

Abstract
Here we present a novel methodology for the identification of the targeted post-translational modifications present in highly modified proteins using mixed integer linear optimization and electron transfer dissociation (ETD) tandem mass spectrometry. For a given ETD tandem mass spectrum, the rigorous set of modified forms that satisfy the mass of the precursor ion, within some tolerance error, are enumerated by solving a feasibility problem via mixed integer linear optimization. The enumeration of the entire superset of modified forms enables the method to normalize the relative contributions of the individual modification sites. Given the entire set of modified forms, a superposition problem is then formulated using mixed integer linear optimization to determine the relative fractions of the modified forms that are present in the multiplexed ETD tandem mass spectrum. Chromatographic information in the mass and time dimension is utilized to assess the likelihood of the assigned modification states, to average several tandem mass spectra for confident identification of lower level forms, and to infer modification states of partially assigned spectra. The utility of the proposed computational framework is demonstrated on an entire LC-MS/MS ETD experiment corresponding to a mixture of highly modified histone peptides. This new computational method will facilitate the unprecedented LC-MS/MS ETD analysis of many hypermodified proteins and offer novel biological insight into these previously understudied systems.
Accurate identification of post-translational modifications (PTMs)1 is a critical and often difficult task in proteomics. Most standard mass spectrometry-based techniques for the identification of protein modifications utilize a “bottom up” approach where the proteins are enzymatically digested into smaller peptides that are subsequently ionized and fragmented via CID to derive their sequence information (1–9). The identification of all the modifications present in a protein hinges on the successful identification of the PTM modifications of its corresponding peptides. This protocol can be limited by (a) insufficient elution and detection of all the peptides that cover the entire sequence of the protein, (b) false or incomplete identifications at the peptide level, and (c) the existence of different modification states of the same protein. Additional complications arise when using CID to study labile PTMs such as phosphorylation, glycosylation, or sulfonation. In these instances, the preferred reaction is often the cleavage of the PTM as opposed to the backbone of the peptide, resulting in a high intensity peak corresponding to the difference of the parent mass and the cleaved modification. The advent of electron capture dissociation (ECD) (10, 11) and electron transfer dissociation (ETD) (12–15) has enabled researchers to address the aforementioned issues associated with bottom up approaches using CID by adopting a complementary top down or middle down analysis strategy.
ECD and ETD both involve the reaction of an electron with a highly protonated cation to form an odd electron peptide. This process induces large amounts of backbone cleavage to yield c and z· ions that are analogous to the b and y ion series typically encountered in CID tandem mass spectra. Unlike CID, ECD/ETD cleavage is weakly affected by the composition and number of amino acids in the peptide and for certain systems can provide more fragmentation coverage than CID alone, especially for bigger peptides with higher charge states. Both ECD and ETD also prevent the cleavage of labile modifications, and thus PTMs are retained on the corresponding c and z· ions. The aforementioned benefits make ECD/ETD particularly well suited for the LC-MS/MS top down and middle down analysis of post-translationally modified proteins. These top down and middle down approaches also enable the approximate inference of protein abundance from the chromatogram and MS1 information because the full protein sequence elutes from the column (16).
In recent years, there has been significant interest in the identification of highly modified proteins, such as histones. Histone proteins are key regulators of many important DNA processes in eukaryotes, and recent studies have elucidated complex relationships between histone modifications and many nuclear events. It has also been shown that differences in global histone modifications in tissues can be used to predict the clinical outcome of cancer patients (17). Early MS or immunoassay studies were only able to analyze these modifications on a site-by-site basis and as a result lost important connectivity information on the molecular level because several modified forms of the same protein exist concurrently. In MS-based applications, the use of traditional reversed phase HPLC for the separation of a highly modified protein results in poor chromatographic resolution because all the modified forms are physically similar. Successful off-line techniques for the separation of highly modified histone forms have been achieved using cation exchange hydrophilic interaction chromatography (HILIC) (18), which separates the modified species primarily by the number of acetyl groups and secondly by the degree of methylation. The separation must be conducted off line because the mobile phase additives used are non-volatile components, and subsequent fractionation is necessary for mass spectrometric analysis. This protocol has made it possible to analyze the first 50 amino acids of the N-terminal tail of histone H3 and provided important insight regarding connectivity information between the modification sites. A major disadvantage of this approach is that the off-line nature of the experimental protocol is extremely time-consuming (on the order of months) and thus prohibits the ability to conduct multiple runs for high throughput studies and statistical validation. Additionally, other off-line techniques have been successful in the extraction and purification of modified histone proteins using acid-urea gel electrophoresis (19) but suffer from similar throughput constraints.
We have recently developed chromatography that is particularly suited for LC-MS ETD analysis of highly modified polypeptides with successful applications to histone proteins (20). The protocol uses a “saltless” pH gradient to elute the various modified forms in a weak cation exchange HILIC. Unprecedented separation of the modified histone forms is achieved within a single LC-MS/MS ETD experiment, thereby introducing important chromatographic information that can be utilized in the subsequent identification and quantification of these post-translational modifications. Although the achieved separation is exceptional in comparison with previous attempts, the complexity and relative similarity of the modified forms still results in minor species co-eluting with similar mass and retention times, thus resulting in multiplexed tandem mass spectra. The term “multiplexed” as used here refers to the fact that several species are dissociated and measured in a single tandem mass spectrum (21) and should not be confused with the multiplex experimental protocols. Computational methodologies that utilize the extensive and complementary information contained within these LC-MS/MS data sets are nonexistent as the technology has only recently been developed.
In this work, we present a novel mixed integer linear optimization (MILP) computational framework for the identification and quantification of highly modified proteins using LC-MS and ETD tandem mass spectrometry. Key concepts of the proposed framework are illustrated using histone H3.2 as an example system. For a given primary sequence, the entire set of post-translational modifications that satisfy a precursor mass are enumerated by solving an MILP feasibility problem. Given this set of PTM forms, an MILP superposition problem is then solved to determine the relative fractions of the modified forms that are present in the multiplexed ETD tandem mass spectrum. An important aspect of the proposed framework is that chromatographic information is used to correlate the modification states as a function of modification position, mass, and time. The proposed computational framework is applied to an entire LC-MS/MS ETD experiment corresponding to a mixture of highly modified histone peptides to demonstrate its utility.
MATERIALS AND METHODS
In this section, we present the mathematical models used to identify and quantify the PTMs present within a highly modified protein mixture. Key concepts are illustrated using histone H3.2, but it is important to note that the same model applies to any highly modified protein. The framework is comprised of 1) a mixed integer linear optimization model for enumerating the entire space of position-targeted modified forms that satisfy a given precursor mass and 2) a superposition problem based on mixed integer linear optimization for determining the relative composition of the modified forms in a multiplexed ETD tandem mass spectrum. In a subsequent section, we demonstrate using an entire LC-MS/MS experiment how chromatographic information is integrated into this MILP framework to ensure the highest confidence identifications.
MILP Model 1: Enumeration of All Position-targeted Modified Forms
Parameter and Set Definitions
We begin with the assumption that the sequence information is known for the peptide or protein under investigation; that is, the sample being analyzed has been purified to contain only the modified forms of the protein of interest (for experimental purification protocols corresponding to histone families, please see Refs. 19 and 22). Throughout this section, we will use histone H3.2, whose sequence is presented in Fig. 1, to motivate the problem.
Fig. 1.

Primary sequence for the first 50 amino acids of histone H3 with labeled potential post-translational modifications. “1Me,” “2Me,” and “3Me” denote single, di-, and trimethylation, respectively; “Ac” denotes acetylation; and “P” denotes phosphorylation.
