

Abstract
Human P450 2A6 displays a small active site that is well adapted for the oxidation of small planar substrates. Mutagenesis of CYP2A6 resulted in an increased catalytic efficiency for indole biotransformation to pigments and conferred a capacity to oxidize substituted indoles (Wu, Z.-L., Podust, L.M. and Guengerich, F.P. (2005) J.Biol.Chem. 49, 41090-41100). Here, we describe the structural basis that underlies the altered metabolic profile of three mutant enzymes, P450 2A6 N297Q, L240C/N297Q and N297Q/I300V. The Asn297 substitution abolishes a potential hydrogen bonding interaction with substrates in the active site, and replaces a structural water molecule between the helix B′-C region and helix I while maintaining structural hydrogen bonding interactions. The structures of the P450 2A6 N297Q/L240C and N297Q/I300V mutants provide clues as to how the protein can adapt to fit the larger substituted indoles in the active site, and enable a comparison with other P450 family 2 enzymes for which the residue at the equivalent position was seen to function in isozyme specificity, structural integrity and protein flexibility.
Keywords: P450 2A6, CYP2A6, monooxygenase, X-ray structure, enzyme specificity, benzyloxyindole, indigo, indirubin
Introduction
Cytochrome P450 (CYP) enzymes are extremely versatile catalysts of diverse oxygenation reactions [1]. Despite the wide range of structurally dissimilar compounds a given P450 can metabolize, they exhibit remarkable stereo- and regio-specificity. Significant research efforts have been directed toward elucidating the function of individual P450s in biosynthesis and metabolism of drugs and other xenobiotics. Mutagenesis studies have often been designed to identify residues critical for function and specificity or to increase the stability and activity of specific P450s. In contrast, the use of modified P450s in biocatalysis and fine chemical synthesis for drug discovery and the generation of commercial products is still in its infancy.
The xenobiotic metabolizing enzyme P450 2A6 is the principal human enzyme responsible for nicotine detoxication and coumarin 7-hydroxylation. The latter biotransformation is considered an isoform specific marker reaction for P450 2A6 in profiling new drug candidate interactions with hepatic P450s [2]. P450 2A6 is expressed in the liver and participates extensively in the metabolism of clinically prescribed medications such as fadrozole and losigamone [3]. In addition to drug metabolism, P450 2A6 also plays a role in the activation of some prodrugs such as tegafur [4], and in the activation of tobacco-specific carcinogens to mutagenic products [5, 6]. Like other human drug metabolizing P450s, there are a number of genetic polymorphisms that alter metabolic activity and/or the expression of P450 2A6. The P450 2A6*5 allele, for instance, encodes an unstable enzyme that lacks coumarin 7-hydroxylase activity, and displays a G479L substitution [7]. A recent study in the Japanese population demonstrated that individuals that had at least one defective CYP2A6 allele, had a significant reduction in tobacco-related lung cancer which was most pronounced in individuals who were homozygous for the P450 2A6*4 deletion allele [8].
Structures of human P450 2A6 in complex with coumarin, methoxsalen [9] and synthetic inhibitors [10] have been solved recently by our group. The P450 2A6 structure exhibits a compact, hydrophobic substrate binding site, with one hydrogen bond donor, Asn297. The identities of the residues that contact the ligand molecules are identical in the different P450 2A6 complex structures, and changes in the contacting amino acids are generally restricted to slight rearrangements of phenylalanine 107 to maximize orthogonal aromatic interactions. Nevertheless, the P450 2A6 4,4′-dipyridyl disulfide complex shows a more drastic repositioning of Phe209 [10]. The structure of the active site cavity demonstrates that P450 2A6 is well adapted for the oxidation of small, planar substrates such as coumarin and indole.
In a cumulative approach using different mutagenesis methods (site-directed, staggered extension process and error-prone PCR) Guengerich and co-workers found several mutants of CYP2A6 with higher affinities towards indole and indole derivatives that produced a variety of biologically active compounds [11, 12]. A first generation of mutants yielded P450 2A6 N297Q, N297H and L240C/N297Q mutants that displayed an enhanced rate of oxidation of indole to pigments such as indigo and indirubin, and a broader capacity to oxidize indole derivatives [11]. Further mutagenesis, aimed to confer oxidation of larger substituted indoles to the enzyme, which are not substrates for the wild-type or the P450 2A6 L240C/N297Q double mutant, led to second generation mutants able to oxidize both 4- and 5-benzyloxyindoles (BOIs). Two substitutions, N297Q and I300V, were identified as critical to achieve the extended specificity [12]. The various substituted indigoids produced by these P450 2A6 mutants are of interest for the production of dyes, and also have potential as drugs, because they show enhanced activities as potent inhibitors of at least two kinases, a cyclin-dependent kinase (CDK5/p25) [13] and glycogen synthase kinase 3 (GSK3) [14].
Here, we describe the structures of the P450 2A6 N297Q, L240C/N297Q and N297Q/I300V mutants, determined to a resolution of 1.95, 2.50 and 2.80 Å, respectively. The mutations induce only minor conformational changes, which underlie the altered metabolic profile of these mutant enzymes. The N297Q substitution alters substrate orienting and binding interactions in the active site, and replaces a conserved, structural water molecule between the helix B′-C region and helix I while maintaining hydrogen bonding interactions that stabilize the conformation of the helix B′-C loop. The volume of the substrate binding cavity is slightly extended in the P450 2A6 N297Q/I300V and provides clues as to how the protein can adapt to fit the larger substituted indoles in the active site. The structural information contributes significantly to our understanding of the altered or newly acquired specificities displayed by the P450 2A6 mutants. Additionally, the structures of the P450 2A6 N297Q mutants enable a comparison with other P450 family 2 enzymes for which the residue at the equivalent position was seen to be important for isozyme specificity, structural integrity as well as protein flexibility.
