

Abstract
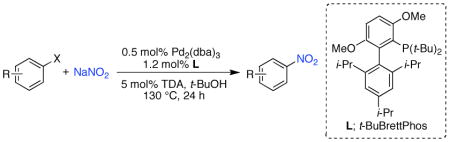
An efficient Pd-catalyst for the transformation of aryl chlorides, triflates and nonaflates to nitroaromatics has been developed. This reaction proceeds under weekly basic conditions and displays a broad scope and excellent functional group compatibility. Moreover, this method allows for the synthesis of aromatic nitro compounds that cannot be accessed efficiently via other nitration protocols. Mechanistic insight into the trasmetallation step of the catalytic process is also reported.
Aromatic nitro compounds are of great importance to a variety of disciplines. They can be found in an array of pharmaceuticals,1 dyes,2 and materials.3 Moreover, they are an important entity in synthesis and can participate in a range of useful transformations.4 Most commonly, nitrobenzenes are synthesized via electrophilic aromatic substitution using HNO3 and a strong acid or with dinitrogen pentoxide.4 However, these methods suffer from issues of poor regioselectivity and imperfect functional group tolerance. Further, the application of these reactions is particularly problematic for the nitration of heteroaromatics. More recently, Prakash and Olah disclosed a method for the conversion of aryl boronic acids to aryl nitro compounds.5 This was followed by a report from Saito disclosing a copper catalyst for the transformation of aryl iodides to nitrobenzenes.6 These processes were important advances in this field and provided complimentary nitration protocols to known chemistry; however, the substrate scope of these reactions was limited and did not include heterocycles.
Herein, we report a general Pd catalyst for the conversion of aryl chlorides, triflates, and nonaflates to nitroaromatics.7 This protocol circumvents issues of regioselectivity and proceeds efficiently under weakly basic conditions, allowing for excellent functional group compatibility. Moreover, it enables the formation of nitroarenes that cannot be accessed effectively by other means, most notably heteroaryl nitro-substituted compounds. Mechanistic insight into the “transmetallation” step of this catalytic process is also provided.
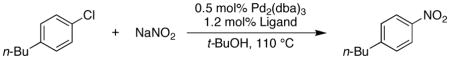
We began our studies by examining the reaction of 4-chloro-n-butylbenzene with commercially available nitrite sources. We hypothesized that a catalyst based on ligand 1 (Figure 1), which we have shown to be effective in amidation reactions of aryl chlorides,8 would be useful for this transformation as we felt that N–Pd bond formation would be difficult as in the latter transformation. Using 0.5 mol% Pd2(dba)3, 1.2 mol% 1, and sodium nitrite in t-BuOH 26% of the desired 4-nitro-n-butylbenzene was obtained (Table 1). Due to the fact that the sodium nitrite is sparingly soluble in t-BuOH, we reasoned that use of a phase transfer catalyst might be beneficial. Adding 5 mol% tris(3,6-dioxaheptyl)amine (TDA) improved the yield to 52%, whereas the use of other phase transfer catalysts did not have a positive influence on the outcome of the reaction.
Figure 1.

Biarylphosphine ligands.
Table 1.
Pd-catalyzed formation of 4-nitro-n-butylbenzene.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | L | 5% PTC | Yielda | Entry | L | 5% PTC | Yielda |
| 1 | 1 | -- | 26% | 6 | 3 | TDA | 10% |
| 2 | 1 | TDA | 52% | 7 | 4 | TDA | 0% |
| 3 | 1 | TBACl | 3% | 8 | 5 | TDA | 0% |
| 4 | 1 | 15-C-5 | 33% | 9 | 1 | -- | 2%b |
| 5 | 2 | TDA | 0% | 10 | 1 | TDA | 99%c |
4-chloro-n-butylbenzene (1 mmol), NaNO2 (2.0 mmol), Pd2(dba)3 (0.5 mol%), ligand (1.2 mol%), t-BuOH (2 mL), 110 °C, 24 h; GC yields.
(TBA)NO2 was used.
Reaction temperature was 130 °C.
Catalysts based on other biarylphosphine ligands (2,8 3,9 4, and 5)10 gave little to no desired product in these reactions. This demonstrates that in ligand 1 the di-t-butylphosphino group, as well as the methoxy substituent ortho to the phosphine are critical to the reactivity of this catalyst system.
We observed that as the amount of TDA was increased from 0 – 5 mol% the yield of the reaction also increased. However, the amount of product generated in these reactions dropped quickly when more than 5% of the TDA was used (Figure 2).11 We reasoned that higher concentrations of nitrite in solution could oxidize the Pd(0) or ligand, which would explain the lower yields at higher loadings of TDA. To test this postulate we ran the reaction with a soluble source of nitrite (tetrabutylammonium nitrite) and obtained only 2% 4-nitro-n-butylbenzene (Table 1, entry 9). These studies show that having the optimal amount of nitrite in solution is essential to the outcome of the reaction.