We also select a number of targeted positions in the sequence. For histone H3, we select 3, 4, 9, 10, 14, 18, 23, 27, 28, and 36, which can take on any of the position-specific post-translational modifications presented in Fig. 1. Specifically, positions 3, 10, and 28 can be phosphorylated; positions 14, 18, and 23 can be acetylated; and positions 4, 9, 27, and 36 can be either methylated, dimethylated, trimethylated, or acetylated. Given this template sequence information, the enumeration model computes the entire space of possible post-translationally modified forms that satisfy the mass balance associated with the modified precursor mass, Mpmod, which is given experimentally in the MS1. Each position on the template sequence is represented by the index k that belongs to the set K as presented in Equation 1,
where L in Equation 1 denotes the length of the template sequence. For instance, we will focus on the 50 N-terminal amino acids of histone H3 (e.g. L = 50) as this subsequence is readily generated by endoproteinase Glu-C digestion, and nearly all known histone H3 modification sites are located within the first 36 residues of this protein (18). It should be noted that only a subset of the template positions in k ∈ K can be post-translationally modified (that is, only certain residues are susceptible to modification for this system). The space of known modifications is encoded by the set M and is presented in Equation 2 for histone H3.
In Equation 2 me1 corresponds to the methylation, me2 corresponds to the dimethylation, me3 corresponds to the trimethylation, ac corresponds to the acetylation, and phos corresponds to the phosphorylation of an amino acid. The monoisotopic masses of the modifications in the set M are given by the parameter ModMass(m) where ModMass(me1) = +14.0157, ModMass(me2) = +28.0314, ModMass(me3) = +42.0471, ModMass(ac) = +42.0106, and ModMass(phos) = +79.9799.
As previously mentioned, only certain positions k in the template sequence can be post-translationally modified, and only a subset of modifications m can exist for a certain position (i.e. m = m(k)). We classify this as position-targeted and PTM type-targeted, respectively. Thus, we define the set modify(m,k) to denote what modifications are possible for a given position k.
 |
For the first 50 amino acids of histone H3, we are interested in the potential position-targeted and type-targeted modifications that are presented in Table I. There are 10 mutatable positions and a total of 36,864 distinct modified histone codes or distinct isomers that are site-specific. These modifications were selected based upon their existence and reported abundance in the literature. We also examined the effects of including lower level modifications (see supplemental material) and found that the modifications presented in Table I best represent this system. The combinations of m and k shown in Table I are the active elements in the parameter set modify(m,k). For example, modify(phos,3) = 1 and modify(me3,4) = 1, but modify(me1,8) = 0. This parameter set is useful for significantly reducing the number of variables needed to formulate the model.
Table I.
List of targeted modification sites and allowed PTM types for the first 50 amino acids of histone H3
| Amino acid position, k | Possible modifications, m |
|---|---|
| 3 | phos |
| 4 | me1, me2, me3, ac |
| 9 | me1, me2, me3, ac |
| 10 | phos |
| 14 | ac |
| 18 | ac |
| 23 | ac |
| 27 | me1, me2, me3, ac |
| 28 | phos |
| 36 | me1, me2, me3, ac |
Analogous to the modified mass of the protein, Mpmod, as determined experimentally in the MS1, we define the parameter Mpunmod to denote the mass of the unmodified protein, which for histone H3 is 5338.09 or 5341.22 Da using the monoisotopic and average masses of the amino acids, respectively. The intensity of an ion peak i in the tandem mass spectrum is defined by the parameter λ(i).
Mathematical Model
The problem of enumerating the entire space of all position- and type-targeted modified forms for a given precursor mass is formulated as an assignment model where binary variables, ym,k, are used to represent the potential assignment of a modification of type m to the amino acid in position k in the template sequence.
 |
To reduce problem complexity, these variables are only defined for the pairs (m,k) such that modify(m,k) = 1. Also note that if ym,k = 0 then amino acid k is unmodified.
We define constraints to ensure that the modifications predicted are physically meaningful. For instance, at most one modification, m, can be assigned to a particular residue in position k in the template sequence. This is modeled by the constraint presented in Equation 5.
 |
Note that this constraint allows for the unmodified alternative (i.e. ym,k = 0) to take place. We can define another set of constraints to ensure that the mass of the protein plus modifications is approximately the mass of the modified protein, Mpmod. Because Mpmod is subject to a certain degree of experimental error, an exact conservation of mass cannot be achieved. Thus, we allow for a variable tolerance of mass error, denoted as toler in Equations 6 and 7, between the experimental and theoretical masses of the modified peptide.
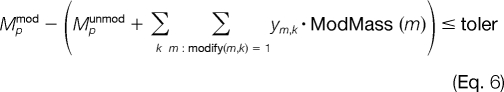 |
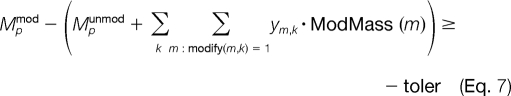 |
One should note that the term in parentheses in Equations 6 and 7 corresponds to the theoretical mass of the modified protein where the unmodified mass, Mpunmod, is adjusted by the weight of the selected post-translational modifications, ym,k·ModMass(m). The tolerance term, toler, is a continuous variable that is allowed to range between 0 and tolerUB (e.g. 0 ≤ toler ≤ tolerUB). The upper bound on the toler variable, tolerUB, is specified by the user based on the protein size and instrument accuracy. For histone H3, we are analyzing a protein of mass ∼5400 daltons or greater using an LTQ ion trap mass analyzer, so an experimental error of ±0.5 for a precursor ion of charge state of +9 (e.g. m/z ≈ 600) translates to a mass error of 4.5 daltons. To accommodate these large mass deviations, we would select tolerUB = 6.5 for that system. A much greater accuracy can be achieved with the use of FTMS (e.g. an OrbiTrap instrument).
The objective function we postulate for this problem is to minimize the mass error between the experimental and theoretical mass of the modified protein, as shown in Equation 8.
 |
Equations 5–8 represent the MILP model for the identification of post-translational modifications for a known template protein sequence. It should be noted that this model can be solved to optimality using existing branch-and-cut solvers, such as CPLEX (23). CPLEX and most other methods for solving MILP problems utilize a branch-and-cut algorithm (24). The essence of this algorithm is to fix a subset of integer variables and allow the remaining integer variables to take on continuous values (which is referred to as relaxing these integer variables) to obtain an easier to solve linear programming (LP) relaxation. A branch-and-bound search tree is used to keep track of which integer variables are being fixed for a given LP relaxation; these variables are denoted as nodes in a search tree. The algorithm traverses this tree and solves an LP relaxation at each node it encounters to generate a theoretical upper bound on the optimal solution to the original problem. Various search techniques, such as depth-first, breadth-first, and best-bound search, are used to specify in which order the subproblems should be considered. There are also several general rules for deciding which variables to branch on or fix. To avoid enumerating all of the subproblems in the tree, three fathoming criteria are applied to each subproblem after solving its linear programming relaxation. This subject is further discussed in several excellent sources (24, 25).
We are interested in (a) finding the modified forms that satisfy the experimental modified mass (within tolerUB) and then (b) relatively ranking them based on supporting c and z· ion peaks observed in the ETD/ECD tandem mass spectrum. To model this explicitly would require an auxiliary set of binary variables to represent the matches between theoretical c and z· ions with the experimental ion peaks in the tandem mass spectrum and would increase the complexity of the MILP problem. However, because we know that there are only a finite number of combinations of possible modified forms, say F, for a given Mpmod, then we can enumerate all F combinations and then subsequently score the potential modified sequences with the experimental tandem mass spectrum. The entire space of possible modifications is generated by solving for the optimal set of modifications and then removing it from the search space using an integer cut as shown in Equation 9 (25),
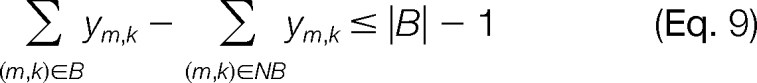 |
where B = {(m,k) : ym,k = 1}, NB = {(m,k) : ym,k = 0}, and |B| is the cardinality of B. After the integer cut is introduced, we can solve the model again to optimality to find the second best sequence according to the objective function in Equation 8. This procedure is repeated until we have enumerated all possible modified forms from the search space that satisfy the given mass balance.
