

Abstract
Peripheral neuropathy is a broad category of disorders with a diverse etiology, grouped together by their common pathogenic effect on the peripheral nervous system (PNS). Because of the heterogeneity observed to be responsible for these disorders, a forward genetics method of gene discovery was used to identify additional affected pathways. In this report, we describe the mutant mouse line 20884, generated by N-ethyl-N-nitrosourea mutagenesis, which is characterized by adult-onset transitory hindlimb paralysis. Linkage mapping revealed that two point mutations are responsible for the phenotype: a partial loss-of-function mutation in the gene for phosphatidate phosphatase Lpin1 and a truncation mutation in the gene that encodes the neuronal cell adhesion molecule NrCAM. To investigate how the 20884 Lpin1 and Nrcam mutations interact to produce the paralysis phenotype, the double mutant and both single mutants were analyzed by quantitative behavioral, histological, and electrophysiological means. The Lpin120884 mutant and the double mutant are characterized by similar levels of demyelination and aberrant myelin structures. Nevertheless, the double mutant exhibits more severe electrophysiological abnormalities than the Lpin120884 mutant. The Nrcam20884 mutant is characterized by normal sciatic nerve morphology and a mild electrophysiological defect. Comparison of the double mutant phenotype with the two single mutants does not point to an additive relationship between the two defects; rather, the Lpin120884 and Nrcam20884 defects appear to act synergistically to produce the 20884 phenotype. It is proposed that the absence of NrCAM in a demyelinating environment has a deleterious effect, possibly by impairing the process of remyelination.
Introduction
Peripheral neuropathy is a class of diverse diseases grouped by their shared affect on the peripheral nervous system (PNS). Diabetes is one of the most common causes of peripheral neuropathy; it is estimated that 45–50% of the growing population with type 2 diabetes will experience neuropathic complications (Yagihashi et al., 2007). Other acquired forms of neuropathy result from exposure to toxic chemicals, including chemotherapeutics, physical injury, nutrient deficiency, HIV, and aberrant immune responses after infection (Péréon et al., 2003).
Additionally, there are numerous inherited neuropathies, which have at their root a remarkable degree of genetic heterogeneity with respect to both the functional diversity of the underlying genes and the allelic variety within these genes (Douglas and Popko, 2009). Causative genes run the gamut from those that have specific nerve-related functions to those that are ubiquitously expressed yet yield exclusively neuropathic phenotypes when altered. Among the former are myelin-specific genes, such as peripheral myelin protein (Pmp22), which, depending on the genetic defect, is responsible for Charcot–Marie–Tooth (CMT) 1A, hereditary neuropathy with liability to pressure palsies, or Dejerine–Sottas neuropathy (Meyer Zu Hörste and Nave, 2006). Similarly, mutations in neuronally specific genes, such as the calcium channel subunit Cacna1a gene (Plomp et al., 2000; Kaja et al., 2007a, 2007b) and the sodium channel gene Scn8a (De Repentigny et al., 2001; Meisler et al., 2004), result in allele-specific neuronal abnormalities. Alternatively, many genes responsible for peripheral nerve disorders are expressed in a wide variety of tissues. For example, the Gars gene product is ubiquitously required for the synthesis of glycyl-tRNA; however, point mutations in this gene are specifically associated with dominant CMT2A and distal hereditary motor neuropathy V (Antonellis et al., 2003; Seburn et al., 2006).
Each of the above genes exemplifies not only the genetic heterogeneity of peripheral neuropathy but also the phenotypic heterogeneity associated with many neuropathy disease genes (Douglas and Popko, 2009). For example, Pmp22 results in different disorders depending on whether the mutant allele is a point mutation, duplication, or deletion. This unpredictable genetic variability made forward genetics an attractive approach to discovering new pathways important to the development, maintenance, and pathogenesis of the PNS.
The N-ethyl-N-nitrosourea (ENU) mutagenized mutant mouse strain 20884 was initially chosen for study because of its dramatic phenotype: adult-onset hindlimb paralysis. Interestingly, the mapping of 20884's underlying mutation revealed that two mutations, linked at a distance of 29 Mb, were responsible: a novel missense mutation in the phosphatidate phosphatase gene Lpin1 and a nonsense mutation in the neural cell adhesion molecule gene Nrcam. Lipin-1 and NrCAM have seemingly unrelated roles in the PNS, yet when deficiencies are introduced into both genes simultaneously, the result is a phenotype much more severe than would be expected from a simple addition of the two mild single mutant phenotypes. Histological, electrophysiological, and morphometric comparisons of the two single mutants with the double mutant suggest that the two mutations act synergistically to result in muscle atrophy and paralysis in the double mutant.
Materials and Methods
Mice.
The strain ENU20884 arose on the C57BL/6J (C57) background during an ENU mutagenesis screen at the Mouse Heart, Lung, Blood and Sleep Disorders Center (The Jackson Laboratory) (Clark et al., 2004). ENU20884 heterozygous mice were maintained by sibling crosses and by breeding with C57BL/6J mice (stock #000664; The Jackson Laboratory). Additional strains BALB/cJ (BALB; stock #000651) and Lpin1fld (Stock #001592) were also obtained from The Jackson Laboratory. Mice were housed under pathogen-free conditions and all animal studies were performed in compliance with the University of Chicago's Institutional Animal Care and Use Committee guidelines.
Positional cloning and genotyping.
The chromosome 12 critical region was identified using a panel of 78 simple sequence length polymorphism (SSLP) markers. Additional markers within this region were added as required. Intercross breeding, genotyping (Traka et al., 2008), and candidate gene sequencing (Chen et al., 2007) were performed as previously described. Initially, mutants presenting by 6 weeks of age were genotyped; later, all F2 progeny were genotyped and observed until at least 10 weeks.
Mice were genotyped for the 20884 alleles by PCR amplification using the following primers: Lpin1-L 5′-CTGTCTCCCCTGCTCACTTC-3′, Lpin1-R 5′-CCACATATCAGCCTTCCCAG-3′, NrCAM-L 5′-CAGATCAATTGTGTAAATAGAAGTGTG-3′, and NrCAM-R 5′-AATGCCTTTTAAATTCCCCG-3′. RsaI digestion of Lpin1 amplicons produces a 222 bp wild-type fragment and a 267 bp 20884 allele fragment. Nrcam amplicons digested by Hpy188I digestion of Nrcam produce a 630 bp wild-type fragment and a 725 bp 20884 allele fragment. Mice were genotyped for the fld allele of Lpin1 by a three-primer reaction using the primers p1, p3, and p4 described previously (Péterfy et al., 2001).
20884 fld interbreeding. Lpin1fld/fld mutant females were bred to Lpin120884/20884 males to generate Lpin1fld/20884 progeny, which were observed for phenotype onset. Additional progeny were generated by Lpin1fld/fld × Lpin120884/+ and Lpin1fld/+ × Lpin120884/+ crosses to generate wild-type littermates for histological comparison.
To generate Lpin1fld/fld Nrcam20884/20884 double mutants, Lpin1fld/fld and Lpin1fld/+ females were crossed with Nrcam20884/+ males. Lpin1fld/+ Nrcam20884/+ progeny were retained and bred to either wild-type C57BL/6J or BALB/cByJ mice to screen for recombinant Lpin1fld/+ Nrcam20884/+ progeny; the two resulting lines were kept separate. The recombinant double heterozygotes were then intercrossed to generate Lpin1fld/fld Nrcam20884/20884 double mutant progeny, which were observed for phenotype onset.
Protein isolation and Western blotting.
Wild-type and Lpin120884 Nrcam20884 double mutant brains were homogenized in 50 mm Tris-HCl, pH 7.2, containing 1% SDS, 1 mm PMSF, and Complete Protease Inhibitor Tablets (Roche Diagnostics), and centrifuged at 10,000 rpm at 4°C for 15 min, and the supernatant was retained. Protein concentrations were measured using the Bio-Rad Protein Assay (Bio-Rad). Thirty micrograms of protein was separated on a 4–20% SDS-PAGE gel, transferred to a nitrocellulose membrane, blocked with Tris-buffered saline–Tween 20 (TBST) buffer containing 5% nonfat dry milk, and incubated overnight with polyclonal anti-NrCAM raised against the extracellular domain of the protein (ab24344, Abcam) diluted 1:500 in blocking buffer. Immunopositive bands were detected using the Enhanced Chemiluminescence detection system (Amersham). Blots were washed with TBST and reincubated with anti-α-actin mouse monoclonal antibody (1:1000 dilution, Sigma) as a loading control.
Behavior, grip strength, and electrophysiology.
Grip strength and electrophysiology measurements were recorded as previously described (Levedakou et al., 2005) except for the following alterations. The grip strength was measured from both the forelimbs and hindlimbs of six pairs of mutant/wild-type littermates (three male, three female) for the three mutant genotypes (Lpin120884 mutant, Nrcam20884 mutant, Lpin120884 Nrcam20884 double mutant) at 2 week intervals from 4 to 16 weeks of age, and then every 4 weeks until 52 weeks of age. At each time point, weight was recorded and gait was observed. Electrophysiological recordings were taken from the sciatic nerve at 4, 8, and 12 weeks from at least five mice per genotype and from the sural nerve at 8 and 12 weeks from at least three mice per genotype.