Figure 2.
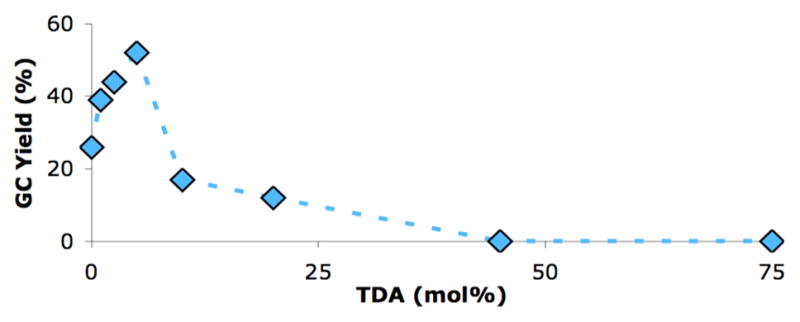
Effect of TDA on the yield for the Pd-catalyzed formation of 4-nitro-n-butylbenzene.
By increasing the reaction temperature, we were able to convert 4-chloro-n-butylbenzene to the desired nitro product in virtually quantitative yield (Table 1, entry 11). Using these conditions we set out to explore the scope of the Pd-catalyzed transformation of aryl chlorides to nitro aromatics using a catalyst based on ligand 1 (Table 2). Ortho substituents, as well as acid-sensitive functional groups, such as an acetal, pyrrole, and free alcohol, were all well tolerated. Nitro arenes containing electron-withdrawing groups or amines in the para position and electron-donating substituents in the meta position were synthesized in high yields with this method; these compounds cannot be efficiently formed through traditional nitration protocols. Nitro heteroaromatics could also be prepared in good to excellent yields. For example, 5- and 7-chloroindole were transformed to the corresponding nitroindoles in good yields without the need of a protecting group. This catalyst system is complimentary to current nitration protocols and allows for the synthesis of nitro aromatic compounds that are not readily accessible via other means.
Table 2.
Conversion of aryl chlorides nitro aromatics.a
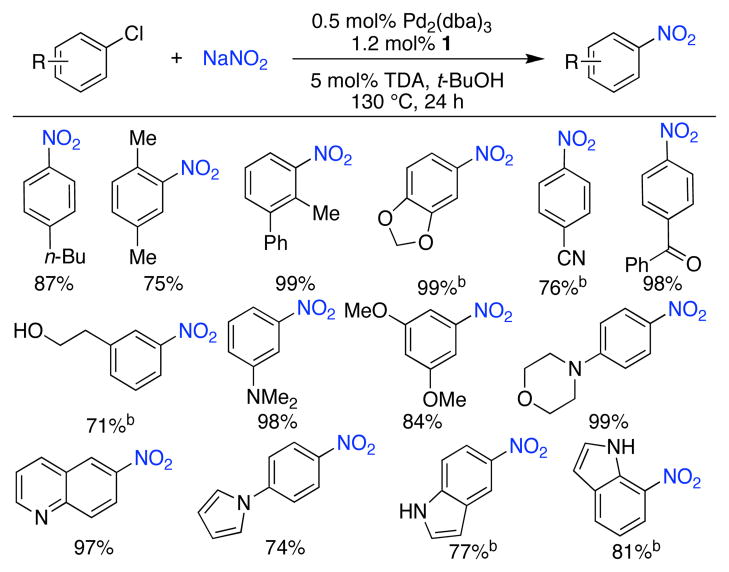 |
ArCl (1 mmol), NaNO2 (2.0 mmol), Pd2(dba)3 (0.5 mol%), 1 (1.2 mol%), t-BuOH (2 mL), 130 °C, 24 h; isolated yields, average of 2 runs.
Pd2(dba)3 (2.5 mol%), 1 (6 mol%).
Based on the broad scope this catalyst displayed for the nitration of aryl chlorides, we decided to explore nitrations of aryl bromides and iodides. Under the optimized reaction conditions 3,5-dimethylbromobenzene (6) and 3,5-dimethyliodobenzene (7) gave 44% and 0% of the desired product, respectively (Figure 3). In order to aid our interpretation of this result a competition experiment between the aryl bromide or iodide and aryl chloride were performed. In experiment A, where a 1:1 mixture of 6 and 8 was subjected to the reaction conditions, 44% of the aryl bromide-derived and none of the aryl chloride-derived coupling products were observed. In experiment B, where a 1:1 mixture of 7 and 8 was used, no nitroaromatic products were formed. These results demonstrate that the catalyst is undergoing oxidative addition with the aryl iodide or bromide faster than with the aryl chloride and that transmetallation is the slow (problematic) step for these processes.12 Moreover, this establishes that the rate of transmetallation follows the order of Cl>Br>I in these reactions.
Figure 3.

Reactions of aryl bromides and iodides.