There are several advantages to enumerating all possible targeted modifications and then subsequently scoring them using the fragmentation information in the tandem mass spectrum. First, evaluating the matches between the theoretical and experimental ion peaks “off line” significantly reduces the number of binary variables required in the problem formulation, and as a result, the scoring function is modular and can take on a variety of functional forms. If we were to introduce binary variables to represent the matches between theoretical and experimental ion peaks, then the scoring function for these peak matches in the MILP model must be a linear function. However, because the peak scoring is done externally to the MILP model, we can utilize any nonlinear scoring function, such as a probabilistic model or cross-correlation. The theoretical singly charged c and z ions are computed using the following formulas,
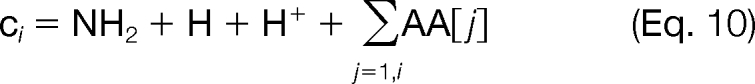 |
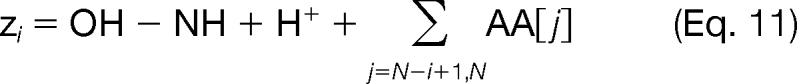 |
where O corresponds to the monoisotopic mass of oxygen (15.9949146), N corresponds to the monoisotopic mass of nitrogen (14.003074), H corresponds to the monoisotopic mass of hydrogen (1.00783), and H+ is the mass of a proton (1.00728).
Another important advantage is that evaluating all possible targeted modifications allows us to normalize the relative abundances of the modified forms because the ratio of individual modification sites is proportional to the abundance of the forms present in the samples (16) for these larger, more charged peptides. For instance, for every possible modified form that was generated from the MILP model for histone H3, there exists a theoretical c9 ion peak, which can take on several different m/z values because the N-terminal residue positions 3, 4, and 9 are modifiable (as specified in Table I) and can shift the mass of the c9 ion by the additive weight of their side chain modifications. In Fig. 2, we illustrate this concept for the c9 ion for histone H3 using a single MS2 scan for a precursor ion of 610.10 m/z.
Fig. 2.

An enlarged m/z region illustrating the observation of several modified forms for the c9 ion for histone H3 in a tandem mass spectrum corresponding to an observed precursor mass of 610.1. Note that in the figure the term “n me” corresponds to the number of methyl equivalents up to that position in the template sequence. For instance, “c91+ 2me” indicates that two methyl equivalents are incurred up to the ninth lysine that may correspond to K4me2K9me0, K4me1K9me1, or K4me0K9me2.
Note that the different c9 ions are characterized based on the number of “methyl equivalents” required to achieve that mass from the unmodified form. For instance, “c91+ 2me” indicates that two methyl equivalents (28 Da) are incurred up to the ninth lysine that may correspond to K4me2K9me0, K4me1K9me1, or K4me0K9me2 where me0 denotes the unmodified residue. Additional ion peak observations toward the N terminus of the protein are required to distinguish which form is most probable. It should be noted that the tandem MS spectra of histone H3 are often multiplexed spectra of several modified forms (i.e. there is some percentage of K4me2K9me0, K4me1K9me1, and K4me0K9me2 actually present). From this full set of modified forms consistent with the parent mass, we construct the superset of all the unique theoretical c9 ions, which we will denote as ĉ9. For example, the ĉ9 for the tandem mass spectrum in Fig. 2 consists of {1058.7, 1072.7, 1086.7, 1100.7, 1114.8, 1128.8, 1142.8} where the peaks corresponding to 1114.8, 1128.8, and 1142.8 were not observed, and so this range was omitted from the figure.
The relative abundance for every element of ĉ9 is determined from information in the experimental tandem mass spectrum using a peak matching algorithm (described in the supplemental material). For now, let us define the peak in ĉ9 with the estimated maximum abundance as c̄9. In Fig. 2, we see that c̄9 most likely corresponds to the peak with three methyl equivalents (m/z = 1100.7). The relative abundance for any of the c9 ions can then be approximated as λ(c9)/λ(c̄9), which we denote by the parameter ψ(c9). This normalization is performed for all theoretical c and z ions in the predicted sequences.
Given this information, we can rank order the modified forms based on the scoring metric presented in Equation 12,
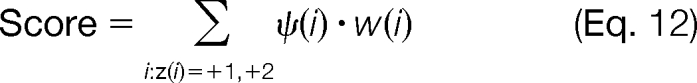 |
where i denotes the backbone position starting from the N terminus and C terminus for the c and z ion series, respectively, and w(i) denotes an optional weighting function that is a function of charge state for reducing random matches to experimental peaks. Note that the scoring function in Equation 12 uses the approximate abundances to estimate the modified forms (16). This relative normalization for each of the ions also prevents the score from being skewed by the higher intensity peaks that are near the N and C termini of the protein in the ETD tandem mass spectrum. The sensitivity of the scoring function presented in Equation 12 for identifying the most abundant modified form is examined in the supplemental material.
We applied the proposed method to an ETD tandem mass spectrum of histone H3 corresponding to a precursor mass of 610.5 m/z to illustrate the application of the MILP model for creating a rank-ordered list of potential modified forms. The model generated and cross-correlated a total of 227 potential modified forms in 18 CPU seconds on an Intel Pentium 4 3.0-GHz Linux-based computer. One should note here that the enumeration of these modified forms can be done off line for analyzing a full LC-MS experiment as precursor ion mass to charge ratios are repetitively observed. Table II presents the top 10 rank-ordered modified forms for this tandem MS spectrum of histone H3 as predicted by the proposed method.
Table II. Top ranked modified histone H3 forms.
A total of 227 modified histone H3 forms were enumerated that satisfied the precursor m/z ratio of 610.5 using the first MILP model. These forms were then rank-ordered by comparing the theoretical ion peaks corresponding to each modified form with the peaks in the experimental tandem mass spectrum using our peak matching algorithm (described in the supplemental material), and the top 10 ranked forms are presented for reference here.
| Rank | Modified residues | Supporting ions in MS2 | Score |
|---|---|---|---|
| 1 | K9me3K14acK27me2K36me2 | 168 | 39.26 |
| 2 | K9me3K14acK27me1K36me3 | 155 | 37.49 |
| 3 | K9me3K18acK27me2K36me2 | 165 | 37.25 |
| 4 | K9me3K14acK27me3K36me1 | 154 | 35.66 |
| 5 | K9me3K18acK27me1K36me3 | 153 | 35.47 |
| 6 | K14acK18acK27me2K36me2 | 161 | 35.34 |
| 7 | K4me1K9me2K14acK27me2K36me2 | 166 | 34.64 |
| 8 | K4me2K9me1K14acK27me2K36me2 | 157 | 33.69 |
| 9 | K4me3K14acK27me2K36me2 | 161 | 33.66 |
| 10 | K9me3K18acK27me3K36me1 | 152 | 33.65 |
In Table II, the top scoring modified form is K9me3K14acK27me2K36me2, which is correct for this tandem mass spectrum. This modified form is strongly supported by the following ion peaks that were observed to be the most abundant in intensity with respect to the other possible forms: c41+ = 474.3, c91+ = 1100.7, c92+ = 550.9, c141+ = 1573.0, c182+ = 1013.7, and z151+ = 1821.1. Two other ion peaks, c232+ = 1284.5 m/z and z282+ = 1523.3 m/z, were not observed to be the most abundant with respect to all species but were strong in intensity and supported the predicted modified form. The change in score between the first and second ranked sequence is 1.79, which is approximately a 5% difference. One should note the overlap in the modifications assigned to the top 10 forms due to the fact that they share a large subset of theoretical ion peaks that match well to experimental ion peaks that are high or moderate in intensity. The correct secondary form for this tandem mass spectrum, K4me1K9me1K14acK27me2K36me3, is ranked 71st in this ordered list, implying that additional analysis is required to detect this modified form.
MILP Model 2: Relative Composition of Modified Forms
Once we have determined the entire space of possible modified forms that satisfy the mass balance constraints in Equations 6 and 7 via the MILP model in the previous section, we formulate a second MILP model to determine the overall fraction of these forms that are present in the multiplexed ETD tandem mass spectrum. In this section, we present an iterative method based on MILP for computing the relative fractions of modified forms based on linear superposition. Important concepts are again illustrated using histone H3.2 as an example.