Histology.
To obtain hindlimb muscle cryosections for hematoxylin and eosin staining, animals were deeply anesthetized by intraperitoneal injection with avertin (0.5 mg/g), and the muscle tissues were harvested and snap-frozen in isopentane precooled in liquid nitrogen. Sciatic nerve cross-sections were processed for toluidine blue staining and EM as described previously (Dupree et al., 1998).
Morphometric analysis.
Sciatic nerve g-ratios were calculated from the nerves of three mice for each genotype (Lpin120884 mutant, Nrcam20884 mutant, Lpin120884 Nrcam20884 double mutant) at four time points (4, 8, 12, and 52 weeks). Axon and fiber circumference and area were measured using NIH ImageJ software from 20 (4–12 weeks) or 25 (52 weeks) 16.52 μm2 images per nerve. The g-ratios were calculated as axon diameter/fiber diameter as described previously (Auer, 1994). The numbers of unmyelinated and hypomyelinated axons were obtained from the same images as the g-ratio data. Hypomyelinated axons were defined as myelinated fibers whose g-ratios fell above the 95th-percentile value of wild-type myelinated fibers at a given age. Unmyelinated and hypomyelinated axon counts were normalized against the total number of axons counted per nerve and expressed as a percentage.
Sample preparation for phosphatidate phosphatase type 1 activity assay.
Tissues (brain, liver, inguinal subcutaneous white adipose, hindlimb muscle, and sciatic nerve) were harvested from 4-week-old wild type, Lpin120884 mutants, and Lpin120884 Nrcam20884 double mutants. Sciatic nerves from three mice were pooled for each sciatic nerve sample. Tissues were homogenized in 0.25 m sucrose containing 2 mm dithiothreitol, 0.15% Tween 20, and Complete Protease Inhibitor Tablets (Roche Diagnostics) and frozen to preserve PAP activity. Total protein concentrations were measured using the Bio-Rad Protein Assay (Bio-Rad). Phosphatidate phosphatase type 1 (PAP1) activity was calculated after subtracting lipid phosphate phosphatase activity from the total measured production of diacylglycerol from phosphatidate. The activity assay and lipin-1 Western blots were performed as described by Manmontri et al. (2008).
Statistical analysis.
All results are expressed as the mean ± SD. Statistical analysis was performed using SigmaStat software (Systat Software Inc.). Grip strength data were analyzed by a two-tailed distribution Student's t test after testing for equal variance; equal variance was not assumed for data sets failing the equal-variance test. In cases in which the data failed the normality test, a Mann–Whitney rank sum test was used. Weight, electrophysiological measurements, and morphometric data were analyzed by one-way ANOVA within age groups. Enzymatic activity data were analyzed by one-way ANOVA by tissue. Post hoc pairwise comparisons were performed using the Holm–Sidak method. In cases in which the normality or equal-variance tests failed, the Kruskal–Wallis one-way ANOVA on ranks was used, followed by post hoc analysis by Dunn's method. In cases in which post hoc analysis failed to specify pairwise differences, the appropriate t test was used; these instances are denoted by a dagger (†). Differences were considered to be statistically significant if p < 0.05.
Results
20884 mutants are characterized by an adult-onset, transitory paralysis phenotype
The 20884 strain arose on the C57 background during a recessive ENU mutagenesis screen. Mutants of the 20884 strain appear normal at 4 weeks but by 5 weeks develop a floppy gait that progresses to hindlimb paralysis by 6 or 7 weeks of age (Fig. 1; supplemental Video 1, available at www.jneurosci.org as supplemental material). This paralysis is characterized by partial to total loss of hindlimb joint mobility (Fig. 1D), the inability of the hindpaws to grip structures (Fig. 1E), and the clenching of the hindlimbs to the body when suspended by the tail (Fig. 1F), a common neuropathic phenotype. The paralyzed mutants are also identifiable by visibly wasted hindquarter muscles that are characterized by widespread atrophic fibers (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Additionally, males do not breed; females are able to produce normal-sized litters but appear incapable of suckling their progeny.
Figure 1.

20884 mutant phenotype. At 6–7 weeks of age, the hindquarters of 20884 mutant mice (D) become noticeably wasted compared with those of the wild type (A), and the hindlimbs become paralyzed, with the joints frozen in a bent position. 20884 mutants are unable to grip structures with their hindpaws (E), compared with the wild type (B), and they clench their hindlimbs to their body when suspended by the tail (F), unlike wild-type mice, which splay their limbs and toes (C). A, D, Seven weeks old; B, C, E, F, 3.5 months old.
Interestingly, when observed over time, 20884 mutants demonstrate an attenuation of the paralysis phenotype (supplemental Video 1, available at www.jneurosci.org as supplemental material). At the onset of paralysis, likely as a consequence of rapid muscle wasting, the hindlimb joints are stiff and restricted to a bent position. After ∼14 weeks of age, however, the wasted appearance of the 20884 mutants gradually becomes less obvious, while the hindlimbs take on a flaccid disposition and progressively regain mobility and strength. Between 8 months and 1 year of age, these mutants regain the ability to both grip structures and walk, although their gait remains floppy.
To elucidate the genetic underpinnings of the 20884 mutant's dramatic paralysis and novel recovery, the strain was subjected to positional cloning.
The 20884 phenotype maps to two distinct mutations in the Lpin1 and Nrcam genes
Because 20884 mutants have difficulty breeding, an intercross breeding scheme was used, using BALB as the outcross strain. A genome-wide panel of SSLP markers was used to localize the genetic defect to the proximal end of chromosome 12, which was consistent with the findings reported by Moran et al. (2006). Increasing marker density refined the critical region to the 29.83 Mb (NCBI Build 37) region between markers D12Mit170 and D12Mit236 (Fig. 2A). Breeding additional 20884 mutants failed to further reduce the size of the critical region.
Figure 2.

Identification of the 20884 mutations. A, Correlation of F2 progeny genotype with phenotype demonstrated that the “mild” phenotype maps to the proximal (top) end of the chromosome 12 critical region, between markers D12Mit170 and D12Mit184. All “severe” mutants required C57 homozygosity at the proximal end of the critical region as well as at the distal end. The existence of both “severe” and “mild” mutants with recombination events between D12Mit235 and D12Mit236 indicated that the modifying genetic lesion was located between these markers. B, C, The sequencing of all coding exons in the implicated proximal and distal regions revealed two point mutations, a T-to-A transversion in the Lpin1 gene (B) and a C-to-T transition in the Nrcam gene (C). All “mild” mutants were confirmed to be homozygous for the Lpin120884 mutation only (red highlighted row), whereas all “severe” mutants were confirmed to be homozygous for both mutations (red and green hightlighted rows) (A). D, The mutation in Lpin1 results in a missense mutation, Y873N (red arrows), in the final exon of both known gene products, lipin-1 a (a) and lipin-1 b (b). N-terminal lipin domain, Light blue; C-terminal lipin domain, dark blue; alternately spliced 33 aa, black. E, The mutation in Nrcam results in a nonsense mutation, Q1033X, that occurs in a region present in all known splice forms, truncating potential gene products before the transmembrane and intracellular domain (green arrow). Ig domain, Yellow; fibronectin domain, brown; transmembrane domain, gray; intracellular domain, orange. F, PAP activity in Lpin120884 and double mutant brain and inguinal subcutaneous white adipose tissue (WAT) was significantly reduced compared with wild type. The reduction in PAP activity in double mutant liver and hindlimb muscle, and in Lpin120884 sciatic nerve, was also statistically significant. For brain, liver, adipose tissue, and hindlimb muscle, n = 3 for each genotype. For sciatic nerve, n = 3 for each genotype, with samples examined for each genotype consisting of nerves pooled from three animals. Significant difference from wild type as determined by one-way ANOVA for each tissue. *p < 0.05. Error bars indicate SD. G, Western blotting demonstrated NrCAM protein is not detectable in Nrcam20884 mutant brains. The anti-NrCAM antibody used was raised against the extracellular domain of the protein. Actin was used as a loading control.
A closer examination of the outcrossed F2 mutant mice revealed two distinct recessive phenotypes. In a majority of cases, the original severe 20884 phenotype, described above, was similar on the outbred background. A second, milder phenotype was characterized by a floppy gait that was subtle in most instances, with a later age of onset, between 6 and 9 weeks. Detection of this phenotype was further complicated by the attenuation of the gait abnormality between 14 and 16 weeks. This mild-gait phenotype is not readily observable on the original inbred C57 background.
By correlating genotype data with phenotypic observations, a clear pattern emerged (Fig. 2A). The mild phenotype mapped to the 377 kb region between D12Mit170 and D12Mit184 at the proximal end of the critical region, whereas the severe phenotype was observed in animals homozygous for the C57 background within both the proximal region and the 2.27 Mb region between D12Mit235 and D12Mit236 at the very distal end of the critical region. Therefore, the original 20884 strain appeared to possess two distinct ENU-derived mutations.
