 (Met1-Tyr92) spliced with the C terminus KCNE1 (Asn79-Pro129), KCNE3N/1C (B). Each KCNE3 construct is shown in Left. Shown are plots of mean current amplitude ± SEM vs. activating pulse voltage (Center) as well as representative current traces (Right) that were obtained by the same protocol as in Figure 2A. The cAMP-induced changes are described by superimposing I–V plots before (control, open squares) and after the cAMP application (0.5 mM cpt-cAMP, filled squares) as in Figure 3B for KCNE2. Notably, currents in the presence of KCNE3N/1C were responsive to the cAMP stimulation. The numbers of experiments were as follows: wild type KCNE3, n = 4, and KCNE3N/1C, n = 5. *p < 0.05 vs. control, Paired Student’s t-test.
Discussion
In this study, we demonstrate that the functional response to cAMP of K+ channels that consist of the alpha subunit KCNQ1 assembled with different members of the KCNE family of beta subunits depends critically on the KCNE1 variant in the expressed channel. We find cAMP-mediated PKA phosphorylation of KCNQ1 (at residue Ser27 ) is not affected by its expression with the KCNE variants KCNE1, KCNE2 or KCNE3. However, only channels that contain either KCNE1 or KCNE2 respond functionally to KCNQ1 phosphorylation. Comparison of the distinct functional cAMP insensitivity of KCNE3 channels with KCNE1 and KCNE2 channels, we were able to identify critical regions of these beta subunits that are necessary for functional responses of the assembled channels. Comparison of amino acid sequences among KCNE1, KCNE2 and KCNE3 revealed a distinct difference in their C-T domains. Deletion analysis of the KCNE1 C-T domain and a chimeric strategy identified a critical region of KCNE1, which is sufficient to transfer a functional cAMP response to KCNQ1 expressed with the KCNE3N/1C chimera. These results clearly indicate that it is the beta subunit of the assembled channel that confers functionality of the channel to post translational PKA-dependent modification of the alpha subunit and it is the C-T domain of the KCNE family members that is necessary for this critically important physiological response. In the case of KCNE3/KCNQ1 channels, it is possible that, because KCNE3/KCNQ1 channel currents are much larger than either KCNQ1_KCNE2 or KCNQ1_KCNE1 channel currents, KCNE3 already induces an enhanced kinetic state of the channel that can no longer be further added to by PKA phosphorylation. However, even if this is the case, our results with the KCNQ1_KCNE3N/C chimera indicate that the KCNE3 C-T domain plays a critical role in this functional property of the expressed channels.
Co-assembly of widely expressed homotetrameric KCNQ1 channels24 with different KCNE peptides may be a major reason for the diverse gating of native K+ currents. In the heart, co-assembly of KCNE1 and KCNQ1 generates a robust functional demonstration as the IKs channel, resulting in a major contributor to cardiac repolarization.4–6 It is clear from our previous studies15,18 that requirement of KCNE1, which is not a substrate of PKA phosphorylation, for cAMP-mediated regulation of the IKs channel has a significant impact on regulation of the cardiac ventricular action potential duration by the SNS stimulation. Here, we unraveled a molecular mechanism underlying the role of KCNE1 in transduction of KCNQ1 phosphorylation into the functional regulation.
KCNQ1 also conducts almost time-independent K+ currents through channels that favor the open state when co-assembled with KCNE2 or KCNE3, and this clearly contributes to various physiological responses in distinct tissues.7–9 Our results indicate that KCNQ1_KCNE2 channels respond to cAMP in a manner that results in an increase in expressed currents. Furthermore, we confirm KCNE2 does not mediate KCNQ1 phosphorylation at S27 but rather mediates the functional consequences of the phosphorylation, similar to the role of KCNE1 in the IKs channel. Because KCNE2 and KCNE3 are expressed in the heart,25 and KCNE2 has been linked to long-QT syndromes (LQT-6),10 and overexpression of KCNE3 alters cardiac repolarization process,12 the results of our study will impact our understanding of K+ channel currents that may contribute to control of cardiac electrical activity in the face of SNS stimulation. Our results clearly indicate that channels formed by KCNQ1_KCNE2 co-assembly will respond to SNS stimulation, but will result in component K+ channel currents that are increased that have almost a time-independent voltage-dependence (see also Fig. 3).26,27 The magnitude of this contribution to current expressed in the heart will, of course, depend on the relative expression of KCNE2, which occurs at lower levels than KCNE1,25 but also their promiscuous interaction with KV alpha subunits. It is interesting that currents blocked by the KCNQ1 blocker azimilide recorded from normal canine cardiomyocytes showed purely IKs-like properties including gating and the cAMP-dependent regulation, but azimilide-sensitive currents recorded from myocytes in epicardial border zone have a time-independent and β-adrenergic receptor-sensitive component.28 Our data suggest that KCNE2-KCNQ1 channels might very well conduct this azimilide-sensitive current in border zone tissue. Clearly additional work is needed to confirm this interesting possibility.