Material and Methods
Protein production and purification
The parent pCW2A6dH expression vector used to produce protein for the crystallization of wild-type P450 2A6 was described previously [9]. The pCW2A6 vector was designed to yield a modified, conditionally soluble enzyme that retained catalytic activity, using approaches employed previously for the crystallization of mammalian microsomal P450s 2C5 [15], 2C8 [16], 2C9 [17, 18], 2B4 [19], 2D6 [20] and 3A4 [21, 22]. Plasmid templates containing CYP2A6 cDNAs for the three mutants were kindly provided by F. Peter Guengerich, Vanderbilt University, School of Medicine, and have been reported [11, 12]. In order to construct the expression plasmids used to produce protein for crystallization, restriction fragments were isolated from CYP2A6 N297Q (SphI-HindIII), CYP2A6 L240C/N297Q (BsrGI-SalI) and CYP2A6 N297Q/I300V (SphI-HindIII) plasmids and ligated into the corresponding restriction sites of the 2A6dH pCW expression vector. The resulting expression constructs were sequenced to confirm the presence of the mutation(s) and the truncated N-terminus. The modified enzymes, designated CYP2A6dH N297Q, CYP2A6dH L240C/N297Q and CYP2A6dH N297Q/I300V, were expressed in Escherichia coli and purified with slight modifications of the procedure described previously [9]. Protein preparations were stored in a 50 mM potassium phosphate (KPi) buffer (pH 7.4) containing 500 mM NaCl, 20% glycerol, and 1 mM EDTA for protein crystallization.
Estimation of binding affinity of CYP2A6dH and mutants for indole
Dissociation constants for the binding of indole to purified CYP2A6dH and CYP2A6dH mutants were determined by titration [23]. The conversion of P450 2A6dH and P450 2A6dH mutants from a low spin to a high spin ferric heme protein upon the addition of indole was monitored by visible difference spectroscopy. Purified proteins were diluted to concentrations of 1 to 1.5 μM using the storage buffer without glycerol. Spectra from 260 nm to 800 nm were recorded at ambient temperature using a CARY 1E UV-visible spectrophotometer, following each addition of indole dissolved in ethanol. The total concentration of ethanol remained under 1.2%. The absorbance changes at the peak (∼384 nm) and trough (∼418 nm) of computed difference spectra were used to estimate a spectral dissociation constant and maximum spectral change, using the program SlideWrite Plus, in which the data were fit to a one-site binding hyperbolic equation using non-linear least-squares regression.
Protein crystallization and structure determination
All crystals were grown by sitting drop vapor diffusion in 2.5 μl drops equilibrated against 700 μl of well solution at 24°C. The P450 2A6 N297Q mutant yielded crystals when the protein solution containing 312 μM CYP2A6dH N297Q, 0.58 mM ellipticine and 2% ANAPOE®-35 was combined with an equal volume of well solution composed of 30% polyethylene glycol (PEG) 3350, 100 mM Tris pH 8.5 and 200 mM ammonium sulfate. The P450 2A6 L240C/N297Q data set was collected from a crystal prepared by mixing a protein solution containing 620 μM CYP2A6dH L240C/N297Q, 7.20 mM indole and 2% ANAPOE®-35 with an equal volume of well solution. The well solution contained 30% PEG 3350, 100 mM Tris pH 8.5 and 200 mM ammonium sulfate. Crystals of P450 2A6 N297Q/I300V were grown by combining equal volumes of protein solution containing 275 μM CYP2A6dH N297Q/I300V, 0.58 mM ellipticine and 2 % ANAPOE®-35 with the well solution composed of 35% PEG MEM 5000 and 200 mM ammonium sulfate in a 100 mM Tris pH 8.5 buffer solution. All substrates and detergents were purchased from Sigma (St. Louis, MO) and Anatrace (Maumee, OH), respectively.
X-ray diffraction data were collected on single crystals cooled to 100 K at the Stanford Synchrotron Radiation Laboratory (SSRL, Palo Alto, California) on beamline 11-1 for the P450 2A6 N297Q structure, beamline 5-1 for the P450 2A6 L240C/N297Q structure and beamline 9-1 for the P450 2A6 N297Q/I300V structure. Prior to harvesting, 2 μl cryoprotectant solution was added to the sitting drop. Crystals were harvested and then flash-cooled to 100 K in liquid N2. The cryoprotectant solution was composed of well solution, storage buffer, ethylene glycol and water, in a 5:1:5:9 ratio. Diffraction data were processed with HKL2000 and Scalepack [24]. Data collection and processing statistics for the three datasets are provided in Table 1. Crystals of the three different P450 2A6 mutants belonged to space group P21, with unit cell parameters that are highly isomorphous with the reported structure of wild-type P450 2A6 in complex with coumarin [9]. Hence, initial phasing information for the structures of the three different P450 2A6 mutants could be obtained by isomorphous replacement, using the P450 2A6 coumarin complex structure (PDB:1Z10) as a template, after omitting water molecules, glycerol and coumarin. Models for the P450 2A6 N297Q and P450 2A6 L240C/N297Q structures were refined against 1.95 Å and 2.50 Å resolution data respectively, using multiple rounds of conjugant gradient least-squares minimization, torsion angle simulated annealing and isotropic individual B-factor refinement using the program CNS [25]. Refinement of the P450 2A6 N297Q/I300V structure followed a similar approach, with the exception that the moderate limiting resolution of the data (2.8 Å) only allowed for isotropic grouped B-factor refinement. Water molecules were added during the final stages of refinement. Alternative cycles of editing and adjustment of the models into σa-weighted 2|Fo|-|Fc|, |Fo|-|Fc| and 2|Fo|-|Fc| composite omit maps were performed using the graphics programs O [26] and Coot [27].
Table 1.
Data collection and refinement statistics.
