

Abstract
Background
The glutathione S-transferase M1 (GSTM1) null variant is a common copy number variant associated with adverse pulmonary outcomes, including asthma and airflow obstruction, with evidence of important gene-by-environment interactions with exposures to oxidative stress.
Objective
To explore the joint interactive effects of GSTM1 copy number and tobacco smoke exposure on the development of asthma and asthma-related phenotypes in a family-based cohort of childhood asthmatics.
Methods
We performed quantitative PCR-based genotyping for GSTM1 copy number in children of self-reported white ancestry with mild to moderate asthma in the Childhood Asthma Management Program. Questionnaire data regarding intrauterine (IUS) and postnatal, longitudinal environmental tobacco smoke exposure were available. We performed both family-based and population-based tests of association for the interaction between GSTM1 copy number and tobacco smoke exposure with asthma and asthma-related phenotypes.
Results
Associations of GSTM1 null variants with asthma (p= .03), younger age of asthma symptom onset (p=.03), and greater airflow obstruction (reduced FEV1/FVC, p=.01) were observed among the 50 children (10% of the cohort) with exposure to IUS. In contrast, no associations were observed between GSTM1 null variants and asthma-related phenotypes among children without IUS exposure. Presence of at least one copy of GSTM1 conferred protection.
Conclusion
These findings support an important gene-by-environment interaction between two common factors: increased risk of asthma and asthma-related phenotypes conferred by GSTM1-null homozygosity in children is restricted to those with a history of IUS exposure.
Keywords: Asthma, GSTM1, copy number variation (CNV), gene by environment, intrauterine smoke exposure, tobacco smoke
INTRODUCTION
Glutathione S-transferase M1 (GSTM1) facilitates the scavenging of oxygen free radicals by catalyzing the reaction between glutathione (GSH) and organic hydroperoxides. GSTM1 is located on chromosome 1p13.3, a region known to be copy-number variable for a common large-scale deletion that spans GSTM1. Among Caucasians the GSTM1-null allele has a frequency of 70%, meaning that the approximately one-half of Caucasian individuals are GSTM1-null (homozygous for the deletion) and lack functional GSTM1.
GSTM1-null status has been implicated in poor outcomes in several auto-immune diseases, including systemic lupus erythematosus and rheumatoid arthritis. It has been clearly established as an important gene in chronic obstructive pulmonary disease (COPD). Its role in defense against oxidative stress also makes it a logical asthma candidate, and in fact, GSTM1 has been extensively studied as an asthma candidate gene, with more than 20 publications to date.
Yet the data for GSTM1 are conflicting. While several studies show increased risk of asthma or lung function decrements in all GSTM1 null subjects,1–8 others have identified such outcomes only in those subjects exposed to increased oxidative stress (tobacco smoke,9–11 or ozone,12–14 for example). Still others have found that absence of GSTM1 does not affect pulmonary outcomes, but rather modulates the effects of other glutathione S-transferases (such as GSTP1).15, 16 Functional data are limited, but also have not supported a role of GSTM1 genotype in asthma pathogenesis. Ercan et al. showed that asthmatics had lower levels of reduced glutathione and higher levels of malondialdehyde, consistent with higher levels of oxidative stress, but found no difference based on GSTM1 presence or absence.17 Similarly, Mak and colleagues found that GST activity was higher in asthmatics but, again, did not vary with GSTM1 genotype.18
Herein we report results from our efforts to characterize the joint interactive effects of GSTM1 copy number and smoke exposure on the development of asthma and related phenotypes in a large cohort of asthmatic children who were participating in the Childhood Asthma Management Program (CAMP), for which detailed smoke exposure data and longitudinal measures of pulmonary outcomes are available.
METHODS
Population
The Childhood Asthma Management Program (CAMP) is a multicenter North American clinical trial designed to investigate the long-term effects of inhaled anti-inflammatory medications in children with mild to moderate asthma. A diagnosis of asthma was based on methacholine hyperreactivity (PC20 [provocative concentration causing a 20% fall in FEV1] no greater than 12.5 mg/ml) and one or more of the following criteria for at least 6 months in the year before recruitment: (1) asthma symptoms at least two times per week, (2) at least two uses per week of an inhaled bronchodilator, or (3) daily asthma medication. Lung function measurements were obtained 13 times over the course of the 4-year follow-up (at time 0, 2, 4, and 8 months and then 3 times yearly throughout the trial). IgE and methacholine PC20 were assessed twice and 5 times respectively. Exposure to environmental tobacco smoke, including intrauterine smoke (IUS, any history of maternal smoking during pregnancy) and postnatal environmental tobacco smoke (ETS, history of smoke exposure in the household by a parent or primary caregiver between birth and enrollment), was assessed at baseline via questionnaire response by a child’s caregiver. ETS exposure was also assessed at each follow-up visit. Protocols for collection of baseline phenotypic data have been described in detail elsewhere.19, 20 Of the 1,041 children enrolled in the original clinical trial, 968 children and 1,518 of their parents contributed DNA samples to the CAMP Genetics Ancillary Study.
The Institutional Review Boards of the Brigham and Women’s Hospital and of the other CAMP study centers approved this study. Informed assent and consent were obtained from the study participants and their parents to collect DNA for genetic studies.