Preprocessing: Building a Consensus
We can utilize redundant information in the data by building a consensus of ion peak observations. It is well known that ETD fragmentation results in an almost continuous series of c and z· ions and that most types of post-translational modifications are retained on the side chain during fragmentation (12, 13). Thus, a PTM observed in one position simply shifts the ion series of subsequent positions by the difference in weight between the modified and unmodified residue side chain. This is illustrated in Fig. 3 for the c4 ion of histone H3 where we see that the ratios of the zero, one, and two methyl equivalent forms for the c4 through c8 ions are consistent and represent redundant observations. It should be noted that c9 ion is not included in this set because it can be post-translationally modified (see Table I), but its modified forms are supported by ions c10 through c13.
Fig. 3.

An enlarged m/z region illustrating the existence of redundant information for modifications that are present on the fourth lysine (Lys-4) in histone H3. Specifically, the c4, c5, c6, c7, and c8 ions all show the existence of zero, one, and two methyl equivalent forms, and the relative ratios between these forms are consistent across these ions (i.e. the “0 me” is the most abundant, the “1 me” form is the second most abundant, and the “3 me” form is the least abundant). This redundant information is then averaged to build a “consensus” observation for these different modifications that exist on the fourth lysine.
To exploit the redundancy in the data, we compute the average and standard deviations of these consistent ions to provide a consensus for the relative levels of the modified forms. For instance, in Fig. 3 for histone H3, the resulting consensus for relative abundance of the modifications on the fourth lysine, based on the c ion series, would be {0.7575, 0.2193, 0.0135, 0.0097} with a standard deviation of {0.0228, 0.0115, 0.0089, 0.0131} for zero, one, two, and three methylations, respectively. Those terms that represent less than 4% of the relative abundance are set to zero as they are indistinguishable from noise. One should note that for higher mass ions the singly and doubly charged ions are used to compute the consensus. To build an accurate consensus, some observations are rejected as outliers if their observed value lies outside of two standard deviations from the mean. This rejection procedure is iterated until all observations lie within two standard deviations of the mean or the consensus has been reduced to only one observation.
A consensus is created for each of the ions that represent the mutatable residues (i.e. in histone H3, c4 for Lys-4, c9 for Lys-9, c14 for Lys-14, c18 for Lys-18, c23 and z28 for Lys-23, z24 for Lys-27, and z15 for Lys-36, just for the lysines). This results in a vector of average relative abundances of the different modified states for each mutatable residue. The confidence associated with an average relative abundance is proportional to the standard deviation of the consensus. Fig. 4 illustrates the overall consensus created when just considering the modifications on the lysines in Table I for m/z = 605.5.
Fig. 4.

An example of the consensus derived from averaging redundant ion series data for m/z = 605.5 of histone H3. The circles denote the value of the average for a particular modification site, and the triangles mark one standard deviation above and below this average value derived from the consensus. The ions corresponding to the modification sites are listed at the bottom of the plot for reference. For instance, the first four averages correspond to zero, one, two, and three methyl equivalents for the c4 ion where one can see that the unmodified form is clearly the most abundant.
Mathematical Model
Given the consensus for the relative abundance of the various modifications as shown in Fig. 4, we would like to determine what fraction of these modified forms are present in the multiplexed tandem mass spectrum. In other words, we would like to determine the linear combination of modified forms that best reconstructs the observed forms in Fig. 4. To accomplish this, we formulate a superposition problem using mixed integer linear optimization where the deviation between the experimental observations and weighted combinations of the “pure” modified forms is minimized.
We first define the set of modified forms to be represented by the index f ∈ F where F denotes the total number of modified forms that satisfy the mass balance of the parent peptide as determined by the MILP for enumerating all forms. We also define the set s ∈ S to denote the unique modification sites as represented by each of the vertical lines in Fig. 4. For instance, in histone H3 the modified form K9me2K23acK27me2 belongs to the set F, and the corresponding modification sites c4 0me, c9 2me, c14 2me, c18 2me, c23 5me, z15 0me, z24 2me, and z28 5me belong to the set S.
Based upon these set definitions, we can define the contributions of the theoretical pure forms for all f ∈ F that exist in the multiplexed tandem mass spectrum based on those identified by the MILP model for modified form enumeration. We model this using the parameter PureForm(f,s) where PureForm(f,s) = 1 if modified form f contributes the modification site s in the theoretical tandem mass spectrum. For histone H3, if f = K9me2K23acK27me2, then PureForm(f,c4 0me) = 1, PureForm(f,c9 2me) = 1, PureForm(f,c14 2me) = 1, PureForm(f,c18 2me) = 1, PureForm(f,c23 5me) = 1, PureForm(f,z15 0me) = 1, PureForm(f,z24 2me) = 1, and PureForm(f,z28 5me) = 1. The consensus based upon the average relative abundance of a modified site s is defined by the parameter avg(s), and its corresponding standard deviation is std(s).
These set and parameter definitions allow us to derive the appropriate variables and constraints for the mathematical model. The first set of variables defines the relative fraction of each modified form, f, present in the experimental tandem mass spectrum.
We also define an analogous set of binary variables to denote the decision of whether or not to include the modified form in the theoretical multiplexed tandem spectrum.
 |
Based upon these variable definitions, we define a constraint to enforce an affine combination of the weights of the modified forms (e.g. the weights sum to one) as shown in Equation 15.
 |
The fraction of a modified form, f, in the tandem mass spectrum should be greater than zero if and only if it is included in the tandem mass spectrum (i.e. uf = 1), which is modeled by Equation 16.
From Equation 16, wtf is only greater than zero when uf = 1 (i.e. form f is included in the tandem mass spectrum) and is zero otherwise. The weighted sum of the different modified forms should match the observed relative abundances in the experimental tandem mass spectrum as displayed in Fig. 4. For an ideal experimental spectrum, some weighted combination of the modified forms would exactly match and explain all of the observed abundances, and the determination of the weights, wtf, would reduce to a simple linear algebra problem as presented in Equation 17 where the only variable is wtf.
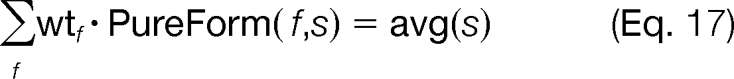 |
However, due to the inherent noise level and varying quality of experimental tandem mass spectra, it is not possible to exactly fit the theoretical weighted combinations of these modified forms to the experimental data. To address this variability, we must relax the equality of the fit in Equation 17 for which we allow one standard deviation above and below the observed relative abundances as shown on the right-hand side of Equations 18 and 19.
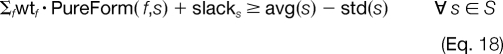 |
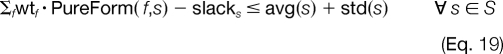 |
If we interpret Equations 18 and 19 without the slack variables, slacks, then these constraints enforce that the linear combination of the pure forms should fall within one standard deviation of the consensus average for that modification site. The incorporation of the slack variables in Equations 18 and 19 allows for the possibility of outliers and deviations to further relax this fit if needed.
The constraint presented in Equation 20 specifies that only N pure forms are to be used in the theoretical multiplexed spectrum.
 |
The logic behind incorporating this constraint is to force the model to only pick a small number of forms, N, because the set of pure modified forms is not linearly independent. In other words, the composition of one form can be made up from some linear combination of other forms.
The objective function for this problem is to minimize the slack variables in Equations 18 and 19; this in turn maximizes the fit between the predicted and the experimental consensus spectrum as shown in Equation 21,
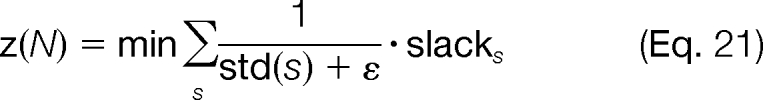 |
where ε is equal to some small constant (we use ε = 1e−4 in our algorithm). The motivation behind weighting the slack variables, slacks, to be inversely proportional to the standard deviation of the consensus for a modification site s is that a small standard deviation is indicative of a better consensus, and thus the corresponding slack variable should have a higher weight to favor this fit, whereas a large standard deviation indicates that the consensus is not in agreement, and thus a perfect fit for these modification sites is not as important.
