

Abstract
Inclusions comprised of fibrils of the microtubule (MT)-associated protein tau are found in the brains of those with Alzheimer’s disease (AD) and other neurodegenerative tauopathies. The pathology that is observed in these diseases is believed to result from the formation of toxic tau oligomers or fibrils, and/or from the loss of normal tau function due to its sequestration into insoluble deposits. Hence, small molecules that prevent tau oligomerization and/or fibrillization might have therapeutic value. Indeed, examples of such compounds have been published but nearly all have properties that render them unsuitable as drug candidates. For these reasons, we conducted quantitative high-throughput screening (qHTS) of ~292,000 compounds to identify drug-like inhibitors of tau assembly. The fibrillization of a truncated tau fragment that contains four MT-binding domains was monitored in an assay that employed complementary thioflavine T fluorescence and fluorescence polarization methods. Previously described classes of inhibitors as well as new scaffolds were identified, including novel aminothienopyridazines (ATPZ’s). A number of ATPZ analogs were synthesized and structure-activity relationships were defined. Further characterization of representative ATPZ compounds showed they do not interfere with tau-mediated MT assembly, and they are significantly more effective at preventing the fibrillization of tau than the Aβ(1–42) peptide which forms AD senile plaques. Thus, the ATPZ molecules described here represent a novel class of tau assembly inhibitors that merit further development for testing in animal models of AD-like tau pathology.
Intracellular accumulations comprised of hyper-phosphorylated forms of the protein tau are found within the somatodendritic regions of neurons in Alzheimer’s disease (AD), certain frontotemporal dementias and a host of additional neurodegenerative disorders that are broadly referred to as “tauopathies” (for review see (1)). These tau lesions correlate with the severity of dementia in AD (2–4) and missense mutations within the tau gene lead to inherited forms of frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) (5;6). Thus, tau has been directly implicated as a causative agent in AD and related neurodegenerative diseases.
Normally, tau binds to tubulin and is believed to promote MT assembly and stabilization (7–9). This role of tau is particularly important in neurons, where the stability of MTs is critical for axonal transport and the delivery of cellular materials to and from synapses (10). Tau is normally phosphorylated and the extent of this post-translational modification is believed to play an important role in regulating MT dynamics (11). Thus, the hyper-phosphorylation of tau that occurs in tauopathies and its sequestration into aggregates could reduce MT binding and stabilization, thereby resulting in an impairment of axonal transport with resulting synaptic dysfunction. Consistent with this loss-of-function hypothesis are data which demonstrate that hyper-phosphorylation of tau can diminish MT binding (12–14) as well as increase its propensity to fibrillize (15;16). Moreover, cell-based studies have shown that alterations of tau phosphorylation affect MT function (17;18) and altered axonal transport has been demonstrated in a transgenic mouse model in which over-expression of human tau leads to neuronal tau inclusions (19). It is also possible that tau accumulations could lead to neuropathology through a gain of one or more functions (1;20). For example, tau oligomers and/or fibrils might cause direct neuronal damage through yet to be defined mechanisms. It should be noted that gain-of-function and loss-of-function explanations of tau-induced neurodegeneration need not be mutually exclusive and it is possible that both mechanisms contribute to disease.
Based on the current understanding of how multimeric tau assemblies might lead to neuron dysfunction and degeneration, several strategies for intervening in disease progression have been proposed. These include identifying drugs that; 1) stabilize brain neuronal MTs (19;21), 2) reduce the effects of tau hyper-phosphorylation through kinase inhibition (11;22;23), 3) enhance tau intracellular degradative pathways (24;25), or 4) prevent tau assembly into oligomers and/or fibrils (22;26). Arguably, this latter approach might abrogate both tau gain-of-function toxicity attributable to the formation of oligomers/fibrils and loss-of-function resulting from diminished tau binding to MTs due to its sequestration into aggregates. Although inhibition of tau assembly is a conceptually appealing approach for treating tauopathies, disruption of macromolecular interactions of this type with small molecule drugs is considered extremely challenging due to the large surface areas involved in protein-protein binding. Further, the molecular details of tau-tau interactions within assembled fibrils are not fully understood, although it has been shown that alteration of a single amino acid in one of the MT binding domains of tau can render the protein fibrillization-incompetent (27). Thus, it may be possible to shield this or other critical sites in tau with a small molecule, thereby blocking tau assembly into oligomers/fibrils.
The tau fibrillization process can be recapitulated in vitro with the aid of anionic co-factors such as lipids or heparin (28–30), using either full-length tau or truncated species of tau containing the three or four MT-binding domains that are found in alternatively spliced human tau isoforms (31). Utilizing such fibrillization assays, prior attempts have been made to identify inhibitors of tau assembly through high-throughput screening (HTS) of compound libraries (32–34) or via rational chemistry approaches (35–37). Several chemical series have been identified that block tau fibril formation, including anthraquinones (33), polyphenols (37), quinoxalines (32), phenothiazines (36) and phenylthiazolyl-hydrazides (35). However, with the exception of the phenothiazine methylene blue (38), no compound has yet been tested in vivo for its effect on tau deposition, neurodegeneration and behavioral impairments, and the majority of the previously described molecules have chemical or biological properties that likely make them unsuitable as CNS-directed therapeutic agents.
To identify prototype inhibitors of tau assembly that exhibit favorable combinations of potency, selectivity and drug-like physical-chemical properties, we performed quantitative HTS (qHTS) of ~292,000 compounds using a truncated form of tau that contains four MT-binding repeats (32;35) and harbors the P301L tau mutation found in FTDP-17 (5). Tau fibrillization was monitored by thioflavine T (ThT) binding and consequent fluorescence emission. In addition, we utilized a fluorescence polarization (FP) measurement to assess tau multimerization, using Alexa Red-tagged tau that can be incorporated into growing tau assemblies. To our knowledge, this is the most comprehensive screen for tau assembly inhibitors reported to date, resulting in the identification of both previously described molecules as well as a new compound series, the ATPZ’s, that effectively inhibit tau fibril formation. A series of ATPZ analogs were synthesized to highlight possible structure-activity relationships (SARs). Furthermore, examination of representative active compounds from this class showed that they do not interfere with tau-mediated MT assembly. In addition, testing of ATPZ compounds in tau and Aβ(1–42) peptide fibrillization assays suggests that this class of inhibitors is largely tau-specific. Finally, the ATPZ identified in these studies exhibit promising drug-like properties with no violations of Lipinski’s rule of five. (39) Collectively, the data presented here suggest that this class of tau assembly inhibitors holds considerable promise for the development of candidate compounds appropriate for in vivo evaluation.
Experimental Procedures
Tau Protein Preparations
Full length tau (Tau40; amino acids 1–441) (40), tau K18 fragment containing the P301L missense mutation (5) (K18PL; amino acids 244–369) (41), and a non-fibrillizing K18 tau construct carrying the K311D missense mutation (K18KD) (27) were cloned into the pRK172 expression vector, expressed and purified as previously described (42). To eliminate batch-to-batch variations that might contribute to assay variability, K18PL from several preparations was pooled so that a single homogeneous preparation was used throughout qHTS.
Alexa Red Labeling of K18PL and K18KD
K18PL and K18KD were labeled with Alexa Red (Invitrogen) according to the manufacturer’s protocol. Labeled protein was determined to contain 1 mol of dye per 3 mol protein, following the quantification method described in the manufacturer’s instructions.
Fibrillization Reactions
Tau fibril formation reactions were performed essentially as described (32). For qHTS in a 1536-well plate format, reagents were scaled to 25% of the amount used in the previously described 384-well plate format (32). K18PL (2 µl of a 30 µM stock that also contained 0.24 µM Alexa Red-labeled K18PL) was dispensed into black 1536-well plates using a solenoid-based dispenser. Following transfer of 23 nL of test compound or DMSO vehicle by a pin tool from compound library plates, 2 µl/well of 40 µM heparin in reagent buffer was dispensed and the plate was centrifuged for 15 s at 1000 rpm in a table-top centrifuge. The fibrillization reaction proceeded for 6 hours at 37°C in a humidified incubator.
