

Abstract.
Understanding the basic biology of human ageing is a key milestone in attempting to ameliorate the deleterious consequences of old age. This is an urgent research priority given the global demographic shift towards an ageing population. Although some molecular pathways that have been proposed to contribute to ageing have been discovered using classical biochemistry and genetics, the complex, polygenic and stochastic nature of ageing is such that the process as a whole is not immediately amenable to biochemical analysis. Thus, attempts have been made to elucidate the causes of monogenic progeroid disorders that recapitulate some, if not all, features of normal ageing in the hope that this may contribute to our understanding of normal human ageing. Two canonical progeroid disorders are Werner’s syndrome and Hutchinson-Gilford progeroid syndrome (also known as progeria). Because such disorders are essentially phenocopies of ageing, rather than ageing itself, advances made in understanding their pathogenesis must always be contextualised within theories proposed to help explain how the normal process operates. One such possible ageing mechanism is described by the cell senescence hypothesis of ageing. Here, we discuss this hypothesis and demonstrate that it provides a plausible explanation for many of the ageing phenotypes seen in Werner’s syndrome and Hutchinson-Gilford progeriod syndrome. The recent exciting advances made in potential therapies for these two syndromes are also reviewed.
Keywords. Ageing/aging, senescence, progeria, Werner’s syndrome, therapy, HGPS, WRN, LMNA
Introduction: why study ageing?
Ageing presents a worldwide social and financial challenge: by the year 2050, more than two billion people will be over the age of 65 [1]. Long-term care costs of the ageing population in the United Kingdom alone are estimated at ≥ £60 billion by 2030. Thus, the effects of the ageing process are predicted to be the major global health care challenge of this century [2]. In humans, ageing is associated with a subset of diseases and crippling conditions including cardiovascular disorders, diabetes, neoplasms, cataract, macular degeneration, osteoporosis and auditory impairment which result in frank ill health and low quality of later life. However, ageing organisms are also frequently subject to severe physiological frailty resulting in tolerable performance under ‘normal’ circumstances but very poor survival chances if the organism is placed under stress. This is most clearly seen in the immune system where adequate protection against infection is normally afforded by the ageing immune system, but where disease susceptibility increases markedly following physical or psychological traumas that do not affect immune function in the young [3 – 5]. Understanding the mechanisms of human ageing may therefore give important insights into the pathogenesis of a range of age-related diseases and also has the potential to allow intervention before overt degenerative pathology has developed.
Progress in understanding the basic biology of ageing has been rapid over the last 10 years and has reached the point where clinical interventions aimed at ameliorating at least some aspects of the ageing process can be envisaged. However, the complexity of ageing requires an understanding of biological processes at multiple levels, from molecules through cells to tissues, organs and the organism, i.e. an integrative biology. We contend that an integrative approach is of particular value in the study of ageing because its evolution contrasts sharply with that of many biochemical pathways.
Approaches to studying ageing
Ageing is essentially an evolutionary side effect of millions of years of selection for reproductive success. Thus, the genetic basis of the process is potentially extremely broad and the ‘ageing’ phenotype observed as a result of gene action may be quite distinct from the primary role which the gene product plays in the life history of the organism [6 – 10]. It has been estimated that thousands of genes could play a role in determining lifespan in Homo sapiens [11]. However, much of the scientific literature within gerontology can be read as though single, simple mechanisms of ageing are pitted against each other as alternative and mutually exclusive explanatory theories for how ageing occurs. Such single-mechanism views appear to us rather naive. Similarly, because so many loci and pathways are involved in the production of an essentially ‘soft’ phenotype (robust survival, as opposed to a ‘hard’ monogenic phenotype such as eye colour), the identification of candidate genes for successful ageing in humans, although probably feasible, is unlikely to occur either quickly or unambiguously.
Animal models of ageing have proven useful in identifying individual genes or biochemical pathways important in longevity, such as the IGF-1 axis in the nematode Caenorhabditis elegans and fruit fly Drosophila melanogaster [12, 13]. Whilst such systems are clearly reflective of many aspects of ageing in higher organisms [R.D.C.Saunders, I. Boubriak, D. J. Clancy, L.S. Cox, unpublished data], they may have less relevance to the senescence of mitotically active adult cells in humans. Rodent models have therefore been developed (discussed further below), but the direct applicability of data from these systems again may be rather limited with respect to humans. Many species of rodent invest more heavily in reproduction and far less in maintenance of the soma than do humans, and as a result susceptibility to oxidative stress and several genome stability and DNA repair pathways differ significantly between different rodent species and between rodents and humans [14, 15]. Thus, these models trade utility for direct physiological relevance to the human situation.
Progeroid syndromes
Analternative approach to understanding how human ageing operates, which complements those briefly outlined above, is the study of heritable genetic diseases which mimic some, but not all, the features of normal ageing, in order to gain insights into how the ageing process functions in normal individuals [11, 16 – 18]. Originally identified by an unbiased appraisal of the number of cardinal signs of normal human ageing, which appear as clinical features within a given genetic disease, these progeroid syndromes are classed as either unimodal disorders (which show only very limited aspects of human ageing such as increased cancer incidence or rapid neurodegeneration) or segmental disorders, which show a wide range of ageing-specific phenotypes across multiple tissue and organ systems.
The study of such progeroid syndromes has the advantage that mutation of only a single gene is usually causative in each case. This renders hypotheses easier to frame and test and allows the power of modern genetics and cell biology to be brought to bear with peculiar force. The disadvantage of studying such disorders is that they are essentially phenocopies of normal ageing rather than the genuine article. We focus upon the two canonical segmental progeroid syndromes, Werner’s syndrome (WS) and Hutchinson- Gilford progeroid syndrome (HGPS), because these are the best understood with regard to their pathology. That pathology is most consistent with the cell senescence hypothesis of ageing which has been proposed to account for many aspects of the ageing of mitotic tissue in normal individuals. We outline this hypothesis below, but wish to sound a note of caution at the outset. The genes mutated in these disorders participate in a wide variety of pathways and processes (which we have attempted to capture). However, simple participation in a biological process does not make a gene product the central player in that process, and being the central player in a cellular process does not make that cellular process the cardinal mechanism by which ‘ageing’ pathology is generated at the level of the whole organism. The relationship between genotype and phenotype in WS in particular illustrates this degree of systemic complexity rather well.
The cell senescence hypothesis of ageing
The cell senescence hypothesis of ageing proposes that the progressive accumulation of senescent cells contributes to, but does not exclusively cause, the ageing of the tissue in which they reside. Senescence (sometimes called replicative senescence for clarity) is a permanent block to division in cells from the mitotic tissue compartments of metazoans [19]. This block can be established in a wide variety of ways which result in a viable but permanently non-dividing cell. Senescent cells display many biochemical features that are distinct from their proliferating counterparts as a result of widespread changes in the transciptome [20, 21]. Overall, the differences between senescent cells and their growing counterparts are as large as those observed during cell differentiation [22].
Although failure to divide in response to a mitotic stimulus is the hallmark of senescent cells, not every non-dividing cell is senescent. Senescence is distinct from quiescence (transient growth arrest), and in systems where it is possible to experimentally separate the two, senescence is distinct from terminal differentiation [23, 24].
Senescence has the potential to contribute to ageing in at least two distinct ways: (i) through simple loss of proliferative capacity; (ii) through alterations in the tissue microenvironment as a result of the accumulation of cells with an altered phenotype. Three types of objection have been made to the idea that senescent cells play a role in the ageing process. These can be simply summarised as:
-
(i)
Senescence does not exist in vivo. It simply results from tissue culture conditions so deficient that the mechanisms by which growth arrest occurs in vitro are unlikely ever to occur in the normal animal.
-
(ii)
Senescence does occur in vivo but so few cells ever become senescent that any effects they might have are so small that they can be discounted.
-
(iii)
Senescent cells do occur in vivo at appreciable frequencies. However, the phenotypic differences between them and their growth-competent counterparts are trivial. Essentially, they are present but they do no harm.
The first of these objections focuses on how senescent cells occur, the second on the frequency with which they occur and the third on whether senescent cells can exert a deleterious effect. Sufficient data have now been accumulated to counteract these objections. These data are derived from the culture dynamics of normal cells grown in vitro, the detection of senescent cells in vivo and the behaviour of such cells both ex vivo and in vivo. It should be noted that none of these lines of evidence requires that cells becomes senescent by a particular pathway, merely that they have entered the senescent state. Some of these findings are touched on below but space precludes a full treatment of the subject.