| Human P450 2A6 construct | N297Q | L240C/N297Q | N297Q/I300V |
|---|---|---|---|
| PDB code | 2PG5 | 2PG6 | 2PG7 |
| Data collection | |||
| Space group | P21 | P21 | P21 |
| Used wavelength (Å) | 0.98 | 0.98 | 0.98 |
| Resolution limit (Å)1 | 1.95 (2.02 – 1.95) | 2.50 (2.59 – 2.50) | 2.80 (2.87 – 2.80) |
| Cell parameters | |||
| a, b, c (Å) | 70.85, 157.97, 103.74 | 70.70, 158.25, 103.76 | 70.89, 159.39, 104.10 |
| β(°) | 92.23 | 92.09 | 91.92 |
| X-ray source | BL11-1 SSRL | BL 1-5 SSRL | BL 9-1 SSRL |
| Total observations1 | 573507 | 221269 | 268802 |
| Unique reflections1 | 163342 (15732) | 70956 (6642) | 56574 (4147) |
| Completeness of all data (%)1 | 99.2 (95.5) | 94.0 (88.3) | 99.6 (99.4) |
| Mean I /σ1 | 28.1 (2.5) | 16.8 (3.4) | 4.5 (1.2) |
| Rsym –value (%)1,2 | 8.9 (38.3) | 9.6 (23.8) | 13.0 (63.0) |
| Refinement | |||
| Resolution range (Å) | 35.0 – 1.95 | 40.0 – 2.53 | 40.0 – 2.80 |
| Reflections used | 163265 | 70898 | 56547 |
| Reflections used in Rfree set | 8012 | 3604 | 2827 |
| Rcryst/ Rfree3 | 0.215/0.239 | 0.209/0.261 | 0.235/0.289 |
| Protein atoms | 15014 | 15019 | 15014 |
| Solvent atoms | 648 | 309 | 91 |
| Heme atoms | 172 | 172 | 172 |
| Ethylene glycol atoms | 16 | 0 | 0 |
| R.m.s. deviation4 | |||
| Bond angles (°) | 1.3 | 1.3 | 1.4 |
| Bond lengths (Å) | 0.007 | 0.008 | 0.009 |
Values in parentheses indicate data in the highest resolution shell.
Rsym = ΣhΣj | <I(h)> - I(h)j| / ΣhΣj<I(h)>, where I(h) is the intensity of an individual reflection and <I(h)> is the mean intensity of that reflection.
Rcryst or Rfree = Σ| |Fo| - |Fc| | / Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
R.m.s. deviations relate to the Engh and Huber parameters [40].
The final model of P450 2A6 N297Q exhibits an R value of 0.22 and an Rfree value of 0.24 with four molecules in the asymmetric unit. The model comprises residues 31-494, 32-495, 32-494, and 31-494 of chains A-D respectively, 648 water molecules and 4 ethylene glycol molecules. An ethylene glycol molecule was defined by 2|Fo|-|Fc| σa weighted electron density maps and found in the active site, above the distal surface of the heme prosthetic group of each molecule in the asymmetric unit. The structure of P450 2A6 L240C/N297Q could be refined to an R value of 0.21 and an Rfree value of 0.26 and includes residues 30-494, 32-495, 31-494, and 31-494 of chains A-D respectively, and 309 water molecules. Refinement of the P450 2A6 N297Q/I300V structure resulted in values of 0.22 and 0.29 for R and Rfree, respectively, and comprises residues 31-494, 32-495, 31-494 and 31-494 of chains A-D respectively, and 91 water molecules. Residual electron density is apparent in the active sites of each protein molecule in the asymmetric unit for both the P450 2A6 L240C/N297Q and the P450 2A6 N297Q/I300V structure. Nevertheless, the presence of ethylene glycol or water molecules was not clearly defined, and hence, no solvent molecules were modelled. The final P450 2A6 mutant models exhibit good stereochemical parameters, with all but one residue in the allowed regions of the Ramachandran plot. Similar to the geometry of the reported P450 2A6 wild-type structures [9, 10], Phe42 of each molecule is situated in a disfavoured region in the Ramachandran plot. Nevertheless, the position of Phe42 is unambigously defined in the electron density maps. Further refinement statistics for the three structures are provided in Table 1.
Cavity Volume Calculations
Structures of the four molecules (A-D) in the asymmetric unit are highly similar, with small differences uniquely located at surface regions connecting secondary structure elements such as the linking regions between helices G′ and G, helices H and I, and helices K and K′. Comparisons of P450 2A6 wild-type and mutant structures are based on molecule A of the asymmetric unit. The wild-type P450 2A6 structure in complex with coumarin (PDB:1Z10) [9] provides the reference structure. In that structure, Leu370 located in the active site is seemingly not confined by substrate binding and is found in two alternative conformations (A and B) in the A molecule. As the position of Leu370 in the three P450 2A6 mutant structures corresponds to conformation A, conformation A of Leu370 was used for active site cavity volume calculations. Volumes of the substrate binding cavities were calculated using a 1.4 Å probe and the probe-occupied volume routine using the program VOIDOO [28].
Ligand Docking
Both 4- and 5-benzyloxyindole (BOI) were manually docked into the determined X-ray structure of the P450 2A6 N297Q/I300V mutant. Initial positions of the substrates in the active site cavity are based on the P450 2A6 coumarin complex and the sites of oxidation inferred from the product profiles [12]. Subsequent energy minimization of the P450 2A6 N297Q/I300V 4- and 5-BOI complexes employed CNS [25] using the model minimization routine. An iron-oxo heme model was used in the minimization of the docked P450 2A6 N297Q/I300V 4- and 5-BOI complexes in order to better mimic the conditions for catalysis. An iron oxygen distance of 1.65 Å was modeled based on quantum mechanical calculations on the heme Compound I structure, the active species of P450 enzymes [29]. Furthermore, a slightly lowered sigma value (2.6 Å) for the non-bonded repulsive energy-term of the oxygen was used, to allow a proper approach of the ligands to the iron-oxo heme structure. The geometry of the minimized structures was analyzed using Procheck.
Results
Substrate binding affinities of truncated enzymes
The binding affinities of P450 2A6dH wild-type and the three mutant enzymes for indole were determined by monitoring the conversion of the enzyme from the low spin to high spin state spectroscopically (Table 2). P450 2A6 N297Q shows the tightest binding (Kd = 14.4 ± 1.1 μM), and the two P450 2A6 double mutants display a slightly decreased binding affinity for indole. The wild-type enzyme exhibited a 3- to 6-fold increase in the apparent Kd for indole binding when compared to the Kd values of the three P450 2A6 mutant enzymes. The estimated Kd-value for the P450 2A6 L240C/N297Q mutant is similar to the value reported earlier for the full-length L240C/N297Q mutant [12]. In general, the modifications to the termini of CYP2A6dH mutants facilitated the crystallization without significantly altering indole binding.
Table 2.