ThT Assay
K18PL fibril formation was quantified with a ThT assay as previously described (32). Briefly, 1 µl/well of 62.5 µM ThT in 100 mM glycine, pH 8.5 (12.5 µM final concentration) was dispensed into 1536-well plates after completion of the fibrillization reaction and incubated at room temperature for 1 hour. Plates were read on an Envision fluorescence plate reader with an excitation of 450 nm and an emission setting of 510 nm.
Tau Fluorescence Polarization (FP) Assay
A tau FP method was developed to quantify Alexa Red-labeled K18PL tau incorporation into multimeric tau assemblies based on a similar methodology that was previously employed to measure the oligomerization and fibrillization of α-synuclein (43). Although Alexa Red-labeled K18PL tau preparations did not readily assemble into fibrils under the conditions described above, the fluorescently-labeled tau could be incorporated into nascent oligomers and fibrils with normal assembly kinetics when mixed with unlabeled K18PL at a 1:62.5 molar ratio. Under these conditions, a fluorescence polarization increase of >100 polarization units (mP) was obtained upon completion of the fibrillization reaction, as determined on an Envision fluorescence plate reader using a Texas Red FP dual mirror, Texas Red FP excitation filter at 555 nm, and Texas Red FP emission filter (P and S channel) at 632 nm. The detector gains for each emission filter were calibrated to obtain maximum mP values. FP values were calculated from the S and P channel readings according to the equation, mP = 1000*(S−G*P)/(S+G*P), with the G value set to one. Total Alexa Red fluorescence emission was calculated using the equation TF = S + 2*P.
Comparison of K18PL Assembly Kinetics by ThT Fluorescence and FP
During assay validation, K18PL fibrillization reactions were concurrently monitored by ThT fluorescence and FP as described above, with the exception that ThT (12.5 µM) was present during the assembly reactions. Prior studies (not shown) revealed that the time-course of fibrillization is similar in the absence or presence of ThT. FP and ThT fluorescence were determined at regular intervals after the initiation of the oligomerization/fibrillization reaction to monitor the formation of ThT-positive and FP-positive K18PL assembly products.
Compound Library
A total of 291,948 molecules were interrogated by qHTS using the K18PL fibrillization assay described above. Compounds were obtained from the NIH Molecular Libraries Small Molecule Repository (222,431 molecules), the Centers for Chemical Methodology and Library Development from Boston University, Kansas University, and the University of Pittsburgh (7,145 molecules), and from private and commercial sources (62,372 molecules). Compounds were prepared for qHTS as described (44).
Quantitative High-Throughput Screening (qHTS) Procedures
The compound library underwent qHTS on an integrated robotic platform (45) at six five-fold dilutions ranging from 56 µM to 18 nM (44). K18PL tau fibrillization reaction components were dispensed into 1536-well plates as described above. Each screening plate also contained 16 two-fold dilutions beginning at 56 µM, in duplicate, of a previously identified inhibitor, oleocanthal (46), as well as 32 wells of non-fibrillizing K18KD tau (containing a 1:62.5 molar ratio of Alexa Red-labeled K18KD) and 32 wells of K18PL tau with DMSO vehicle. FP analysis was conducted after ThT readings following the methods described above.
Screening Data Analysis
Compound effects on tau assembly were determined for the ThT and FP assays using the formula 1-(Fcompound− F̄ DMSO)/( F̄ control− F̄ DMSO), where F̄ control denotes the median plate fluorescence values (ThT assay) or mP values (FP assay) of the non-fibrillizing K18KD control wells, F̄ DMSO denotes the median value of the DMSO control wells, and F̄compound denotes the compound well values. The resulting values were multiplied by 100 to obtain the percent inhibition of tau fibril formation. These normalized activity values were then corrected by applying a pattern correction algorithm using DMSO-only plates placed at 24 plate intervals in the screen, as well as the beginning and start. Concentration-response curves (CRC’s) were fit and classified as described (44). Four major curve classes (1–4) were created based on the completeness of the curve, goodness of fit, and efficacy. The compounds with class 1.1, 1.2, and 2.1 curves are statistically the most reliable, while the compounds with class 2.2 or 3 curves are less reliable, and Class 4 compounds show no concentration response and little (<30%) efficacy. Alexa Red total fluorescence (TF) measurements were expressed as a percentage of the DMSO control value, with values >30% indicating potential fluorescence interference. An in-house auto scaffold detection program was used to cluster 2989 FP actives (curve classes 1 and 2.1, as well as class 2.2 curves with >50% efficacy) yielding 514 structural series containing at least three compounds of which at least one was active in addition to 755 singletons. All of the screening data were deposited into PubChem (AIDs 1460, 1463 and 1468).
Centrifugation and Electron Microscopy (EM) Assays
The determination of compound-induced effects on the amount of tau that remains soluble upon centrifugation, and EM visualization of tau species following incubation with test compounds, were as previously described (32).
MT Assembly Assay
The effect of compounds on tau-mediated MT assembly was essentially as described (42), adapted to a 384-well plate. Wild-type K18 tau was used for MT assembly because K18PL has a decreased ability to promote MT assembly. Lyophilized bovine brain tubulin was reconstituted in RAB (100 mM MES, pH 6.9; 1 mM EDTA; 0.5 mM MgSO4) at a concentration of 10 mg/ml. Compounds (50 µM) were added to K18 tau (40 µM) in RAB and pre-incubated at room temperature for 60 minutes. To initiate the MT assembly reaction, 8.25 µl per well of 10 mg/ml tubulin was dispensed on a UV-clear 384-well plate, followed by 1.0 µl of 100 mM GTP in RAB and 41.25 µl of the compound:tau mixture. This resulted in a final reaction mixture of 30 µM tubulin, 40 µM K18, 50 µM compound and 2 mM GTP. The plate was incubated in a Spectramax M5 plate reader at 37°C and the absorbance at 340 nm was read every minute for 45 minutes.
Aβ1–42 Fibrillization Assay
Synthetic Aβ(1–42) peptide was resuspended in 1,1,1,3,3,3- hexafluoroisopropanol (HFIP) at a concentration of 5 mg/ml for 30 minutes and then air dried in small aliquots, followed by storage at −80°C. For fibrillization assays (47), HFIP-treated Aβ(1–42) aliquots were reconstituted to 2 mg/ml in DMSO and then diluted to 15 µM in 25 mM Tris, pH 7.0 buffer to which test compound was added at 50 µM final concentration, or at several concentrations ranging from 0.16–40 µM. The reaction mixtures were dispensed at 25 µl/well into a 384-well plate and then incubated at 37°C for 4 hrs. Upon completion of the reaction, 25 µl of 25 µM ThT was added to each well followed by ThT fluorescence readings as described above for K18PL.
Caspase-1 Assay
Caspase-1, kindly provided by Dr. James Wells (University of California, San Francisco), was purified as described (48) and assayed at 100 nM in buffer containing 50 mM HEPES pH 7.5, 50 mM KCl, 200 mM NaCl, and 0.1% CHAPS. Caspase-1 was dispensed at 3 µL/well into black solid 1536-well plates and incubated 5 min with various concentrations of test compound, with subsequent addition of 1 µl/well Ac-WEHD-AFC substrate (20 uM final concentration). Within 1 minute of substrate addition, fluorescence intensity (405 nm excitation and 525 nm emission) was measured on a ViewLux (PerkinElmer) every 30 s for 10 min. The first 3 minutes of fluorescence values were linear and used to calculate the slope of substrate conversion as a measure of enzyme activity.