Perhaps the best evidence against the idea of senescence as a tissue culture artefact is the body of data which demonstrates that primary cell populations are mixtures of clones with very variable intrinsic growth potentials which co-exist in the same medium. The thrust of these experiments is exemplified in a series of studies in which the two daughter cells resulting from a single mitotic event were separated immediately following cytokinesis and their replicative capacity determined. Such daughter cells differ by up to 28 in their proliferative capability [25]. Such large differences in proliferation between identical daughter cells are difficult to reconcile with issues of establishment of cell culture or simply ‘poor tissue culture’. This divisional behaviour also leads to the appearance of senescent cells with very limited capacity for cell division, consistent with their appearance over the life course of a human [26 – 28]. However, this is not proof that such cells exist in tissue.
Fortunately, several studies using different methodologies designed to detect senescent cells in vivo have now directly demonstrated their appearance and accumulation with age in a variety of human, primate and rodent tissues [29 —31]. The most recent of these [32, 33] has shown that at least 15%of all dermal cells in very old baboons can be classed as senescent (by immunocytochemical detection of makers of the senescent state). Whilst there will always be arguments about the number of cells required to exert an effect, it is not unreasonable to conclude that changes on this scale could produce degenerative effects if senescent cells are capable of exerting them. Indeed, senescent cells have been shown to adopt a secretory phenotype that adversely impacts on their local environment [34].
Direct measurement of senescent cells in vivo is always superior to attempts to infer their presence through the growth analysis of biopsies. However such studies occasionally appear in the literature and thus need to be considered. The classic studies in this area reported an inverse correlation between donor age and the number of population doublings achieved in vitro when cells from donors of undefined health status were used [35], or a sharp decline in proliferative capacity of cells from subjects who were either diabetic or pre-diabetic combined with no loss of proliferative capacity in healthy controls [36].
Similar studies have been conducted more recently using fibroblast cultures derived from the Baltimore Longitudinal Survey on ageing and the Leiden 85-Plus study [37, 38]. These demonstrate no statistically significant decline in the replicative potential of fibroblasts derived from healthy donors (the Leiden study also failed to show a proliferation reduction in a small number of donors with diabetes).
At first sight, these observations seem difficult to reconcile with the idea that there is a relationship between cell senescence and ageing, and it is a great pity that no large-scale studies have yet been performed which combine detection of senescent cells in vivo with the growth capacity of the same biopsy material in vitro. It is of course possible to invoke immediate technical difficulties (e.g. a selection process that occurs during establishment of cultures from biopsies, or limited powers of the study) as an explanation for the failure to observe a reduction in growth capacity in old material. However the failure to observe differences in proliferative capacity could, in fact, be quite consistent with the cell senescence hypothesis. An explicit prediction of the theory is that senescent cells are causal agents of ageing and disease. It would be close to direct disproof of the theory if the mitotic tissues of the elderly were full of senescent cells but showed no diminution in physiological function. This has never been shown to occur and could only arise if senescent cells were incapable of exerting degenerative effects.
Perhaps the best evidence that senescent cells can really exert significant ‘ageing’ effects is provided by a study in which human dermal populations aged in vitro were incorporated into reconstituted human skin equivalents. Dermal fragility and subepidermal blistering increased in this system with increasing numbers of senescent cells [39]. A still more striking demonstration that senescent cells can produce life-threatening pathology was provided by experimental induction of senescence in living rat carotid arteries. This produced severe vascular inflammation and changes consistent with the development of atheroma [40]. This study should be regarded as a landmark in the relationship between cell senescence and organismal ageing.
Molecular mechanisms of cellular senescence
Replicative senescence exists as a mechanism for tumour suppression [41 – 43], balancing the requirement for cells to proliferate and the inherent risk of neoplastic transformation with the loss of proliferative capacity which leads to tissue dysfunction. As a result of its importance in carcinogenesis, there is a large literature on the mechanisms by which cells enter a senescent state. A comprehensive treatment of this is beyond the scope of our review, so only a brief and broad overview of the area is given below.
The role of senescence in tumour suppression is probably best illustrated by the observation that both rodent and human cells (of several different types) will enter a state of senescence in the presence of high-grade stimulation of the proto-oncogenic ras pathway (typically achieved by ectopic expression of oncogenic K-ras V12) [44]. Acute exposure to ceramide [45] or DNA-damaging agents can also trigger this reactive style of senescence which probably contributes to the presence of at least some senescent cells in vivo.
Cells will also enter senescence via a series of intrinsic constitutive pathways that do not require the presence of exogenous agents. Many normal human cell types enter ‘telomere-dependent senescence’ as a result of the loss of chromosomal ends (telomeres) at each round of DNA replication. Eventually this progressive telomere attrition triggers a p53-dependent cell cycle arrest leading to senescence. p53 is a driver of senescence as determined from the premature ageing phenotypes observed in mice expressing ‘hyper-active’ p53 [46], although p53-independent telomere-dependent senescence has been reported in cells with experimentally induced telomere instability [47]. p53 is also a significant factor in maintaining the senescent state: p53 ablation by ubiquitin-mediated degradation (through ectopic HPV E6 expression) or neutralisation by antibody microinjection releases cells from senescence [48]. However, not all human cell types use this pathway as an initial proliferative lifespan barrier. Some, notably keratinocytes and pancreatic β cells, primarily enter senescence through the p16-pRb pathway [49] (so-called ‘telomere-independent senescence’). Human glial cells enter a senescent state via the activation of p19ARF, leading to downstream induction of both p53 and its transcriptional target p21CDKN1 [50– 52]. This state has been termed ‘p53-dependent telomere-independent senescence’ [53]. Significantly, p19ARF seems to provide a response to oncogenic stress through the p53 pathway [reviewed in ref. 54]. Thus p53 and its activators and effectors are intimately linked to the onset and establishment of senescence, which may account, at least in part, for its potent activity as a tumour suppressor.
Cross-species studies also complicate the mechanistic picture. Rodents in particular do not show telomere-dependent senescence, and fibroblasts from these animals normally enter senescence despite long telomeres and the presence of high levels of the telomere maintenance enzyme, telomerase. Normal rodent fibroblast senescence is thus more closely akin to that induced by the p19ARF pathway than the classical telomere-p53 axis seen in human fibroblasts. Mice lacking the catalytic component of telomerase (terc-/-) show premature ageing after five to six generations [55]. The primary phenotypes observed in this system seem to be driven by apoptosis as result of chromosomal uncapping rather than senescence (though apoptosis-driven proliferation to repopulate depleted tissues may in itself accelerate the onset of senescence, particularly in the absence of telomerase). Rescue of fertility and germ cell viability in terc-/- mice with short telomeres by additional loss of p53 serves to highlight the importance of p53 in triggering the response to telomere loss in such transgenic rodent systems [56]. Very recently, p21CDKN1 deficiency has been shown to increase lifespan and rescue proliferative defects of both haemopoietic stem cells and gut epithelium in mice with pathologically short telomeres [57], without increasing cancer susceptibility. These results strongly suggest that p21 is involved in the response of rodent cells to aphysiologically short telomeres. These observations underscore the close relationship between the senescent state and apoptosis, suggesting that whether a given cell will die or senesce in response to pathological stimuli is highly cell type and context dependent.
Werner’s syndrome
WS, first described by Otto Werner in 1904 [58], provides perhaps the best current model of normal human ageing. Cardinal features include bilateral cataracts, grey hair, skin abnormalities, short stature and hyaluronic acid excretion. In addition to the cardinal signs, a range of clinical problems are widely penetrant, from hoarse voice, alopecia, diabetes, soft-tissue calcification with ulceration, atherosclerosis, arteriosclerosis, osteoporosis, T cell atrophy (leading to immunodeficiency) and a greatly elevated risk of developing a subset of cancers, predominantly sarcomas. Patients are generally of small stature and very low body weight with little subcutaneous fat, but have an unusual redistribution of fatty deposits around the abdominal region reminiscent of morbid obesity and correlating with greatly increased risk of myocardial infarction [59] (see also International Registry Of Werner Syndrome: http://www.pathology.washington.edu/research/werner/registry/registry.html). Their increased atherosclerosis and hyperlipidaemia predisposes to transient ischaemic attacks with possible neurological consequences [60]. Schizophrenia and senile dementia are also elevated in Werner’s patients [61, 62], but there is no association of WRN protein variants with Alzheimer’s disease [63]. With greater clinical recognition of the syndrome, together with improvements in cancer detection and treatment, and health screening programmes, the median age of death of Werner’s patients has increased from 47 to 57 years [64, 65]. Remarkably, this wide range of clinical phenotypes that provide a fairly comprehensive phenocopy of normal ageing results from mutation of the single WRN gene. How can this be accounted for?