To identify the two mutations, all coding sequences from the three known genes within the 377 kb proximal region and the six known and predicted genes within the 2.27 Mb distal region were sequenced from the genomic DNA of a severe-phenotype mutant and compared with those sequenced from a C57 wild-type mouse, as well as with the online genome sequence. Within the proximal region, a T-to-A transversion was found in exon 20 of Lpin1 (Fig. 2B), resulting in a tyrosine-to-asparagine missense mutation (Y873N) affecting a residue that is present in both lipin-1 isoforms (Fig. 2D) (Péterfy et al., 2005) and conserved among mammals. Within the distal region, 29.13 Mb from the Lpin1 mutation, a C-to-T transition was discovered in exon 36 of Nrcam (Fig. 2C), which results in a premature stop codon (Q1033X) (Fig. 2E). Thus, the mild phenotype appeared to result from a mutation in the Lpin1 gene alone, whereas the more severe phenotype appeared to result from a mutation in the Nrcam gene together with the mutation in the Lpin1 gene.
The Lpin120884 missense allele is a partial loss-of-function allele with reduced PAP activity
The availability of other mutations in the Lpin1 gene enabled confirmation, by complementation analysis, that the Lpin120884 mutation is responsible for the “mild” phenotype. The fatty liver dystrophy (fld) mouse carries a spontaneous null mutation in the Lpin1 gene on the BALB/cByJ background (Péterfy et al., 2001) that results in, preweaning, an enlarged, fatty liver that is visible through the skin, delayed hair growth, and reduced body size (Langner et al., 1989), as well as a progressive peripheral neuropathy that presents at postnatal day 10 (P10) with a floppy gait and tremor (Langner et al., 1991). Lpin120884/Lpin1fld compound heterozygous mice that lacked the NrCAM20884 mutation were generated. The mild 20884 and fld phenotypes failed to complement, indicating the two mutations are allelic. Whereas Lpin1fld/20884 compound heterozygotes, like Lpin120884/20884 mutants, lack the fatty liver, delayed hair growth, and small size seen in preweaning fld mutants, they do develop a peripheral neuropathy phenotype at 6 weeks of age similar to that seen in the F2 outcrossed mutants. This peripheral neuropathy is characterized by a low, floppy gait, a tendency to clench the hindlimbs in toward the body when suspended by the tail (Fig. 3B), and abnormal sciatic nerve morphology, visualized by toluidine blue-stained cross-sections (Fig. 3D), that is less severe than fld mutant de/dysmyelination (Langner et al., 1991). Therefore, Lpin120884, a new allele of Lpin1, is responsible for the mild 20884 phenotype.
Figure 3.

A–D, Genetic confirmation of the 20884 mutations. Complementation testing confirmed the identity of the Lpin120884 mutation. Lpin1fld/20884 compound heterozygotes clench their hindlimbs to their bodies when suspended by the tail (B), whereas heterozygous and wild-type littermates splayed their hindlimbs and toes (A), as commonly seen in wild-type mice. Histological observation of sciatic nerve cross-sections from 8-week-old compound heterozygotes (D) revealed abnormalities, such as thin myelin sheaths and darkly stained debris, not seen in wild-type sections (C). Scale bars, 10 μm. E, Additionally, the Nrcam20884 mutation is able to modify the fld phenotype. Compared with their fld littermates, who demonstrate a floppy gait, fld Nrcam20884 double mutants are greatly reduced in size and develop total hindlimb paralysis and partial forelimb paralysis at ∼2.5 weeks.
Lipin-1 was recently demonstrated to be one of a three-member family of proteins possessing PAP activity in mammals; these enzymes function in lipid metabolism and signal transduction by catalyzing the removal of a phosphate group from phosphatidate (Donkor et al., 2007; Harris et al., 2007). The milder phenotype of the Lpin120884 mutant, compared with the fld mutant, suggested that the 20884 allele is a partial loss-of-function allele. To quantitate the PAP activity of the Lpin120884 gene product, protein extracts were collected from the brain, liver, inguinal subcutaneous white adipose tissue, hindlimb muscle, and sciatic nerve of Lpin120884 Nrcam20884 double mutants, Lpin120884 single mutants, and wild-type animals and analyzed for PAP activity as described by Donkor et al. (2007) and Manmontri et al. (2008).
In all tissues examined, PAP activity is present but similarly reduced in both the double mutants and Lpin120884 single mutants as compared to wild-type levels, with one or both mutants showing statistically significant reductions for each tissue (Fig. 2F). Other lipin family members are expressed in brain and liver, so a full loss of PAP activity would not be expected in these tissues, even in the absence of lipin-1. Meanwhile, Lpin1 is the major lipin gene expressed in skeletal muscle and adipose tissue, and fld mutant mice lack all or nearly all PAP activity in these tissues (Donkor et al., 2007; Harris et al., 2007). In contrast, PAP activity was measured at 20% of wild type in double and Lpin120884 mutant adipose tissue, and at 25 and 33% of wild type in double and Lpin120884 mutant muscle, respectively. The incomplete loss of PAP activity in Lpin120884 mutant tissues demonstrates that the 20884 allele is a hypomorph with respect to PAP function. Quantitation of protein levels by Western blot demonstrated that lipin-1 is expressed at similar levels in the Lpin120884 mutant as in wild type in all tissues examined except for adipose tissue, in which mutant protein expression was twice as high as that of the wild type (data not shown). Therefore, the loss of PAP activity in Lpin120884 is likely attributable to an impairment of enzymatic function rather than a reduction in expression.
The Nrcam20884 null allele's interaction with Lpin1 is not allele specific
NrCAM is a membrane-bound protein found on the cell surface of Schwann cells and neurons during development, as well as at the nodes of Ranvier in mature axons (Grumet, 1997; Arroyo and Scherer, 2000), where it is involved in the clustering of Na+ channels to the node (Susuki and Rasband, 2008). The 20884 nonsense mutation, which, based on previous analysis of the human and chicken homologs, falls within a region common to all splice forms (Grumet et al., 1991; Kayyem et al., 1992; Lane et al., 1996; Dry et al., 2001), is predicted to result in a truncated protein that lacks the transmembrane and intracellular domains (Fig. 2E). Therefore, it is expected that the mutant gene product, if expressed, would be secreted from the cell.
Nrcam mRNA is detectable in Nrcam20884/20884 mice and semiquantitative PCR analysis did not show a strong decrease in the amount of mRNA present (data not shown). Nevertheless, the truncated protein (predicted molecular weight of 114 kDa) is not detectable by Western blot (Fig. 2G) or immunohistochemical staining of teased nerve fibers (data not shown) using an antibody raised against the extracellular domain of murine NrCAM. The 20884 allele can therefore be considered a null allele.
Lpin1fld/fld Nrcam20884/20884 double mutants were generated to determine whether the modifying effect that the Nrcam mutation has on the Lpin1 phenotype is allele-specific. Indeed, the combination of the 20884 allele of Nrcam with the fld mutation produced an explicit increase in phenotype severity compared with the fld mutation alone. Although initially similar in phenotype to their fld littermates, by 2.5 weeks after birth, fld Nrcam20884 double mutants develop severe muscle wasting and demonstrate delayed body growth. As a result, fld Nrcam20884 mutants are dramatically smaller than their fld littermates (Fig. 3E). Additionally, fld Nrcam20884 mutant hindlimbs become fully paralyzed, although some hip function may be retained. The forelimbs also show partial loss of function. In contrast, the fld littermates develop the expected floppy, weak gait without wasting and paralysis. Therefore, the fld Nrcam20884 double mutant phenotype reveals that the modifying effect of NrCAM absence on the Lpin1 mutant phenotype is not allele specific and that the effect does not require the presence of the lipin-1 protein.
The 20884 double mutant paralysis and the milder Lpin120884 mutant weakness are transient
To understand better the role that interaction between Lpin1 and Nrcam plays in peripheral myelin development and maintenance, the appearance and subsequent amelioration of the phenotype that were observed in 20884 mutant mice were quantified. Grip strength and body weight were measured in mice that were homozygous for the Lpin20884 and NrCAM20884 alleles, alone and in combination. Six mice, three male and three female, of each of the mutant genotypes were examined in conjunction with age- and gender-matched controls. Animals were examined for both hindlimb and forelimb grip strength every 2 weeks from 4 weeks until 16 weeks of age, and then every 4 weeks thereafter (Fig. 4A–F). At 4 weeks of age, before the appearance of a gait abnormality, the Lpin120884 Nrcam20884 (double) mutants demonstrated significantly reduced hindlimb grip strength compared with the wild type (Fig. 4B). By 6 weeks, half of the double mutant cohort had lost the ability to grip with their hindpaws; by 8 weeks, the hindlimbs of all six double mutants were paralyzed. After gradually recovering from hindpaw paralysis, the first of the six regained the ability to grip at 32 weeks, the last by 48 weeks. It was observed that some of these recovering double mutants clearly regained hindpaw mobility before recording a return of grip strength because of an apparent inability to sense the grip strength apparatus, suggesting that a sensory deficit may sometimes linger beyond the return of motor function. Meanwhile, forelimb weakness did not appear in the double mutant until 6 weeks (Fig. 4A). Although forelimb grip strength never returned to wild-type levels, the double mutant measurements show a clear pattern of progressive weakness followed by an attenuation of the phenotype beginning at ∼24 weeks of age.