β-Lactamase Assay
The β-lactamase promiscuous inhibitor assay was adapted from a previously published method (49). Reactions (0.15 ml final volume) were performed with test compounds dissolved in DMSO or with DMSO alone (2% final v/v) in 50 mM potassium phosphate, pH 7.0 containing 1.6 U/ml β-lactamase (Sigma P4399). Reactions were conducted either in the absence or presence of 0.01% Triton X-100 to determine whether detergent-sensitive compound aggregates inhibited β-lactamase. After incubation for 5 min at room temperature, CENTA β-lactamase substrate (Calbiochem 219475) was added to a final concentration of 5 µM. Enzyme activity was monitored by changes in optical density at 405 nm in 96-well clear plates (Fisher 12-565-501) using a Spectramax M5 reader at 15 s intervals for 5 min. Substrate background was subtracted from the readings as was the initial reading to account for any compound absorbance. The slope of the increase in optical density was calculated by linear regression (Graphpad Prism) and considered the initial velocity of the reaction. The percent of β-lactamase inhibition was determined from the ratio of initial velocity in the presence of compound to the initial velocity of the vehicle control. The percent inhibition in the absence or presence of 0.01% Triton X-100 was compared and if the compound inhibited β-lactamase by >25% in the absence of detergent but by <10% in the presence of detergent it was considered a promiscuous inhibitor. Rottlerin (Sigma 5648) was used as a positive control (53% inhibition in the absence of Triton X-100) and Furosemide (Sigma F4381) as a negative control (3% inhibition in the absence of Triton X-100).
Size-Exclusion Chromatography (SEC) of Tau40 Fibrillization Reaction Post-Centrifugation Supernatants
Tau40 (50 µM) and heparin (20 µM) were incubated in a PCR tube with or without the compound MLS000062428 (100 µM) at 37°C for 5 days in a final volume of 100 µl of 100 mM sodium acetate buffer, pH 7.0. In the absence of compound, tau40 fibrillization is complete under these conditions, as determined by ThT fluorescence and centrifugation assays. A non-assembly control reaction was also prepared as above with tau40 and MLS000062428 in the absence of heparin. The completed reactions were centrifuged at 40,000 g for 30 minutes to sediment fibrils and the supernatants were applied to a Superdex 200 10/300 column (GE Healthcare) employing an Äkta Basic FPLC unit (GE Healthcare) with a flow rate of 0.4 ml/minute. Tau elution was monitored by absorbance at 280 nm. Under these chromatography conditions, monomeric tau can be separated from oligomeric species (27).
Synthesis of ATPZ’s
NCGC00031883 (5a), NCGC00053250 (5b), NCGC00183206 (5c), NCGC00183204 (5d), NCGC00183205 (5e), NCGC00183195 (5f), NCGC00183326 (5g), NCGC00183325 (5h), NCGC00183199 (6a), and NCGC00182500 (6b) were synthesized (Scheme 1) following a modified literature procedure (50). Commercially available anilines 1a–h were converted to aryl diazonium salts 5, which reacted with β-ketoesters to form hydrazones 6 as a mixture of E/Z isomers. Condensation reactions of the hydrazones with ethyl cyanoacetate gave pyridazines 7. Subsequently reactions of pyridazines 7 with sulfur under Gewald conditions generated 5a–h. Saponification of 5a–b gave the corresponding 6a–b.
Scheme 1.

NCGC00182501 (11d) was synthesized by a DMC-mediated coupling reaction between acid 9b and diethyl amine. NCGC00183201 (11a), NCGC00183202 (10b), and NCGC00183207 (10c) were prepared by a reaction sequence starting from 7a as depicted in Scheme 2. Saponification of 7a under acidic condition yielded 10a. Amide 10b was formed by reacting 7a with ammonium. Coupling reactions of acid 10a with appropriate amines followed by Gewald reaction yielded the desired amides 11a–c.
Scheme 2.

NCGC00182502 (12a), NCGC00183197 (12b), and NCGC00183198 (12c) were prepared by reactions of 8a–b with appropriate acid chlorides, as shown in Scheme 3.
Scheme 3.

The preparation of NCGC00183207 (18) is illustrated in Scheme 4. EDC-mediated coupling reaction of acid 10a with tert-butylcarbazate followed by Boc-deprotection gave hydrazide 14. The hydrazide reacted with nitrous acid in acetic acid produced carboazide 15. Curtius rearrangement of 15 in ethanol gave ethyl carbamate 17. Under basic condition, the carbamate was converted to the desired amine 18. NCGC00183196 (19) was prepared by LiAlH4 reduction of ester 8a, Scheme 5.
Scheme 4.

Scheme 5.

Compound Identity and Purity Determination
All compounds underwent purity and mass determination using reverse-phase HPLC/MS (ESI+) method with elution monitoring by UV absorbance (220 nm) and evaporative light scatter (ELS). Analyses were performed at a flow rate of 0.5 ml/min on a Waters Acquity UPLC system using a Phenomenex 2.5 µm Luna C18(2)-HST 100×2 mm column at 45° C. A linear 2–100% acetonitrile (0.025% TFA)/H2O (0.05% TFA) gradient was utilized over 2.2 min, with a total run time of 3.0 min. Purity was assessed by integration of chromatograms (UV220 nm and ELSD).
Results
Compound Screening and Analysis
We previously described a screen, conducted in a 384-well plate format, in which fibril formation by the tau K18 fragment that contains four MT-binding domains was monitored by the binding and fluorescence of thioflavine T (ThT) (32). This methodology was modified here to allow screening in a 1536-well plate format, utilizing a modified K18 fragment containing the P301L mutation (K18PL) found in patients with FTDP-17 (5;6). The K18PL fragment forms fibrils more rapidly than wild-type K18 (51), thereby allowing faster screening times. Dithiothreitol (DTT) was eliminated from the reaction because this reagent could chemically interact with certain compounds, resulting in tau oxidation and false-positive activities (32;52). Finally, Alexa Red-labeled K18PL was included at a 1:62.5 molar ratio to unlabelled K18PL to assess differential FP caused by the formation of tau oligomers/fibrils. We previously developed a comparable FP assay to assess the oligomerization and fibrillization of α-synuclein (43), which is found within Lewy body inclusions that are a signature of Parkinson’s disease. Importantly, and in contrast to the ThT assay, the FP readout does not rely on a cross-β-fibril structure to elicit a positive signal and is instead dependent on the formation of multimeric species that can slow the rotational freedom of incorporated Alexa Red-labeled K18PL.
Whereas multimeric species of α-synuclein could be detected by FP prior to the formation of ThT-positive fibrils (43), K18PL tau assembly results in essentially identical FP and ThT kinetics (Figure 1). As the fibrillization of amyloid proteins is believed to proceed only after the formation of nucleation cores (53), this result suggests there is a considerably shorter lag time between the assembly of nucleating cores and subsequent fibril formation with K18PL tau compared with α-synuclein. This is in keeping with the observation that maximal fibril formation occurs within ~6–8 hours with K18PL (Figure 1) compared to 60–80 hours for α-synuclein (43). Although the time-course of FP and ThT changes are similar with K18PL tau, utilization of the FP measurement in conjunction with ThT fluorescence allows for the differentiation of compounds that block the initial stages of tau assembly from those that preferentially interfere with the growth of fibrils. The former would be expected to elicit similar inhibition of both ThT and FP signals, whereas the latter should inhibit ThT fluorescence while having a substantially lesser effect on FP.
Figure 1. Comparison of K18PL oligomerization/fibrillization kinetics as determined by ThT fluorescence and FP.

K18PL fibrillization reactions were conducted in the presence of ThT and Alexa Red-labeled K18PL, as described in Materials and Methods. Data are normalized to the maximum ThT and FP signal, and represent the mean of quadruplicate wells with error bars representing standard deviations.