The WRN gene
The human gene responsible for WS is located on chromosome 8p12-p11.2 [66–68]. It encodes a protein of 1432 amino acids with homology to the RecQ family of helicases [reviewed in ref. 69), and the DEAH box family of RNA helicases [70], and it shows 3′–5′ helicase activity in vitro [71]. WRN shares ATPase and single-strand (ss) DNA strand annealing activities with other members of the RecQ family. Conserved HRDC and RQC domains are located C terminal to the helicase region. Enzymatically, WRN is unique amongst the RecQ family in that it also possesses an amino-terminal 3′–5′ exonuclease domain [72]. At the C terminus is a nuclear localisation sequence (aa 1358–1432) and a nucleolar localisation sequence ([73]; either at amino acids 949–1092 [74], or R1403 and K1404 [75]). Consistent with this, WRN is generally sequestered in the nucleolus [76], colocalising with nucleolin (inhibition of its helicase activity by nucleolin has been suggested [77]).
The vast majority of patient-derived WRN mutations encode a truncated protein lacking the C-terminal nuclear localisation sequence, and hence are not targeted to the nucleus [78]. Additionally, the stability of WRN mRNAs bearing such mutations is lower than that of intact WRN message [79], suggesting that RNA surveillance and nonsense-mediated decay are responsible for maintaining the very low levels of WRN mRNA in patient cells [80]. By contrast, two missense mutations have recently been described where the mRNA is stable but the WRN protein product is unstable [65]. These mutations lie beyond the exonuclease active site and are likely to impact on folding in this domain rather than enzymatic activity per se [81]. A further polymorphism/mutation has been reported that abolishes helicase and helicase-associated nuclease activity (polymorphism R834C [82]). Various other polymorphisms have been described [83–85]. Of these, there is some controversy over R1367C, which shows no association with longevity in Finnish centenarian studies [86], although it has been reported to be protective against myocardial infarction in Japanese populations [87]. However, this finding was not supported by data from the Baltimore longitudinal study, and no impact of the R1367C polymorphism on WRN enzyme activity was detected [88]. Whilst it is not at first sight surprising that an enzymatically intact WRN variant has no detectable effect on human disease incidence, WRN may also function by recruiting proteins via its non-enzymatic RQC or HRDC domains. We cannot at this stage rule out the possibility that genetic background contributes to the impact of particular WRN polymorphisms. Indeed, significant differences between WRN polymorphisms in different racial groups are highlighted by the HapMap project (http://egp.gs.washington.edu/gty_-data/wrn/, also www.hapmap.org), though it is too early to determine which, if any, differences might impact on human lifespan.
WRN in DNA metabolism
The domain structure and nuclear/nucleolar localisation of the WRN protein suggest a role in DNA metabolism (replication, repair, recombination or transcription; see Fig. 1), and at least in vitro, WRN helicase and exonuclease activities are directed to-wards DNA structures mimicking those found in DNA replication (forks), DNA recombination (four-way junctions), transcription (loops) and telomere maintenance (G4 tetraplexes) [89; reviewed in ref. 77]. A hallmark phenotype of WS cells is high levels of genome instability [90 – 92], and it is significant that epigenetic inactivation of human WRN can lead to genomic instability and human cancer [93]. However, the complex phenotype of cells from WS patients (see below) does not reveal directly the mode of action of WRN. It is possible that this complexity is the result of enormous pleiotropy of WRN action in many or all aspects of DNA metabolism. Since it is not yet clear which of these processes may be the primary driver of premature senescence in WS, we review below all aspects in which WRN has been implicated experimentally.
Figure 1.

Pleiotropy of WRN action in DNA metabolism. WRN affects all key aspects of DNA metabolism (boxed), either directly through its helicase and exonuclease activities, or mediated through multiple protein-protein interactions (grey ovals — for clarity only a subset of known interacting proteins are shown) Inappropriate execution of these processes in the absence of WRN leads to the molecular outcomes shown below the arrows.
DNA replication
WRN is important in, though not essential for, DNA replication, as shown by a wide range of studies, from in vitro biochemistry to whole-cell analysis. First, purified recombinant WRN protein assayed in vitro for helicase activity shows a marked preference for replication fork-like oligonucleotide templates [reviewed in ref. 77].Moreover, WRN protein extracted from cultured cells co-purifies with a very large replication complex containing the essential DNA polymerase processivity factor, PCNA [94]. Interaction with PCNA occurs through a classical PCNA-binding motif [95, 96] within the exonuclease domain of WRN [97]. Additionally, the C-terminal domain of WRN binds to an essential replication nuclease, FEN1 [98, 99] required for processing Okazaki fragments on the lagging strand of the replication fork [100], stimulating Fen endonuclease cleavage of 5′-flap or nicked substrates [101]. Notably, Fen1 has been implicated in processing stalled replication forks [102], an activity also suggested for WRN [103 – 105]. As the WRN-binding site on Fen1 is adjacent to but does not overlap that of PCNA, it is possible that all three proteins can act co-ordinately [106].
Consistent with its interaction with replication factors, WRN moves to replication foci in S phase and colocalises with replication factors such as PCNA [97] and RPA [107]. Although WRN is present at a large subset of replication foci (60% in normal primary human fibroblasts [104]), it is not needed to establish such sites, as formation and progressive morphological changes in S phase foci are indistinguishable between WS and normal fibroblasts [104]. Phenotypically, cells have an apparent problem with DNA replication: cultured WS lymphoblastoid cells and fibroblasts have an extended cell cycle with a delayed S phase [104, 108, 109] which is not a feature of normal senescent cells [110].
DNA fibre autoradiography studies of whole-cell populations suggest that replication origins are more widely dispersed in WS fibroblasts than in normal controls [111, 112], consistent with a defect at the level of origin firing. Confocal microscopy of combed DNA from synchronised WS fibroblasts demonstrated that replication fork progression is aberrant, with the majority of bidirectional origins showing significant fork asymmetry [104], suggesting an accumulation of stalled replication forks in WS fibroblasts. This hypothesis has very recently been tested by resolving the four-way DNA junctions predicted to form at stalled forks, which rescues the characteristic WS phenotypes of poor proliferation, low S phase populations and hypersensitivity to camptothecin [105]. Moreover, in vitro studies have shown that WRN can regress replication fork-like structures [113].
Further support for a role of WRN at the replication fork comes from the finding that WS fibroblasts and lymphoblastoid cells are hypersensitive to the topoisomerase I poison camptothecin [114 – 116]; attempts to replicate over camptothecin-induced single-strand breaks is thought to lead to replication fork collapse [117]. Moreover, a functional association of WRN with TopoI has been demonstrated [118].
Taken together, these results suggest that WRN may play a role in restarting stalled replication forks or in preventing the accumulation of recombination intermediates such as Holiday junctions at collapsed forks [105]. This may be of particular significance during the replication of repetitive DNA or genomic regions containing replication fork barriers such as the rDNA [119, 120]. It is noteworthy that yeast cells mutant for the WRN homologue Sgs1 show excessive recombination of rDNA, resulting in extrusion of rDNA circles [121]. Accumulation of such extrachromosomal rDNA circles strongly correlates with senescence [122].
Telomere maintenance
WRN may be involved in telomere maintenance as well as during global DNA replication. Purified recombinant WRN protein can act on G4 tetraplex and D-loop structures analogous to telomeric DNA [123, 124]. Not only does WRN bind to the telomere-capping protein TRF2, but this interaction also regulates the exonuclease and helicase activities of WRN on telomere-like DNA [125 –127]. WRN colocalises with telomeres in ALT cells [127], telomere replication appears aberrant in cells expressing a dominant negative WRN helicase [128], and telomere abnormalities have been described in WS cells [129, 130], though single telomere length measurements on the X chromosome have shown roughly normal rates of telomeric attrition in WS [131]. Additionally, the very low proliferative capacity of WS fibroblasts can be overcome by ectopic expression of human telomerase [132]. These findings are suggestive that telomere defects may be causal in WS senescence [133, 134], supported by very recent data indicating that altered telomere dynamics can lead to genomic instability in WS [135, 136].