Genotyping Methods
2003 individuals (including 708 CAMP probands) were genotyped for GSTM1 copy number by real-time PCR with the Applied Biosystems Taqman copy number assay on a 7900HT instrument as previously described,21 using off-the-shelf GSTM1 (FAM-labeled) and RNaseP (VIC-labeled, serving as endogenous 2-copy reference) probes. Because of the small number of samples from non-white ethnic groups and concerns regarding genetic heterogeneity, we restricted this analysis to the self-described white subjects and their available parents (n=1630, including 517 white probands). Each sample was run in quadruplicate, with the ΔCt averaged over the 4 runs. Continuous copy number values are assigned according to the ΔΔCt method by ABI SDS software. Ambiguous and failed calls were re-run. Continuous CNV measures were binned into discrete CNV genotype calls (i.e. 0 copies, 1, 2, 3). 90 subjects were genotyped twice to assess reproducibility.
Statistical Analysis
Population-based analysis was performed using R v.2.4.1 and SAS v.9.1 (Cary, NC). We tested for CNV association with lung function phenotypes including post-bronchodilator forced expiratory volume in one second (FEV1), forced vital capacity (FVC), the ratio of FEV1/FVC, and log-transformed total serum IgE level and methacholine PC20. We tested for association with CNV both at baseline (adjusting for age, gender, and height as appropriate) and longitudinally using repeated measures (additionally adjusted for ongoing tobacco smoke exposure and CAMP treatment assignment (inhaled budesonide, nedocromil, or placebo)). For the longitudinal analysis, we constructed a linear mixed-effect model assuming an exponential decay in covariance using the PROC MIXED procedure in SAS. We tested for GSTM1 interaction with IUS and ETS exposure variables. Data were stratified for further analysis when significant interactions were noted.
Family-based testing was performed with PBAT v.5.3.0 (Bozeman, MT) for asthma affection status and all variables assessed in the population-based analyses above, using both baseline and longitudinal measures.22, 23 Haploview was used to obtain counts of allele transmission.24
Our primary analyses were run with a dominant model for GSTM1 (consistent with previous association results). We secondarily tested for association using an additive genetic model. Power calculations were performed using QUANTO version 1.2.25, 26
RESULTS
Genotyping
Over 98% of subjects were successfully genotyped by quantitative PCR. Reproducibility was excellent, as 89 of the 90 subjects had the same binned genotype call on repeat genotyping. Because of concerns for population stratification, we restricted analysis to the Caucasian individuals. As shown in Figure 1, the continuous CNV estimates obtained were easily binned into discrete genotype calls. In total, 282 probands were GSTM1 null, 191 were hemizygotes (i.e. had one copy of GSTM1), and 38 were homozygous for GSTM1 (i.e. had 2 or more functional copies of GSTM1). The null allele frequency was 71%, similar to the findings of other groups among Caucasian individuals.
Figure 1. GSTM1 genotyping among white probands, using Taqman qPCR.
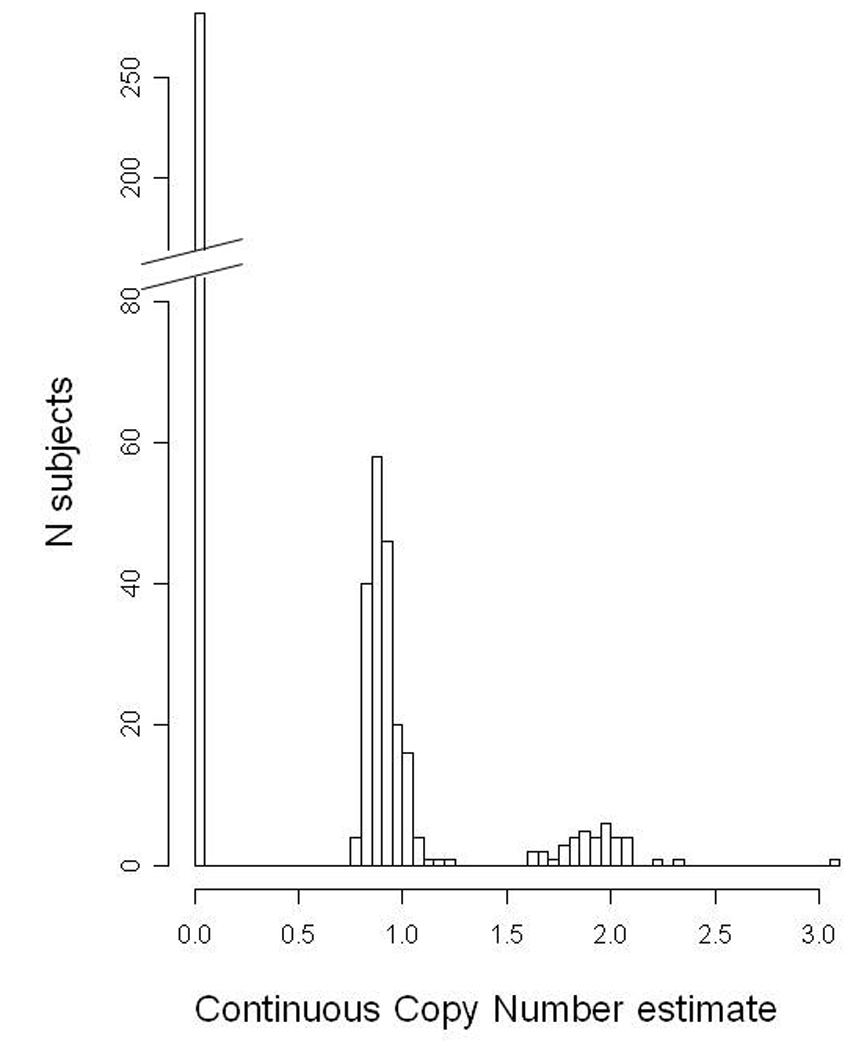
The 511 probands fall into several distinct bins, with 282 GSTM1 null, 191 with 1 copy, 37 with 2 copies and, 1 outlier subject with 3 copies of GSTM1, confirmed on repeat genotyping. Please note that Y axis has a break for improved visibility of low-frequency CNV bins.