The combined ThT/FP assay was optimized through trial HTS using 1536-well test plates to ensure that automated reagent dispensing would provide low coefficients of variation (CV) and Z values (54) exceeding 0.5. Subsequently, a ~292,000 compound library was screened at six concentrations ranging from 56 µM to 18 nM for each compound. The combination of good signal-to-background and low CV values resulted in average plate Z-scores of 0.85 ± 0.12 and 0.79 ± 0.03 for the ThT and FP analyses, respectively. All of the compound data were computationally analyzed following the completion of the screen, with an evaluation of compound concentration-dependency using both endpoint measurements. Each compound was categorized as belonging to one of four possible CRC classes, as summarized in Figure 2a. Although some compounds were identified that appeared to enhance K18PL fibril formation, as evidenced by an increase in ThT fluorescence and/or FP, only compounds that caused an apparent decrease in tau assembly were deemed of interest. Furthermore, class 3 compounds, which are defined as showing inhibition of ThT or FP signal at a single concentration or poorly fit curves, were excluded. As depicted in Figure 2b, a total of 7,094 compounds (2.4%) showed either a class 1.1, 1.2 or 2.1 concentration-dependent inhibition of ThT fluorescence. A smaller number of compounds (1,011 or 0.3%) were identified that caused a similar inhibition of FP signal, and 285 of these overlapped with the ThT-positive cohort (Figure 2b). Relaxation of the stringency to include class 2.2 FP active compounds with >50% maximal inhibition increased the overlap with class 1.1–2.1 ThT active molecules to 967 compounds. Compounds with this profile were of particular interest, as meaningful inhibition in both the FP and ThT assay suggested that these molecules were affecting the earlier steps in tau assembly. The observation that there were significantly more compounds that were class 1.1–2.1 inhibitors of ThT fluorescence than FP likely reflects both an abundance of compounds that are competitive inhibitors of ThT binding to tau fibrils (34) and compounds that preferentially affect fibril growth rather than initial oligomer assembly.
Figure 2. Description of CRC’s and number of screening hits.

(A) Fitted CRC’s obtained from qHTS were fit and classified as belonging to one of four major curve classes based on curve-fit and efficacy metrics. Class 1 and 2 compounds were further defined as sub-class X.1 or X.2, depending on the compound total efficacy (maximal inhibition based on curve fit data). (B) The number of compounds from the K18PL qHTS campaign that were classified as class 1 or 2.1 using the ThT and FP readouts. The number of compounds active in ThT only (red), FP only (yellow) and both ThT and FP (orange) are indicated.
All FP class 1 and 2.1 compounds, as well as those from class 2.2 with >50% efficacy (2989 total), were grouped into clusters comprised of shared core structural elements. This resulted in 514 series for which there were at least 3 related compounds within the screening library. After structural clustering, only compounds that were active in both the FP and ThT assays were considered further. These compounds were examined for their activities in other PubChem screens, and those found to be non-specific (as evidenced by an unusually large number of interactions with other targets) were de-prioritized. In addition, compounds that showed a large (>50%) reduction of Alexa Red total fluorescence were removed from further consideration as they were likely attenuating fluorescence emission and affecting the accuracy of the FP and ThT measurements. Finally, compound chemical structures were carefully examined to segregate unwanted series bearing reactive or unstable functional groups.
After triaging the structural series by the above criteria, compounds of interest were analyzed for proper molecular mass and purity. A small subset of seven compounds with >85% purity and a diversity of structures was selected for follow-up analyses as summarized in Table 1. The compounds had comparable IC50 values with both the ThT and FP primary screen readouts, although the maximal inhibition observed in the ThT assay was typically much greater than that observed with FP. This result suggests that these compounds cause nearly quantitative inhibition of the cross-β fibril structures that are required for ThT binding and fluorescence, but incomplete inhibition of assembly into multimeric structures that contribute to the FP signal. In all cases, the IC50 values clustered between 1–10 µM for both the ThT and FP endpoints. Since 15 µM tau was utilized in the screen, it is likely that the inhibitory activity is limited by compound-to-tau stoichiometry such that ~1:1 compound:tau molar ratio is required for maximal inhibition.
Table 1.
Activities of selected tau fibrillization inhibitors from the primary qHTS, with repeat analyses and secondary assays.
| 1° ThT Screen1 | 1° FP Screen1 | Total Fluor2 |
Repeat ThT Assay3 |
2° Assays4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Structure | Structural Class |
Curve class |
Log IC50 |
% Inhib | Curve class | Log IC50 | % Inhib | % Inhib | Log IC50 |
% Inhib | Centrifugation % Soluble |
EM | Tau40 % Inhib |
| MLS000062428 | 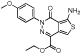 |
Aminothieno- pyridazine |
2.1 | −5.0 | 94 | 2.2 | −5.0 | 46 | 49 | −4.5 | 97 | 80 | 2 | 64 |
| MLS000034832 | 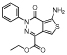 |
Aminothieno- pyridazine |
2.1 | −5.0 | 80 | 1.2 | −5.3 | 35 | 41 | −5.1 | 93 | 73 | 2 | 62 |
| NCGC00093795 | Catecholamine | 1.1 | −5.5 | 96 | 1.2 | −5.7 | 58 | 36 | −5.0 | 103 | 70 | 2 | 89 | |
| MLS000339510 | thiourea | 1.1 | −5.7 | 93 | 1.1 | −5.5 | 73 | 33 | −5.0 | 70 | 51 | 2 | 74 | |
| MLS000530738 | 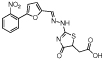 |
Hydrazinyl- dihydrothiazole |
1.1 | −5.7 | 97 | 1.2 | −5.8 | 48 | 36 | −5.0 | 92 | 58 | 4 | 83 |
| MLS000530726 | 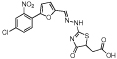 |
Hydrazinyl- dihydrothiazole |
1.1 | −6.0 | 99 | 1.2 | −5.8 | 66 | 34 | −5.8 | 95 | 74 | 3 | 84 |
| MLS000058666 | 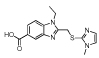 |
Benzimidazole | 1.2 | −5.4 | 65 | 1.2 | −5.1 | 41 | <10 | −3.1 | 84 | 56 | 3 | 81 |
Data from primary qHTS with K18PL tau. Curve class, IC50 values and maximum % inhibition for ThT fluorescence and FP are as described in Experimental Procedures.
Percent inhibition of Alexa Red total fluorescence as described in Experimental Procedures.
IC50 values and maximum percent inhibition in repeat testing of K18PL tau ThT fluorescence.
Secondary testing of compounds in the K18PL tau centrifugation assay followed by EM analysis and Tau40 ThT fluorescence assay, as described in Experimental Procedures.
Screening Hit Confirmation
The compounds listed in Table 1 were re-tested in the K18PL ThT assay to confirm that the primary screening results were reproducible. Upon repeat analysis, all of the compounds caused at least 70% maximal inhibition in the ThT assay (Table 1). The majority of the compounds showed maximal inhibition values that were comparable to those observed in the primary qHTS, but there was a fairly uniform shift in the IC50 values to lower potency. It should be noted that the primary qHTS was conducted with single determinations per compound concentration while the repeat testing was performed with duplicate samples at each of the tested concentrations. Thus, variations in curve fitting may have contributed to differences in the IC50 values between analyses, with the duplicate values of the repeat analyses likely resulting in more accurate determinations.