DNA recombination
There are many lines of evidence suggesting that WRN is important in homologous recombination (HR), not least its functional or direct association with recombination proteins such as Rad51 [137] and Rad52 [138]. Recombination is aberrant in cells lacking functional WRN [139], WRN can suppress illegitimate recombination in yeast cells defective for the homologue Sgs1 [140], and the Schistosaccharomyces pombe homologue Rqh1 is required for suppression of homologous recombination, permitting transient S phase arrest on damage [141]. All these lines of data further support a role for WRN in restraining recombination. Moreover, a hypomorphic mutation of the Drosophila WRN exonuclease results in extremely high levels of somatic recombination [Saunders et al., unpublished data]. The prokaryotic Holliday junction resolvase, RusA, can complement WS cells to permit recombination and survival following cis-platin damage to DNA, apparently acting downstream from Rad51 [137]. In support of this idea, WRN interacts with the DSB response complex MRN, probably via Nbs1 which acts either upstream or downstream of Rad51 [142– 144]. Moreover, RusA expression restores S phase progression and proliferative capacity to WS fibroblasts [105], phenotypes that are presumed to result from excessive accumulation of Holliday junctions, at least in part at stalled replication forks. This strongly suggests either that WRN acts directly in HR, or that it prevents the accumulation of substrates that are otherwise resolved via recombinational mechanisms. The hyper-recombination seen in WS patient cells [92] is more supportive of the latter hypothesis. Interestingly, dissolution of double Holliday junctions by BLM, in combination with BLAP and TopoIIIα [145, 146], requires the HRDC domain [147]; domain swap experiments with WRN HRDC should prove interesting.
In addition to accurate HR, WRN is implicated in the error-prone recombinational pathway of non-homologous end joining (NHEJ) [148]. In a plasmid-based assay for NHEJ in transfected WS cells, large deletion products were formed [149], with products differing according to whether DNA had 5′ overhang, 3′ overhang or blunt ends, and a ‘balance’ between exonuclease and helicase activities was postulated to be important in mediating correct processing without excessive deletion [150]. Interestingly, the Ku heterodimer, an essential factor for NHEJ, is a known protein partner of WRN, stimulating its exonuclease activity [151, 152], while double deletion of WRN and Ku leads to lower sensitivity to camptothecin [153]. It is possible that WRN may act exonucleolytically in NHEJ when its helicase activity is repressed. Surprisingly, NHEJ in WS cells can be complemented by a mutant variant of WRN that lacks both helicase and exonuclease function (complementation by each single-point mutant form was partial) [150]. This suggests that WRN may recruit factors necessary for NHEJ rather than, or in addition to, acting enzymatically.
DNA repair
WS cells are hypersensitive to a specific subset of DNA-damaging agents including 4NQO [105, 154] and accumulate a large number of oxidative lesions on exposure to H2O2 [155] or under normal culture conditions [156]. These findings are suggestive of problems in dealing with oxidative damage through the base excision repair (BER) pathway. Recently, it has been demonstrated that both patient-derived WS and RNAi-WRN-knockdown cells are also hypersensitive to methylating agents [157, 158]. Alkyl and oxidative lesions in DNA are normally processed through the BER pathway, of which there are two distinct biochemical routes. The first allows simple removal of the damaged base by a glycosylase, with cleavage of the phosphodiester backbone by an AP endonuclease at the resulting apurinic/apyrimidic site. Replacement synthesis by DNA pol β then ligation by DNA ligase 1 repairs the lesion quickly and precisely. WRN physically interacts with and stimulates DNA polymerase β, in vitro [159, 160], and WRN helicase activity is inhibited by AP1 endonuclease, with inhibition relieved by DNA pol β [159, 161].
An alternative ‘long patch’ BER pathway also exists [162], requiring the action of the same enzymes employed in Okazaki fragment processing: interestingly, WRN interacts with many of these components, including PCNA [94, 97], RPA [163] and Fen1 [98]. Furthermore, WRN interacts with the poly(ADP-ribosyl) polymerase (PARP1) [164, 165], important in damage signalling. Such binding is thought to modulate the helicase and exonuclease activities of WRN and occurs shortly after oxidative and alkylating DNA damage and prior to caspase cleavage of PARP [Cox, unpublished data]. Taken together, it is highly likely that WRN is involved both in single-nucleotide (‘short-patch’) and long-patch BER.
Transcriptional regulation
Global transcription, as assayed by 3H-uridine incorporation, is decreased in WS compared with normal controls, and this is seen especially in the nucleolus [166], suggesting an impact of WRN on RNA polymerase I transcription that may be direct or through provision of an accessible template. rRNA transcription is enhanced by WRN interaction with RNA polymerase I [167]. Additionally, a role for WRN in RNA polymerase II-mediated transcription has been shown in permeabilised cells and in vitro studies [168]; furthermore, loss of WRN may result in lower basal levels of transcription through inability to resolve unusual DNA or chromatin structures. It is interesting that WRN is required to prevent the accumulation of overt DNA double-strand breaks upon treatment with the chromatin-modifying drugs chloroquine and trichostatin [169], supporting a role for WRN in maintaining a template competent for both transcription and replication.
It is possible that WRN may play further regulatory functions in controlling gene expression, given the action of WRN homologues in evolutionarily distant species. For example, a RecQ in Neurospora crassa and Mut-7 in C. elegans are both involved in RISC-dependent RNA silencing [170, 171], and the WRN exonuclease homologue in Arabidposis thaliana, WEX, mediates post-transcriptional gene silencing [172]. Human WRN has not at present been shown to possess similar activities, though experimental difficulties in obtaining stable RNAi knockdown of WRN [L. S. Cox, R. G. A. Faragher unpublished data] are provocative.
Chromosome segregation and stability
Correct segregation of mitotic chromosomes is essential to maintain gene and chromosome copy number and genome stability. Yeast models of WS demonstrate that chromosome segregation is aberrant when Sgs1 or Rqh1 are mutated [173, 174]; this may reflect an indirect (via checkpoint activation) or a direct action of these homologues on chromosome segregation mechanisms. It is therefore possible that WRN may play a role in sister chromatid cohesion or release of cohesion at mitosis. Interestingly, cohesion is established in S phase [175], a time when WRN migrates to the nucleoplasm and sites of DNA synthesis [97, 104, 107]. It will be interesting to explore more rigorously a possible role of WRN in chromatid segregation.
It is equally possible that WRN (or its yeast homologues) is important in chromosome segregation through the spindle checkpoint. By comparison, another human RecQ protein, BLM, is phosphorylated during mitosis by the MPS-1 kinase, and phosphorylated BLM then interacts with polo-like kinase 1. A BLM point mutation (S144A) that cannot be phosphorylated by MPS1 supports normal rates of sister chromatid exchange (SCE) (Bloom’s syndrome (BS) is characterised by excessive SCEs) but cannot maintain mitotic arrest on MPS1 activation through the spindle checkpoint [176].
Impact of WRN loss on cells and tissues
Probably the most interesting question which can be asked when considering WS is how loss-of-function mutations in WRN produce the pleiotropic ‘ageing’ phenotype seen in the patient. This requires the application of integrative biology, moving beyond the gene to the cell and the tissue.
At the cellular level, WS fibroblasts show probably the most limited divisional capacity of any human cells. Ninety percent of all WS fibroblast cultures enter senescence after proliferating for fewer than 20 population doublings [177]. This premature senescence results from very rapid rates of exit from the cell cycle compared to normal controls [178]. The multiple roles of WRN in DNA metabolism and the known hypersensitivity of WRN null cells to agents such as camptothecin immediately suggest that elevated DNA damage (essentially occurring at random across the genome) may cause the poor ability to grow. The alternative to arrest due to this sort of dispersed damage would be that WS fibroblasts show premature senescence as a result of an acceleration of telomere-driven senescence. Elevated telomeric deletion from single sister chromatids and telomeric dysfunction have been reported recently and several groups have shown that Werner’s syndrome fibroblasts can be immortalised by ectopic expression of telomerase [132, 179, 180]. This suggests a primary role for telomere shortening as the driver of the short lifespan; however attempts to demonstrate a consistently increased rate of telomere loss have been unsuccessful [131], suggesting that it is not going on! How are these data to be interpreted?