Binding affinities of wild-type P450 2A6 and mutants for indole.
| P450 2A6 construct | Kd (μM)1 | Δε max1 (mM-1cm-1) |
|---|---|---|
| Wild-type P450 2A6 | 81.2 ± 13.7 | 54.8 ± 0.6 |
| P450 2A6 N297Q | 14.4 ± 1.1 | 58.3 ± 2.8 |
| P450 2A6 L240C/N297Q | 22.8 ± 3.6 | 59.0 ± 5.4 |
| P450 2A6 N297Q/I300V | 17.7 ± 1.8 | 52.7 ± 2.3 |
Mean and standard errors are shown for at least three titrations for each enzyme
Structure of P450 2A6 N297Q
The tertiary structures of P450 2A6 wild-type and the P450 2A6 N297Q mutant are highly homologous, with a root mean squared (rms) deviation of only 0.15 Å for all equivalent Cα atoms. Furthermore, with the exception of residue 297, the side chain position of the residues that constitute the substrate binding cavity are very similar, indicating that there are essentially no conformational changes between both structures. The P450 2A6 N297Q protein was crystallized in the presence of ellipticine. Ellipticine is reported to be a competitive inhibitor of human P450 2A6 with an estimated Ki of 7 μM [30]. The absence of ellipticine in the P450 2A6 N297Q structure most probably results from the limited solubility of ellipticine, which is not sufficient for complex formation under the high protein concentration conditions used for crystallization. In contrast, the presence of one ethylene glycol molecule originating from the cryoprotectant solution, and one water molecule in the P450 2A6 N297Q active site cavity was well defined by σa-weighted 2|Fo|-|Fc| composite omit electron density maps calculated for a model that did not include these solvent molecules (Fig. 1A). The ethylene glycol atoms as well as the water molecule that is coordinated to the heme iron reside in one plane, that is nearly co-planar with the position of coumarin in the wild-type P450 2A6 coumarin complex structure. The Gln297 side chain extends in the same orientation as the wild-type P450 2A6 Asn297 side chain. The longer side chain of Gln297 resides in the space in a turn of the helix B′-C loop that is normally occupied by a water molecule in the wild-type structure. The Gln297 Oε atom maintains a hydrogen bond donated by the main chain N atom of Val117 that is observed for Asn297 Oδ in the wild-type protein. Additionally, the Gln297 Nε atom forms a hydrogen bond with the carbonyl O atom of Tyr114, which is hydrogen bonded to the water molecule in the wild-type structure (Fig. 1B). Nevertheless, the hydrogen bonding interaction between the backbone N atom of Phe117 and the water molecule is lost in the N297Q mutant. In its position, the Gln297 Nε atom is buried too deeply to provide a hydrogen-bonding interaction with substrates such as coumarin, bound in the active site cavity. Mutation of asparagine to glutamine at position 297 hardly changes the substrate binding cavity volume, from ∼250 Å3 as observed in the P450 2A6 coumarin complex [9] to ∼265 Å3.
Figure 1.

A. Detailed view of the P450 2A6 N297Q active site cavity. The position of the heme prosthetic group, ethylene glycol and a water molecule are defined by σa-weighted 2|Fo|-|Fc| electron density maps contoured at 1σ (blue mesh). The amino acid residues constituting the active site cavity are depicted in sticks, with portions of the protein backbone represented as a cartoon. Ethylene glycol and the heme prosthetic group are shown in sticks, with carbon atoms colored yellow and pink, respectively. Oxygen, nitrogen and iron are colored red, blue and orange, and a water molecule is represented as a red sphere. B. Comparison of the interaction between the helix I residue at position 297 and the B′-C region for P450 2A6 wild-type (left panel) and the P450 2A6 N297Q mutant. Residues constituting the B′-C region are shown in sticks. For clarity, only the backbone atoms of Val116 and Phe118 are depicted. Atom color code is the same as in panel A, with sulfur colored yellow. Dashed lines indicate hydrogen bonds. C. Detailed view on the L240C mutation in the structure of the P450 2A6 L240C/N297Q mutant (marine), overlayed with the same portions of the P450 2A6 wild-type structure (white). The L240C mutation does evoke a conformational change of the Ile208 side chain on helix F and a slight repositioning of the Phe107, Phe111, Phe118, Phe209 and Phe480 side chains that line the roof of the active site cavity is also evident. D. Comparison between the active site cavity volume (blue mesh) of the P450 2A6 wild-type structure (white, left panel) and the P450 2A6 N297Q/I300V mutant crystal structure (green, right panel). The amino acid residues constituting the active site cavity are depicted in sticks, with portions of the protein backbone represented as a cartoon. For clarity, part of the B′-C region with Phe118 is not displayed. The active site cavity is extended in the P450 2A6 N297Q/I300V mutant structure to include residues Ile208, Leu241, Leu244 and Leu296 at the roof of the cavity. Heme color code is the same as in panel A. E. Overlays of the crystal structure of the P450 2A6 N297Q/I300V mutant (green sticks) and the P450 2A6 N297Q/I300V energy-minimized structures (beige sticks) in which 4-BOI and 5-BOI were docked in positions compatible with 2-(left panel), 7- (middle panel) and 2-oxidation (right panel), respectively. The iron-oxo heme model used in the minimization is depicted as a dumbbell. Substrate contacting residues within 4 Å of 4-BOI or 5-BOI are displayed.
Structure of P450 2A6 L240C/N297Q
P450 2A6 mutants obtained from random mutagenesis of substrate recognition subsites (SRS) 3 and 4 [31] displayed an altered catalytic efficiency for indole oxidation [11]. Additionally, the P450 2A6 L240C/N297Q showed a significantly broader capacity to utilize indoles bearing one or two small substituents such as halogens, methyl or methoxy groups among others [12]. Curiously, the P450 2A6 wild-type structure [9] indicates a compact active site cavity in which L240C does not directly contribute. The latter structure superimposes very well on the P450 2A6 L240C/N297Q mutant structure, with an rms deviation of 0.20 Å for the equivalent Cα atoms. Not surprisingly, in the P450 2A6 L240C/N297Q structure, Cys240 is not part of the substrate binding cavity residues, and is found on the surface of helix G. Whereas the cysteine for leucine mutation at position 240 does not introduce changes in the protein backbone, the L240C mutation does evoke a conformational change of the Ile208 side chain on helix F (Fig. 1C) reflecting the additional space available with the smaller cysteine side chain. A slight repositioning of the Phe107, Phe111 and Phe118 side chains that line the roof of the active site cavity is also evident (Fig. 1C). Gln297 is found in exactly the same position and is involved in identical contacts as described for the P450 2A6 N297Q mutant structure. The volume (∼245 Å3) of the substrate binding cavity of the P450 2A6 L240C/N297Q structure is nearly unaffected by both mutations. The apparent binding affinity (Kd) of P450 2A6 L240C/N297Q for indole was estimated to be 0.022 mM (Table 2). Nevertheless, the addition of indole to the crystallization setup did not result in the presence of indole in the P450 2A6 L240C/N297Q crystal.