Association of GSTM1 copy number with asthma phenotypes
Baseline characteristics of the 511 CAMP subjects are noted in Table 1. Among all subjects, we found no evidence of association between GSTM1 null status and asthma affection, spirometric measures of lung function (FEV1, FVC, and FEV1/FVC), total serum IgE levels, or airway hyperresponsiveness to methacholine (Table 2). Repeated measures analysis also showed no association with any of these phenotypes (all p >.1). Though GSTM1-null status was associated with younger age of onset of asthma in family-based testing (p=.01), similar associations were not observed in population-based testing.
Table 1.
Baseline Characteristics of 511 white CAMP probands
| Characteristic | Value |
|---|---|
| Age (years) | 8.8 (2.1) |
| Male gender (%) | 316 (62%) |
| Age of onset of asthma | 3 (2.4) |
| FEV1 % predicted | 103 (13) |
| FVC % predicted | 107 (13) |
| FEV1/FVC | 85 (6) |
| Total IgE (ng/mL) | 405 (159, 1086) |
| Methacholine PC20 | 1.1 (.5, 2.7) |
| Intrauterine smoke exposure (%) | 50 (10%) |
| History of post-natal exposure to environmental tobacco smoke (%) |
175 (34%) |
All values shown are at baseline, off all controller medication for asthma. Mean (SD) or Median (IQR) are shown as appropriate for normality of the data. 489 of the 511 probands had complete trio data available for family-based testng; characteristics of this subset were similar.
Table 2.
GSTM1 null status is associated with multiple asthma phenotypes among the subset exposed to IUS
| All subjects | Smoke exposed only | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GSTM1- null (n=282) |
GSTM1 present (n=229) |
Association p-value | GSTM1- null (n=29) |
GSTM1 present (n=21) |
Association p-value | ||||
| Population- based |
Family- based |
Population- based |
Family- based |
||||||
| Asthma | N/A | N/A | N/A | .2 | N/A | N/A | N/A | .032 | |
| FEV1 (% predicted) | 103.6 | 103.1 | .6 | 1 | 104.2 | 102.9 | .4 | .8 | |
| FVC (% predicted) | 107.4 | 106.5 | .5 | .7 | 110.5 | 104.7 | .041 | .1 | |
| FEV1/FVC | 85.4 | 85.7 | .7 | .7 | 83.8 | 86.9 | .031 | .012 | |
| PC20 (mg/dl) | .09 | .10 | .9 | .7 | .05 | −.04 | .7 | .3 | |
| IgE (IU/ml) | 2.6 | 2.6 | .5 | .9 | 2.5 | 2.4 | .9 | .3 | |
| Age of Onset (yrs) | 3.0 | 3.0 | .8 | .012 | 2.5 | 4.1 | .04 | .032 | |
P values represented here are for baseline association, using linear regression for population-based testing, and FBAT for family-based testing. Age of onset of asthma adjusted only for gender. IgE and LnPC20 also adjusted for age. Lung function variables additionally adjusted for height and height squared.
FEV1/FVC and FVC were additionally significant in longitudinal analysis with p= .02 and .003 respectively. Longitudinal analyses for FEV1, PC20, and IgE showed no association (p>.05)
Transmission of the GSTM1 null allele was associated with asthma, earlier age of onset of asthma, and a lower FEV1/FVC ratio.
In contrast, we found evidence for consistent gene-by-environment interactions between GSTM1 and smoke exposure in association with asthma and related outcomes. Stratified analysis revealed that GSTM1-null individuals with a history of IUS exposure had earlier age of onset of asthma, higher FVC, and a lower FEV1/FVC (Table 2) than IUS-exposed individuals with at least one copy of GSTM1. Transmission of the GSTM1-null allele was associated with asthma affection status, (p=.03) with a more than 2-fold increased ratio of transmitted to untransmitted alleles (T:U ratio of 23:10). The age of onset of asthma was 1.5 years lower among IUS-exposed subjects who were GSTM1-null (mean age 2.5 vs. 4.1, p<.05 in both population and family-based testing) than those with at least one copy of GSTM1. GSTM1-null status was also associated with lung function, with higher FVC and lower FEV1/FVC. This was noted both at baseline (Table 2) and with repeated measures analysis over the 4-year trial (p=.003 and .02 respectively). As shown in Figure 2, among IUS-exposed GSTM1-null subjects, the mean FEV1/FVC remained approximately 3% lower throughout the follow-up period (p=.02). GSTM1 status was not associated with any of the asthma phenotypes among subjects not exposed to IUS (data not shown). Formal testing for interaction between GSTM1 genotype and smoke exposure in population-based models confirmed significant interaction terms for FEV1/FVC and age-of-onset (p<.05).
Figure 2. FEV1/FVC reduction among GSTM1 null, IUS-exposed subjects persists over the 4-year trial period.