The ability of the selected compounds to inhibit K18PL fibril formation was also examined by determining whether they would cause an increase in the percentage of tau that remained in the soluble fraction upon centrifugation, as previously described (32). Tau fibrils readily sediment upon centrifugation and all of the compounds in Table 1 caused >50% reduction of K18PL in the pellet fraction when tested at a single concentration of 100 µM (see Figure 3a and Table 1). The percentage of soluble tau in the centrifugation assay was somewhat less than the maximal percent inhibition observed in the ThT assay for all compounds. This presumably reflects the presence of multimeric tau species that lack sufficient cross-β fibril structure to allow ThT binding, yet which have sufficient density to sediment during centrifugation. To confirm that compound-mediated reductions of K18PL sedimentation after centrifugation resulted from an inhibition of fibril formation, the pellet fractions obtained after centrifugation of compound-tau reaction mixtures were examined by transmission electron microscopy (EM) and were graded for the presence of fibrils using a semi-quantitative four-point scale (Figure 3b and 3c). All of the compounds disrupted fibril structure as evidenced by a score ≥2 (Table 1).
Figure 3. Representative examples of K18PL fibrillization reactions performed in the absence and presence of test compounds as assessed by the centrifugation assay and EM analysis.

(A) SDS-PAGE analysis of supernatant (s) or pellet (p) samples obtained following centrifugation of K18PL fibrillization reactions conducted in the presence of DMSO vehicle, 100 µM MLS00062428 or 100 µM MLS000034832. The gel was stained with Coomassie Blue. (B) Representative electron micrographs of objects visible at the endpoint of K18PL fibrillization reactions containing 100 µM of a) MLS000062428 or b) MLS000034862. The micrograph is shown at 100,000x with the bar representing 100 nm. (C) EM grading system used to judge fibril content in Table 1: a) grade=0, all fibrils in clumps, no fragments; b) grade=1, small fibril clumps, free filaments and some pieces; c) grade=2, filaments and pieces but no clumps; d) grade=3, short filaments and tiny pieces; e) grade=4, only tiny pieces; f) no objects.
Although previous studies have demonstrated that tau fragments such as K18PL form fibrils which resemble those observed in tauopathies (32), it was important to demonstrate that the compounds could disrupt assembly of full-length tau40. As summarized in Table 1, all of the compounds caused at least a 60% inhibition of ThT fluorescence in the tau40 assay when tested at 50 µM concentration. These data thus confirm that the selected compounds identified in the K18PL screen affect full-length tau in addition to the truncated tau fragment.
Characterization of Novel ATPZ Inhibitors of Tau Assembly
Upon completion of confirmation testing of the selected compounds, secondary analyses were conducted with the two compounds containing the ATPZ scaffold. Compounds with this core scaffold have not previously been reported to inhibit tau fibrillization, and thus these molecules appear to represent a novel class of tau assembly inhibitors. Moreover, these compounds showed drug-like structural features meriting their further characterization. We assessed whether these molecules interfered with the ability of tau to bind and stabilize MTs. As depicted in Figure 4a, neither of the ATPZ’s had a meaningful effect on tau-mediated tubulin polymerization. In contrast, methylene blue, a promiscuous compound active in a high percentage of PubChem screens (data not shown), caused a significant diminution of tau-facilitated MT formation. The ATPZ’s were also tested for their ability to block the fibrillization of Aβ(1–42), an amyloid peptide found within senile plaques in AD brain (55). The compounds were less effective in blocking Aβ(1–42) fibril formation, as judged by ThT fluorescence, than they were in inhibiting tau fibrillization (Figure 4b). Thus, these data suggest that the ATPZ’s preferentially block tau assembly, but the extent to which this selectivity extends to other amyloidogenic proteins awaits further testing.
Figure 4. Effects of ATPZ tau fibrillization inhibitors on tau-mediated MT assembly and Aβ(1–42) fibril formation.

(A) Representative traces of tau-induced microtubule assembly in the presence of 50 µM test compounds or DMSO, as described in Materials and Methods. (B) Comparison of K18PL tau and Aβ(1–42) fibrillization in the presence of ATPZ’s.
The observation that the ATPZ’s caused greater ThT than FP inhibition in qHTS (Table 1) suggested that they might be more effective in preventing tau fibrillization than oligomerization. To further investigate the nature of the tau that remained in solution after incubation with an ATPZ inhibitor, aliquots of the post-centrifugation supernatants from full-length tau40 fibril assembly reactions conducted in the presence or absence of 50 µM MLS000062428 were analyzed by size-exclusion chromatography (SEC) to allow for the separation of monomeric tau40 from larger oligomeric species (27). As shown in Figure 5, inclusion of the ATPZ caused an appreciable increase within the post-centrifugation supernatant of the amount of monomeric tau relative to the untreated fibrillization reaction and also led to detectable oligomeric species that eluted in the column void volume. For comparison, the amount of monomeric tau in the supernatant fraction from a non-fibrillizing mixture of tau40 and MLS000062428 incubated in the absence of heparin is also depicted in Figure 5. These data suggest that MLS000062428 prevents the transition of tau oligomers to full fibrils, which could have beneficial consequences by preventing the formation of potentially neurotoxic fibrils and by increasing the pool of tau monomer that would be available to stabilize MTs.
Figure 5. Inhibition of tau assembly by MLS000062428 increases both monomericand oligomeric tau content.

Tau40 fibrillization reactions were performed in the presence or absence of MLS000062428, and a non-fibrillizing control reaction was conducted with tau40 and MLS000062428 in the absence of heparin. Following incubations as described in Materials and Methods, the reaction mixtures underwent centrifugation and the resulting supernatant fractions were analyzed by SEC to determine the relative content of tau monomer and oligomer. The chromatographs shown correspond to the non-fibrillizing control (grey trace) and fibrillization in the absence (dashed trace) or presence (black trace) of MLS000062428. The column void volume (Ve) was defined by the elution of blue dextran and the elution volumes of bovine serum albumin (BSA; 66 kD) and cytochrome c (Cyt. c; 12 kD) are also indicated. Tau is a highly unstructured protein that is known to elute upon SEC with an aberrantly high apparent molecular weight (7).
The promising properties of the ATPZ class of tau assembly inhibitors led to the synthesis of additional analogs with the aim of evaluating possible SAR. As listed in Table 2, a series of 21 analogs were synthesized which exhibited a variety of substitutions of the ATPZ core structure. In addition, the previously tested compounds MLS000034832 and MLS000062428 were re-synthesized to confirm batch-to-batch reproducibility. All synthesized compounds were characterized by NMR and LC-MS analyses, and in all cases purity level was >85% as determined by LC-MS. As summarized in Table 2, all of the ATPZ compounds had physicochemical properties that are consistent with prevailing guidelines for drug like compounds (39).
Table 2.
Summary of novel ATPZ compounds.