Equations 15, 16, and 18–21 comprise the mixed integer linear optimization model for determining the corresponding fractions of the modified forms that are present in the multiplexed tandem mass spectrum. To start the algorithm, we select N = 2 and require that the most abundant form, as determined by the highest scoring modification from the MILP framework presented under “MILP Model 1: Enumeration of All Position-Targeted Modified Forms,” is used in the tandem mass spectrum (by specifying that the appropriate uf = 1). As a result, the remaining form selected by the model corresponds to the second most abundant form in the spectrum. We then fix these two forms to be used again (by activating the appropriate uf variables), set N = 3, and resolve the model to find the third most abundant form. This procedure of incrementing N is repeated until the relative difference between the objective functions (defined in Equation 21) when using N − 1 and N forms is less than 15% (e.g. (z(N − 1) − z(N))/z(N − 1) < 0.15) or no additional modification sites s are explained when going from N − 1 to N forms.
The overall algorithmic framework for identifying and quantifying the various modified forms for highly modified proteins using LC-MS ETD tandem mass spectrometry is presented in Fig. 5. The automated framework takes as input an mzXML file corresponding to an entire LC-MS/MS ETD run. A central control element (represented by the leftmost circle in Fig. 5) reads the individual scans from the mzXML file and stores the predictions of the algorithm. In the first stage of the algorithm, the entire space of PTMs that satisfy the given precursor mass of a tandem mass spectrum are enumerated by solving an MILP feasibility problem (represented by the upper right rectangle in Fig. 5). A score for each modified form is computed by incorporating observed ion peak information from the tandem mass spectrum using the peak matching algorithm (represented by the oval in Fig. 5 and described in the supplemental material), which predicts the theoretical isotopic profiles and then cross-correlates the experimental and theoretical ion profiles to determine the individual quality of fit. Also computed at this step are the relative abundances for each modification site, which are used to create a consensus of observations that is fed as input into the MILP superposition problem (represented by the lower right rectangle in Fig. 5) to determine the relative compositions of the modified forms present in the tandem mass spectrum. These fractions are used to estimate the abundance of each modified form using the precursor ion intensity.
Fig. 5.

A flow diagram of the two-stage mixed integer linear optimization framework for identifying and quantifying the post-translationally modified forms of highly modified proteins using LC-MS/MS ETD data.
It should be noted that the first MILP feasibility model (the upper right rectangle in Fig. 5) can be evaluated prior to analyzing the entire LC-MS experiment to enumerate all the modified forms that satisfy the set of precursor masses consistent with the allowed set of targeted PTMs. These modified forms are then stored for quick access at run time instead of resolving the MILP model for each tandem mass spectrum. This approach is currently used in our implementation and significantly speeds up the overall analysis time.
RESULTS
In this section, we demonstrate the utility of the proposed MILP-based framework depicted in Fig. 5 for an LC-MS/MS ETD experiment of a highly modified histone H3.2 mixture. The data were acquired using a novel on-line LC-MS protocol for the high throughput characterization of highly modified proteins (20). The method utilizes a pH gradient to elute the various modified forms in a weak cation exchange HILIC separation; the eluate from this separation is directly sprayed into a benchtop linear quadrupole ion trap mass spectrometer equipped with ETD (20). The on-line HILIC chromatography primarily separates the modified histone proteins with respect to several dimensions: 1) the number of acetylations, 2) the position of the acetylations, 3) the number of methylations, and 4) the position of the methylations. Even with this unprecedented degree of separation, the eluting precursor ion masses can still correspond to several isobaric modification states, which result in multiplexed tandem mass spectra. It is of significant importance to be able to identify all of the modified forms that are co-eluting as lower abundance forms can be chromatographically buried under higher abundance forms. Fig. 6 illustrates separation achieved for three precursor ion m/z values using the novel on-line chromatography method.
Fig. 6.

Illustration of the separation of the modified forms for histone achieved in a single LC-MS run. Note that each precursor ion frequently corresponds to several modified forms, which can only be identified using the fragmentation information from the tandem mass spectrum.
We applied the proposed methodology to the LC-MS/MS ETD experiment to gain insight into the dynamics and provide an approximate quantification of the various eluting modified forms. A full LC-MS analysis also enables us to incorporate important chromatographic information into our annotations and also quantify the degree of chromatographic resolution achieved experimentally. To analyze the full LC-MS run, we converted the Thermo RAW data into mzXML format (26) using the program ReAdW (SourceForge, Inc.). The mzXML data are then read over the entire range of scan events using the ramp data parser available in the Trans-Proteomic Pipeline (SourceForge, Inc.). We individually analyzed each MS2 scan event corresponding to the chromatogram shown at the top of Fig. 6 and reported the top N modified forms predicted to be present. If the most abundant modified form did not contain at least 50 supporting ions in the tandem mass spectrum, then the spectrum was rejected on the basis of poor fragmentation. The relative fraction of each modified form, as determined by the MILP superposition problem, is multiplied by the intensity of the corresponding precursor ion in the MS1 to provide an estimate of the approximate abundance of each modified form. It should be noted here that the annotations presented in this section were determined using single scan MS2 events and not averages over multiple MS2 scan events.
The corresponding annotations for the entire LC-MS experiment were computed in under 2 CPU hours on an Intel Pentium 4 3.0-GHz Linux-based computer and are presented in Fig. 7 for reference. One should note that in Fig. 7 the highly modified histone H3 forms are the first to elute, and the less modified forms are the last to elute from the column. The approximate abundance for most of the modified forms assumes a parabolic peak shape with respect to time, which is similar to the shapes of the chromatographic profiles of the individual precursor ions presented in Fig. 6. To elucidate the various modified forms present in the single LC-MS run, Fig. 7 was dissected into four time ranges, and the findings will be discussed for each.
Fig. 7.

Approximate abundance of several modified forms of histone H3 as a function of elution time. The most abundant forms are annotated in the figure. It is observed that the highly modified forms are the first to elute, and the less modified, unacetylated forms are the last to elute from the column. The symbol “*” corresponds to the modifications K14acK18acK23ac.
DISCUSSION
The approximate abundance of the modified forms that eluted during the time range of 80–122 min are presented in Fig. 8. One should note that the lysines in positions 14, 18, and 23 are acetylated (e.g. K14acK18acK23ac, which is denoted as “-*-” in Fig. 8) for all of the modified forms detected in this time range. This is chromatographically consistent because the separation of the modified forms is expected to occur primarily with respect to the number of acetylations. The modified forms with a greater degree of methylation were the first to elute from the column, and it was observed that the lysines in positions 9 and 27 were trimethylated in the forms eluting before 95 min.
Fig. 8.

Approximate abundance of several modified forms of histone H3 for the elution time window of 80–122 min. All observed modified forms in this time range have acetylated lysines in positions 14, 18, and 23 (K14acK18acK23ac, which is denoted by -*-).
An interesting feature of these forms is that some are observed to elute twice within the time interval presented in Fig. 8. For instance, the modified form K4me1K9me3-*-K27me2K36me2 has a large elution peak at about 90 min, but there also exists a smaller peak corresponding to the same annotation that elutes at about 98 min. These two peaks are chromatographically resolved, suggesting that they correspond to the elution of two distinct modified forms. One hypothesis that explains the difference between these two modified forms is that the assigned trimethylation of the lysine in position 9, K9me3, actually corresponds to an acetylation (i.e. K9ac). Recall that the monoisotopic mass of a trimethylation and acetylation is 42.0471 and 42.0106 daltons, respectively, which is not resolvable on a linear ion trap mass spectrometer. However, because the chromatography separates primarily based on the number of acetyl groups, we can hypothesize the interchange of a trimethylation with an acetylation and then test it experimentally with a high precision instrument, such as an OrbiTrap mass spectrometer. This is chromatographically consistent if the larger peak at 90 min corresponds to K9ac and the smaller peak at 98 min corresponds to K9me3 as a modified form with four acetylations (K9ac, K14ac, K18ac, and K23ac) is expected to elute before one with three acetylation sites. Furthermore, all of the forms before 95 min have a K9me3 annotation that could be interpreted as K9ac, and thus all the modified forms within this chromatographic region would exhibit four sites of acetylation. This example illustrates why chromatographic information, which is inherent in an on-line LC-MS experiment, is so valuable in the interpretation of the modified forms.