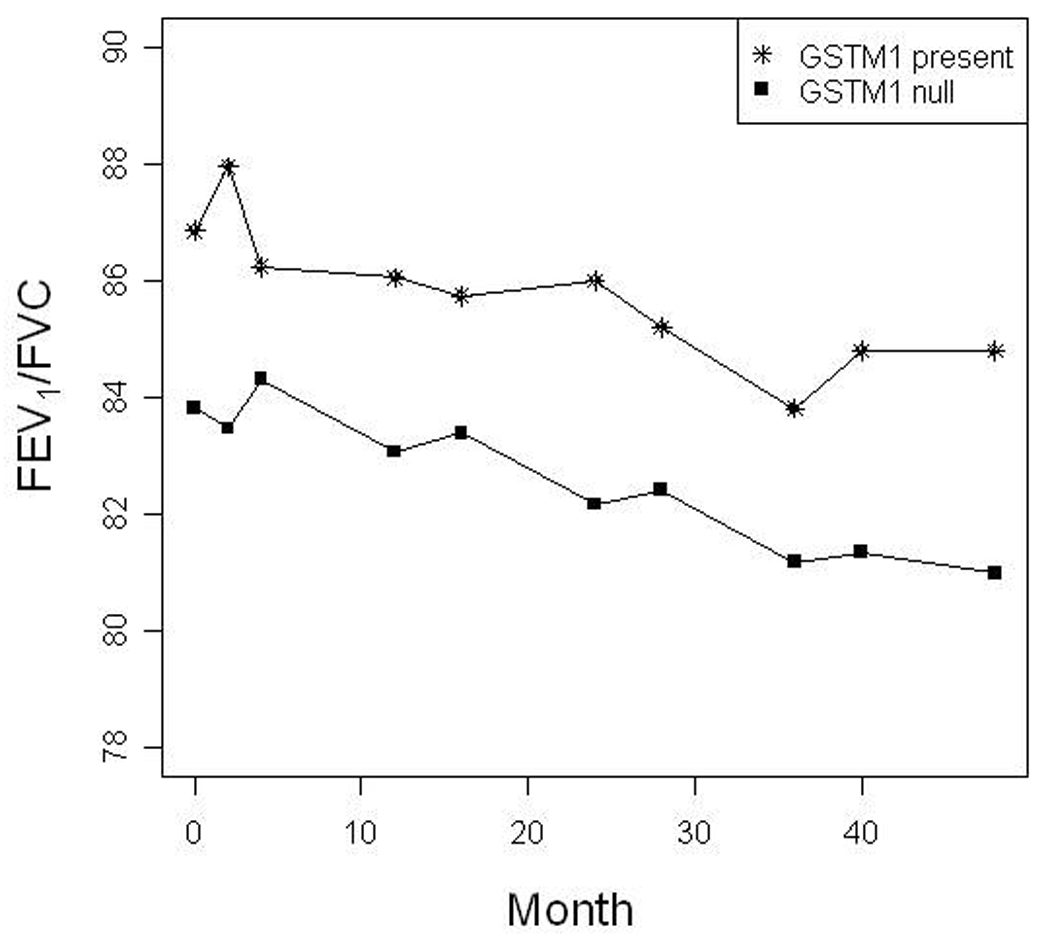
Mean FEV1/FVC values over the 48-month trial are depicted
Interaction between GSTM1 and postnatal environmental tobacco smoke (ETS) exposure was similar but of lower magnitude than the IUS interaction; GSTM1-null status is associated with lower baseline FEV1/FVC, and younger age of onset (p=.08 and .05 in population-based testing, for example, among the 175 subjects with ETS exposure). IUS is highly correlated with ETS, however, with 46 of the 50 IUS-exposed subjects reporting ETS as well. When analysis is limited only to the 129 ETS-exposed subjects without IUS exposure, we find no evidence of association between GSTM1 copy number and any of the variables tested.
One plausible explanation for association in the IUS-exposed subset is that the child’s GSTM1 copy number is simply a surrogate for maternal GSTM1 copy number (i.e. smoking causes higher oxidative stress in utero among mothers who lack GSTM1, which leads to long-term effects on the child’s risk for asthma and lung function). This was not the case in this cohort; the GSTM1 associations with asthma and lung function were noted only with the child’s genotype, not the mother’s. Testing outcomes in an additive model (as opposed to dominant) did not reveal any additional associations.
DISCUSSION
We demonstrate evidence for association of the GSTM1 null variant with asthma affection, age of asthma symptom onset, and pulmonary function among children with a history of passive smoke exposure. Though our results are compelling because of their consistency in both population- and family-based analysis (the latter rules out cryptic population stratification as a biasing confounder), our findings are best understood in the context of previously published studies. Table 3 summarizes all prior studies that assessed asthma affection status. Though statistically significant evidence of increased asthma risk among GSTM1-null carriers was observed in only 7 of 20 populations previously studied, trends of similar association (i.e., OR >1) were observed in 12 of the 14 publications in which an OR was either reported or could be calculated from the published data. Given the large sample sizes typically required to demonstrate statistically significant modest genetic effects, the consistency in the direction of association across most of these studies suggests that the effects observed across populations are likely meaningful.
Table 3.