A helpful observation is that not all types of WS cells senesce prematurely, though all of them show features consistent with aberrant DNA metabolism (see above). The best characterised example of this is found in T cells derived from WS patients. These show no reduction in growth capacity compared to normal controls [181] but are hypersensitive to DNA-damaging agents with aberrations consistent with being null for WRN [177, 182]. Fibroblasts and T cells differ in their expression levels of telomerase (it is essentially absent in fibroblasts but is induced upon activation in T cells). These differences have been generalised into models which suggest that the effects of WRN loss on the ability to grow will depend on the controls on replicative lifespan of the cell type in question [133, 134, 183]. The best working model for how WS cells become senescent is therefore a mixture of irreversible cell cycle exit and accelerated telomere-driven senescence. These molecular insights provide important clues to potential therapeutic routes (see below). The observation that premature replicative senescence is not a universal response to a lack of WRN provides a satisfying explanation for the most obvious clinical difference between WS patients and normal ageing humans. WS patients show multiple but tissue-specific decline in functional capacity. Some tissues (such as the dermal layer of the skin or the bones) are very severely affected by the disease whilst some others, notably the immune system, are essentially normal. In contrast, normal older people show a pattern of consistent decline across multiple organ systems. We do not see geriatric humans with the bones of a centenarian and the T cells of a teenager. Something close to this is observed in WS.
Perhaps the best evidence that replicative senescence plays a major role in the clinical presentation of the syndrome comes from Wrn knockout mice. Animals lacking WRN alone show few premature ageing phenotypes despite the fact that their cells show a mutator phenotype characteristic of the disease and increased sensitivity to the effects of several DNA-damaging agents [184, 185]. Most notably, the premature replicative senescence seen in human WS fibroblasts was not recapitulated in this model. Thus simple genomic instability is not in itself sufficient to replicate the WS phenotype in rodents. In contrast Wrn/Terc double mutants develop age-dependent pathologies very similar to those seen in WS humans (including grey hair, osteoporosis, type II diabetes, cataracts, an elevated frequency of non-epithelial malignancies and premature death). Fibroblasts from these animals show both sensitivity to DNA-damaging agents and accelerated replicative senescence in vitro [186].Thus, loss of WRN appears only to give a profound phenotype if senescent cells are generated at accelerated rates.
The accumulation of senescent cells with their proinflammatory phenotype provides a good potential explanation for the development of many of the pathologies seen in the disease, but is this mechanism the whole story? At least one clinical feature of the disease seems unlikely to be caused simply by senescent cells. This is the observation that WS patients have elevated cancer frequencies for a selected subset of mesenchymal neoplasms [187]. The increased frequency of these unusual tumours is more consistent with the genomic instability caused by loss of WRN than with an excessive generation of senescent cells. Provocatively, this is also one of the aspects of the disease which is most at variance with the clinical presentation of normal elderly people.
In whole organisms, different tissue systems interact to maintain physiological homeostasis. Thus a serious perturbation in one tissue may have equally serious effects on another even though the primary cause of that perturbation is absent. Based on what we know about cell division counting mechanisms, the cells of the cardiovascular system (endothelium and vascular smooth muscle) are not especially strong candidates for a severe lifespan deficit in the absence of WRN. However, cardiovascular disease is a striking feature of WS, suggesting either that these cells will senescence prematurely or that some other mechanism is at work. Although the replicative lifespans of vascular cells from WS patients have yet to be formally measured, there are at least some data to suggest that systemic perturbations are playing a major role in this aspect of the phenotype.
WS patients accumulate visceral fat by an unknown mechanism (probably due to altered cell turnover in the pre-adipocyte compartment) and show a robust phenotype of insulin resistance which is mediated by high circulating levels of plasma tumour necrosis factor (TNF)-α and low levels of adiponectin [188, 189]. Thus, although not morbidly obese, WS patients have cytokine profiles akin to those seen in metabolic syndrome. Since severe cardiovascular disorders (including atherosclerosis) are common in metabolic syndrome, it is at least plausible that these systemic factors are driving the disease in WS patients. This pattern of pathology would be difficult to explain simply in terms of a RecQ helicase mutation, but an integrative approach allows the ramifications of such a mutation to be conceptualised. This also gives insights into potential treatments [see ref. 190 and below].
From genotype and phenotype to therapy in WS
The most important factors in increasing life expectancy and quality of life for WS patients are early diagnosis, regular monitoring and effective treatment, particularly of neoplasia. The efficacy of such an approach can be seen in the Japanese population, where life expectancy of WS patients has increased markedly over the past decade [64]. In addition to these, an understanding of the key roles of WRN in DNA metabolism (Fig. 1) may be central to providing an effective therapy for those with the disease (see also Fig. 3). In particular, the idea that the S phase deficit results from accumulation of stalled replication forks, with concomitant accumulation of unusual DNA structures such as Holliday junctions, has led to the development of an experimental reversal of the WS phenotypes of poor cell cycle progression and drug sensitivity, by supplying an ectopic Holliday junction-cleaving enzyme [105, 137]. Whilst not a treatment option per se, these findings have highlighted the HR pathway components as potential targets for intervention in WS.
Figure 3.

Drivers of senescence and therapeutic opportunities in WS and HGPs. Drivers, factors leading to the accumulation of DNA damage, are boxed at the top of the diagram, with causative factors shown in red. All result in DNA damage if unchecked; however, several therapeutic or modulatory options are now available. Blocking pre-lamin A farnesylation with farnesyl transferase inhibitors (FTIs), or depleting lamin A using either morpholinos or RNAi leads to a significant reduction in aberrant morphological changes of the nucleus usually associated with HGPS, and a reduction in resultant DNA damage. Mouse cells and mice models of HGPS treated with FTIs show significant improvement in proliferation, and the entrie organism shows better bone structure and increased longevity. FTIs are already licensed for treatment of malaria and various cancers. Statins also reduce farnesylation and may prove useful in HGPS. Telomere attrition leading to signalling into the DNA damage pathway can be overcome in cell cultures by ectopically supplying telomerase, whilst the problems with DNA replication and cell proliferation resulting from loss of WRN can be overcome using a Holliday junction resolving enzyme, resolvase. As far as Signal transduction is concerned, during adipose differentiation, SRBEP1 binds and stimulates the PPARγ transcription factor; SREBP1 sequestration by progerin may prevent this in HGPS. Use of the PPARγ agonist pioglitazone has proven valuable in diminishing several aspects of lipodystrophy and metabolic syndrome in WS patients and may play an equally important role in the treatment of HGPS. Once DNA damage has occurred, through whatever route, there is still scope to modulate the response of the cell by inhibiting the stress signal transducing kinase p38 with the inhibitor SB203580, a drug already licensed for use in diabetes. Moving on to effectors; inhibition of the tumour suppressor p53 may not be ideal as it would be expected to lead to increase in cancer incidence; however, interfering with its downstream effector p21CDKN1 appears to reduce excess cell cycle exit and senescence in mouse models, without increasing cancer incidence. Gene deletion in people is not yet a viable option as in experimental animals, but RNAi may prove useful in this context. The flip side of preventing senescence using drugs is the possibility of causing senescence by design, with major benefits in cancer therapy. In particular, hyperactivating the p21CDKN1 pathway through small-molecule drugs (e.g. nucleoside analogue CYC102) may prove a fruitful therapeutic route.
Knowledge of the protein interactions of WRN and importantly the signal transduction pathways that transmit the signal from stalled replication forks or broken DNA has led to an alternative strategy for WS therapy. Inhibition of the p38 stress kinase, which is involved in signal transduction from DNA damage to cell cycle arrest, using the small-molecule compound SB203580, appears to reverse not only the proliferative deficits of cultured WS fibroblasts but also their gross morphological abnormalities, including a restructuring of the actin cytoskeleton [191]. WS fibroblasts grow well in the presence of the drug and although there are legitimate concerns that continued proliferation may pose an increased cancer risk in WS patients, correcting the proliferative defect may bring real clinical benefit.
Lastly, the systemic effects of WRN mutations are also amenable to treatment. Fat distribution, serum adiponectin levels and insulin resistance can all be improved in WS patients by pioglitazone treatment [188 – 190, 192], suggesting that this drug may be one effective route to improving patient life expectancy by decreasing risk of cardiovascular accidents. It is likely that a combination of therapies targeting various pathologies will be most effective in terms of clinical outcome.
Hutchinson-Gilford progeroid syndrome
This is a very rare, severe infant-onset progeria in which children manifest marked signs of ageing within their first year, with significant growth retardation throughout the truncated lifespan [193]. Like WS patients, they suffer from alopecia, lipodystrophy, osteoporosis, arteriosclerosis and atherosclerosis. The major cause of death is myocardial infarction at median age 11, due to coronary artery atherosclerosis. Unlike WS, HGPS patients do not suffer from cataracts or show neurological problems.