Based on these results and previous findings in our group,10 we postulated that reactions of aryl triflates and nonaflates would proceed with an accelerated rate of transmetallation and be suitable substrates for these reactions. Using a catalyst comprised of 1 and Pd2(dba)3, an array of aryl nonaflates and triflates were converted to product in good to excellent yields (Table 3), including those containing esters and nitriles; heteroaryl triflates were also transformed in high efficiencies.
Table 3.
Conversion of aryl triflates and nonaflates to nitro aromatics.a
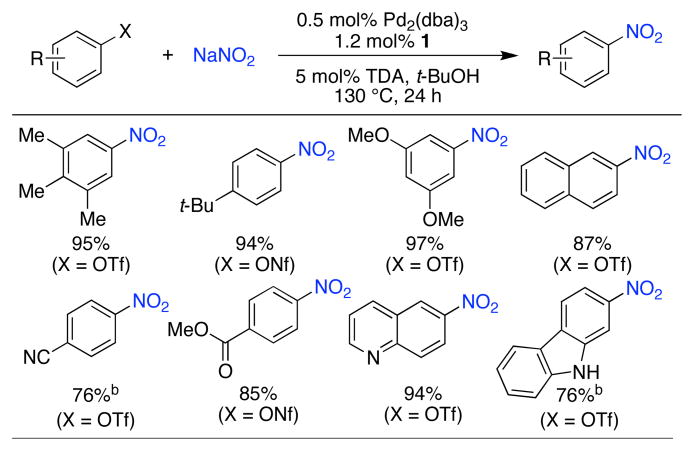 |
ArX (1 mmol), NaNO2 (2.0 mmol), Pd2(dba)3 (0.5 mol%), 1 (1.2 mol%), t-BuOH (2 mL), 130 °C, 24 h; isolated yields, average of 2 runs.
Pd2(dba)3 (2.5 mol%), 1 (6 mol%).
In summary, an effective catalyst for the conversion of aryl chlorides, triflates, and nonaflates to nitroaromatics has been developed. This method is complimentary to previous nitration protocols and evades issues of regioselectivity and functional group compatibility. It was also shown that the rate of transmetallation follows the order of Cl>Br>I in these reactions. Further studies to better understand the mechanism of the reaction and extend its scope to aryl iodides, bromides, and tosylates are currently underway in our laboratories.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIH) for financial support of this project (GM-58160). We thank Merck, BASF, and Nippon Chemical for additional support. B.P.F. thanks Merck and the William Asbornsen Albert Memorial Fund for fellowships.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Procedural and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Muller WE. The benzodiazepine receptor. Cambridge University Press; New York: 1988. [Google Scholar]; (b) Belciug M, Ananthanarayanan VS. J Med Chem. 1994;37:4392. doi: 10.1021/jm00051a017. [DOI] [PubMed] [Google Scholar]
- 2.Zollinger H. Color Chemistry. Wiley-VCH; New York: 1987. p. 161. [Google Scholar]
- 3.Fan FF, Yao Y, Cai L, Cheng L, Tour JM, Bard AJ. J Am Chem Soc. 2003;126:4035. doi: 10.1021/ja0359815. [DOI] [PubMed] [Google Scholar]
- 4.Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH; New York: 2001. [Google Scholar]
- 5.(a) Prakash GKS, Panja C, Mathew T, Surampudi V, Petasis NA, Olah GA. Org Lett. 2004;6:2205. doi: 10.1021/ol0493249. [DOI] [PubMed] [Google Scholar]; (b) Salzbrunn S, Simon J, Prakash GKS, Petasis NA, Olah GA. Synlett. 2000:1485. [Google Scholar]
- 6.Saito S, Koizumi Y. Tetrahedron Lett. 2005;46:4715. [Google Scholar]
- 7.For Pd-catalyzed amination reactions, see: Surry DS, Buchwald SL. Angew Chem, Int Ed. 2008;47:6338. doi: 10.1002/anie.200800497.Hartwig JF. Synlett. 2006:1283.
- 8.Fors BP, Dooleweerdt K, Zeng Q, Buchwald SL. Tetrahedron. 2009;65:6576. doi: 10.1016/j.tet.2009.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fors BP, Watson DA, Biscoe MR, Buchwald SL. J Am Chem Soc. 2008;130:13552. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikawa T, Barder TE, Biscoe MR, Buchwald SL. J Am Chem Soc. 2007;129:13001. doi: 10.1021/ja0717414. [DOI] [PubMed] [Google Scholar]
- 11.For another example of a phase transfer catalyst having a detrimental effect on the reaction after a certain point, see: Kuwano R, Utsunomiya M, Hartwig JF. J Org Chem. 2002;67:6479. doi: 10.1021/jo0258913.
- 12.It is well known that the rate of oxidative addition for I>Br>Cl, see: Barrios-Landeros F, Carrow BP, Hartwig JF. J Am Chem Soc. 2009;131:8141. doi: 10.1021/ja900798s.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.