Literature review: Effects of GSTM1-null on asthma affection status
| Paper | N | Children vs. Adults |
Race/ Nation |
Asthma def. | Main Effect1 | Gene by Environment Interaction? |
Interaction with other GST’s? |
|---|---|---|---|---|---|---|---|
| Fryer 200033 | 127/44 | Adult | White | Wheeze, cough, dyspnea, or chest tight + BDR 15% + atopy | OR 1.13 (CI .6, 2.3)* | ||
| Ivaschenko 20024 | 109/90 | Mixed | Russian | Specialist-dx asthma + recurrent sx’s +PF variability >30% | OR 3.49 (1.9, 6.4) | Additive effects with GSTT1 | |
| Gilliland 20029 | 2950 pop (451 asthma) | Children | Mixed | MD-dx asthma | - | IUS: early-onset asthma, wheezing, active asthma, asthma treatment, ER visits among GSTM1− & IUS+ No ETS effects | |
| Tamer 20046 | 101/103 | Adult | Turkish | Clinical asthma hx + BDR reversible | OR 2.34 (1.3, 4.2) | Several GSTM1/GSTT1/ GSTP1 interactions sig | |
| Saadat 20045 | 85/85 | Adult | Iranian | 2 of 3: Typical sx’s, + BDR >15%, >20% PF variability | OR 3.2 (1.7, 6.1) | GSTM1* GSTT1 are additive | |
| Kabesch 200410 | 3054 population (268 asthma) | Children | German white | MD-dx asthma or MD-dx “recurrent spastic or asthmatic bronchitis” | OR 1.19 (.9, 1.5)* | GSTM1−/ETS+ higher asthma OR than GSTM1+/ETS− | - interaction with GSTT1 |
| Brasch-Andersen 20041 | 160 families (296 asthmatics + parents) | Mixed | Danish | Recurrent cough, wheeze, dysp + methacholine PC20 | TDT - for asthma (+ for asthma + RAST in recessive model) | ||
| 86 families (156 asthma + parents) | Children | Danish | Clinical asthma sx’s & effect of std. asthma meds | TDT + for asthma | |||
| Lee 200534 | 82/184 | Children | Taiwan | MD-dx asthma | OR 1.37 (.8, 2.4) | GSTP1 effects noted if GSTM1 + | |
| Holla 20068 | 306/331 | Adult | Caucasian | Current MD-dx asthma + sx + anti-asthma meds | OR 1.18 (.9, 1.6) | No interaction with GSTT1 | |
| Romieu 200613 | 151 | Children | Mexico City | Clinical sx & response to rx | OR of mod-severe asthma 2.05 (vs mild, no control group) | Ozone. Ozone → more difficulty breathing | Additive ozone interaction effects with GSTP1 val/val |
| Palmer 200611 | 504 asthmatics | Children | white | MD-dx asthma | 1.34 (.9, 2) | ETS: Younger subset higher OR of asthma & ETS. Age- dept changes in FEV1, PEFR vary w/ ETS | |
| Ercan 200617 | 196 mild/ 116 mod- sev/ vs. ctrls: 187 and 68 | Children | White | Intermittent wheeze + 12% BDR or response to rx or + PC20 | OR 1.05 (.7, 1.5)* | - interaction with GSTP1 | |
| Salam 200735 | 3124 pop (476 asthma) | Children | Hispanic and Non-hispanic white | MD-dx | OR 1.21 (.98, 1.5) | - interaction between GSTP1 & EPHX1 | |
| Hanene 20072 | 105/112 | Children | Tunisia | NR | OR 2.35 (1.3, 4.3) | ||
| Kamada 200715 | 391 kids, 462 adult/ 639 ctrl | Children & adult | Japanese | MD-dx asthma | - | GSTP1 effects only in GSTM1 + | |
| 115 kids /184 ctrl | Children | Japanese | MD-dx asthma | - | GSTP1 effects only in GSTM1 + | ||
| Mak 200718 | 315/315 | Adult | Chinese | Expert Panel criteria + cough/wheeze + FEV1varies +/−15% | OR 0.75 (.5, 1.03)* | ||
| Imboden 200836 | 4426 pop; 144 develop asthma | Adult | Swiss, “Predominan tly Caucasian” | New-onset, MD-dx asthma | OR .93 (.7, 1.3)* | No interaction with GSTP1 | |
| Islam 20093 | 1610 CHS; 152 develop asthma | Children | Hispanic or non-Hispanic white | MD-dx asthma | + HR of 1.6 for new onset asthma during adolescence | - No change in HR when adj for ozone or PM25 | No interaction with GSTP1 effects |
| Rogers 2009 | 511 white asthmatic probands in 489 trios | Children | White | MD-dx + methacholine PC20 | − TDT for asthma + Early age of onset | +IUS exposed: asthma, early age of onset, ↑ FVC, ↓ FEV1/FVC |
Abbreviations: MD-dx = doctor diagnosed; Sx = symptoms; Rx = treatment; BDR = bronchodilator response to albuterol; PF = peak flow; TDT = transmission disequilibrium test.
Literature search included the HuGENavigator site for GSTM1 and asthma, supplemented with a PubMed search for the terms “GSTM1 and Asthma”. Included here are English-language studies with a primary outcome diagnosis of asthma.
Bold = statistically significant
Main effects presented:
OR or HR for asthma calculated by authors if no asterisk present (adjusted results presented if performed).
OR presented was calculated based on data from paper, where possible.
negative for asthma in all subjects; unable to calculate OR from data presented in publication.
Though asthma is known to be highly heritable, with relative risk to siblings of an asthmatic of ~ 3, the role of environmental exposures in asthma pathogenesis is critical and is often underappreciated in genetic association studies. We note that, though many potential asthma genes are emerging from candidate gene and genome-wide association studies,27, 28 the vast majority of even the most promising asthma candidate genes have failed to show association in multiple other populations. One explanation for such disparate results in different populations is a failure to account for gene-by-environment interaction. Our data and others suggest that defining the importance of GSTM1 in asthma requires careful consideration of such interactions. It is certainly plausible that the cohorts that showed GSTM1 effects in all subjects simply had higher levels of exposure to environmental tobacco smoke, ozone, or other sources of increased oxidative stress.