Identification of the HGPS gene
In 2003, the gene on human chromosome 1q21 responsible for HGPS was identified as LMNA [194, 195]. It is large gene (24 kb) with 12 exons and encodes at least four distinct splice variant products [196, 197]. Of these, lamin A (75 kDa) and lamin C (64 kDa), together with lamin B, which is encoded on a separate locus, make up the 20- to 50-nm-thick intermediate filament network of the nuclear lamina that underlies and supports the nuclear envelope [reviewed in refs. 198, 199]. Unlike the cytoplasmic and extracellular intermediate filaments, the lamins undergo rapid and reversible depolymerisation at every cell division, mediated through phosphorylation by the G2/M phase cyclin-dependent kinases [200, 201]; lamin repolymerisation promotes nuclear envelope reassembly after mitosis [reviewed in refs 198, 202, 203]. Lamin A is initially synthesized as a precursor prelamin A that undergoes farnesylation on a conserved CAAX motif (cysteine-aliphatic-aliphatic-variable), with subsequent cleavage in two stages to form a mature laminA (see Fig. 2) [reviewed in ref. 204]. The most common mutation in HGPS occurs in exon 11 leading to activation of an otherwise cryptic splice donor site. Incorrect splicing then results in formation of an aberrant mRNA lacking the last 150 nucleotides of exon 11 [194, 195, 205, 206]. The encoded protein is 50 amino acids shorter than wild-type lamin A, and lacks the cleavage sites usually recognised by the NER-associated Zmpste24 metalloprotease, resulting in accumulation of incorrectly processed pre-lamin A, also known as ‘progerin’ or Δ50 lamin A. There are at least two possible steps in the processing pathway that involve proteolysis; disruption of either would lead to accumulation of pre-lamin A (Fig. 2 [see also ref. 204]. From this it can be seen that the classic LMNA G608G mutation in HGPS results in formation of a truncated pre-lamin A, whilst Zmpste24 mutation leads to accumulation of wild-type pre-lamin A (the usual lamin A precursor). Interestingly, other mutations in lamin A cause human disease but with a different phenotypic spectrum from HGPS [reviewed in refs 197, 207, 208; see also OMIM entry 150330].
Figure 2.
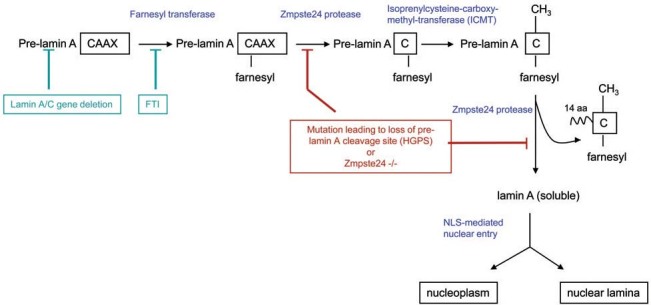
LaminA-processing pathway. Pre-laminA mRNA is encoded by the lamin A/C gene (lamin C is a shorter splice variant lacking the CAAX motif) It is translated by cytosolic ribosomes and then becomes farnesylated by the action of farnesyl transferase, providing it with a hydrophobic anchor by which it associates with the cytosolic side of the rough endoplasmic reticulum membrane. Proteolytic cleavage of the two aliphatic and adjacent residue of the CAAX motif leaves pre-lamin A with a farnesylated cysteine residue. Further cleavage by the Zmpste24 protease releases mature lamin A protein into the cytosol, from where it is transported into the nucleus. Once inside the nucleus, lamin A can associate with membrane-bound lamin B to form the nuclear lamina, or it can remain soluble in the nucleoplasm. Experimental or therapeutic interventions to relieve accumulation of pre-lamin A are shown in green, whilst deleterious impacts on the pathways (naturally occurring or experimentally induced mutations) are shown in red.
The main mutation involved in HGPS, G608G, is a de novo mutation occurring on a paternal inherited allele in the majority of cases [194, 195, 205], although one case of somatic mosaicism in an asymptomatic mother carrying the G608G mutation, and transmitting pro-geria, has been reported [209]. There is strong evidence that accumulated farnesylated prelamin A (progerin) exerts toxic effects [210], suggesting that only one mutant allele is required to cause disease. Expression of progerin in the presence of wild-type lamin A impacts significantly on the structure and organisation of the nucleus [211], consistent with a dominant negative action disrupting normal lamin processing, assembly and turnover. Similarly, heterozygous Lmna HG/+ mice show HGPS-like cellular defects [212]. These findings all support the contention that the common HGPS mutation (G608G) exerts a dominant effect. However, compound heterozygous mutations have also been reported [213], which may result in atypical HGPS, and homozygous mutation has been mooted to cause HGPS [214]. It is noteworthy that some cases of the related laminopathy, neonatal lethal restrictive dermopathy (RD) actually result from LMNA mutation [215 – 217; reviewed in ref. 206], rather than the more usual mutation in the Zmpste24 metalloprotease [216, 218]. It is possible that these may be more properly considered as atypical, neonatal and severe progeria. Moreover, syndromes in which Zmpste24 is mutant show autosomal recessive inheritance.
Cellular phenotype in HGPS
HGPS cells show altered population dynamics, abnormal nuclear architecture and problems with DNA replication, DNA repair and transcription as a consequence of mutations in LMNA.
Abnormal nuclear morphology including deformation of the nuclear envelope and a loss of the usual spherical shape of the nuclei is the major cellular feature of the disease and is associated with the appearance of micronuclei, where the genome has been fragmented and recompartmentalised. Such fragmented genomes are incapable of correct replication or segregation at mitosis, and may trigger apoptosis. The fraction of cells with such abnormal nuclear morphology increases as the population proliferates in culture, as does the proportion of cells with a missing or abnormal A-type lamin [219], consistent with the idea that such morphological abnormalities arise as a consequence of cell replication and division. In addition to abnormal lamina staining, HGPS nuclei show histone demethylation (specifically loss of methyl-K9 on histone H3) and loss of the heterochromatin protein HP1a [220], which contributes to the observed loss of peripheral heterochromatin [221] and further impacts on chromosome stability. Since the nuclear lamina penetrates through the nucleus and provides a scaffold for DNA spooling during replication and transcription [222], loss of lamina integrity can impact not only on nuclear structure but also on nucleic acid processing. This is borne out by experimental data demonstrating defects in DNA replication [223 – 225] and transcription [226, 227] in nuclei with defective lamins. In addition, lamin A has been proposed to recruit DNA repair proteins to sites of damage, based on the aberrant distribution of Rad51 foci and the persistence of γH2AX foci in irradiated HGPS cells compared with wild-type fibroblasts [228].
In a mouse model of HGPS, which is null for the Zmpste24 protease (hence with accumulation of wildtype pre-lamin A), similar problems with Rad51 redistribution are observed, together with persistence of damage foci [228]. Additionally, nuclear morphology is aberrant in embryonic fibroblasts derived from Zmpste24-null mice [229], similar to that observed in mice null or mutant for Lmna [230]. A high frequency of micronuclei and loss of peripheral heterochromatin is detected, entirely consistent with the phenotype observed for HGPS patient fibroblasts. However, there is no increase in apoptosis in the Zmpste24-null mice [231], unlike the very high rates of cell death seen in Lmna mutant mice [230]. The phenotypes in Zmpste24 mutant mice were reversed by creation of lamin A heterozygotes in an Zmpste24-null background [210], suggesting that at least in the presence of mature lamin A, there is a threshold limit of prelamin A that cells can tolerate before pathological changes in nuclear architecture develop. However, wild-type pre-lamin A and progerin may be differentially toxic.
Consistent with high levels of DNA damage and other stresses resulting from aberrant nuclear and chromatin architecture, the p53 pathway appears to be hyperactivated in HGPS cells [231]. Upregulation of many p53 transcriptional targets including p21CDKN1 and Gadd45 is observed, though oddly without detectable increase in p53 protein or post-translational modification of p53 usually associated with DNA damage responses [231]. Most notably, many of the HGPS phenotypes are fully rescued by a decrease in lamin A (i.e. Lmna+/-) and partially rescued by deletion of p53; double-knockout p53-/- Lmna-/- mouse cells proliferate more readily than single knockdowns and show a normal nuclear morphology, while the lifespan of the mice is extended [231]. This result is strongly suggestive that p53 or its downstream effectors such as p21 drive the senescence of HGPS cells. Cultures of fibroblasts from some HGPS patients show a reduced proliferative capacity, with aberrant population dynamics. However, rather than the very rapid rates of exit from the cell cycle into senescence seen in WS, HGPS cells show a hyerproliferative phenotype in conjunction with high rates of apoptosis [219].