Figure 4.

Grip strength and body weight phenotype progression. A, Beginning at 6 weeks, double mutant forelimb grip (blue) became progressively weaker until 20 weeks, after which it steadily improved but remained significantly weaker than wild type (white). B, Double mutant hindlimb grip strength (blue) was already significantly reduced at 4 weeks. At 6 weeks, half of the double mutants could no longer perform the grip strength test; by 8 weeks, all six double mutants were paralyzed. As early as 32 weeks, mutants regained the ability to grip; all could grip by 48 weeks. C, No grip strength abnormalities were observed in the Lpin120884 mutant forelimbs (red). D, A transitory weakness in Lpin120884 mutant hindlimbs (red) was observed at ∼8–12 weeks, demonstrating significance at 10 weeks. The modest increase in grip strength of older Lpin20884 mice is attributable to mechanical stiffness of their paws. E, Nrcam20884 mutants (green) demonstrated an inconsistent forelimb weakness beginning at 14 weeks. F, No hindlimb weakness was observed in Nrcam20884 mutants (green). Cohorts consisted of six mutant mice and six wild-type littermate controls (white) for each mutant genotype. Significance was determined by Student's t test. *p < 0.05. G, The same mice analyzed for grip strength were observed for abnormalities in body weight. The Lpin1 and Nrcam mutants were indistinguishable from the wild type. Double mutants, however, were significantly smaller than wild-type mice beginning at 8 weeks, because of their obvious hindquarter muscle atrophy. By 32 weeks, double mutant size was no longer significantly different from wild type. Significance was determined by one-way ANOVA at each time point. *p < 0.05. Error bars indicate SD.
Unlike the double mutant, Lpin120884 mutant forelimb grip strength was normal (Fig. 4C); however, a transitory defect was observed in the hindlimbs. A mild weakness was observed in the hindlimbs from ∼8 to 12 weeks of age, although this weakness reached statistical significance only at 10 weeks of age (Fig. 4D). This hindlimb weakness did not reoccur later in life; however, an increase in hindlimb grip strength over wild type at the later time points, found to be significant at 48 weeks, was associated with observed stiffness in the joints of the paw.
Whereas an inconsistently significant weakness in the forelimbs was observed in the Nrcam20884 mutants beginning at 14 weeks (Fig. 4E), these mutants never displayed weakness in their hindlimbs (Fig. 4F).
Because visible hindquarter muscle wasting was observed in the double mutants, body weight was also recorded for all mice undergoing grip strength analysis (Fig. 4G). As expected, reduced body weight in double mutants, compared with wild-type littermates, was associated with the onset of paralysis, when muscle mass becomes visibly decreased. Improvement in relative body weight compared with wild type was associated with the return of hindlimb function, as well as a visible recovery of muscle mass. Both of the single mutant cohorts remained similar in size to their wild-type littermates. These observations indicate that both the severity and transient nature of the double mutant phenotype are attributable to the interaction of the two mutant alleles, rather than simply an additive effect of the mutant phenotypes of each allele alone.
The Lpin120884 and Nrcam20884 mutant electrophysiological abnormalities interact in a nonadditive manner in the double mutant
To examine the effect of the Lpin120884 and Nrcam20884 mutations on peripheral nerve function, mutant and wild-type animals were subjected to electrophysiological examination. Recordings were made from the sciatic nerve at 4 weeks, before the onset of wasting and paralysis in the double mutant, and from both the sciatic and sural nerves at 8 and 12 weeks, time points during the progression of the double mutant paralysis (Fig. 5; supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 5.

Electrophysiological analysis. CMAPs were recorded from the sciatic nerves of double, Lpin120884, and Nrcam20884 mutants and wild-type littermates at 4, 8, and 12 weeks (n ≥ 5). A, At 4 weeks, the double mutant exhibited a significant 45% decrease in conduction velocity. By 8 weeks, the conduction velocities of all three mutants were significantly lower than wild type; additionally, those of the double and Lpin120884 mutants were significantly lower than the Nrcam20884 mutant (as indicated by the purple bracket). At 12 weeks, the double and Lpin120884 mutant conduction velocities were significantly different from wild type, but the Nrcam20884 mutant conduction velocity was not. B, The distal CMAP amplitudes were significantly reduced by 70% in double mutants at 8 and 12 weeks, and by 50% in Lpin120884 mutants at 12 weeks. Significant difference from wild type; significance was determined by one-way ANOVA at each time point. *p < 0.05. Error bars indicate SD.
At 4 weeks, double mutant conduction velocity is significantly reduced to 45% of wild type (Fig. 5A). The 30% reductions in Lpin120884 and Nrcam20884 mutant conduction velocities are not significant. By 8 weeks, however, the conduction velocities of all three mutants are significantly different from wild type; furthermore, the double mutant (65% reduced) and Lpin120884 mutant (60% reduced) are significantly more affected than the Nrcam20884 mutant (40% reduced). As the Lpin120884 and double mutants show similar levels of dysfunction, the combined effect of the two 20884 mutations does not appear to be additive during the onset of muscle wasting and paralysis. By 12 weeks, the double mutant and the Lpin120884 single mutant remain significantly reduced compared with wild type, although the Nrcam20884 single mutant does not. Additionally, the double mutant (80% reduced) displays a far more severe deficit than the Lpin120884 single mutant (50% reduced). These results are suggestive of a myelin defect in the absence of lipin-1, NrCAM, or both. This defect is apparent in the sensory (sural nerve) as well as the motor pathway, as conduction velocities are significantly reduced at both the 8 and 12 week time points in all three mutant genotypes in a manner similar to that observed in the sciatic nerve (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). A sensory component to the 20884 phenotype is consistent with the apparent lack of sensation observed in recovering double mutants during the grip strength examination.
The recorded compound muscle action potentials (CMAPs) from the double and Lpin120884 mutants demonstrate that amplitude, as well as velocity, is reduced in these mice (Fig. 5B). The distal CMAP amplitudes are significantly reduced in the double mutant at 8 weeks (by 70%) and in both the double mutant and the Lpin120884 mutant at 12 weeks (by 70 and 45%, respectively) (Fig. 5B). In contrast, CMAP amplitudes are not significantly reduced in mice that possess the Nrcam20884 mutation alone. Therefore, lipin-1, or lipin-1 in conjunction with NrCAM, is required for the maintenance or connectivity of motor fibers. The loss of muscle mass in the double mutant is likely attributable to the greater impairment of this activity in the double mutant. Interestingly, the action potential amplitudes were reduced to similar extents in the sural nerves of all three mutants at 8 weeks, although the data did not demonstrate statistical significance. At 12 weeks, the double and Lpin120884 mutant sural nerve amplitudes were significantly reduced (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
The molecular organization of the nodes of Ranvier, as visualized by the immunohistochemical staining of teased fibers using antibodies against nodal (pan-sodium channel), paranodal (Caspr/paranodin), and juxtaparanodal (potassium channel) markers, was observed to be normal in all three mutant genotypes, indicating that abnormalities in this organization do not play a role in the observed electrophysiological defects (data not shown).
Lpin120884 and double mutant sciatic nerves are characterized by similar myelin abnormalities
Sciatic nerve morphology was examined to determine whether the peripheral nerve pathology correlates with the single and double mutant behavioral and physiological phenotypes. Previous work on null alleles of Lpin1 has demonstrated that the loss of Lpin1 results in severe myelin abnormalities in the sciatic nerve (Langner et al., 1991; Nadra et al., 2008). To observe the morphological phenotype of the 20884 mutations, toluidine blue-stained cross-sections of nerves from the three mutant genotypes, at 4, 8, 12, and 52 weeks, were compared with age-matched wild-type sections (Fig. 6). At 4 weeks, toluidine blue-stained sciatic nerves of all four genotypes appear normal (Fig. 6, first column). By 8 weeks, however, abnormalities are visible in the Lpin120884 and double mutant nerves (Fig. 6, second column). Demyelinated axons are present (arrows), the nerves appear qualitatively less organized when compared with nerves from wild-type and Nrcam20884 animals, and some darkly stained debris appear to be present, although the nature of the debris cannot be identified at this magnification. At 12 weeks, the Lpin120884 and double mutant nerves (Fig. 6, third column) appear similar to those taken at 8 weeks, although, in addition to axons lacking myelin (arrows), some axons appear to have thin myelin sheaths (asterisks). At 52 weeks, after amelioration of the mutant phenotype, some thinly myelinated (asterisks) and demyelinated (arrows) fibers are still present in the Lpin120884 and double mutant nerves (Fig. 6, fourth column), although the general state of myelination seems to have improved compared with the sciatic nerves of 12-week-old animals. At all time points, Nrcam mutant nerves (Fig. 6, third row) were similar in appearance to wild type (Fig. 6, first row). Therefore, the behavioral and electrophysiological abnormalities observed in Lpin120884 homozygous and Lpin120884 Nrcam20884 double mutant mice correlate partially with structural changes in peripheral nerve morphology.
Figure 6.