Our cohort, with 511 asthmatics and 489 complete trios, is among the largest asthmatic cohorts tested for GSTM1 association (see Table 3). The study was thus powered to appreciate clinically meaningful differences in the entire cohort (with 80% power to find a change in FEV1 of 4%, or onset of asthma that was 7 months earlier for example); adequate power makes our findings of no association between GSTM1 and various asthma phenotypes in the entire cohort more meaningful. In contrast to the findings in all subjects, we found strong evidence of association with asthma among subjects exposed to IUS (with 23:10 Transmitted: Untransmitted ratio of the GSTM1-null allele). Similarly, GSTM1-null individuals who were exposed to IUS had a younger age of asthma onset and a lower FEV1/FVC ratio than those with at least one copy of GSTM1; GSTM1 status did not influence these phenotypes in subjects without IUS exposure. We also note that four of the five studies that assessed GSTM1 in the context of high oxidative stress found significant association (Table 3); similarly, all studies that evaluated GSTM1 copy number and environmental effects on lung function were significant (Table 4). Thus, the literature strongly supports a role for environmental modulation of GSTM1 effects.
Table 4.
GSTM1 effects on lung function in asthma
| Paper | N | Race | Asthma Diagnosis |
Children vs. Adults |
Lung Function Effect of GSTM1 null |
Gene by Environment interaction? |
Interaction with other GST’s? |
|---|---|---|---|---|---|---|---|
| Gilliland 027 | 1940pop (287 asthma) | Mixed | MD-dx asthma | Children | ↓ FEV1& FVC growth over 4y; esp. in asthmatics | GSTM1*GSTP1 effects additive | |
| Carroll 200537 | 224 families with 418 children | Caucasian | MD-dx asthma, + wheeze in past year | Children | - in probands ↑ FEV1, FVC in siblings | Additive GSTP1, GSTM1 | |
| Holla8 | 675 (~50% asthma) /331 | Caucasian | Current MD-dx asthma + sx + anti-asthma meds | Adult | ↓ FEV1/FVC; do not report FEV1 or FVC | ||
| Romieu14 | 158 asthmatics | Mexico City | Sx and response to rx | Children | - | ↓FEF 25–75 with ↑ozone exposure, esp. in more severe asthma | |
| Palmer11 | 504 asthmatics | white | MD-dx asthma | Children | NR | Age 3–12, no ETS: ↓ FEV1 Age 13–21 + ETS: ↓ PEFR | |
| Rogers | 511 white asthmatic probands in 489 trios | MD-dx + methacholine challenge | Children | - | IUS exposed: ↑FVC, ↓ FEV1/FVC |
Abbreviations: MD-dx = doctor diagnosed; Sx = symptoms; Rx = treatment; BDR = bronchodilator response to albuterol; PF = peak flow NR = not reported
Literature search included the HuGENavigator site for GSTM1 and asthma, supplemented with a PubMed search for the terms “GSTM1 and Asthma” and “GSTM1 and Lung Function”. Included here are English-language studies focused on asthmatics (as opposed to COPD or the general population) that evaluated pulmonary function effects.
Our results are most similar to those of Gilliland et al., who found that in a population-based cohort of 2590 children, associations of the GSTM1-null variant with active asthma, medication use, and earlier age of asthma onset were restricted entirely to children with a history of in utero smoke exposure.9 We note that early onset of asthma was assessed only in Gilliland’s work and ours, and both studies identified significant GSTM1 effects in the context of IUS exposure. Given the putative mechanism of GSTM1 as a defense against oxidative stress, exposure to IUS or ETS early in life could logically result in an earlier age of asthma onset among those who lack GSTM1, suggesting that this phenotype may be of particular interest for studying genetic interactions with early-life exposures.
GSTM1-null status was associated with a lower FEV1/FVC and higher FVC in IUS-exposed subjects in our cohort. These differences were present at baseline and sustained over the 4-year trial; in contrast, the slope of FEV1/FVC and FVC did not change with GSTM1 status. These lung function findings are most consistent with dysanapsis, in which airway size is relatively smaller than lung parenchyma.29 Nicotine exposure has been shown to stimulate lung branching morphogenesis and increase lung size in embryonic murine explants,30 while IUS exposure is associated with increased airway collagen deposition in rhesus monkeys31 and with increased airway thickness in human infants.32 While our results are consistent with the hypothesis that the anti-oxidant effects of GSTM1 prevent such changes in lung vs. airway growth in utero, this has not been previously examined.
A strength of this study is our use of quantitative PCR to genotype GSTM1 for 0, 1, or 2 copies. Most previous publications genotyped only GSTM1 presence or absence, and therefore are unable to distinguish whether the associations observed are driven by GSTM1 hemizyogtes or homozygotes. Our results demonstrate that a single copy of GSTM1 is sufficient to confer protective effects. Due to the relatively small number of 2-copy individuals in the IUS-exposed cohort (N=4), we are underpowered to make firm inferences regarding additional effects of a second copy of GSTM1 in asthma.
We note that our findings were significant in the IUS-exposed subjects (n=50) but not significant in the much larger subset exposed to ETS (n=175). Yet 46 of the 50 subjects exposed to IUS were also exposed to ETS later in life, and tended to have more ongoing smoke exposure throughout the 4-year follow-up period. Due to this very high degree of correlation between IUS and ETS, we are unable to discern whether the observed associations with IUS reflect specific gene-by-environment interactions that are established primarily in utero, or whether IUS is simply a marker for more extensive and sustained ETS exposure later in life. We acknowledge that our findings were significant only in a small subset of subjects, with correspondingly marginal p values; though this subgroup analysis was planned given the previous GSTM1 literature, confirmation of the importance of the interactions of GSTM1 and smoke exposure in additional asthma cohorts is necessary.