To summarise, lamin A loss and accumulation of progerin has three profound consequences: (i) loss of nuclear integrity and architecture, leading to changes in chromatin structure and aberrant gene expression; (ii) frank and persistent DNA damage resulting from both aberrant sequestration by progerin of essential repair proteins (e.g. Rad51) and lack of targeting of such proteins by mature lamin A to sites of damage; (iii) elevated rates of cell turnover driven primarily by apoptosis. How do these features of the mutation link to the progeroid features of the patient?
Our own view is that the fundamental nuclear instability at the level of individual cells leads to elevated rates of cellular apoptosis. These elevated rates of cellular apoptosis lead to increased rates of cell turnover within a population of HGPS cells. Such increased rates of turnover generate increased numbers of senescent cells, which probably mediate much of the pathology. In this sense, HGPS would be analogous to WS at the cell population level but with elevated senescent production being driven by a failure to retain cells rather than an elevated tendency to enter the senescent state.
Impact of defects in lamin A processing on tissues and cells
Abnormal fat metabolism is a clinically significant feature of HGPS, with atherosclerosis leading to early morbidity and mortality. By analogy to a related laminopathy, Dunnigan-type familial partial lipodystrophy [232], we suggest that the abnormalities in fat metabolism in HGPS may be caused by progerin binding to and sequestering SREBP1 (Fig. 3), an adipogenic transcription factor important in fat metabolism and adipose differentiation. In normal adipose differentiation, intranuclear SREBP1 binds and activates the transcription factor PPARγ [233]; it is possible that this does not occur adequately in HGPS, and that the characteristic lipodystrophy is a consequence of such sequestration of important transcription factors. Lipodystrophy is a common feature in many laminopathies, but structural differences in the molecular conformation and stability of mutant lamins in other laminopathies [234] suggest that the causative mechanism may differ in each — for example, Dunnigan-type familial partial lipodystrophy results from a stable lamin A that can no longer form appropriate protein associations [234], though later studies have shown the presence of peripheral prelamin A in this syndrome [232]; whether proteinprotein interactions are similarly aberrant for progerin is less clear.
From genotype and phenotype to therapy in HGPS
A-type lamins are differentially expressed in human cells [196], so it is possible that therapies directed at HGPS could aim to decrease overall levels of LMNA without deleterious effects. To this end, lentiviral expression of short hairpin RNAi (shRNAi) targeted against pre-spliced and mutant LMNA mRNA has been carried out in HGPS fibroblasts [235]. This treatment led to significant decreases in SA-β-gal staining (a marker of senescent cells). Moreover, such cells showed marked increases in proliferative capacity determined both by clonogenic assays and cumulative population doublings. Most surprisingly, these cells showed reversion of nuclear morphology to a more rounded and regular appearance, with loss of micronuclei, even though they are significantly lacking in full length and Δ50 lamin A/progerin [235]. These authors suggest that shRNAi may be useful as a tool in restricting arterial wall stenosis and thus eventually be employed as gene therapy for HGPS once better delivery systems targeting the arterial wall have been developed [235]. This is obviously of huge importance given that the major cause of death in HGPS is from coronary artery atherosclerosis, and deserves rapid follow-up.
Whilst RNAi has significant potential for in vivo therapy, morpholinos (modified oligonucleotides) have been reported to cause fewer ‘off-target’ effects in gene expression knock down studies. Morpholinos targeted to the cryptic splice site of progerin have been used successfully to reverse phenotypes in HGPS cells including nuclear morphology, lamin A levels and distribution, heterochromatin distribution and transcription [220]. This is a proof of principle that deserves rapid translation into the clinic.
Since gene delivery systems and in vivo gene therapy in humans are still in their infancy, alternative strategies are necessary if HGPS is to be treated in any meaningful way in the near future. The loss of heterochromatin observed in HGPS has been targeted by use of the histone deacetylase inhibitor trichostatin A; together with mevinolin, this was found not only to lead to restoration of heterochromatin organisation, but also to reduce progerin levels markedly [221]. Such drugs therefore provide important new leads for development of therapies for HGPS.
The most promising of current possible therapies is the use of farnesyl transferase inhibitors to prevent the formation of farnesylated pre-lamin A, a step that normally targets pre-lamin A to the nuclear envelope (see Fig. 2). The rationale is based on the assumption that abnormalities in nuclear architecture and nuclear blebbing resulting from incorporation of abnormal pre-lamin A are causative of the pathology in HGPS. This is based on data from HeLa cells bearing mutations that result in either pre-lamin A that could not be farnesylated, or farnesylated lamin A that could not be cleaved [236]. Nuclei appeared morphologically normal without lamin A, but HGPS-like aberrant architecture was observed in cells expressing the uncleavable variant [236]. Thus it is the presence of progerin rather than the absence of lamin A that is important in abnormal nuclear structure in HGPS. Although FTIs would also interfere with isoprenylation of B-type lamins (and possibly other proteins such as c-ras), it was felt that this would be less deleterious than accumulation of incorrectly processed lamin A. To test this hypothesis, a mouse model of HGPS was created by gene targeting to produce an Lmna HG allele that encodes progerin [212, 237]. Embryonic fibroblasts from such mice showed nuclear blebbing and abnormal lamin staining characteristic of HGPS, together with high levels of progerin. Upon treatment with the farnesyl transferase inhibitor PB-43, there was a very marked decrease in nuclear blebbing in the HG mouse fibroblasts [212], and improved growth and bone structure in the mice [237]. Whilst these studies were conducted in a mouse model, there is clear hope that FTIs will prove of benefit to HGPS patients. As drugs, they are already approved for use in treating malaria and cancers such as ductal breast carcinomas. In terms of treating very sick children, it is also fortunate that FTIs are stable and can be given orally, with generally good tolerance. Given the severity and inevitable lethality of the condition, fast-track approval of FTIs for clinical trials in HGPS patients is a vital goal.
Conclusions
The last decade has produced an exponential rise in our understanding of the molecular pathology of both WS and HGPs. This growth in our understanding of the disorders has also driven the development of potential therapies which deserve to enter clinical trials in the near future. The relationship between disease mechanisms and routes to therapy is shown in Figure 3. The successful treatment of a human premature ageing syndrome by any of these means would represent a tremendous medical advance.
Our knowledge of the pathology of these progeroid syndromes may very well also bring benefits outside the immediate arena of disease treatment. In this review we have tried to show that there is a solid body of data suggesting that the accumulation of senescent cells is critical to many of the primary premature-ageing phenotypes seen in WS and HGPS. This is an important step in our understanding of the normal ageing process itself, because at the time the disorders were selected as useful potential ageing models, virtually nothing was known about how the mutations causing each disorder resulted in the phenotype of the patients. In the case of HGPS, even the mode of inheritance of the disease was the subject of intense debate. Both WS and HGPS could, in principle, have been caused by a very wide variety of mechanisms which had no relevance to normal ageing. However it is now clear that the most likely cause of the progeroid phenotype in both types of disorder is cell senescence, a mechanism that was independently proposed to cause ageing in normal individuals. Thus, if we can successfully treat the premature-ageing diseases, it is at least possible that we can ameliorate some aspects of normal ageing as well. This is an exciting prospect.
Acknowledgement
We are grateful to the BBSRC for financial support (grant numbers ERA16310 and BB/E000924/1 to LSC).
Footnotes
Received 12 March 2007; received after revision 1 June 2007; accepted 21 June 2007
References
- 1.Lutz W., Sanderson W., Scherbov S. Doubling of world population unlikely. Nature. 1997;387:803–805. doi: 10.1038/42935. [DOI] [PubMed] [Google Scholar]
- 2.Mills H. In: Fit for the future: the prevention of dependency in later life. Prophet H., editor. London: Report of the Continuing Care Conference; 1998. [Google Scholar]
- 3.Butcher S. K., Lord J. M. Stress responses and innate immunity: aging as a contributory factor. Aging Cell. 2004;3:151–160. doi: 10.1111/j.1474-9728.2004.00103.x. [DOI] [PubMed] [Google Scholar]
- 4.Butcher S. K., Killampalli V., Lascelles D., Wang K., Alpar E. K., Lord J. M. Raised cortisol:DHEAS ratios in the elderly after injury: potential impact upon neutrophil function and immunity. Aging Cell. 2005;4:319–324. doi: 10.1111/j.1474-9726.2005.00178.x. [DOI] [PubMed] [Google Scholar]
- 5.Pawelec G. Immunity and ageing in man. Exp. Gerontol. 2006;41:1239–1242. doi: 10.1016/j.exger.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Medawar, P. B., An unsolved problem of biology. Lewis, London.