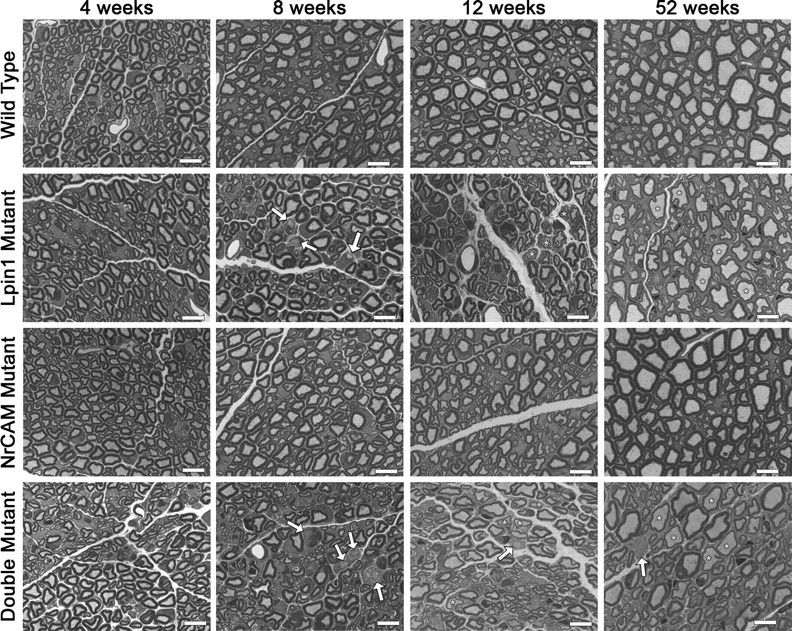
Sciatic nerve histology. Toluidine blue-stained semithin sections were taken from the medial region of the sciatic nerves of mice of all four genotypes at 4, 8, 12, and 52 weeks. At 4 weeks, Lpin120884, Nrcam20884, and double mutant sciatic nerves appear similar to wild type. At 8 weeks, however, demyelinated axons are visible (arrows) in the sciatic nerves of Lpin120884 and double mutants. At 12 weeks, demyelinated axons (arrows) are still visible in Lpin120884 and double mutant nerves. There also appear to be thinly myelinated axons (asterisks). By 52 weeks, some demyelinated axons (arrow) are present in the Lpin120884 and double mutant nerves, but the predominant feature is thinly myelinated axons (asterisks). The Nrcam20884 mutant nerves appeared similar to wild type at all time points. Scale bars, 10 μm.
Examination by electron microscopy provided insight into the debris-like structures observed in the toluidine blue-stained sections but failed to identify differences between the Lpin120884 and double mutants that could explain the extreme disparity in phenotype severity. In the nerves of 4-week-old Lpin120884 and double mutants, membranous debris can be seen infrequently beside myelinated axons, located within the surrounding Schwann cell (data not shown). By 8 weeks, in addition to membranous debris similar to that seen at 4 weeks (Fig. 7B,D,F, arrows), compact myelin structures can be seen beside axons within Schwann cells (Fig. 7D, arrowhead), either singly or in multiples. Additionally, expanded regions of membranous debris can be found within Schwann cells surrounding both myelinated and unmyelinated fibers (Fig. 7C,E, respectively, arrows). Finally, demyelinated axons with no myelin debris are present (Fig. 7B,F, bullets).
Figure 7.
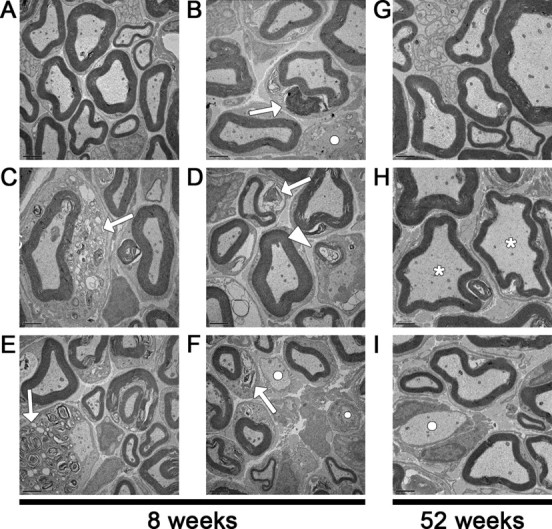
Sciatic nerve abnormalities visualized by electron microscopy. A, Representative sciatic nerve cross-section from an 8-week-old wild-type mouse. B–F, These sections from 8-week-old Lpin120884 mice are representative of the similar myelin abnormalities observed in both Lpin120884 and double mutant mice. These abnormalities include myelin debris within Schwann cells surrounding both myelinated (B–D, F, arrows), and demyelinated (E, arrow) axons. Compact myelin structures can also observed within Schwann cells surrounding demyelinated axons (D, arrowhead). Myelin debris such as that visualized in B, D, and F is observable at all time points, whereas the more extensive debris in C and E are limited to the 8 week time point. Fully demyelinated axons are also present at 8 weeks (B, F, bullets). G, Representative sciatic nerve cross-section from a 52-week-old wild-type mouse. H, I, These sections from 52-week-old Lpin120884 mice are representative of both Lpin120884 and double mutants at this age. The predominant feature is thinly myelinated axons (H, asterisks), although unmyelinated axons are still present (I, bullet). Scale bars, 2 μm.
By 12 weeks, the compact myelin structures and expanded regions of membranous debris observed in the Lpin120884 and double mutants at 8 weeks are rare (data not shown). Nevertheless, the smaller clumps of debris are still present, as are demyelinated axons. Additionally, as with the toluidine blue sections, thinly myelinated fibers are observed. The reduction in compact myelin structures and extensive debris, coupled with the presence of thin myelin sheaths, which are indicative of remyelination, suggests that the 12-week-old nerve is undergoing a process of recovery from the myelin insult caused by the Lpin120884 mutation.
At 52 weeks, the predominant feature in Lpin120884 and double mutant nerves is a visibly increased presence of thinly myelinated fibers (Fig. 7H, asterisks), which are likely the result of remyelination. Moreover, demyelinated axons (Fig. 7I, bullet) are present but not common. Consistent with the toluidine blue findings, the morphology of Nrcam20884 mutant nerve fibers, when observed via electron microscopy, was indistinguishable from that of the wild type at all ages. This ultrastructural analysis suggests that myelin repair is a component of both the Lpin120884 and double mutant phenotypes and that the phenotypic differences between these mice do not result from differences in peripheral nerve pathology.
Lpin120884-induced demyelination peaks at 8 weeks and affects a small fraction of myelinated fibers
The sciatic nerves of the double, Lpin120884, and Nrcam20884 mutants were analyzed morphometrically to quantify the extent of myelin loss in the Lpin120884 and double mutants, to identify any subtle abnormalities that may be present in the Nrcam20884 mutant, and to determine whether axonal loss contributes to the mutant phenotypes. The four genotypes examined displayed no significant difference in g-ratio or average myelin thickness at 4, 8, or 12 weeks (Table 1). At 52 weeks, however, the qualitative observation of thinner myelin sheaths was confirmed; the average g-ratios of the double and Lpin120884 mutants were significantly increased by 5.0 and 4.8%, respectively, compared with wild type, and the average myelin thicknesses were reduced by 19.4 and 24.2%, respectively. When the number of unmyelinated axons, expressed as a percentage of the total number of axons measured for each nerve, was analyzed, differences among the mutant and wild-type mice could be observed. At 8 weeks, but not at 4, 12, or 52 weeks, one-way ANOVA analysis reported a statistical difference among the genotypes. Although post hoc analysis failed to specify pairwise differences, comparison by t test confirmed that the Lpin120884 mutant nerves contained a significantly elevated percentage of unmyelinated axons compared with Nrcam20884 mutant and wild-type animals (19.0- and 5.1-fold greater than Nrcam20884 mutant and wild type, respectively) (Fig. 8A). The lack of significance in the double mutant is likely due to greater variation among the samples. The double and Lpin120844 did not significantly differ from each other; nevertheless, a difference between the double and Lpin120884 mutants cannot be rejected because of the small sample size.
Table 1.