In summary, we found that lack of GSTM1 is associated with asthma, an early onset of asthma, and low FEV1/FVC among asthmatic children exposed to IUS. One copy of GSTM1 appears to be sufficient to confer protective effects. Our findings support the growing literature on the importance of GSTM1 in asthmatics exposed to tobacco smoke.
Acknowledgements
We thank all subjects for their ongoing participation in this study. We acknowledge the CAMP investigators and research team, supported by the National Heart, Lung, and Blood Institute (NHLBI), for collection of CAMP Genetic Ancillary Study data.
The CAMP Genetics Ancillary Study is supported by the NHLBI, N01 HR16049. Additional support for this research came from grants R01 HL093076, R01 HL086601, U01 HL065899, P01 HL083069, and T32 HL07427 from the National Institutes of Health and the NHLBI. All work on data from the CAMP Genetic Ancillary Study was conducted at the Channing Laboratory and the Brigham and Women’s Hospital under appropriate CAMP policies and human subjects protections.
References
- 1.Brasch-Andersen C, Christiansen L, Tan Q, Haagerup A, Vestbo J, Kruse TA. Possible gene dosage effect of glutathione-S-transferases on atopic asthma: using real-time PCR for quantification of GSTM1 and GSTT1 gene copy numbers. Hum Mutat. 2004;24:208–214. doi: 10.1002/humu.20074. [DOI] [PubMed] [Google Scholar]
- 2.Hanene C, Jihene L, Jamel A, Kamel H, Agnes H. Association of GST genes polymorphisms with asthma in Tunisian children. Mediators Inflamm. 2007;2007:19564. doi: 10.1155/2007/19564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Islam T, Berhane K, McConnell R, Gauderman WJ, Avol E, Peters JM, et al. Glutathione-S-transferase (GST) P1, GSTM1, exercise, ozone and asthma incidence in school children. Thorax. 2009;64:197–202. doi: 10.1136/thx.2008.099366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivaschenko TE, Sideleva OG, Baranov VS. Glutathione- S-transferase micro and theta gene polymorphisms as new risk factors of atopic bronchial asthma. J Mol Med. 2002;80:39–43. doi: 10.1007/s001090100274. [DOI] [PubMed] [Google Scholar]
- 5.Saadat M, Saadat I, Saboori Z, Emad A. Combination of CC16, GSTM1, and GSTT1 genetic polymorphisms is associated with asthma. J Allergy Clin Immunol. 2004;113:996–998. doi: 10.1016/j.jaci.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Tamer L, Calikoglu M, Ates NA, Yildirim H, Ercan B, Saritas E, et al. Glutathione-S-transferase gene polymorphisms (GSTT1, GSTM1, GSTP1) as increased risk factors for asthma. Respirology. 2004;9:493–498. doi: 10.1111/j.1440-1843.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- 7.Gilliland FD, Gauderman WJ, Vora H, Rappaport E, Dubeau L. Effects of glutathione-S-transferase M1, T1, and P1 on childhood lung function growth. Am J Respir Crit Care Med. 2002;166:710–716. doi: 10.1164/rccm.2112065. [DOI] [PubMed] [Google Scholar]
- 8.Holla LI, Stejskalova A, Vasku A. Polymorphisms of the GSTM1 and GSTT1 genes in patients with allergic diseases in the Czech population. Allergy. 2006;61:265–267. doi: 10.1111/j.1398-9995.2006.01000.x. [DOI] [PubMed] [Google Scholar]
- 9.Gilliland FD, Li YF, Dubeau L, Berhane K, Avol E, McConnell R, et al. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med. 2002;166:457–463. doi: 10.1164/rccm.2112064. [DOI] [PubMed] [Google Scholar]
- 10.Kabesch M, Hoefler C, Carr D, Leupold W, Weiland SK, von Mutius E. Glutathione S transferase deficiency and passive smoking increase childhood asthma. Thorax. 2004;59:569–573. doi: 10.1136/thx.2003.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer CN, Doney AS, Lee SP, Murrie I, Ismail T, Macgregor DF, et al. Glutathione S-transferase M1 and P1 genotype, passive smoking, and peak expiratory flow in asthma. Pediatrics. 2006;118:710–716. doi: 10.1542/peds.2005-3030. [DOI] [PubMed] [Google Scholar]
- 12.Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM. Diabetes. 1994;43:613–621. doi: 10.2337/diab.43.5.613. [DOI] [PubMed] [Google Scholar]
- 13.Romieu I, Ramirez-Aguilar M, Sienra-Monge JJ, Moreno-Macias H, del Rio-Navarro BE, David G, et al. GSTM1 and GSTP1 and respiratory health in asthmatic children exposed to ozone. Eur Respir J. 2006;28:953–959. doi: 10.1183/09031936.06.00114905. [DOI] [PubMed] [Google Scholar]
- 14.Romieu I, Sienra-Monge JJ, Ramirez-Aguilar M, Moreno-Macias H, Reyes-Ruiz NI, Estela del Rio-Navarro B, et al. Genetic polymorphism of GSTM1 and antioxidant supplementation influence lung function in relation to ozone exposure in asthmatic children in Mexico City. Thorax. 2004;59:8–10. [PMC free article] [PubMed] [Google Scholar]
- 15.Kamada F, Mashimo Y, Inoue H, Shao C, Hirota T, Doi S, et al. The GSTP1 gene is a susceptibility gene for childhood asthma and the GSTM1 gene is a modifier of the GSTP1 gene. Int Arch Allergy Immunol. 2007;144:275–286. doi: 10.1159/000106316. [DOI] [PubMed] [Google Scholar]