Clarification of a specific role of each KCNE family beta subunit other than KCNE1 in cardiac physiology and pathophysiology needs further investigation with such approaches as targeted genetic deletion and/or RNAi approaches. Recently, KCNE2 knockout mouse has been introduced to demonstrate that the KCNE2-KCNQ1 channel is essential for cAMP-dependent (histaminergic) gastric acid secretion without redundancy.9 Although the cardiac phenotype has not been indicated in the KCNE2 knockout mice, it would be intriguing to consider involvement of KCNE2-mediated transduction of phosphorylation in gastric secretion. But, parietal cells do not have detectable expression of Yotiao (unpublished data by Kurokawa), although our study shows that KCNE2-mediated transduction occurs in the presence of Yotiao. It is possible that other A-kinase anchoring protein-mediated regulation29 is involved, but this remains unsolved. Although mutations in KCNE3 associate with an inherited human disease, periodic paralysis in skeletal muscle,30 cardiac phenotypes have not been reported.
The experiments we report here extend our previous observations that demonstrated the requirement of KCNE1-KCNQ1 co-assembly for the well-known functional response of IKs channels to cAMP,18 by demonstrating a specific region of the KCNE1 C-T domain which is necessary for this functional transduction in the IKs channel and sufficient to transfer the functional response to a normally unresponsive KCNQ1_KCNE3 channel. This region of KCNE1 is physiologically critical that is reflected by number of LQT-5 mutations reported in it.19–22 The present results are also consistent with our previous finding that the D76N LQT-5 mutation in the KCNE1 C-T domain ablates the functional response of the resulting IKs channels to cAMP.18
How the KCNE1 C-T domain senses a phosphorylation-induced change in charge at KCNQ1residue S27 and then modifies gating of the assembled channel remains to be determined. Clearly our results imply a close proximity of the KCNQ1 N-T and the KCNE1 (and KCNE2) C-T domains, but our results do not prove interactions between the subunits nor do they provide evidence for proximity of these channel structures. However, others have provided evidence for KCNE1 modulation of KCNQ1 through the KCNE1 C-T domain.23,31 Multiple studies have provided evidence either for direct interactions between KCNE1 and KCNQ1 or for sufficiently close arrangements to permit spontaneous cross linking between cysteines substituted into the two subunits.32–34 Thus it is very likely that the phosphorylation-induced change in charge at KCNQ1 S27, either directly or indirectly alters interactions between the KCNQ1 N-T and KCNE1 C-T which, in turn cause multiple effects on channel activity including altered gating, through allosteric interactions that affect channel function through multiple interaction sites.
Experimental Procedures
Molecular biology
The human cDNA clone KCNE2 and KCNE3 were a gift from Dr. G.W. Abbott (Cornell University, New York, NY). Insertion of stop codon either at Tyr94 (Y94stop) or at Asn79 (N79stop) in KCNE1 was performed by plaque-forming unit-based mutagenesis (Quick Change™ site-directed mutagenesis kit; Strategene). KCNQ1 mutants at Ser27 were constructed previously.16 A chimeric KCNE3N/1C fragment was constructed using a PCR-based approach by joining a part of the C terminus of KCNE1 to the N terminus of KCNE3 at an XbaI site at Ser82 and Arg83 in the cytosolic C terminus of KCNE3. Pairs of primers used for construction of the KCNE3N/1C cDNA are as follows (restriction sites underlined): 5′-primer (sense), ACT TGG ATC CAT GGA GAC TAC CAA TGGA, sequence beginning with KCNE3 Met1 including a BamHI restriction site, and 3′-primer (antisense), AGT TTC TAG AGC GGG TGT ATC CCAG, sequence ending with KCNE3 C terminal Arg83 including an XbaI restriction site; 5′-primer (sense), ATT CTC TAG AAA AGT GGA CAA GCG TAG TGA CCC CTA TAA CGT CTA CAT CGA GTCC, sequence overlapping the C terminus of KCNE3 ending with Tyr92 and the C terminus of KCNE1 beginning with Asn79 including an XbaI restriction site, and 3′-primer (antisense), ATC CGA ATT CTC ATG GGG AAG GCT TCGT, sequence ending with KCNE1 Pro129 including an EcoRI restriction site. All cDNA clones for transfection were subcloned into the mammalian vector pcDNA 3.1 (Invitrogen). All sequence manipulations were confirmed by using the chain termination method in the DNA Sequencing Facilities either at Tokyo Medical and Dental University or at Columbia University.