Morphometric analysis
| Age (weeks) | Genotype | g-Ratio | Myelin thickness (μm) | Axon diameter (μm) | Axon numbera |
|---|---|---|---|---|---|
| 4 | Double mutant | 0.751 ± 0.009 | 0.592 ± 0.029 | 3.38 ± 0.19 | 146.7 ± 19.0 |
| Lpin1 mutant | 0.749 ± 0.001 | 0.629 ± 0.017 | 3.56 ± 0.02 | 131.3 ± 15.1 | |
| Nrcam mutant | 0.737 ± 0.016 | 0.655 ± 0.067 | 3.52 ± 0.12 | 138.3 ± 24.0 | |
| Wild type | 0.730 ± 0.019 | 0.647 ± 0.054 | 3.31 ± 0.07 | 137.0 ± 9.6 | |
| 8 | Double mutant | 0.758 ± 0.009 | 0.624 ± 0.052 | 3.79 ± 0.26 | 111.7 ± 21.4 |
| Lpin1 mutant | 0.746 ± 0.018 | 0.607 ± 0.048 | 3.46 ± 0.11† | 115.3 ± 13.8 | |
| Nrcam mutant | 0.746 ± 0.035 | 0.753 ± 0.115 | 4.31 ± 0.16 | 89.0 ± 5.2 | |
| Wild type | 0.765 ± 0.012 | 0.633 ± 0.008 | 3.92 ± 0.29 | 119.0 ± 20.0 | |
| 12 | Double mutant | 0.781 ± 0.007 | 0.606 ± 0.052 | 4.40 ± 0.19 | 101.7 ± 14.2 |
| Lpin1 mutant | 0.782 ± 0.006 | 0.612 ± 0.034 | 4.36 ± 0.46 | 101.7 ± 32.5 | |
| Nrcam mutant | 0.753 ± 0.017 | 0.730 ± 0.046 | 4.26 ± 0.43 | 94.3 ± 18.0 | |
| Wild type | 0.766 ± 0.024 | 0.660 ± 0.092 | 4.15 ± 0.16 | 112.0 ± 10.6 | |
| 52 | Double mutant | 0.790 ± 0.006* | 0.684 ± 0.011* | 5.37 ± 0.15 | 75.2 ± 5.6 |
| Lpin1 mutant | 0.789 ± 0.003* | 0.643 ± 0.026* | 5.13 ± 0.33 | 79.2 ± 9.8 | |
| Nrcam mutant | 0.763 ± 0.021 | 0.814 ± 0.042 | 5.20 ± 0.29 | 69.6 ± 0.8 | |
| Wild type | 0.752 ± 0.008 | 0.849 ± 0.039 | 5.17 ± 0.12 | 68.0 ± 5.0 |
*Significant difference from wild type, p < 0.05.
†Significant difference from Nrcam mutant, p < 0.05.
aAxons per 20 × 16.52 μm2 section.
Figure 8.

Percentage of unmyelinated and thinly myelinated axons. To quantify the extent of demyelination and remyelination at each time point, the percentages of unmyelinated and thinly myelinated axons were calculated using the data obtained from morphometric analysis of electron micrographs (n = 3 for each genotype at each time point). For the purposes of this analysis, thinly myelinated axons were defined to be any axon with a g-ratio that fell above the 95th-percentile value of wild-type myelinated fibers at a given age. A, Both the double and Lpin120884 mutants are characterized by increased percentages of unmyelinated axons compared with wild type at 8 weeks of age (6.4- and 5.1-fold greater than wild type, respectively). Although a one-way ANOVA determined there was a significant difference among the genotypes at 8 weeks, post hoc analysis failed to specify which pairs were different. †Significant difference from wild type as determined by t test, p < 0.05. B, The number of thinly mylinated axons was increased by >2.5-fold at 12 weeks and by 4-fold at 52 weeks in double and Lpin120884 mutants compared with wild type. The increase in thinly myelinated axons coincides with a decrease in demyelinated axons, indicating remyelination is occurring. Significance was determined by one-way ANOVA at each time point. *p < 0.05. Error bars indicate SD.
Additionally, the percentage of hypomyelinated axons was calculated for each nerve examined (Fig. 8B). For this purpose, hypomyelinated axons were defined as axons with g-ratios falling above the 95th-percentile value of wild-type myelinated fibers at a given age. No differences were observed at 4 or 8 weeks, but at 12 weeks, a >2.5-fold increase in the percentage of thinly myelinated axons compared with wild type was observed in both double and Lpin120884 mutant nerves. Notably, this increase in hypomyelinated axons is concurrent with the reduction in the percentage of unmyelinated axons, suggesting that the hypomyelinated axons are newly remyelinated. As expected given the significant elevation in g-ratio, the percentage of hypomyelinated fibers was further increased at 52 weeks to percentages fourfold higher than wild type in the double and Lpin120884 mutants. It is possible that the elevated frequency of thin myelin sheaths is attributable to a continuous low-level process of segmental demyelination followed by remyelination that never achieves wild-type sheath thickness.
No significant differences between Nrcam20884 mutant and wild-type nerves were noted. Finally, axon loss does not appear to be a common feature in sciatic nerve sections of either the Lpin1 or double mutant, as no difference in axon numbers was observed at any time point (Table 1). However, a distal axonal loss cannot be excluded given the presence of atrophic fibers in muscle pathology (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these data suggest that myelin repair contributes to the attenuation of the 20884 mutant phenotype and that the phenotypic differences between the Lpin120884 and double mutant mice are not attributable to ultrastructural differences in their peripheral nerves.
Discussion
Investigation of the ENU-induced 20884 mutant, characterized by transient adult-onset paralysis, has demonstrated an unexpected interaction between two genes, Lpin1 and Nrcam, that is important to PNS health. Previous work examining peripheral neuropathy in Lpin1 mutants focused solely on null alleles (Langner et al., 1991; Péterfy et al., 2001; Nadra et al., 2008). Lpin120884 is a partial loss-of-function allele resulting in a significant reduction in PAP activity but not the grossly visible adipose deficiency or preweaning fatty liver seen in fld and fld2J mutants (Péterfy et al., 2001). Homozygous Lpin20884 mice display a 78% reduction in PAP activity in adipose tissue, which lacks all PAP activity in the absence of lipin-1 protein (Donkor et al., 2007). The spontaneous Lpin1 mutation, fld2J, was found to retain 20% PAP activity in an in vitro study (Harris et al., 2007); however, fld2J mutants share the same severe adipose and hepatic phenotypes as fld mice, possibly because of reduced protein expression from the fld2J allele or impaired nuclear localization of the fld2J protein (Péterfy et al., 2001, 2005), which likely interferes with its function as a transcriptional coactivator in hepatic cells (Finck et al., 2006) and its role in inducing adipogenesis in adipose cells (Phan et al., 2004; Péterfy et al., 2005). Because Lpin120884 mutants do not demonstrate the severe hepatic and adipose defects seen in fld and fld2J mutant mice, it is possible that the 20884 allele is deficient only in PAP activity and experiences little or no reduction in transcriptional coactivation activity. This might produce a defect in phosphatidylcholine synthesis, required for myelin turnover, or it might affect the relative signaling effects of phosphatidate versus diacylglycerol (Brindley et al., 2009). The latter proposal would be consistent with the recent study by Nadra et al. (2008), which demonstrated that Schwann cells predominantly express the cytoplasmic isoform of lipin-1 and that accumulation of phosphatidate, the PAP substrate, is responsible for activating the MEK–ERK pathway, which is capable of both interfering with myelination and causing demyelination. If the 20884 allele of Lpin1 does preserve its transcriptional regulatory functions, it may prove useful for separating out the downstream effects of the two lipin-1 functions.
The differences between the fld and 20884 alleles may also offer a clue to the mechanism behind the transient phenotype of the Lpin120884 mouse. The 20884 mutant protein has reduced activity, whereas the fld allele produces no protein. Lpin3 is upregulated in fld livers in response to the absence of Lpin1 (Donkor et al., 2007), whereas this study demonstrates that Lpin120884 protein expression is upregulated in mutant adipose tissue. It is therefore possible that the ability of the Lpin120884 mutant to increase expression of the mutant protein to compensate for lower enzymatic efficiency may account for phenotype amelioration not seen in the null. Additionally, should peripheral nerve cells lack the ability to compensate for reduced PAP activity by upregulating the other two lipin family members, the absence of lipin-1 in fld mutant mice may explain why fld neuropathy is progressive, as it lacks the ability to compensate for reduced PAP activity.
In humans, LPIN1 mutations have been found to result in recurrent myoglobinuria or statin-induced myopathy; however, these patients were not examined for peripheral neuropathy (Zeharia et al., 2008). Further investigation is required to determine whether the reported difference in pathology between the human and mouse disorders represents allelic or diagnostic differences. Additionally, the study of Lpin1 may lead to a better understanding of the high incidence of peripheral neuropathy among diabetics (Yagihashi et al., 2007). Several studies in humans have associated LPIN1 expression with obesity (Miranda et al., 2007), the metabolic syndrome (van Harmelen et al., 2007), and insulin sensitivity (Donkor et al., 2008; Reue and Brindley, 2008). Natural genetic variation in LPIN1 has also been found to associate with diabetes, hypertension (Wiedmann et al., 2008), and insulin levels (Suviolahti et al., 2006; Loos et al., 2007). Furthermore, a recent study in mouse has shown that TORC2, which is upregulated in liver during insulin resistance, upregulates lipin-1, which intensifies insulin resistance by increasing DAG signaling (Ryu et al., 2009). Additionally, loss of Lpin1 expression in Schwann cells is sufficient to cause peripheral neuropathy (Nadra et al., 2008). Thus, hyperinsulinemia could inhibit Schwann cell expression of lipin-1, thereby inhibiting myelin maintenance and repair; this suggests that neuropathy may not be a secondary effect of diabetes. Further investigation of lipin-1, in particular the Lpin20884 mutation, which has little or no effect on hepatic and adipose tissues, may provide new targets for the treatment of diabetic neuropathy.
The second mutation identified from the 20884 strain is a truncation mutation in Nrcam. Two Nrcam null allele knock-outs have previously been described (Moré et al., 2001; Sakurai et al., 2001). Consistent with the wild-type appearance of the Nrcam20884 null mutant, the Nrcam knock-out mice do not demonstrate an observable phenotype. In the PNS, a brief delay in Na+ channel clustering to the nodes of Ranvier before P10 was observed, but conduction velocity was found to be similar to wild type (Custer et al., 2003). The lack of an electrophysiological defect in the Nrcam knock-out, however, is not inconsistent with the mild conduction velocity reduction recorded in the Nrcam20884 mutant. In the knock-out study, conduction velocity was measured in explants at P2, when nerve conduction is not yet expected to function by means of mature saltatory conduction.