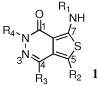 |
Tau ThT Assay 3 |
Tau Cent Assay 4 |
Aβ42 ThT Assay 5 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound | R1 | R2 | R3 | R4 | PCP 2 | Log IC50 |
% Inhib. |
% Soluble |
% Inhib. |
| 1 | MLS000062428 | H | H | -COOEt | MW:345.4 PSA: 94.2 ClogP: 2.6 |
−5.3 | 86 | 64 | 28 | |
| 2 | NCGC00182502 | 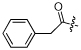 |
H | -COOEt | MW: 463.5 PSA: 97.3 ClogP: 4.3 |
NC | 31 | 0 | ND | |
| 3 | NCGC00183197 | Ac- | H | -COOEt | Ph | MW: 357.4 PSA: 88 ClogP: 2.6 |
NC | 27 | 11 | ND |
| 4 | NCGC00183198 | 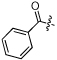 |
H | -COOEt | Ph | MW: 419.5 PSA: 88.1 ClogP: 4.3 |
NC | 8 | 2 | −7 |
| 5 | NCGC00183210 | Ac | Me | H | Ph | MW: 299.3 PSA: 61.8 ClogP: 2.9 |
NC | 11 | 5 | −11 |
| 6 | NCGC00183195 | H | Me | -COOEt | Ph | MW: 329.4 PSA: 85 ClogP: 2.8 |
NC | 35 | 4 | 21 |
| 7 | NCGC00182501 | H | H | 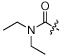 |
MW: 372.5 PSA: 88.2 ClogP: 2.5 |
−5.0 | 95 | 99 | 35 | |
| 8 | NCGC00183196 | H | H | Ph | MW: 273.3 PSA: 78.9 ClogP: 1.9 |
−5.2 | 91 | 81 | 10 | |
| 9 | NCGC00183200 | H | H | Ph | MW: 286.3 PSA: 101.8 ClogP: 1.3 |
−5.2 | 93 | 89 | 16 | |
| 10 | NCGC00183201 | H | H | 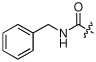 |
Ph | MW: 376.4 PSA: 87.8 ClogP: 3.5 |
−5.1 | 80 | 54 | 27 |
| 11 | NCGC00183202 | H | H | 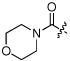 |
Ph | MW: 356.4 PSA: 88.2 ClogP: 1.4 |
−4.8 | 81 | 89 | 0 |
| 12 | NCGC00183207 | H | H | -NH2 | Ph | MW: 258.3 PSA: 84.7 ClogP: 2.1 |
−5.2 | 92 | 86 | 21 |
| 13 | NCGC00183199 | H | H | -COOH | Ph | MW: 286.3 PSA: 98.8 ClogP: 2.2 |
−5.1 | 92 | 84 | 20 |
| 14 | NCGC00182500 | H | H | -COOH | MW: 316.3 PSA: 108 ClogP: 2.1 |
−5.6 | 94 | 95 | 1 | |
| 15 | MLS000034832 | H | H | -COOEt | Ph | MW: 315.3 PSA: 85 ClogP: 2.7 |
−5.2 | 80 | 55 | −1 |
| 16 | NCGC00182344 | H | H | -COOEt | MW: 349.8 PSA: 85 ClogP: 3.3 |
−5.4 | 84 | 62 | 41 | |
| 17 | NCGC00182326 | H | H | -COOEt | 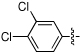 |
MW: 384.2 PSA: 85 ClogP: 3.9 |
NC | 29 | 12 | 6 |
| 18 | NCGC00182325 | H | H | -COOEt | MW: 329.4 PSA: 85 ClogP: 3 |
−3.9 | 51 | 35 | 12 | |
| 19 | NCGC00183206 | H | H | -COOEt | 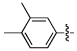 |
MW: 343.4 PSA: 85 ClogP: 3.3 |
NC | 48 | 28 | 7 |
| 20 | NCGC00182503 | H | H | -COOMe | MW: 319.3 PSA: 85 ClogP: 2.5 |
−5.3 | 86 | 64 | 29 | |
| 21 | NCGC00183204 | H | H | -COOMe | MW: 319.3 PSA: 85 ClogP: 2.5 |
NC | 28 | 20 | −5 | |
| 22 | NCGC00182343 | H | H | -COOMe | 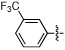 |
MW: 369.3 PSA: 85 ClogP: 3.3 |
−5.0 | 66 | 41 | ND |
| 23 | NCGC00183205 | H | H | -COOEt | MW: 387.4 PSA: 111.3 ClogP: 2.9 |
NC | 29 | 23 | 4 | |
The R1-R4 substitutions correspond to the drawing above.
Calculated physical-chemical properties, including molecular weight (MW), polar surface area (PSA) and calculated log permeability (cLogP).
IC50 values for K18PL tau ThT fluorescence inhibition was as described in Experimental Procedures. The % inhibition represents that obtained with the highest tested concentration (80µM) of compound. NC = not calculated due to insufficient activity.
Secondary testing of compounds in the K18PL tau centrifugation assay as described in Experimental Procedures.
Analysis of the compounds in the Aβ(1–42) fibrillization assay as described in Experimental Procedures. The percent inhibition represents that obtained with the highest tested concentration (80 µM) of compound. ND=not determined.
The various ATPZ analogs were evaluated in the K18PL ThT and sedimentation assays to determine their IC50 values and percent maximal inhibition (Table 2). As expected, re-synthesized MLS000034832 and MLS000062428 showed nearly comparable activity to the samples from the original screening library (compare values in Table 1 and Table 2). The results summarized in Table 2 reveal certain emerging SAR trends. For example, while N-acylations (R1; entries 2–5) as well as substitution in the C-5 position (R2; entries 5 and 6) can result in a dramatic loss of activity in the biochemical assays, structural modifications in the fragment linked at C-4 (R3; entries 7–13) appear to be generally well tolerated. Indeed, compounds bearing R3 modifications have been identified that appear to cause greater maximal inhibition in the ThT and centrifugation assays than the original hits identified during qHTS (compare entries 1 and 15 with entries 7–9, 13). Interestingly, these data reveal that the activity of the ATPZ is not dependent upon the presence of the ester moiety at C-4 (compare respectively 1 and 14, 15 and 7), thereby suggesting that the ATPZ’s do not inhibit tau fibrillization via an acylation mechanisms. Furthermore, although the correlations between biological activity and the stereo-electronic properties of phenyl moieties (R4) are not entirely clear, the nature, number and position of the substituents in the aromatic ring appeared to modulate ATPZ activity. Thus, among the mono-substituted phenyl derivatives, para-Me and para-COOEt clearly had reduced activity, whereas other para-substitutions (e.g., -OMe, -Cl, -F; entries 1, 16 and 20, respectively)) were well tolerated. In contrast, ortho- and meta -substituted analogs, as well as 3,4-disubstituted derivatives, were comparatively less potent.
The ATPZ analogs were also examined in the Aβ(1–42) fibrillization assay to determine whether the relative lack of activity observed with the representatives from the screening library (Figure 4b) was characteristic of the series as a whole. As summarized in Table 2, none of the ATPZ compounds caused >45% inhibition of Aβ(1–42) fibril formation when tested at 80 µM, and thus IC50 values were not determined. Although there was some variation in the extent of maximal inhibition of Aβ(1–42) fibrillization within the series, it appears that in general the ATPZ’s are significantly more effective at preventing the multimerization of tau than Aβ(1–42).
The observed SAR within the ATPZ series, coupled with the relative lack of activity in the MT assembly and Aβ fibrillization assays, suggests that compounds from this class are preventing tau fibril formation through a selective interaction. This is further borne out by the observation that the two active ATPZ compound from the screening deck (MLS000034832 and MLS000062428) do not show signs of promiscuity as evidenced by a general inactivity in other screens conducted at the NIH Chemical Genomics Center. Moreover, all of the ATPZ compounds in Table 2 were shown to be inactive in a β-lactamase enzymatic assay (49) that is sensitive to promiscuous molecules that tend to form aggregates (data not shown). To further confirm that the ATPZ’s act in a selective manner, the compounds in Table 2 were tested for activity in a caspase-1 assay that is known to be sensitive to non-specific oxidizing agents because of an active site cysteine that must remain in the reduced state for enzymatic activity (56). None of the ATPZ’s caused inhibition of caspase-1 activity when tested at concentrations up to 10 µM (data not shown). Thus, the ATPZ’s do not appear to be oxidizing agents and their ability to prevent tau assembly presumably relates to a specific molecular interaction.
Discussion
A body of evidence suggests that somatodendritic accumulations of hyper-phosphorylated tau lead to neurodegeneration in a variety of diseases that are broadly referred to as tauopathies (reviewed in (1)). This provides an impetus to identify disease modifying interventions that will mitigate the effects of tau aggregates in hopes of improving clinical outcome in AD and related diseases. Although there has been a vigorous effort within the pharmaceutical sector to identify small molecule drugs that will reduce the senile plaque burden in AD, there appear to be significantly fewer programs directed to the discovery of compounds that abrogate the effects of tau pathologies. To date, most tau-directed drug discovery efforts appear to be focused on the identification of inhibitors of the kinases responsible for tau hyper-phosphorylation (23;57) as enhanced phosphorylation of tau increases its propensity to form fibrils (15;16) and decrease its normal interaction with MTs (14;58;59).