With increasing time in Fig. 8, it is observed that the degree of methylation for the lysines in positions 4 and 36 decreases, and eventually they become unmodified. For all of the modified forms detected in this time range, the lysine in position 27 has at least a single methylation. The lysine in position 4 is mostly observed to be unmodified or singly methylated in its most abundant forms, but a dimethylated species is also detected as a secondary form at around 90 and 97 min.
It is interesting to note the positional relationships of the methylations between primary and secondary modified forms. For instance, the primary form at 105 min in Fig. 8 is detected to be K9me2-*-K27me2K36me2 with a secondary form of K4me1K9me2-*-K27me2K36me1 where the only difference between these two modified forms is that a single methylation on Lys-36 has “moved” to Lys-4. Similarly, for positions 9 and 27, the primary modified form at 115 min is K9me2-*-K27me2 and the secondary form is K9me1-*-K27me3 where the difference between these two modified forms is that a single methylation in position 9 has moved to the lysine in position 27. This slight difference between the primary and secondary modified forms is consistent with the chromatography as we would expect these forms to co-elute.
In Fig. 9, we present the approximate abundances for the modified forms that eluted within the time range of 122–168 min in the chromatogram. The most abundant modified forms detected in Fig. 9 have acetylated lysines in positions 14 and 23. This particular pairing of acetylations is consistent with earlier findings regarding the observed partial ordering dependence of acetylations in histone H3 (18). Namely, when considering the lysines in positions 14, 18, and 23, a single acetylation was observed to primarily occur in either position 14 or position 23 (and not in position 18), and two acetylations were most commonly detected on positions 14 and 23 and less so on positions 14 and 18 or positions 18 and 23. Consistent with the observations in Fig. 8, the primary and secondary modified forms differ only by the degree of methylation on the lysines in positions 9 and 27. For instance, during the time range of 155–160 min, the most abundant modified form is initially K9me2K14acK23acK27me1, then K9me1K14acK23acK27me2, and finally K14acK23acK27me3 where the successive difference between each most abundant form corresponds to the shift of a single methylation from Lys-9 to Lys-27. There is a strong presence of secondary and even tertiary modified forms in the majority of these spectra.
Fig. 9.

Approximate abundance of several modified forms of histone H3 for the elution time window of 122–168 min. The majority of the modified forms within this time range contain K14acK23ac, which is consistent with earlier findings regarding the dominance of this particular diacetyl pair (18).
During the time range of 168–212 min, the detected modified forms all exhibit a single acetylation modification as shown in Fig. 10. It is also observed in this time range that the chromatography effectively separates each of the acetylations based on the position of the acetylated lysine. Namely, modified forms containing K14ac eluted over the range of 168–187 min, modified forms containing K18ac eluted subsequently in a narrow window around 184 min, and modified forms containing K23ac were the last to elute over the range of 183 min to 212 min. The secondary modified forms are strong in abundance and frequent for the species presented in Fig. 10. For instance, in the time range of 193–202 min, where the primary modified form is K9me2K23acK27me2, the more abundant secondary form is initially K9me3K23acK27me1 but then changes over to K9me1K23acK27me3 after 198 min. Consistent with the previous secondary modified forms and the chromatography, the primary difference between these three forms corresponds to the shift of a single methylation from Lys-9 to Lys-27.
Fig. 10.

Approximate abundance of several modified forms of histone H3 for the elution time window of 168–212 min. The majority of the modified forms in this time range have a single acetylation of the lysine in either position 14, 18, or 23. It is interesting to note the chromatographic resolution with respect to the acetylated position where K14ac elutes first, K18ac elutes second, and K23ac elutes last.
The dominance of the modified forms with either K14ac or K23ac is again consistent with the partial ordering previously observed for these acetylations (18). There is also strong evidence for K18ac, as depicted by the sharp but consistent peak at about 184 min in Fig. 10, that corresponds to the modified form K9me2K18acK27me2. The detection of this modified form can be attributed to the resolution of the chromatography and its resulting ability to enhance the signal of lower level forms.
The last chromatographic section corresponds to 212–268 min, and the approximate abundance of the modified forms over this time range is presented in Fig. 11. The majority of the modified forms in Fig. 11 do not have any acetylation modifications and are dominated by species with two or three sites of methylation. An interesting observation in this time range is the existence of modified forms that elute over multiple time instances. For example, at a time of 214 min, the modified forms K9me2K14acK23acK27me3 and K9me3K14acK23acK27me2 are observed as the primary and secondary forms, respectively, but recall that they were previously observed to elute with a much larger abundance at a retention time of about 137 min (see Fig. 9). This also occurs for the modified forms K9me2K27me2 and K9me3K27me1, which elute during the time ranges of 217–219 min and 250–265 min in Fig. 11.
Fig. 11.

Approximate abundance of several modified forms of histone H3 for the elution time window of 212–268 min. The majority of these modified forms are unacetylated and exhibit isobaric methylations.
One explanation for the observation of multiple elution times for predicted modified forms could be the presence of an acetylation instead of a trimethylation (or vice versa) in one of the modified residues as observed previously in Fig. 8 for the lysine in position 9. For instance, the existence of a trimethylation in place of a single acetylation for K9me2K14acK23acK27me3 (i.e. K9me2K14me3K23acK27me3 or K9me2K14acK23me3K27me3) at about 214 min would be a more chromatographically consistent explanation as all of the singly acetylated modified forms are observed to elute just prior to this time (see Fig. 10). Specifically, the modified form K9me2K14me3K23acK27me3 is the most consistent with the chromatogram as the forms containing K23ac elute close to a time of 214 min as shown in Fig. 10.
We also compared the modified forms identified in our analysis with those reported in the literature (18) and found the results to be consistent (see supplemental material). However, our approach was able to identify a higher percentage of di- and trimethylation modifications because of the enhanced ionization efficiency and sensitivity of the proposed chromatography approach.
Averaging Multiple Tandem MS Scans
The proposed identifications from the single MS2 scan events presented in the previous section can also be verified by averaging several of the individual tandem MS scans to increase the signal to noise ratio and enhance the confidence of observed ion peaks. The general approach of averaging tandem mass spectra is also a useful technique for enhancing the signal on lower level forms that were not statistically distinct from the noise level in the single scans. It is important that the averaged spectra correspond to approximately the same relative distribution of modified forms or else actual ion peak signals will be smoothed out in the resulting composite spectrum.
The strategy we use for averaging tandem mass spectra is to perform a single pass through the chromatogram using the proposed MILP framework and identify all possible forms, even storing the modified forms that are not fully annotated (that is, some of the modification sites do not have any supporting ions in the tandem mass spectrum). Then for every partially and fully annotated form, we identify the largest consecutive subregion in time over which the modified form does not change in relative rank. Within this time range, we fit a gaussian distribution to the approximate abundance of this modified form and determine the tandem MS scans that lie with the full-width at half-maximum (i.e. ±1.17741 σ) of this chromatographic peak. These scans are then averaged to yield a single tandem mass spectrum, which is analyzed using the proposed MILP framework.
This approach for averaging tandem mass spectra is very effective in validating several lower level (i.e. rank 3 or 4) forms and also identified lower abundance forms that were not detected in the first pass through the chromatogram. For instance, the averaging of two scans around a retention time of 85.5 min revealed the presence of K4me3K9me3K14acK18acK23acK27me2K36me2 as the third most abundant form. This form is particularly interesting as it is highly modified and contains a trimethylation on Lys-4.
Integration of Chromatographic Information
An important feature of the proposed framework is that chromatographic information can be integrated into the identification of the modified forms. We have already shown examples for how the observation of multiple elution times of the same modified form can help distinguish between trimethylation and acetylation post-translational modifications in histone H3. In this section, we demonstrate how this information can be automatically incorporated into the annotation procedure, thereby utilizing all of the information encoded in the entire LC-MS/MS experiment in a complementary fashion to ensure the highest confidence identifications. The LC-MS/MS data set corresponding to histone H3 is used to illustrate the approach.
Recall that the chromatogram in the retention time dimension for the histone H3 LC-MS/MS data assumes an approximately gaussian distribution for a particular m/z value as shown in Fig. 6. One should note that these distributions are generally observed in any LC-MS chromatogram. For the histone H3 data, each of the gaussian distributions corresponds to a particular state of acetylation where for m/z = 605.4 in Fig. 6 we observe a bimodal distribution corresponding to K14ac and K23ac, respectively. An important question to investigate is how these distributions behave as a function of both retention time and m/z value.