Biochemistry
Chinese Hamster Ovary (CHO) cells were transfected with cDNAs for KCNQ1 (0.4 μg per flask) and Yotiao (2.0 μg per flask) as well as KCNE1, KCNE2 or KCNE3 (0.4 μg per flask). Forty-eight hours after transfection, cells were incubated with 300-μM membrane preamble cpt-cAMP plus 0.2-μM okadaic acid (OA) for 10 minutes to induce PKA-phosphorylation. Control experiments were performed without using cpt-cAMP and OA. Cells were lysed in a 2× sample buffer (5% SDS, 75 mM urea, 300 mM sucrose, 50 mM TrisCl (pH 7.4) and 200 mM DTT). The lysates were then incubated at 50°C for 5 min before being fractionized on 4–20% SDS/PAGE gels. Dual western blots were performed using a rabbit antibody that was raised to recognize the phosphorylated S27 residue on KCNQ1, and a commercial goat anti-KCNQ1 antibody (Santa Cruz Biotechnology). An IRDye 800-conjugated donkey anti-rabbit IgG antibody (Rockland) and an Alexa Fluor 680-conjugated donkey anti-goat IgG antibody (Invitrogen) were used as secondary antibodies. Fluorescence signals from both phosphorylated KCNQ1 and total KCNQ1 were detected simultaneously by an Odyssey infrared imaging system (Li Cor Bioscience). Western blot results were quantified and analyzed using the Odyssey Software (Li Cor Bioscience).
Cell culture and transfection
Chinese hamster ovary (CHO) cells (American Type Cell Culture; USA, Riken; Japan) were cultured in Ham’s F12 medium and transiently transfected with cDNAs for KCNQ1, KCNE1–3, CD8 and Yotiao (0.4 μg, 0.4 μg, 0.4 μg and 2 μg, respectively) using LipofectAMINE with LipofectAMINE-PLUS reagents (Invitrogen) as previously reported by us.18 Transfected cells were plated in culture dishes and cultured in an incubator with 5% CO2 until use (<48 h). Dynabeads M-450 anti-CD8 beads (Dynal ASA, Oslo, Norway) were used to visually identify transfected cells.
Electrophysiology
Currents were recorded by either using the whole-cell patch-clamp technique for KCNE1 or using a perforated configuration of patch-clamp technique to minimize run-down of currents for KCNE2 or KCNE3.18,35 Cells in culture dishes were placed on the stage of an inverted microscope (IMT-2 or IX-71, Olympus), and the culture medium was replaced by Tyrode’s solution (132 mM NaCl/4.8 mM KCl/2 mM CaCl2/1.2 mM MgCl2/5 mM glucose/10 mM HEPES, pH 7.4) including Dyna-beads M-450 before measurements of currents. All measurements were made at the room temperature (22 ± 2°C). Pipette resistance of borosilicate glass electrodes was 2–4 MΩ when filled with the internal solution composed of 110 mM K-aspartate, 5 mM ATP-K2, 11 mM EGTA, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES (pH 7.3). In whole-cell patch-clamp experiments, series resistance was 3.5–8 Mω. Perforated patch-clamp experiments were performed by adding amphotericin B (0.3 mg/mL; Nacalai, Japan) to the internal solution.35,36 Adequate series resistance (<15 MΩ) were usually attained within 3-min of the GΩ seal formation.
Currents were recorded with Axopatch 200A or 200B amplifiers (Axon Instruments). Signals were low-pass filtered at 2 kHz, sampled at 1 kHz, and compensated for cell capacitance. Series resistance was not compensated, and the liquid junction potential was not corrected. KCNQ1/KCNE1 currents were studied by analysis of peak deactivating tail currents recorded at −40 mV following a broad range of 2-s activating pulses. These isochronal activation curves were fit to a Boltzmann curve using the equation:
where I is the measured tail current; V is the applied activating voltage; V1/2 is the apparent voltage of half activation; and k is the slope factor. Deactivating tail currents were fit with functions containing single exponential time components. KCNQ1/KCNE2, KCNQ1/KCNE3 and KCNQ1/KCNE3N/1C currents were studied by analysis of currents recorded at the end of a broad range of 2-s activating pulses. Pulse frequency was 0.067 Hz unless otherwise noted.