Structure of P450 2A6 N297Q/I300V
The N297Q and I300V mutations were seen to be critical in gaining activity for the hydroxylation of bulky, substituted indoles such as 4- and 5-BOI, by human P450 2A6 [12]. The active site cavity volume in the P450 2A6 N297Q/I300V mutant is indeed increased to ∼310 Å3. The tertiary structures of the P450 2A6 wild-type [9] and the P450 2A6 N297Q/I300V mutant are still very similar, with an rms deviation of 0.30 Å for all equivalent Cα atoms. The contacts and position of Gln297 are the same as described above for the P450 2A6 N297Q mutant. The I300V mutation results in an extension of the distal end of the active site cavity by opening the space between Phe111 and the side chain of residue 300, reflecting the absence of the Cδ atom of Ile300. The shortest spanning distance between helix B′ and helix I at the roof of the cavity is lengthened from 4.43 Å (distance between Phe111 Cζ and Ile300 Cδ) to 6.06 Å (Phe111 Cζ and Val300 Cγ). Concomitantly, the side chains of Phe107 and Phe118 are repositioned slightly. Similar to the P450 2A6 N297Q mutant, crystallization of the P450 2A6 N297Q/I300V protein in the presence of ellipticine did not result in complex formation.
Substrate docking and minimization of P450 2A6 N297Q/I300V
Structures of P450 2A6 in complex with coumarin, methoxsalen and several inhibitors [9, 10] indicate that the positions of the ligands are constrained to be co-planar by the narrow P450 2A6 substrate binding cavity. Detailed characterization of products and intermediates of 4- and 5-BOI oxidation by the P450 2A6 N297Q/I300V mutant has been reported [12]. Structures and numbering of 4- and 5-BOI are illustrated in Figure 2. Oxidation of 5-BOI mainly resulted in the production of 5,5′-BOI-indirubin (Fig. 2), indicating indole oxidation in positions 2 or 3. In contrast, 4-BOI oxidation by the P450 2A6 N297Q/I300V mutant primarily resulted in a non-indigo blue pigment in which a pyrrole ring and a benzene ring are connected (Fig. 2), demonstrating oxidation of both the pyrrole and benzene moieties of indole [12]. Therefore, starting positions for docking of 5-BOI were compatible with indole 2 or 3 hydroxylation, and 4-BOI was initially modelled according to indole hydroxylation at positions 2 or 7. In all cases the starting positions of the substituted indoles in the P450 2A6 N297Q/I300V active site, following the planarity revealed by the P450 2A6 complex structures, could be achieved without steric clashes.
Figure 2.
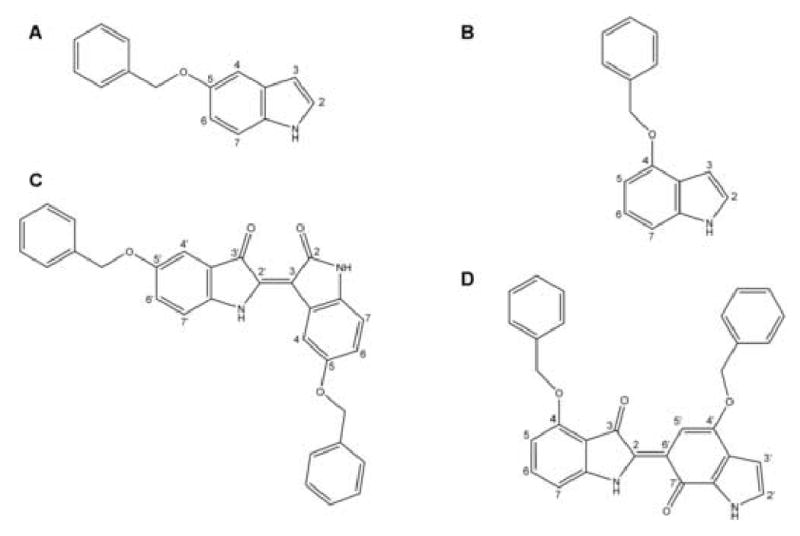
Structure and numbering of 5-Benzyloxyindole (A), 4-Benzyloxyindole (B) and two examples of pigments derived from them [12], 5,5′-BOI-indirubin (C) and 4-Benzyloxy-2-(4′-Benzyloxy-1′,7′-dihydro-7′-oxo-6′H-indol-6′-ylidene)indolin-3-one (D).
Minimization of non-bonded interaction energies of P450 2A6 N297Q/I300V with 4- or 5-BOI docked in the active site does not interfere with the good geometry displayed by the crystal structure, and yields reasonable protein-ligand interaction distances. Overall the conformational changes are minor and restricted to contacting residues that change side chain dihedral angles in order to accommodate the ligand molecule, without introducing further rearrangements in regions that are not part of the active site cavity. In the P450 2A6 coumarin complex structure, 8 active site residues are positioned within a 4 Å radius around the substrate [9]. The number of contacting residues increases to 10 and 11 in energy-minimized structures of the P450 2A6 4-BOI and P450 2A6 5-BOI complexes, respectively, to include Ile208 and Leu296 (4-BOI), and Leu244 (5-BOI). The substrates in the minimized complex structures reside in positions compatible with regio-specific oxidation. The closest distances to the iron-oxo heme structure are in the range 3.4 to 3.7 Å. The minimal conformational perturbations in the minimized structures of the P450 2A6 N297Q/I300V 5-BOI complexes result in a significant extension of the substrate binding cavity along helix I. The distance between Phe111 Cζ and Val300 Cγ is lengthened by only 0.57 Å to 6.63 Å to accommodate 5-BOI in both docked positions. These conformational changes increase the active site cavity volume to ∼380 Å3 by expanding the hydrophobic roof of the active site cavity to also include Ile208, Leu244 and Leu296 (Fig. 1E). The structure of 4-BOI is slightly more condensed than the 5-BOI structure. The longest intramolecular distance in 4-BOI is 8.84 Å versus 10.52 Å in 5-BOI. Therefore, the conformational changes to the P450 2A6 N297Q/I300V structure required to fit 4-BOI in the two docked positions, are highly similar but somewhat less pronounced than the ones described for 5-BOI. The Phe111 Cζ and Val300 Cγ distance is 6.49 Å, and the substrate binding cavity volume is extended to ∼370 Å3 The more compact shape and structure of 4-BOI allows the indole to bind in a position compatible with oxidation of the benzene ring at position 7, an orientation that can not be achieved for 5-BOI (Fig. 1E).