- 16.Lee YL, Lee YC, Guo YL. Associations of glutathione S-transferase P1, M1, and environmental tobacco smoke with wheezing illness in school children. Allergy. 2007;62:641–647. doi: 10.1111/j.1398-9995.2007.01380.x. [DOI] [PubMed] [Google Scholar]
- 17.Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, Soyer OU, et al. Oxidative stress and genetic and epidemiologic determinants of oxidant injury in childhood asthma. J Allergy Clin Immunol. 2006;118:1097–1104. doi: 10.1016/j.jaci.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Mak JC, Ho SP, Leung HC, Cheung AH, Law BK, So LK, et al. Relationship between glutathione S-transferase gene polymorphisms and enzyme activity in Hong Kong Chinese asthmatics. Clin Exp Allergy. 2007;37:1150–1157. doi: 10.1111/j.1365-2222.2007.02704.x. [DOI] [PubMed] [Google Scholar]
- 19.Childhood Asthma Management Program Research Group. The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Control Clin Trials. 1999;20:91–120. [PubMed] [Google Scholar]
- 20.Long-term effects of budesonide or nedocromil in children with asthma. The Childhood Asthma Management Program Research Group. N Engl J Med. 2000;343:1054–1063. doi: 10.1056/NEJM200010123431501. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM. Using the noninformative families in family-based association tests: a powerful new testing strategy. Am J Hum Genet. 2003;73:801–811. doi: 10.1086/378591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lange C, Laird NM. On a general class of conditional tests for family-based association studies in genetics: the asymptotic distribution, the conditional power, and optimality considerations. Genet Epidemiol. 2002;23:165–180. doi: 10.1002/gepi.209. [DOI] [PubMed] [Google Scholar]
- 24.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 25.Gauderman WJ. Candidate gene association analysis for a quantitative trait, using parent-offspring trios. Genet Epidemiol. 2003;25:327–338. doi: 10.1002/gepi.10262. [DOI] [PubMed] [Google Scholar]
- 26.Gauderman WJ, Morrison JM. Quanto 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies. 2006 [Google Scholar]
- 27.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006 doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 28.Rogers AJ, Raby BA, Lasky-Su JA, Murphy A, Lazarus R, Klanderman BJ, et al. Assessing the Reproducibility of Asthma Candidate Gene Associations Using Genome-wide Data. Am J Respir Crit Care Med. 2009 doi: 10.1164/rccm.200812-1860OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mead J. Dysanapsis in normal lungs assessed by the relationship between maximal flow, static recoil, and vital capacity. Am Rev Respir Dis. 1980;121:339–342. doi: 10.1164/arrd.1980.121.2.339. [DOI] [PubMed] [Google Scholar]
- 30.Wongtrakool C, Roser-Page S, Rivera HN, Roman J. Nicotine alters lung branching morphogenesis through the alpha7 nicotinic acetylcholine receptor. Am J Physiol Lung Cell Mol Physiol. 2007;293:L611–L618. doi: 10.1152/ajplung.00038.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sekhon HS, Jia Y, Raab R, Kuryatov A, Pankow JF, Whitsett JA, et al. Prenatal nicotine increases pulmonary alpha7 nicotinic receptor expression and alters fetal lung development in monkeys. J Clin Invest. 1999;103:637–647. doi: 10.1172/JCI5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elliot J, Vullermin P, Robinson P. Maternal cigarette smoking is associated with increased inner airway wall thickness in children who die from sudden infant death syndrome. Am J Respir Crit Care Med. 1998;158:802–806. doi: 10.1164/ajrccm.158.3.9709055. [DOI] [PubMed] [Google Scholar]
- 33.Fryer AA, Bianco A, Hepple M, Jones PW, Strange RC, Spiteri MA. Polymorphism at the glutathione S-transferase GSTP1 locus. A new marker for bronchial hyperresponsiveness and asthma. Am J Respir Crit Care Med. 2000;161:1437–1442. doi: 10.1164/ajrccm.161.5.9903006. [DOI] [PubMed] [Google Scholar]
- 34.Lee YL, Hsiue TR, Lee YC, Lin YC, Guo YL. The association between glutathione S-transferase P1, M1 polymorphisms and asthma in Taiwanese schoolchildren. Chest. 2005;128:1156–1162. doi: 10.1378/chest.128.3.1156. [DOI] [PubMed] [Google Scholar]
- 35.Salam MT, Lin PC, Avol EL, Gauderman WJ, Gilliland FD. Microsomal epoxide hydrolase, glutathione S-transferase P1, traffic and childhood asthma. Thorax. 2007;62:1050–1057. doi: 10.1136/thx.2007.080127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imboden M, Rochat T, Brutsche M, Schindler C, Downs SH, Gerbase MW, et al. Glutathione S-transferase genotype increases risk of progression from bronchial hyperresponsiveness to asthma in adults. Thorax. 2008;63:322–328. doi: 10.1136/thx.2007.085555. [DOI] [PubMed] [Google Scholar]
- 37.Carroll WD, Lenney W, Jones PW, Strange RC, Child F, Whyte MK, et al. Effects of glutathione S-transferase M1, T1 and P1 on lung function in asthmatic families. Clin Exp Allergy. 2005;35:1155–1161. doi: 10.1111/j.1365-2222.2005.02313.x. [DOI] [PubMed] [Google Scholar]