Let us consider the raw, unannotated LC-MS chromatogram as a function of retention time and m/z value, as shown in Fig. 12, for the histone H3 data set. Note that the entire LC-MS chromatogram, as presented at the top of Fig. 6, can be reconstructed from these data marginalizing out the m/z dimension. From the data in Fig. 12, it is easy to see how the gaussian distributions in the retention time dimension vary with changing m/z values. In particular, it is observed that the mean value of these gaussian distributions in retention time follows a sigmoidal curve as a function of m/z. It is important to note that each of these sigmoidal curves corresponds to a distinct acetyl modification state (i.e. a distinct number and positioning of acetylation modifications) and that these curves are clearly separable in the m/z and retention time dimension. The existence of this sigmoidal shape is due to the elution of various methylated forms within a given acetyl modification state (see supplemental material).
Fig. 12.

LC-MS chromatogram as a function of retention time and m/z value. The axis perpendicular to the page corresponds to the intensity of the signal.
To utilize this information, we fit sigmoidal functions to the raw chromatogram data in Fig. 12 by solving a weighted regression problem for each curve where we aim to minimize the squared error between the experimentally observed retention time, RTexp, and the predicted retention time as a function of m/z, RT(m/z), as shown in Equation 22,
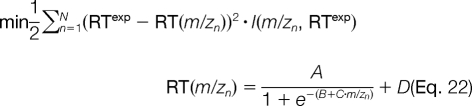 |
where I(m/zn, RTexp) is the precursor intensity for a given m/z and RTexp, and this weight biases the regression to pass through the mean of the gaussian distributions in the retention time dimension; that is, the resulting value of RT(m/z) is the mean value of the gaussian distribution in the retention time dimension for a particular m/z value. Note that the objective function in Equation 22 is a nonlinear function of the continuous variables A, B, C, and D. This unconstrained optimization problem is solved by analytically computing the gradient and hessian matrix of the objective function and using the Newton-Raphson method to converge to a local minimum of Equation 22.
Once the gaussian distributions in the retention time dimension are characterized as a function of m/z (e.g. using RT(m/z)), we can then determine the corresponding acetylation state for each sigmoidal curve (let us say that there are S such sigmoidal curves, corresponding to RTS(m/z)) based on our current annotations. This is accomplished by first postulating that every trimethylation is actually an acetylation and then grouping the resulting modified forms by their resulting acetylation state, say AS. For instance, examples of elements in the set AS for histone H3 would include K14ac, K23ac, K14acK23ac, K9acK14acK18acK23acK27ac, and so forth. Now for every set of modified forms that have an acetylation state of AS, we can compute the likelihood that each of the S sigmoidal curves corresponds to this particular state of acetylation. We solve this problem for each value of m/z in AS where we use a gaussian mixture model in the retention time dimension that has a fixed mean at RTS(m/z) for that particular m/z and curve S; that is, we have S gaussian distributions in the retention time dimension at a fixed mean of RTS(m/z) for the given m/z slice, but we allow for their standard deviations to vary, and we compute the posterior probability distribution that each modified form belongs to a particular curve S. The mixture model is solved using the classic expectation maximization algorithm to provide the standard deviation and mixture proportion for each sigmoidal curve, and the curve S with the largest mixture proportion is then said to contribute to the acetylation state corresponding to AS. This procedure is repeated for all m/z values of the acetylation state AS, and the sigmoidal curve with the highest frequency of observation is then assigned the acetylation state under investigation. This method also provides us with the standard deviation for each of the gaussian distributions as a function of m/z, and the process is repeated for all possible AS acetylation states.
The end result of this approach is a map that correlates the various acetylation states as a function of m/z and time. In particular, we know how the mean (given by RTS(m/z)) and standard deviation of each acetyl state varies with changing m/z values. Furthermore, each of these curves is completely separable in the m/z and retention time domain enabling us to confidently associate annotations of a given m/z and retention time with a particular state of acetylation. This information can be incorporated into the MILP framework for the identification of modified forms in several ways.
One approach involves reanalyzing the tandem mass spectra whose annotations are not chromatographically consistent with the acetylation state map as a function of time and m/z. For these spectra, we resolve the superposition problem by constraining the acetylation state to be consistent with the acetyl state map. We can then compare the objective functions of the original superposition fit with the fit after imposing the most likely acetylation state for the particular retention time and m/z. If the relative change in objective function (e.g. quality of superposition) is not substantial, then the chromatographically imposed modified form is accepted; otherwise the originally annotated form is kept. The resulting annotations for this approach and the corresponding acetyl map are presented in Fig. 13 where we observe an excellent chromatographic separation of the acetylation states. In particular, the acetylation modifications corresponding to K9ac and K27ac are clearly delineated from K9me3 and K27me3 modifications by this approach! We also observe superior separation between the K14ac and K23ac forms. It is easy to see from Figs. 13 and 6 (m/z = 605.4) that these two acetylation states overlap in the retention time dimension but are cleanly separated when projected into the m/z dimension. In fact, this deconvolution of the overlap in retention time is observed for almost all acetyl state species that are adjacent in time. Another interesting feature of this map is that the K18acK23ac forms lie on the right shoulder of the K14acK23ac distribution and that these acetyl states are completely indistinguishable when collapsed onto the retention time domain. One should note that such a separation of acetylation modifications is not possible without these chromatographic correlations.
Fig. 13.

Resulting annotations for various acetyl states as a function of retention time and m/z value after imposing the most likely acetyl state using the underlying acetyl map.
This information also allows us to accurately infer the modification states for partially interpreted tandem mass spectra due to noise or low abundance forms. The N- and C-terminal modification sites are the most commonly annotated as the intensity of the fragment ion peaks typically assumes an inverse parabolic distribution as a function of m/z; that is, the ion peaks in the high and low mass regions of the tandem mass spectra have the largest intensity values, and these regions are sparsely populated, whereas the ion peaks toward the center of the spectra are of significantly lower signal and are often interfered by multiple ion peak observations. For histone H3, this can be particularly problematic because the ion peaks corresponding to Lys-14, Lys-18, and Lys-23 are found near the center of the tandem mass spectra in highly populated peak regions, and as a result, these modification sites are often unannotated, especially in noisy spectra. However, the acetyl map information enables us to resolve these issues as we can simply infer the acetyl state of a particular modified form based upon its retention time and m/z value and then examine the tandem mass spectrum to determine the modifications on the remaining sites. If the resulting mass difference between the inferred form and the experimentally determined precursor mass is less than some tolerance, then we can postulate with high confidence that the inferred modified form is correct. This approach enabled us to identify several highly modified instances of histone H3, including the fully occupied modified form K4me3K9acK14acK18acK23acK27acK36me3, which was a novel form prior to this work.
Automated Annotation of Modified Forms
An additional feature of the proposed algorithm is that it can automatically annotate tandem mass spectra for subsequent user validation if desired. For every modified form detected in the LC-MS run, the tandem mass spectrum with the highest corresponding score, as given by Equation 12, is annotated by explicitly labeling the theoretical ion peaks that matched to experimental ion peaks. For each theoretical ion peak, the corresponding ion type, index, charge state, and matching m/z value are provided, and an arrow is drawn from the annotation label to the matching experimental ion peak in the tandem mass spectrum for clarity. The secondary forms (i.e. rank 2, rank 3, etc.) are also annotated in the same spectrum for reference. The m/z range is divided into four regions, 1) 100–500, 2) 500-1000, 3) 1000–1500, and 4) 1500–2000, to minimize potential overlap of the annotation labels, and the intensity range for each region is adjusted according to the highest intensity peak in that range. A separate figure is generated for each of the four regions using PostScript, and captions are provided to indicate the modified form, the corresponding scan number and retention time of the tandem MS spectrum, and the rank of the modified form for this tandem mass spectrum (i.e. first, second, or third). The four annotated regions are then presented in a two-page PDF document, which is named according to the corresponding modified form, and two annotated regions are provided per page. Because the annotations were created using PostScript and the experimental spectra are presented in profile mode, the user can zoom in as far as they wish without any loss of figure resolution to manually validate individual peak assignments. An example of an automatically annotated tandem MS for the modified forms K14acK23acK27me3 (rank 1) and K9me1K14acK23acK27me1K36me1 (rank 2) is provided in the supplemental material for reference.