Discussion
We have determined crystal structures of the human P450 2A6 N297Q, L240C/N297Q and N297Q/I300V mutants. These three P450 2A6 mutants originated from biased random mutagenesis libraries and were selected on the basis of differences in their abilities to produce colored pigments from indole and substituted indoles [11, 12]. The structures of the three P450 2A6 mutants display a compact, hydrophobic active site cavity, and superimpose very well on structures of wild-type human P450 2A6 in complex with substrate and inhibitors [9, 10]. In these complex structures, only two polar residues were observed in the active site, Asn297 and Thr305. Nδ from the Asn297 side chain can serve as hydrogen bond donor to polar substrates such as coumarin or methoxsalen [9]. The Oδ atom of the Asn297 side chain is positioned to accept hydrogen bonds from the backbone N atom of Val117 and a conserved water molecule normally found stabilizing the turn following helix B′ (Fig. 1B). The water molecule can accept a hydrogen bond from the peptide N of Phe118 and donate a hydrogen bond to the backbone carbonyl of Tyr114. The longer side chain of glutamine displaces the water molecule in the structures of the three mutants. The Nε atom of Gln297 is positioned to donate a hydrogen bond to the carbonyl oxygen of Tyr114, and Oε of Gln297 can accept a hydrogen bond from the main chain N atom of Val117. Thus, the displacement of the water molecule results in the loss of a hydrogen bond acceptor for the amide nitrogen of Phe118. In contrast to the hypothesis that an N297Q mutation might introduce a new hydrogen bond with an O atom in substrates such as 4- and 5-BOI, raised by Wu et al [12], the Gln297 side chain in the structures of the three P450 2A6 mutants is too buried to directly donate a hydrogen bond to a ligand bound in the substrate binding cavity (Fig. 1B). Thus, the implications of the N297Q mutation are two-fold, in that a substrate binding and orienting interaction in the active site cavity is lost, and the protein folding interactions between the helix B′-C region and helix I are slightly altered. The loss of the H-bonding capacity in the substrate binding cavity is likely to be the most important influence on the increase in Km and the decrease in kcat that was observed for coumarin 7-hydroxylation upon mutation of Asn297 in P450 2A6 [11]. In contrast, indole is not a hydrogen bond acceptor, and the apparent binding affinity for indole is increased in the mutants.
The presence of the two mutations in the P450 2A6 L240C/N297Q double mutant does not result in a significant increase in binding affinity for indole (Table 2) but the double mutant shows a 2-3 fold enhanced catalytic efficiency for the oxidation of indole when compared to the N297Q mutant [12]. It was also reported that pigment production was evident for a much larger number of indole derivatives in E. coli expressing P450 2A6 L240C/N297Q when compared to the number observed for E. coli expressing either of the single mutants P450 2A6 L240C and N297Q [11]. The structure of the P450 2A6 L240C/N297Q mutant displays only minimal conformational changes with the wild-type structure. Nevertheless, the introduction of the smaller cysteine side chain allows the Ile208 side chain to adopt an alternate conformation and a repositioning of Phe107, Phe209 and Phe480. Conformational changes in this region were observed in the minimized structures of the P450 2A6 N297Q/I300V 5-BOI complex. Additionally, it is interesting to note that the residues affected by the L240C mutation (Ile208, Phe107, Phe111, Phe118 and Phe209) all reside in the major substrate entry and exit pathway that was identified using steered molecular dynamics simulations [32, 33] with mammalian P450 family 2 enzymes. This pathway is located between the helix B′-C region and helices F/G and I [33]. This suggests that the reduction in size of the residue 240 may increase the flexibility of the roof of the active site facilitating the oxidation of a wider variety of substituted indoles of modestly increased sizes.
The ability to oxidize bulky, substituted indoles such as 4-and 5-BOI, was first observed for the P450 2A6 I140M/L240C/N297Q/I300V/I366V mutant, constructed using error-prone PCR using P450 2A6 L240C/N297Q as a template. The mutations N297Q and I300V were then identified as critical in gaining the hydroxylation activity for 4- and 5-BOI [12]. Predicted, minimized structures of P450 2A6 N297Q/I300V in complex with 4- or 5-BOI illustrate that only slight substrate-induced adjustments to the enzyme active site are required to accommodate the substituted indoles. Similar slight rearrangements to the P450 2A6 active site were observed in structures of complexes with three 3-pyridyl heteroaromatic analogues of nicotine and a more drastic induced fit was seen in the structure of the P450 2A6 4,4′-dipyridyl disulfide complex in which Phe209 swings ∼ 3 Å away from the active site [10]. Nevertheless, these inhibitors coordinate to the heme iron, bind with submicromolar affinity and do not require an increase in active site volume, whereas 4- and 5-BOI binding affinity primarily stems from Van der Waals and orthogonal aromatic interactions, and does require a significant increase in substrate binding cavity volume. These findings are most probably the origin of the relatively low catalytic efficiency and the multiplicity of the products observed in the biotransformation of 4- and 5-BOI by the P450 2A6 N279Q/I300V mutant. Thus, the extended functionality displayed by this P450 2A6 double mutant compromises the binding affinity and stereospecificity.
Despite evidence presented here that glutamine at position 297 is easily accommodated in the structure of P450 2A6, a blast search of the protein sequence database at NCBI did not detect the presence of a glutamine in family 2 P450s for the equivalent position. For P450 subfamilies 2A and 2F, an asparagine is highly conserved at this position, whereas an aspartic acid is generally found for subfamilies 2C, 2D, 2E and 2J. The presence of an aspartic acid could change the nature of substrate interactions due to its capacity to function as a hydrogen bond donor or acceptor, depending on its ionization state. The structures of P450 subfamily 2C enzymes (PDBs: 1DT6, 1PQ2 and 1R9O) indicate that the aspartate residue supports a similar pattern of hydrogen bonding with the helix B′-C loop and with a structural water that is conserved in these structures. Interestingly, a longer residue glutamic acid is found in P450 2R1, a vitamin D3 25-hydroxylase. In the P450 2R1 structure (PDB:2OJD), the glutamic acid residue occupies a position similar to that of glutamine in the structures of the P450 2A6 N297Q mutants. In contrast to the latter structures, the conserved water molecule is present in P450 2R1 and the helix B′-C loop has moved away from helix I. The extra space thus created contributes to the active site volume and the P450 2R1 structure in complex with vitamin D3 (PDB:2OJD) demonstrates binding of the substrate in a substrate binding cavity that extends above the apolar part of the glutamate side chain.