An alternative strategy to reduce the consequences of pathologic tau is to prevent its assembly into oligomers and/or fibrils. Although small molecules are typically viewed as being ineffective at blocking protein-protein interactions such as would occur during tau fibril assembly, there is evidence that tau-tau interactions can indeed be inhibited by small molecules. A number of compounds have been reported to inhibit tau fibrillization (32–35;60), and although many of these do not show drug-like properties, they nonetheless provide proof that small molecules can block tau fibril assembly. Furthermore, it has been demonstrated that a single amino acid change within a region of tau that is critical for fibrillization renders it fibril-incompetent (27). Thus, further exploration of small molecule chemical space could identify drug-like compounds that prevent tau multimeric assembly, and such compounds might be further refined as potential therapeutic agents. With this goal in mind, we developed and optimized an assay that monitors the formation of tau oligomers and fibrils, utilizing a tau construct (K18PL) (32;41) that contains all four of the known MT-binding domains as well as the P301L mutation that has been described in FTLD-17 patients (5;6). This assay was utilized in qHTS of ~292,000 compounds at six concentrations to identify novel inhibitors of tau multimer assembly.
The two assay readouts that were employed in the screen, FP and ThT fluorescence, were robust and reproducible. Analysis of the qHTS results revealed that a larger number of compounds showed clear concentration-dependent inhibition of ThT fluorescence than FP. This was likely due to compounds that competed with ThT binding to tau fibrils (34) or inhibited fibril formation but not the assembly of tau into smaller FP-positive species. Of particular interest were compounds that caused a concentration-dependent inhibition of both FP and ThT fluorescence, and additional studies focused on a small subset of such compounds comprised of several chemical series, including scaffolds that had previously been described. All of the selected compounds showed repeatable activity in the ThT assay and inhibited K18PL fibril assembly as detected in a sedimentation assay. In addition, these compounds inhibited full-length tau (tau40) fibrillization, as assessed by ThT fluorescence.
Among the compounds that were confirmed to reduce tau fibrillization were members of a novel ATPZ scaffold. Further examination of active examples from this series revealed an apparent selectivity for inhibiting tau fibril formation, as they were less effective in preventing Aβ(1–42) fibirillization. Importantly, these compounds did not appreciably affect the ability of tau to promote MT assembly, in contrast to methylene blue, suggesting that in a cellular setting ATPZ’s would not negatively impact the important role that tau plays in stabilizing MTs. A number of ATPZ analogs were subsequently synthesized and tested in tau fibrillization assays to highlight possible SAR within the series. Interestingly, these studies revealed that modifications of the C-4 position of the core structure were not only generally well tolerated, but occasionally resulted in analogs with improved activity, thus indicating that further exploration of series of analogs modified in this position should be examined. In contrast, modifications of the C-5 position and/or the amino group at C-7 resulted in a dramatic loss of potency.
The nature of the tau that remained in the soluble fraction of a fibrillization reaction conducted in the presence of the prototype ATPZ, MLS000062428, was characterized by SEC analysis. As expected, the majority of the tau in the control reaction was converted to fibrils, and only a small amount of monomeric tau remained in the supernatant fraction after centrifugation. In contrast, the MLS000062428-containing reaction had a significantly greater amount of monomeric tau and some oligomeric tau species in the post-centrifugation supernatant. This suggests that the compound either stabilizes non-seeding tau oligomers that are not competent to support fibril growth, or blocks the further addition of tau monomers to oligomeric species that would normally mature into fibrils. It is also possible that the ATPZ’s bind directly to tau monomers to prevent multimerization and that the formation of oligomeric species reflects incomplete compound interaction with monomeric tau within the reaction mixture.
It is unclear whether compounds that prevent tau fibril formation with a resulting increase of monomeric and oligomeric tau would be beneficial in treating tauopathies. There is some disagreement whether tau fibrils or oligomers are toxic to neurons (20;61). For example, neurodegeneration can only be observed when bone fide tau tangles are present in tau transgenic mouse models, thereby raising questions about a potential role for tau oligomers (62). On the other hand, improvements in behavioral endpoints can be observed in tau tangle-bearing transgenic mice upon the cessation of further tau synthesis, suggesting that non-fibrillar tau species may be important (63). The nature of putative harmful tau oligomers has not been elucidated, so it is possible that only a certain type of tau oligomer might be toxic and that other oligomeric structures might be benign. Notwithstanding these unresolved issues, it is likely that tau-mediated neurodegeneration does not result solely from a gain-of-function activity attributable to oligomeric or fibrillar tau, but rather is also caused by the sequestration of tau into fibrils such that there is insufficient monomeric tau available to properly stabilize MTs. Thus, a compound such as MLS000062428 which causes a significant increase in tau monomer concentration might confer a beneficial effect in overcoming this loss-of-function toxicity. Ultimately, elucidation of the relative importance of tau gain-of-function and loss-of-function toxicities, and whether it is oligomeric or fibrillar tau that is more detrimental, will likely depend on the testing in tau transgenic mouse models of small molecule drugs that block tau assembly at different steps. Accordingly, the continued development of drug-like molecules that affect tau-tau interactions is critical to furthering our understanding of how tau aggregates lead to neuropathology in tauopathies such as AD.
Acknowledgements
We thank Sam Michael for automation assistance, Paul Shinn for compound management, Bill Leister and Jeremy Smith for analytical chemistry, and James Wells for the gift of caspase-1. We also thank our CNDR colleagues for their contributions to the work reported here. Moreover, we are indebted to our patients and their families whose commitment to research has made our work possible.