The presence of a mild conduction velocity defect in adult Nrcam20884 mutants indicates that NrCAM's presence at mature nodes is of functional significance. NrCAM interacts with Na+ channels via interaction with the voltage-gated sodium channel subunit β1 (McEwen and Isom, 2004). NrCAM also shares with Na+ channels an intracellular interaction with ankyrin G, which may mediate NrCAM's ability to cluster Na+ channels during node formation (Lustig et al., 2001; Custer et al., 2003). Therefore, as NrCAM is an integral component of the peripheral node with multiple indirect physical interactions with Na+ channels, the loss of NrCAM may negatively impact the stability and electrophysiological properties of the peripheral node.
To investigate the contributions of the Lpin120884 and Nrcam20884 mutations to the 20884 double mutant phenotype, the behavioral, electrophysiological, and histological features of the single and double mutants were examined. The Lpin120884 and double mutants share similar histological features in the sciatic nerve: normal initial development of myelin sheaths followed by a process of demyelination that appears by 8 weeks and shows signs of amelioration by 12 weeks, although the significant increase in hypomyelinated axons by 52 weeks suggests that a cycle of demyelination and remyelination continues at a subclinical level.
Unlike the Lpin120884 mutant, however, double mutants exhibit muscle wasting in the hindquarters, resulting in hindlimb paralysis. This dramatic behavioral phenotype is accompanied by a reduction in sciatic nerve conduction velocity and CMAP amplitude that is of earlier onset and greater severity than that seen in the Lpin120884 mutant. At 8 weeks, the double mutant is distinguished by a CMAP amplitude significantly reduced by 70%, a reduction indicating distal axonal loss or distal conduction block. Although there is not a statistically significant difference when compared with the Lpin120884 mutant's 50% amplitude reduction, the reduced level of stimulation received by the double mutant muscle may fall beneath a threshold required to prevent atrophy, whereas the Lpin120884 mutant muscle receives enough stimulation to experience only mild weakness (Meisler et al., 2004).
The modifying effect of the Nrcam20884 mutation on the Lpin120884 mutation appears unrelated to myelin stability, because of the similar histological features of the Lpin120884 and double mutants. Perhaps there is a synergistic effect, whereby the relevant consequences of NrCAM's absence in the double mutant may be observable only because of the demyelinating environment resulting from the Lpin120884 mutation. It is likely that NrCAM plays the same role in Na+ channel clustering during remyelination that it does during development. As a result, affected axons in the double mutant may experience a slower reassembly of the node during remyelination. Before the attenuation of the Lpin120884 defect, the constant cycle of demyelination and remyelination coupled with this relatively mild NrCAM impairment could be responsible for the further drop in electrophysiological function that results in muscle atrophy and paralysis. Alternatively, NrCAM is known to be present on Schwann cells during development (Grumet, 1997), and Nrcam mRNA expression is upregulated in the sciatic nerve after nerve crush (Nagarajan et al., 2002). Therefore, it is possible that NrCAM is present in remyelinating Schwann cells and its absence in the double mutant may result in abnormal signal transduction during remyelination (Falk et al., 2005) that could be further exacerbated by deficiencies in lipin-1-related signaling.
In summary, although the underlying mechanism of the 20884 double mutant phenotype remains unclear, the benefits of the concurrent appearance of the Lpin1 and Nrcam mutations are twofold: the identification of a novel Lpin1 allele that further emphasizes the effect of lipin-1 dysfunction on the PNS and may provide insight into lipin-1's multiple functions, and the demonstration of the functional importance of NrCAM in the adult nerve undergoing a demyelinating event, which indicates that NrCAM plays a greater role in maintaining nerve health than previously recognized.
Footnotes
The study was funded by grants from the National Institutes of Health (NIH) (NS027336) and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to B.P., the NIH (NS40751) to J.R.B. Jr, the Canadian Institutes of Health Research (MOP 81137) to D.N.B., and the National Institute of Child Health and Human Development (UO1 HD43430) to D.R.B. We thank Danuta Dukala for her assistance in recording electrophysiological data, Erdong Liu for preparing the toluidine blue and electron microscopy sections, Yujuan Yun for her contributions to mouse husbandry and genotyping, Jay Dewald for performing the phosphatidate phosphatase assays and the Western blots for lipin-1, Robert L Wollmann for advice on nerve pathology, and Chuanhong Liao of the Biostatistics Clinic for assistance with statistical analysis.
References
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo EJ, Scherer SS. On the molecular architecture of myelinated fibers. Histochem Cell Biol. 2000;113:1–18. doi: 10.1007/s004180050001. [DOI] [PubMed] [Google Scholar]
- Auer RN. Automated nerve fibre size and myelin sheath measurement using microcomputer-based digital image analysis: theory, method and results. J Neurosci Methods. 1994;51:229–238. doi: 10.1016/0165-0270(94)90015-9. [DOI] [PubMed] [Google Scholar]
- Brindley DN, Pilquil C, Sariahmetoglu M, Reue K. Phosphatidate degradation: phosphatidate phosphatases (lipins) and lipid phosphate phosphatases. Biochim Biophys Acta. 2009;1791:956–961. doi: 10.1016/j.bbalip.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AT, Goldowitz D, Takahashi JS, Vitaterna MH, Siepka SM, Peters LL, Frankel WN, Carlson GA, Rossant J, Nadeau JH, Justice MJ. Implementing large-scale ENU mutagenesis screens in North America. Genetica. 2004;122:51–64. doi: 10.1007/s10709-004-1436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer AW, Kazarinova-Noyes K, Sakurai T, Xu X, Simon W, Grumet M, Shrager P. The role of the ankyrin-binding protein NrCAM in node of Ranvier formation. J Neurosci. 2003;23:10032–10039. doi: 10.1523/JNEUROSCI.23-31-10032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Repentigny Y, Côté PD, Pool M, Bernier G, Girard S, Vidal SM, Kothary R. Pathological and genetic analysis of the degenerating muscle (dmu) mouse: a new allele of Scn8a. Hum Mol Genet. 2001;10:1819–1827. doi: 10.1093/hmg/10.17.1819. [DOI] [PubMed] [Google Scholar]
- Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem. 2007;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- Donkor J, Sparks LM, Xie H, Smith SR, Reue K. Adipose tissue lipin-1 expression is correlated with peroxisome proliferator-activated receptor alpha gene expression and insulin sensitivity in healthy young men. J Clin Endocrinol Metab. 2008;93:233–239. doi: 10.1210/jc.2007-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas DS, Popko B. Mouse forward genetics in the study of the peripheral nervous system and human peripheral neuropathy. Neurochem Res. 2009;34:124–137. doi: 10.1007/s11064-008-9719-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dry K, Kenwrick S, Rosenthal A, Platzer M. The complete sequence of the human locus for NgCAM-related cell adhesion molecule reveals a novel alternative exon in chick and man and conserved genomic organization for the L1 subfamily. Gene. 2001;273:115–122. doi: 10.1016/s0378-1119(01)00493-0. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Blight A, Suzuki K, Popko B. Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci. 1998;18:1642–1649. doi: 10.1523/JNEUROSCI.18-05-01642.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk J, Julien F, Bechara A, Fiore R, Nawabi H, Zhou H, Hoyo-Becerra C, Bozon M, Rougon G, Grumet M, Püschel AW, Sanes JR, Castellani V. Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron. 2005;48:63–75. doi: 10.1016/j.neuron.2005.08.033. [DOI] [PubMed] [Google Scholar]
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Grumet M. Nr-CAM: a cell adhesion molecule with ligand and receptor functions. Cell Tissue Res. 1997;290:423–428. doi: 10.1007/s004410050949. [DOI] [PubMed] [Google Scholar]
- Grumet M, Mauro V, Burgoon MP, Edelman GM, Cunningham BA. Structure of a new nervous system glycoprotein, Nr-CAM, and its relationship to subgroups of neural cell adhesion molecules. J Cell Biol. 1991;113:1399–1412. doi: 10.1083/jcb.113.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TE, Huffman TA, Chi A, Shabanowitz J, Hunt DF, Kumar A, Lawrence JC., Jr Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J Biol Chem. 2007;282:277–286. doi: 10.1074/jbc.M609537200. [DOI] [PubMed] [Google Scholar]