Cyclic AMP stimulation was performed differently in perforated patch-clamp experiments and in whole-cell patch-clamp experiments. In perforated patch-clamp experiments for KCNQ1/KCNE2 or /KCNE3 channels, cpt-cAMP (Sigma) was dissolved with the Tyrode’s solution, and was applied from the extracellular side of the membrane. After achieving the patch perforation, channel activity was monitored by analysis of currents recorded by test pulses to 0 mV. In whole-cell patch-clamp experiments for the KCNQ1/KCNE1 channel, after rupture of the cell membrane, internal solutions without cAMP (control) or with 0.2 mM cAMP plus 0.2-μM okadaic acid (OA) (Calbiochem), a phosphatase inhibitor, were dialyzed for 12 min before measurements were made. During the dialysis, channel activity was monitored by analysis of tail currents recorded by test pulses to +60 mV.
Data analysis
All values are presented as mean ± SEM. PCLAMP software Ver. 8 or 9 (Axon Instruments) was used both to acquire and analyze data for the patch-clamp experiments. Graphical (fitting) and statistical analyses were carried out using Origin 7.0 software (Microcal, Northhampton, MA) and InStat program (GraphPad), respectively. Statistical significance was assessed with Student’s t test for simple comparisons or ANOVA followed by Bonferroni’s post test for multiple comparisons: differences at p < 0.05 were considered to be significant.
Acknowledgments
We thank Dr. GW Abbott (Cornell University) for KCNE2 and KCNE3 cDNAs, Ms. Jenny Rao (Columbia University) and Ms. Eri Ozaki (Tokyo Medical and Dental University) for technical assistance.
This work was supported in part by a Grant from the Ministry of Education, Science, Culture, Sports and Technology of Japan (17790167 and 19689006, Junko Kurokawa), Grant-in-Aid for Scientific Research on Priority Areas (17081007, Tetsushi Furukawa), research grants from the Nagai Foundation Tokyo (Junko Kurokawa), and the Naito Foundation (Junko Kurokawa) and NIH grant 5R01HL44365-11 (Robert S. Kass).
Abbreviations
- IKs
a slowly-activated delayed rectifier potassium current
- SNS
sympathetic nervous system
- PKA
cAMP-dependent protein kinase
- APD
action potential duration
- CHO
chinese hamster ovary
- OA
okadaic acid
- C-T
carboxyl terminus
- N-T
amino terminus
References
- 1.Jenkinson DH. Potassium channels—multiplicity and challenges. Br J Pharmacol. 2006;147:63–71. doi: 10.1038/sj.bjp.0706447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 3.Kurokawa J, Abriel H, Kass RS. Molecular basis of the delayed rectifier current IKs in heart. J Mol Cell Cardiol. 2001;33:873–82. doi: 10.1006/jmcc.2001.1377. [DOI] [PubMed] [Google Scholar]
- 4.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–3. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 5.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KVLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 6.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 7.Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 2000;19:6326–30. doi: 10.1093/emboj/19.23.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, et al. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403:196–9. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- 9.Roepke TK, Anantharam A, Kirchhoff P, Busque SM, Young JB, Geibel JP, Lerner DJ, Abbott GW. The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J Biol Chem. 2006;281:23740–7. doi: 10.1074/jbc.M604155200. [DOI] [PubMed] [Google Scholar]
- 10.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1 and KCNE2. Circulation. 2000;102:1178–85. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 11.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–4. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 12.Mazhari R, Nuss HB, Armoundas AA, Winslow RL, Marban E. Ectopic expression of KCNE3 accelerates cardiac repolarization and abbreviates the QT interval. J Clin Invest. 2002;109:1083–90. doi: 10.1172/JCI15062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll SJ, Kurokawa J, Kass RS. chapter 17 in Cardiac Electrophysiology from Cell to Bedside. Philadelphia: Sanders; 2004. [Google Scholar]
- 14.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 15.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–9. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 16.Kurokawa J, Motoike HK, Rao J, Kass RS. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc Natl Acad Sci USA. 2004;101:16374–8. doi: 10.1073/pnas.0405583101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Kurokawa J, Kass RS. Phosphorylation of the A-kinase-anchoring protein Yotiao contributes to protein kinase A regulation of a heart potassium channel. J Biol Chem. 2005;280:31347–52. doi: 10.1074/jbc.M505191200. [DOI] [PubMed] [Google Scholar]