The high degree of conservation within P450 family 2 subfamilies for residues that correspond to Asn297 of P450 2A6 most probably reflects a critical role of this residue in function and tertiary structure. The folding of the helix B′-C loop plays a key role in substrate recognition by positioning residues corresponding to Phe117, Val 116 and helix B′ residues in the active site cavities of family 2 P450s. In some cases, as described for P450s 2R1 and 2A6 the residue corresponding to Asn297 may be directly involved in substrate interactions, while in others such as P450 2B4, larger residues hinder access to this position. Like most P450 subfamily 2B enzymes, P450 2B4 displays a serine at the equivalent position for Asn297 in P450 2A6. Access to Ser294 in P450 2B4 is restricted by Phe297, and thus Ser294 is not in an optimal position to form direct hydrogen bond interactions with a ligand. The Ser294 side chain is involved in identical hydrogen bonding to the conserved structural water molecule, as described above for the Asn297 side chain in P450 2A6, but does not provide a hydrogen bond acceptor for the hydrogen of the backbone N of the residues corresponding to Val117 of P450 2A6. The network of interactions around Ser294 is believed to contribute in the stabilization of the closed conformation of P450 2B4, and is completely abolished in the structure of an open conformation of the enzyme [34]. Together with helices F-G and the β-sheet 4, both the helix B′-C region and the N-terminal part of helix I are the regions that differ most dramatically between the structures of the open and closed conformation of P450 2B4. Studies of P450 2B Ser294 mutant enzymes [32, 35, 36] invariably showed profound effects on catalytic efficiencies and binding affinities for P450 2B substrates. The S294A mutation in P450 2B1 abolishes the 16β-hydroxytestosterone activity, and also shows a significant reduction of kcat for the oxidation of 7-ethoxy-4-trifluoromethylcoumarin, 7-benzyloxyresorufin [32], and several hydroxylated 7-butoxycoumarins [36]. Mutation of P450 2D6 residue Asp301, which corresponds with Asn297 in P450 2A6, to a basic residue was reported to interfere with proper heme binding and overall folding of the protein. Neutral residues at position 301 in P450 2D6 yielded mutant enzymes with a decreased catalytic activity [37, 38]. Finally, mutation of the equivalent residue Asp293 in P450 2C9 to either aspargine or valine led to large decreases in catalytic function and substantial reductions in thermal stability relative to wild-type P450 2C9 [39]. Taken together, the P450 2A6 N297Q mutation results in the loss of a potential hydrogen bond donor in the substrate binding cavity, and a loss of a structurally conserved water molecule to alter secondary structure folding interactions between the helix B′-C region and helix I, that may contribute to the observed lower catalytic activities towards P450 2A6 substrates such as coumarin and that could result in enhanced protein flexibility.
In conclusion, the availability of crystal structures of the P450 2A6 N279Q, L240C/N297Q and N297Q/I300V mutants provides a structural explanation for the altered specificities displayed by the modified enzymes. Substitution of Asn297 in the active site abolishes a binding and orienting interaction for substrates such as coumarin, but the binding affinity of less polar substrates such as indole is only slightly altered. At the same time, the N297Q mutation displaces a conserved water molecule that provides interactions connecting secondary structure elements that shape the hydrophobic, compact P450 2A6 active site. Minimized structures of the P450 2A6 N297Q/I300V 4- and 5- BOI complexes illustrate how mutations such as I300V, might provide an extension in active site cavity volume that enables binding and oxidation of more bulky substrates. The L240C mutation demonstrates that a mutation in a region distant from the active site can indirectly change the composition and conformation of residues that line the active site cavity and possibly influence catalytic efficiency by enhancing substrate entry and product egress. Iterative rounds of mutagenesis on proteins displaying new or altered catalytic specificities, coupled with structure determination of promising enzymes can be a very powerful strategy for the in-depth study of P450 structure-function relationships, and can be further exploited in the engineering of modified P450s for biocatalysis and fine chemical synthesis. The structures described in this report could be used to design a better complementarity between the ‘new’ substrates and the shape and chemical properties of the substrate binding cavity, and lead to new mixtures of dyes, or kinase inhibitors with enhanced activity. Given the profound effect of Asn297 substitution, it would be interesting to investigate further mutations at this position, solely and in combination with the residues 240 and 300, described with this study. The amino acids serine or aspartic acid, seen at this position for other family 2 P450s, could be good candidates because they maintain the potential to provide a hydrogen-bonding interaction and may result in a similar water-bridged contact between the helix B′-C region and helix I.
Acknowledgments
The authors would like to recognize Fred Guengerich's extensive contributions to our knowledge of P450 monooxygenases and their roles in xenobiotic metabolism. The structural studies described in this manuscript are based on protein engineering studies performed by Fred and his colleagues, who kindly provided cDNAs for the construction of the expression vectors used to produce the mutant proteins for crystallization. The authors also wish to thank Keith Griffin and Qiping Zhao for excellent technical assistance.