Abbreviations
- AD
Alzheimer’s disease
- ATPZ
aminothienopyridazine
- CRC
concentration-response curves
- DTT
dithiothreitol
- FP
fluorescence polarization
- FTDP-17
frontotemporal dementia with Parkinsonism linked to chromosome 17
- MT
microtubule
- qHTS
quantitative high-throughput screening
- SAR
structure-activity relationship
- SEC
size-exclusion chromatography
- ThT
thioflavine T
Footnotes
This work was supported by grants from the National Institutes of Health (P01 AG09215, P30 AG10124, P01 AG11542, P01 AG14382, P01 AG14449, P01 AG17586, PO1 AG19724, P01 NS-044233, UO1 AG24904), the Marian S. Ware Alzheimer Program, NIH Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
References
- 1.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annual Review of Neuroscience. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 2.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathological changes in nondemented elderly individuals matches the pattern in Alzheimers disease. Neurology. 1992;42:1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 3.Arriagada PV, Growdon JH, Hedleywhyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimers disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 4.Wilcock GK, Esiri MM. Plaques, tangles and dementia - A quantitative study. Journal of the Neurological Sciences. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 5.Goedert M. Tau gene mutations and their effects. Movement Disorders. 2005;20:S45–S52. doi: 10.1002/mds.20539. [DOI] [PubMed] [Google Scholar]
- 6.Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochimica et Biophysica Acta-Molecular Basis of Disease. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. Journal of Molecular Biology. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- 8.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and role of tau in microtubule assembly. Journal of Molecular Biology. 1977;116:227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 9.Cleveland DW, Connolly JA, Kalnins VI, Spiegelman BM, Kirschner MW. Physical properties and cellular localization of tau, a microtubule-associated protein which induces assembly of purified tubulin. Journal of Cell Biology. 1977;75:A283. [Google Scholar]
- 10.Higuchi M, Lee VMY, Trojanowski JQ. Tau and axonopathy in neurodegenerative disorders. Neuromolecular Medicine. 2002;2:131–150. doi: 10.1385/NMM:2:2:131. [DOI] [PubMed] [Google Scholar]
- 11.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nature Reviews Drug Discovery. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 12.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VMY. Abnormal tau phosphorylation at Ser(396) in Alzheimers disease recapitulates development and contributes to reduced microtubule-binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 13.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Molecular Biology of the Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alonso AD, GrundkeIqbal I, Iqbal K. Abnormally phosphorylated tau from Alzheimer disease brain depolymerizes microtubules. Neurobiology of Aging. 1994;15:S37. [Google Scholar]
- 15.Alonso AD, GrundkeIqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nature Medicine. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 16.Necula M, Kuret J. Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. Journal of Biological Chemistry. 2004;279:49694–49703. doi: 10.1074/jbc.M405527200. [DOI] [PubMed] [Google Scholar]
- 17.Merrick SE, Trojanowski JQ, Lee VMY. Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons (NT2N cells) Journal of Neuroscience. 1997;17:5726–5737. doi: 10.1523/JNEUROSCI.17-15-05726.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner U, Utton M, Gallo JM, Miller CCJ. Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. Journal of Cell Science. 1996;109:1537–1543. doi: 10.1242/jcs.109.6.1537. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VMY, Trojanowski JQ. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunden K, Trojanowski JQ, Lee VMY. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimers Dis. 2008;14:393–399. doi: 10.3233/jad-2008-14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trojanowski JQ, Smith AB, Huryn D, Lee VMY. Microtubule-stabilising drugs for therapy of Alzheimer's disease and other neurodegenerative disorders with axonal transport impairments. Expert Opinion on Pharmacotherapy. 2005;6:683–686. doi: 10.1517/14656566.6.5.683. [DOI] [PubMed] [Google Scholar]
- 22.Lee VMY, Trojanowski JQ. Progress from Alzheimer's tangles to pathological tau points towards more effective therapies now. J. Alzheimer's Disease. 2006;9:257–262. doi: 10.3233/jad-2006-9s328. [DOI] [PubMed] [Google Scholar]
- 23.Bhat RV, Haeberlein SLB, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. Journal of Neurochemistry. 2004;89:1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- 24.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. Journal of Clinical Investigation. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo WJ, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9511–9516. doi: 10.1073/pnas.0701055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nature Reviews Neuroscience. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 27.Li WK, Lee VMY. Characterization of two VQIXXK motifs for tau fibrillization in vitro. Biochemistry. 2006;45:15692–15701. doi: 10.1021/bi061422+. [DOI] [PubMed] [Google Scholar]
- 28.Chirita CN, Necula M, Kuret J. Anionic micelles and vesicles induce tau fibrillization in vitro. Journal of Biological Chemistry. 2003;278:25644–25650. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- 29.Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides - In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. American Journal of Pathology. 1997;150:2181–2195. [PMC free article] [PubMed] [Google Scholar]
- 30.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 31.Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 32.Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VMY. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochemical and Biophysical Research Communications. 2007;358:1–6. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang YP, Hascher A, Mandelkow EM, Biernat J, Mandelkow E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. Journal of Biological Chemistry. 2005;280:3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- 34.Honson NS, Johnson RL, Huang WW, Inglese J, Austin CP, Kuret J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiology of Disease. 2007;28:251–260. doi: 10.1016/j.nbd.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, Schmidt B, Mandelkow E. Phenylthiazolyl-hydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry. 2007;46:10016–10023. doi: 10.1021/bi700878g. [DOI] [PubMed] [Google Scholar]
- 36.Wischik CM, Edwards PC, Lai RYK, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. Journal of Biological Chemistry. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 38.Harrington CR, Ricard JE, Horsley D, Harrington KA, Hindley KP, Riedel G, Theuring F, Seng KM, Wischik CM. Methylthioninium chloride (MTC) acts as a tau aggregation inhibitor in a cellular model and reverses tau pathology in transgenic mice models of Alzheimer's disease. International Conference on Alzheimer's Disease Abstracts. 2008 [Google Scholar]
- 39.Lipinski C. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 40.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing 4 tandem repeats - differential expression of tau protein messenger-RNAs in human brain. Embo Journal. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- 42.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VMY. Mutation-specific functional impairments in distinct Tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 43.Luk KC, Hyde EG, Trojanowski JQ, Lee VMY. Sensitive fluorescence polarization technique for rapid screening of (X-Synuclein Oligomerization/Fibrillization inhibitors. Biochemistry. 2007;46:12522–12529. doi: 10.1021/bi701128c. [DOI] [PubMed] [Google Scholar]
- 44.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michael S, Auld D, Klumpp C, Jadhav A, Zheng W, Thorne N, Austin CP, Inglese J, Simeonov A. A robotic platform for quantitative high-throughput screening. Assay Drug Dev Technol. 2008;6:637–658. doi: 10.1089/adt.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beauchamp GK, Keast RSJ, Morel D, Lin JM, Pika J, Han Q, Lee CH, Smith AB, Breslin PAS. Phytochemistry - Ibuprofen-like activity in extra-virgin olive oil. Nature. 2005;437:45–46. doi: 10.1038/437045a. [DOI] [PubMed] [Google Scholar]
- 47.Crystal AS, Giasson BI, Crowe A, Kung MP, Zhuang ZP, Trojanowski JQ, Lee VMY. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. Journal of Neurochemistry. 2003;86:1359–1368. doi: 10.1046/j.1471-4159.2003.01949.x. [DOI] [PubMed] [Google Scholar]
- 48.Scheer JM, Wells JA, Romanowski MJ. Malonate-assisted purification of human caspases. Protein Expression and Purification. 2005;41:148–153. doi: 10.1016/j.pep.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 49.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nature Protocols. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferguson GN, Valant C, Horne J, Figler H, Flynn BL, Linden J, Chalmers DK, Sexton PM, Christopoulos A, Scammells PJ. 2-aminothienopyridazines as novel adenosine A(1) receptor allosteric modulators and antagonists. Journal of Medicinal Chemistry. 2008;51:6165–6172. doi: 10.1021/jm800557d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- 52.Johnston PA, Soares KM, Shinde SN, Foster CA, Shun TY, Takyi HK, Wipf P, Lazo JS. Development of a 384-well colorimetric assay to quantify hydrogen peroxide generated by the redox cycling of compounds in the presence of reducing agents. Assay and Drug Development Technologies. 2008;6:505–518. doi: 10.1089/adt.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris AM, Watzky MA, Agar JN, Finke RG. Fitting neurological protein aggregation kinetic data via a 2-step, Minimal/"Ockham's Razor" model: The Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry. 2008;47:2413–2427. doi: 10.1021/bi701899y. [DOI] [PubMed] [Google Scholar]
- 54.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 55.Hardy J, Selkoe DJ. Medicine - The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 56.Smith GK, Barrett DG, Blackburn K, Cory M, Dallas WS, Davis R, Hassler D, McConnell R, Moyer M, Weaver K. Expression, preparation, and high-throughput screening of caspase-8: Discovery of redox-based and steroid diacid inhibition. Archives of Biochemistry and Biophysics. 2002;399:195–205. doi: 10.1006/abbi.2002.2757. [DOI] [PubMed] [Google Scholar]
- 57.Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends in Pharmacological Sciences. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VMY. Abnormal tau phosphorylation at Ser(396) in Alzheimers disease recapitulates development and contributes to reduced microtubule-binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 59.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Molecular Biology of the Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow EM, Mandelkow E, Waldmann H. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angewandte Chemie-International Edition. 2007;46:9215–9219. doi: 10.1002/anie.200704051. [DOI] [PubMed] [Google Scholar]
- 61.Ferreira ST, Vieira MNN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. Iubmb Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- 62.Lee VMY, Kenyon TK, Trojanowski JQ. Transgenic animal models of tauopathies. Biochimica et Biophysica Acta-Molecular Basis of Disease. 2005;1739:251–259. doi: 10.1016/j.bbadis.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 63.SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]