- Kaja S, van de Ven RC, Broos LA, Frants RR, Ferrari MD, van den Maagdenberg AM, Plomp JJ. Characterization of acetylcholine release and the compensatory contribution of non-Ca(v)2.1 channels at motor nerve terminals of leaner Ca(v)2.1-mutant mice. Neuroscience. 2007a;144:1278–1287. doi: 10.1016/j.neuroscience.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Kaja S, van de Ven RC, van Dijk JG, Verschuuren JJ, Arahata K, Frants RR, Ferrari MD, van den Maagdenberg AM, Plomp JJ. Severely impaired neuromuscular synaptic transmission causes muscle weakness in the Cacna1a-mutant mouse rolling Nagoya. Eur J Neurosci. 2007b;25:2009–2020. doi: 10.1111/j.1460-9568.2007.05438.x. [DOI] [PubMed] [Google Scholar]
- Kayyem JF, Roman JM, de la Rosa EJ, Schwarz U, Dreyer WJ. Bravo/Nr-CAM is closely related to the cell adhesion molecules L1 and Ng-CAM and has a similar heterodimer structure. J Cell Biol. 1992;118:1259–1270. doi: 10.1083/jcb.118.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RP, Chen XN, Yamakawa K, Vielmetter J, Korenberg JR, Dreyer WJ. Characterization of a highly conserved human homolog to the chicken neural cell surface protein Bravo/Nr-CAM that maps to chromosome band 7q31. Genomics. 1996;35:456–465. doi: 10.1006/geno.1996.0385. [DOI] [PubMed] [Google Scholar]
- Langner CA, Birkenmeier EH, Ben-Zeev O, Schotz MC, Sweet HO, Davisson MT, Gordon JI. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J Biol Chem. 1989;264:7994–8003. [PubMed] [Google Scholar]
- Langner CA, Birkenmeier EH, Roth KA, Bronson RT, Gordon JI. Characterization of the peripheral neuropathy in neonatal and adult mice that are homozygous for the fatty liver dystrophy (fld) mutation. J Biol Chem. 1991;266:11955–11964. [PubMed] [Google Scholar]
- Levedakou EN, Chen XJ, Soliven B, Popko B. Disruption of the mouse Large gene in the enr and myd mutants results in nerve, muscle, and neuromuscular junction defects. Mol Cell Neurosci. 2005;28:757–769. doi: 10.1016/j.mcn.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Loos RJ, Rankinen T, Perusse L, Tremblay A, Despres JP, Bouchard C. Association of lipin 1 gene polymorphisms with measures of energy and glucose metabolism. Obesity (Silver Spring) 2007;15:2723–2732. doi: 10.1038/oby.2007.324. [DOI] [PubMed] [Google Scholar]
- Lustig M, Zanazzi G, Sakurai T, Blanco C, Levinson SR, Lambert S, Grumet M, Salzer JL. Nr-CAM and neurofascin interactions regulate ankyrin G and sodium channel clustering at the node of Ranvier. Curr Biol. 2001;11:1864–1869. doi: 10.1016/s0960-9822(01)00586-3. [DOI] [PubMed] [Google Scholar]
- Manmontri B, Sariahmetoglu M, Donkor J, Bou Khalil M, Sundaram M, Yao Z, Reue K, Lehner R, Brindley DN. Glucocorticoids and cyclic AMP selectively increase hepatic lipin-1 expression, and insulin acts antagonistically. J Lipid Res. 2008;49:1056–1067. doi: 10.1194/jlr.M800013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen DP, Isom LL. Heterophilic interactions of sodium channel beta1 subunits with axonal and glial cell adhesion molecules. J Biol Chem. 2004;279:52744–52752. doi: 10.1074/jbc.M405990200. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Plummer NW, Burgess DL, Buchner DA, Sprunger LK. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica. 2004;122:37–45. doi: 10.1007/s10709-004-1441-9. [DOI] [PubMed] [Google Scholar]
- Meyer Zu Hörste G, Nave KA. Animal models of inherited neuropathies. Curr Opin Neurol. 2006;19:464–473. doi: 10.1097/01.wco.0000245369.44199.27. [DOI] [PubMed] [Google Scholar]
- Miranda M, Chacón MR, Gómez J, Megía A, Ceperuelo-Mallafré V, Veloso S, Saumoy M, Gallart L, Richart C, Fernández-Real JM, Vendrell J, Vendrell J. Human subcutaneous adipose tissue LPIN1 expression in obesity, type 2 diabetes mellitus, and human immunodeficiency virus–associated lipodystrophy syndrome. Metabolism. 2007;56:1518–1526. doi: 10.1016/j.metabol.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Moran JL, Bolton AD, Tran PV, Brown A, Dwyer ND, Manning DK, Bjork BC, Li C, Montgomery K, Siepka SM, Vitaterna MH, Takahashi JS, Wiltshire T, Kwiatkowski DJ, Kucherlapati R, Beier DR. Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome Res. 2006;16:436–440. doi: 10.1101/gr.4563306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moré MI, Kirsch FP, Rathjen FG. Targeted ablation of NrCAM or ankyrin-B results in disorganized lens fibers leading to cataract formation. J Cell Biol. 2001;154:187–196. doi: 10.1083/jcb.200104038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadra K, de Preux Charles AS, Médard JJ, Hendriks WT, Han GS, Grès S, Carman GM, Saulnier-Blache JS, Verheijen MH, Chrast R. Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev. 2008;22:1647–1661. doi: 10.1101/gad.1638008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan R, Le N, Mahoney H, Araki T, Milbrandt J. Deciphering peripheral nerve myelination by using Schwann cell expression profiling. Proc Natl Acad Sci U S A. 2002;99:8998–9003. doi: 10.1073/pnas.132080999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péréon Y, Nguyen The Tich S, Guihéneuc P. Peripheral neuropathy: a picture is worth a thousand words. Neurophysiol Clin. 2003;33:31–32. doi: 10.1016/s0987-7053(03)00004-2. [DOI] [PubMed] [Google Scholar]
- Péterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- Péterfy M, Phan J, Reue K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem. 2005;280:32883–32889. doi: 10.1074/jbc.M503885200. [DOI] [PubMed] [Google Scholar]
- Phan J, Péterfy M, Reue K. Lipin expression preceding peroxisome proliferator-activated receptor-gamma is critical for adipogenesis in vivo and in vitro. J Biol Chem. 2004;279:29558–29564. doi: 10.1074/jbc.M403506200. [DOI] [PubMed] [Google Scholar]
- Plomp JJ, Vergouwe MN, Van den Maagdenberg AM, Ferrari MD, Frants RR, Molenaar PC. Abnormal transmitter release at neuromuscular junctions of mice carrying the tottering alpha(1A) Ca(2+) channel mutation. Brain. 2000;123:463–471. doi: 10.1093/brain/123.3.463. [DOI] [PubMed] [Google Scholar]
- Reue K, Brindley DN. Thematic Review Series: Glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism. J Lipid Res. 2008;49:2493–2503. doi: 10.1194/jlr.R800019-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Oh KJ, Jo HY, Hedrick S, Kim YN, Hwang YJ, Park TS, Han JS, Choi CS, Montminy M, Koo SH. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab. 2009;9:240–251. doi: 10.1016/j.cmet.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Lustig M, Babiarz J, Furley AJ, Tait S, Brophy PJ, Brown SA, Brown LY, Mason CA, Grumet M. Overlapping functions of the cell adhesion molecules Nr-CAM and L1 in cerebellar granule cell development. J Cell Biol. 2001;154:1259–1273. doi: 10.1083/jcb.200104122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Susuki K, Rasband MN. Molecular mechanisms of node of Ranvier formation. Curr Opin Cell Biol. 2008;20:616–623. doi: 10.1016/j.ceb.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suviolahti E, Reue K, Cantor RM, Phan J, Gentile M, Naukkarinen J, Soro-Paavonen A, Oksanen L, Kaprio J, Rissanen A, Salomaa V, Kontula K, Taskinen MR, Pajukanta P, Peltonen L. Cross-species analyses implicate lipin 1 involvement in human glucose metabolism. Hum Mol Genet. 2006;15:377–386. doi: 10.1093/hmg/ddi448. [DOI] [PubMed] [Google Scholar]
- Traka M, Wollmann RL, Cerda SR, Dugas J, Barres BA, Popko B. Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci. 2008;28:11537–11549. doi: 10.1523/JNEUROSCI.1490-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harmelen V, Rydén M, Sjölin E, Hoffstedt J. A role of lipin in human obesity and insulin resistance: relation to adipocyte glucose transport and GLUT4 expression. J Lipid Res. 2007;48:201–206. doi: 10.1194/jlr.M600272-JLR200. [DOI] [PubMed] [Google Scholar]
- Wiedmann S, Fischer M, Koehler M, Neureuther K, Riegger G, Doering A, Schunkert H, Hengstenberg C, Baessler A. Genetic variants within the LPIN1 gene, encoding lipin, are influencing phenotypes of the metabolic syndrome in humans. Diabetes. 2008;57:209–217. doi: 10.2337/db07-0083. [DOI] [PubMed] [Google Scholar]
- Yagihashi S, Yamagishi S, Wada R. Pathology and pathogenetic mechanisms of diabetic neuropathy: correlation with clinical signs and symptoms. Diabetes Res Clin Pract. 2007;77(Suppl 1):S184–S189. doi: 10.1016/j.diabres.2007.01.054. [DOI] [PubMed] [Google Scholar]
- Zeharia A, Shaag A, Houtkooper RH, Hindi T, de Lonlay P, Erez G, Hubert L, Saada A, de Keyzer Y, Eshel G, Vaz FM, Pines O, Elpeleg O. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet. 2008;83:489–494. doi: 10.1016/j.ajhg.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]