Abbreviations
- P450 or CYP
a generic term for a cytochrome P450 enzyme, individual P450s are identified using a number-letter-number format based on amino acid sequence relatedness
- BOI
benzyloxyindole
- EDTA
ethylenediaminetetraacetic acid
- KPi
potassium phosphate
- PEG
polyethylene glycol
- Fo
observed structure factor
- Fc
calculated structure factor
- PDB
protein data bank
- rms
root mean squared
- SRS
substrate recognition site
Footnotes
This work was supported by National Institutes of Health Grant GM031001 (to E. F. J.). DNA sequencing and the synthesis of oligonucleotides were supported in part by the Sam and Rose Stein Charitable Trust. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The atomic coordinates and structure factors for the human P450 2A6 mutant proteins N297Q (code 2PG5), L240C/N297Q (code 2PG6) and N297Q/I300V (code 2PG7) have been deposited with the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
References
- 1.Guengerich FP. Cytochrome P450: Structure, mechanism, and biochemistry. In: de Montellano PR Ortiz., editor. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 377–530. [Google Scholar]
- 2.Fernandez-Salguero P, Hoffman SMG, Cholerton S, Mohrenweiser H, Raunio H, Rautio A, Pelkonen O, Huang JD, Evans WE, Idle JR, Gonzalez FJ. Am J Hum Genet. 1995;57:651–660. [PMC free article] [PubMed] [Google Scholar]
- 3.Torchin CD, McNeilly PJ, Kapetanovic IM, Strong JM, Kupferberg HJ. Drug Metab Dispos. 1996;24:1002–1008. [PubMed] [Google Scholar]
- 4.Ikeda K, Yoshisue K, Matsushima E, Nagayama S, Kobayashi K, Tyson CA, Chiba K, Kawaguchi Y. Clin Cancer Res. 2000;6:4409–4415. [PubMed] [Google Scholar]
- 5.Patten CJ, Smith TJ, Friesen MJ, Tynes RE, Yang CS, Murphy SE. Carcinogenesis. 1997;18:1623–1630. doi: 10.1093/carcin/18.8.1623. [DOI] [PubMed] [Google Scholar]
- 6.Fujita K, Kamataki T. Environ Mol Mutagen. 2001;38:339–346. doi: 10.1002/em.10036. [DOI] [PubMed] [Google Scholar]
- 7.Oscarson M, McLellan RA, Gullstén H, Agúndez JA, Benítez J, Rautio A, Raunio H, Pelkonen O, Ingelman-Sundberg M. FEBS Lett. 1999;460:321–327. doi: 10.1016/s0014-5793(99)01364-2. [DOI] [PubMed] [Google Scholar]
- 8.Fujieda M, Yamazaki H, Saito T, Kiyotani K, Gyamfi MA, Sakurai M, Dosaka-Akita H, Sawamura Y, Yokota J, Kunitoh H, Kamataki T. Carcinogenesis. 2004;25:2451–2458. doi: 10.1093/carcin/bgh258. [DOI] [PubMed] [Google Scholar]
- 9.Yano JK, Hsu MH, Griffin KJ, Stout CD, Johnson EF. Nat Struct Mo l Biol. 2005;12:822–823. doi: 10.1038/nsmb971. [DOI] [PubMed] [Google Scholar]
- 10.Yano JK, Denton TT, Cerny MA, Zhang X, Johnson EF, Cashman JR. J Med Chem. 2006;49:6987–7001. doi: 10.1021/jm060519r. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura K, Martin MV, Guengerich FP. Arch Biochem Biophys. 2001;395:25–31. doi: 10.1006/abbi.2001.2569. [DOI] [PubMed] [Google Scholar]
- 12.Wu ZL, Podust LM, Guengerich FP. J Biol Chem. 2005;280:41090–41100. doi: 10.1074/jbc.M508182200. [DOI] [PubMed] [Google Scholar]
- 13.Hoessel R, Leclerc S, Endicott JA, Nobel ME, Lawrie A, Tunnah P, Leost M, Damiens E, Marie D, Marko D, Niederberger E, Tang W, Eisenbrand G, Meijer L. Nat Cell Biol. 1999;1:60–67. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 14.Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, Eisenbrand G, Meijer L. J Biol Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 15.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mol Cell. 2000;5:121–132. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 16.Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:9497–9503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 17.Williams PA, Cosme J, Ward A, Angove HC, Matak VD, Jhoti H. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 18.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. J Bio l Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 19.Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR. J Biol Chem. 2004;279:27294–27301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 20.Rowland P, Blaney FE, Smyth MG, Jones JJ, Leydon VR, Oxbrow AK, Lewis CJ, Tennant MG, Modi S, Eggleston DS, Chenery RJ, Bridges AM. J Biol Chem. 2006;281:7614–7622. doi: 10.1074/jbc.M511232200. [DOI] [PubMed] [Google Scholar]
- 21.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 22.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 23.Schenkman JB, Remmer H, Estabrook RW. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.Jones TA, Kjeldgaard M. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Kleywert GJ, Jones TA. Acta Cryst. 1994;D50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 29.de Visser SP, Shaik S. J Am Chem Soc. 2003;125:7413–7424. doi: 10.1021/ja034142f. [DOI] [PubMed] [Google Scholar]
- 30.Draper AJ, Madan A, Parkinson A. Arch Biochem Biophys. 1997;341:47–61. doi: 10.1006/abbi.1997.9964. [DOI] [PubMed] [Google Scholar]
- 31.Gotoh O. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 32.Scott EE, Liu H, Qun HY, Li W, Halpert JR. Arch Biochem Biophys. 2004;423:266–276. doi: 10.1016/j.abb.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 33.Wade RC, Winn PJ, Schlichting I, Sudarko J Inorg Biochem. 2004;98:1175–1182. doi: 10.1016/j.jinorgbio.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD. Proc Natl Acad Sci U S A. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szklarz GD, He YQ, Kedzie KM, Halpert JR, Burnett VL. Arch Biochem Biophys. 1996;327:308–318. doi: 10.1006/abbi.1996.0127. [DOI] [PubMed] [Google Scholar]
- 36.Domanski TL, He YQ, Scott EE, Wang Q, Halpert JR. Arch Biochem Biophys. 2001;394:21–28. doi: 10.1006/abbi.2001.2504. [DOI] [PubMed] [Google Scholar]
- 37.Ellis SW, Hayhurst GP, Smith G, Lightfoot T, Wong MMS, Simula AP, Ackland MJ, Sternberg MJE, Lennard MS, Tucker GT, Wolf CR. J Biol Chem. 1995;270:29055–29058. doi: 10.1074/jbc.270.49.29055. [DOI] [PubMed] [Google Scholar]
- 38.Guengerich FP, Miller GP, Hanna IH, Martin MV, Leger S, Black C, Chauret N, Silva JM, Trimble LA, Yergey JA, Nicoll-Griffith DA. Biochemistry. 2002;41:11025–11034. doi: 10.1021/bi020341k. [DOI] [PubMed] [Google Scholar]
- 39.Dickmann LJ, Locuson CW, Jones JP, Rettie AE. Mol Pharmacol. 2004;65:842–850. doi: 10.1124/mol.65.4.842. [DOI] [PubMed] [Google Scholar]
- 40.CCP. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]