Conclusions
Here we presented a novel MILP-based framework for the identification of highly modified proteins using LC-MS and ETD tandem mass spectrometry. For a given primary sequence, the entire set of post-translational modifications that satisfy a precursor mass are enumerated by solving a mixed integer linear optimization feasibility problem. Given the set of PTM forms, a MILP superposition problem is then solved to determine the relative fractions of the modified forms present in the multiplexed ETD tandem mass spectrum. The proposed computational framework is applied to an entire LC-MS/MS ETD experiment corresponding to a mixture of highly modified histone peptides. The method is able to confidently identify hundreds of modified forms that are present in the complex sample, including many lower level forms that are chromatographically buried by species present in significantly greater abundance. Several of the modified forms identified in this analysis have not been previously reported in the literature, thus providing biological insight regarding the connectivity of modification sites at the molecular level. An important aspect of the proposed framework is that chromatographic information is used to correlate the modification states as a function of modification position, mass, and time. This information facilitates 1) the correct averaging of single tandem MS scans to increase the signal to noise ratio and enhance the confidence of observed ion peaks, 2) the accurate inference of modification states for partially interpreted tandem mass spectra due to noise or low abundance forms, and 3) the ability to assign confidence levels to the individual assignments. To this extent, the proposed algorithm utilizes all of the information encoded in the entire LC-MS/MS experiment in a complementary fashion to ensure the highest confidence identifications. An additional feature of the method is that it automatically generates annotated tandem mass spectra for subsequent user validation. The automated framework is able to identify, quantify, and annotate the modified forms present in an entire LC-MS/MS in a time scale comparable to the experiment. This framework can be easily extended for the analysis of the other histone proteins and other highly modified proteins that have not been previously studied because of technological, analytical, and data analysis limitations.
Supplementary Material
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grant R01LM009338 (to C. A. F.) and National Science Foundation (CBET-0941143) (to C. A. F. and B. A. G.).
 The on-line version of this article (available at http://www.mcponline.org) contains supplemental material including Figs. S1–S4.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material including Figs. S1–S4.
¶ Supported by Princeton University.
1 The abbreviations used are:
- PTM
- post-translational modification
- MILP
- mixed integer linear optimization
- LP
- linear programming
- ETD
- electron transfer dissociation
- ECD
- electron capture dissociation
- HILIC
- hydrophilic interaction liquid chromatography
- me1
- methylation
- me2
- dimethylation
- me3
- trimethylation
- ac
- acetylation
- phos
- phosphorylation
- CID
- collision-induced dissociation.
REFERENCES
- 1.Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem 67, 1426– 1436 [DOI] [PubMed] [Google Scholar]
- 2.Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching databases using mass spectrometry data. Electrophoresis 20, 3551– 3567 [DOI] [PubMed] [Google Scholar]
- 3.Craig R., Beavis R. C. (2003) A method for reducing the time required to match sequences with tandem mass spectra. Rapid Commun. Mass Spectrom 17, 2310– 2316 [DOI] [PubMed] [Google Scholar]
- 4.Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectrometry. Anal. Chem 77, 4626– 4639 [DOI] [PubMed] [Google Scholar]
- 5.Matthiesen R., Trelle M. B., Højrup P., Bunkenborg J., Jensen O. N. (2005) VEMS 3.0: algorithms and computational tools for tandem mass spectrometry based identification of post-translational modifications in proteins. J. Proteome Res 4, 2338– 2347 [DOI] [PubMed] [Google Scholar]
- 6.Kim S., Na S., Sim J. W., Park H., Jeong J., Kim H., Seo Y., Seo J., Lee K. J., Paek E. (2006) Modi: a powerful and convenient web server for identifying multiple post-translational peptide modifications from tandem mass spectra. Nucleic Acids Res 34, W258– W263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimaggio P. A., Floudas C. A. (2007) A mixed-integer optimization framework for de novo peptide identification. AIChE J 53, 160– 173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiMaggio P. A., Jr., Floudas C. A. (2007) De novo peptide identification via tandem mass spectrometry and integer linear optimization. Anal. Chem 79, 1433– 1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiMaggio P. A., Jr., Floudas C. A., Lu B., Yates J. R., 3rd (2008) A hybrid methodology for peptide identification using integer linear optimization, local database search, and QTOF or OrbiTrap tandem mass spectrometry. J. Proteome Res 7, 1584– 1593 [DOI] [PubMed] [Google Scholar]
- 10.Zubarev R. A., Kelleher N. L., McLafferty F. W. (1998) Electron capture dissociation of multiply charged protein cations: a nonergodic process. J. Am. Chem. Soc 120, 3265– 3266 [Google Scholar]
- 11.Bakhtiar R., Guan Z. (2006) Electron capture dissociation mass spectrometry in characterization of peptides and proteins. Biotechnol. Lett 28, 1047– 1059 [DOI] [PubMed] [Google Scholar]
- 12.Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Nat. Acad. Sci. U.S.A 101, 9528– 9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mikesh L. M., Ueberheide B., Chi A., Coon J. J., Syka J. E., Shabanowitz J., Hunt D. F. (2006) The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta 1764, 1811– 1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Udeshi N. D., Shabanowitz J., Hunt D. F., Rose K. L. (2007) Analysis of proteins and peptides on a chromatographic timescale by electron-transfer dissociation MS. FEBS J 274, 6269– 6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molina H., Horn D. M., Tang N., Mathivanan S., Pandey A. (2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Nat. Acad. Sci. U.S.A 104, 2199– 2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pesavento J. J., Mizzen C. A., Kelleher N. L. (2006) Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal. Chem 78, 4271– 4280 [DOI] [PubMed] [Google Scholar]
- 17.Seligson D. B., Horvath S., Shi T., Yu H., Tze S., Grunstein M., Kurdistani S. K. (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435, 1262– 1266 [DOI] [PubMed] [Google Scholar]
- 18.Garcia B. A., Pesavento J. J., Mizzen C. A., Kelleher N. L. (2007) Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods 4, 487– 489 [DOI] [PubMed] [Google Scholar]
- 19.Shechter D., Dormann H. L., Allis C. D., Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc 2, 1445– 1457 [DOI] [PubMed] [Google Scholar]
- 20.Young N. L., DiMaggio P. A., Plazas-Mayorca M. D., Baliban R. C., Floudas C. A., Garcia B. A. (2009) High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics 8, 2266– 2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masselon C., Anderson G. A., Harkewicz R., Bruce J. E., Pasa-Tolic L., Smith R. D. (2000) Accurate mass multiplexed tandem mass spectrometry for high-throughput polypeptide identification from mixtures. Anal. Chem 72, 1918– 1924 [DOI] [PubMed] [Google Scholar]
- 22.Thomas C. E., Kelleher N. L., Mizzen C. A. (2006) Mass spectrometric characterization of human histone H3: a bird’s eye view. J. Proteome Res 5, 240– 247 [DOI] [PubMed] [Google Scholar]
- 23.ILOG (2008) ILOG CPLEX C++ API 11.1 Reference Manual, ILOG, Gentilly, France [Google Scholar]
- 24.Nemhauser G. L., Wolsey L. A. (1988) Integer and Combinatorial Optimization, John Wiley and Sons, Inc., New York [Google Scholar]
- 25.Floudas C. A. (1995) Nonlinear and Mixed-Integer Optimization, Oxford University Press, Oxford [Google Scholar]
- 26.Pedrioli P. G., Eng J. K., Hubley R., Vogelzang M., Deutsch E. W., Raught B., Pratt B., Nilsson E., Angeletti R. H., Apweiler R., Cheung K., Costello C. E., Hermjakob H., Huang S., Julian R. K., Kapp E., McComb M. E., Oliver S. G., Omenn G., Paton N. W., Simpson R., Smith R., Taylor C. F., Zhu W., Aebersold R. (2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol 22, 1459– 1466 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.