- 18.Kurokawa J, Chen L, Kass RS. Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc Natl Acad Sci USA. 2003;100:2122–7. doi: 10.1073/pnas.0434935100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abitbol I, Peretz A, Lerche C, Busch AE, Attali B. Stilbenes and fenamates rescue the loss of IKs channel function induced by an LQT5 mutation and other IsK mutants. EMBO J. 1999;18:4137–48. doi: 10.1093/emboj/18.15.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–40. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 21.Lai LP, Su YN, Hsieh FJ, Chiang FT, Juang JM, Liu YB, et al. Denaturing high-performance liquid chromatography screening of the long QT syndrome-related cardiac sodium and potassium channel genes and identification of novel mutations and single nucleotide polymorphisms. J Hum Genet. 2005;50:490–6. doi: 10.1007/s10038-005-0283-3. [DOI] [PubMed] [Google Scholar]
- 22.Bianchi L, Shen Z, Dennis AT, Priori SG, Napolitano C, Ronchetti E, et al. Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum Mol Genet. 1999;8:1499–507. doi: 10.1093/hmg/8.8.1499. [DOI] [PubMed] [Google Scholar]
- 23.Tapper AR, George AL., Jr MinK subdomains that mediate modulation of and association with KVLQT1. J Gen Physiol. 2000;116:379–90. doi: 10.1085/jgp.116.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwake M, Jentsch TJ, Friedrich T. A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 2003;4:76–81. doi: 10.1038/sj.embor.embor715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lundquist AL, Manderfield LJ, Vanoye CG, Rogers CS, Donahue BS, Chang PA, et al. Expression of multiple KCNE genes in human heart may enable variable modulation of IKs. J Mol Cell Cardiol. 2005;38:277–87. doi: 10.1016/j.yjmcc.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 26.Wu DM, Jiang M, Zhang M, Liu XS, Korolkova YV, Tseng GN. KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of IKs current amplitude in the heart. Heart Rhythm. 2006;3:1469–80. doi: 10.1016/j.hrthm.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 27.Melman YF, Krummerman A, McDonald TV. KCNE regulation of KVLQT1 channels: structure-function correlates. Trends Cardiovasc Med. 2002;12:182–7. doi: 10.1016/s1050-1738(02)00158-5. [DOI] [PubMed] [Google Scholar]
- 28.Dun W, Boyden PA. Diverse phenotypes of outward currents in cells that have survived in the 5-day-infarcted heart. Am J Physiol Heart Circ Physiol. 2005;289:667–73. doi: 10.1152/ajpheart.00180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potet F, Scott JD, Mohammad-Panah R, Escande D, Baro I. AKAP proteins anchor cAMP-dependent protein kinase to KVLQT1/IsK channel complex. Am J Physiol Heart Circ Physiol. 2001;280:2038–45. doi: 10.1152/ajpheart.2001.280.5.H2038. [DOI] [PubMed] [Google Scholar]
- 30.Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, Goldstein SA. MiRP2 forms potassium channels in skeletal muscle with KV3.4 and is associated with periodic paralysis. Cell. 2001;104:217–31. doi: 10.1016/s0092-8674(01)00207-0. [DOI] [PubMed] [Google Scholar]
- 31.Gage SD, Kobertz WR. KCNE3 truncation mutants reveal a bipartite modulation of KCNQ1 K+ channels. J Gen Physiol. 2004;124:759–71. doi: 10.1085/jgp.200409114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung DY, Chan PJ, Karlin A, Marx SO, Liu G, Kass RS. Cysteine substitution reveals novel inter-subunit interactions in the IKs potassium channel. Biophys J. 2008;94:82. [Google Scholar]
- 33.Nakajo K, Kubo Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J Gen Physiol. 2007;130:269–81. doi: 10.1085/jgp.200709805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu X, Jiang M, Hsu KL, Zhang M, Tseng GN. KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J Gen Physiol. 2008;131:589–603. doi: 10.1085/jgp.200809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai CX, Namekata I, Kurokawa J, Tanaka H, Shigenobu K, Furukawa T. Role of nitric oxide in Ca2+ sensitivity of the slowly activating delayed rectifier K+ current in cardiac myocytes. Circ Res. 2005;96:64–72. doi: 10.1161/01.RES.0000151846.19788.E0. [DOI] [PubMed] [Google Scholar]
- 36.Kurokawa J, Tamagawa M, Harada N, Honda S, Bai CX, Nakaya H, et al. Acute effects of oestrogen on the guinea pig and human IKr channels and drug-induced prolongation of cardiac repolarization. J Physiol. 2008;586:2961–73. doi: 10.1113/jphysiol.2007.150367. [DOI] [PMC free article] [PubMed] [Google Scholar]