- 7.Kirkwood T. B. Human senescence. Bioessays. 1996;18:1009–1016. doi: 10.1002/bies.950181211. [DOI] [PubMed] [Google Scholar]
- 8.Partridge L., Gems D. The evolution of longevity. Curr. Biol. 2002;12:R544–546. doi: 10.1016/S0960-9822(02)01048-5. [DOI] [PubMed] [Google Scholar]
- 9.Chapman T., Liddle L. F., Kalb J. M., Wolfner M. F., Partridge L. Cost of mating in Drosophila melanogaster females is mediated by male accessory gland products. Nature. 1995;373:241–244. doi: 10.1038/373241a0. [DOI] [PubMed] [Google Scholar]
- 10.Herndon L. A., Schmeissner P. J., Dudaronek J. M., Brown P. A., Listner K. M., Sakano Y., Paupard M. C., Hall D. H., Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans . Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- 11.Martin G. M. Syndromes of accelerated aging. Natl. Cancer Inst. Monogr. 1982;60:241–247. [PubMed] [Google Scholar]
- 12.Kimura K. D., Tissenbaum H. A., Liu Y., Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans . Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 13.Clancy D. J., Gems D., Harshman L. G., Oldham S., Stocker H., Hafen E., Leevers S. J., Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 14.Harper J. M., Salmon A. B., Leiser S. F., Galecki A. T., Miller R. A. Skin-derived fibroblasts from long-lived species are resistant to some, but not all, lethal stresses and to the mitochondrial inhibitor rotenone. Aging Cell. 2007;6:1–13. doi: 10.1111/j.1474-9726.2006.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andziak B., Buffenstein R. Disparate patterns of age-related changes in lipid peroxidation in long-lived naked mole-rats and shorter-lived mice. Aging Cell. 2006;5:525–532. doi: 10.1111/j.1474-9726.2006.00246.x. [DOI] [PubMed] [Google Scholar]
- 16.Martin G. M. Genetics and aging: the Werner syndrome as a segmental progeroid syndrome. Adv. Exp. Med. Biol. 1985;190:161–170. doi: 10.1007/978-1-4684-7853-2_5. [DOI] [PubMed] [Google Scholar]
- 17.Martin G. M. The Werner mutation: does it lead to a ‘public’ or ‘private’ mechanism of aging? Mol. Med. 1997;3:356–358. [PMC free article] [PubMed] [Google Scholar]
- 18.Martin G. M. Genetic modulation of senescent phenotypes in Homo sapiens . Cell. 2005;120:523–532. doi: 10.1016/j.cell.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 19.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 20.Shelton D. N., Chang E., Whittier P. S., Choi D., Funk W. D. Microarray analysis of replicative senescence. Curr. Biol. 1999;9:939–945. doi: 10.1016/S0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- 21.Doggett D. L., Rotenberg M. O., Pignolo R. J., Phillips P. D., Cristofalo V. J. Differential gene expression between young and senescent, quiescent WI-38 cells. Mech. Ageing Dev. 1992;65:239–255. doi: 10.1016/0047-6374(92)90039-G. [DOI] [PubMed] [Google Scholar]
- 22.James C. G., Appleton C. T., Ulici V., Underhill T. M., Beier F. Microarray analyses of gene expression during chondrocyte differentiation identifies novel regulators of hypertrophy. Mol. Biol. Cell. 2005;16:5316–5333. doi: 10.1091/mbc.E05-01-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norsgaard H., Clark B. F., Rattan S. I. Distinction between differentiation and senescence and the absence of increased apoptosis in human keratinocytes undergoing cellular aging in vitro. Exp. Gerontol. 1996;31:563–570. doi: 10.1016/0531-5565(96)00011-3. [DOI] [PubMed] [Google Scholar]
- 24.Adams J. C., Watt F. M. An unusual strain of human keratinocytes which do not stratify or undergo terminal differentiation in culture. J. Cell Biol. 1988;107:1927–1938. doi: 10.1083/jcb.107.5.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones R. B., Whitney R. G., Smith J. R. Intramitotic variation in proliferative potential: stochastic events in cellular aging. Mech. Ageing Dev. 1985;29:143–149. doi: 10.1016/0047-6374(85)90014-4. [DOI] [PubMed] [Google Scholar]
- 26.Smith J. R., Whitney R. G. Intraclonal variation in proliferative potential of human diploid fibroblasts: stochastic mechanism for cellular aging. Science. 1980;207:82–84. doi: 10.1126/science.7350644. [DOI] [PubMed] [Google Scholar]
- 27.Ponten J., Stein W. D., Shall S. A quantitative analysis of the aging of human glial cells in culture. J. Cell Physiol. 1983;117:342–352. doi: 10.1002/jcp.1041170309. [DOI] [PubMed] [Google Scholar]
- 28.Thomas E., al Baker E., Dropcova S., Denyer S., Ostad N., Lloyd A., Kill I. R., Faragher R. G. Different kinetics of senescence in human fibroblasts and peritoneal mesothelial cells. Exp. Cell Res. 1997;236:355–358. doi: 10.1006/excr.1997.3760. [DOI] [PubMed] [Google Scholar]
- 29.Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O., Peacocke M., Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minamino T., Miyauchi H., Yoshida T., Tateno K., Kunieda T., Komuro I. Vascular cell senescence and vascular aging. J. Mol. Cell. Cardiol. 2004;36:175–183. doi: 10.1016/j.yjmcc.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Li Y., Yan Q., Wolf N. S. Long-term caloric restriction delays age-related decline in proliferation capacity of murine lens epithelial cells in vitro and in vivo. Invest Ophthalmol Vis. Sci. 1997;38:100–107. [PubMed] [Google Scholar]
- 32.Herbig U., Ferreira M., Condel L., Carey D., Sedivy J. M. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 33.Jeyapalan J. C., Ferreira M., Sedivy J. M., Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007;128:36–44. doi: 10.1016/j.mad.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krtolica A., Parrinello S., Lockett S., Desprez P. Y., Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl. Acad. Sci. USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin G. M., Sprague C. A., Epstein C. J. Replicative life-span of cultivated human cells: effects of donor's age, tissue, and genotype. Lab. Invest. 1970;23:86–92. [PubMed] [Google Scholar]
- 36.Goldstein S., Moerman E. J., Soeldner J. S., Gleason R. E., Barnett D. M. Chronologic and physiologic age affect replicative life-span of fibroblasts from diabetic, prediabetic, and normal donors. Science. 1978;199:781–782. doi: 10.1126/science.622567. [DOI] [PubMed] [Google Scholar]
- 37.Cristofalo V. J., Allen R. G., Pignolo R. J., Martin B. G., Beck J. C. Relationship between donor age and the replicative lifespan of human cells in culture: a reevaluation. Proc. Natl. Acad. Sci. USA. 1998;95:10614–10619. doi: 10.1073/pnas.95.18.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maier A. B., le Cessie S., de Koning-Treurniet C., Blom J., Westendorp R. G., van Heemst D. Persistence of high-replicative capacity in cultured fibroblasts from nonagenarians. Aging Cell. 2007;6:27–33. doi: 10.1111/j.1474-9726.2006.00263.x. [DOI] [PubMed] [Google Scholar]
- 39.Funk W. D., Wang C. K., Shelton D. N., Harley C. B., Pagon G. D., Hoeffler W. K. Telomerase expression restores dermal integrity to in vitro-aged fibroblasts in a reconstituted skin model. Exp. Cell Res. 2000;258:270–278. doi: 10.1006/excr.2000.4945. [DOI] [PubMed] [Google Scholar]
- 40.Minamino T., Yoshida T., Tateno K., Miyauchi H., Zou Y., Toko H., Komuro I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation. 2003;108:2264–2269. doi: 10.1161/01.CIR.0000093274.82929.22. [DOI] [PubMed] [Google Scholar]
- 41.DePinho R. A., Wong K. K. The age of cancer: telomeres, checkpoints, and longevity. J.Clin. Invest. 2003;111:S9–14. [PubMed] [Google Scholar]
- 42.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11:S27–31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- 43.Beausejour C. M., Campisi J. Ageing: balancing regeneration and cancer. Nature. 2006;443:404–405. doi: 10.1038/nature05221. [DOI] [PubMed] [Google Scholar]
- 44.Serrano M., Lin A.W., McCurrach M. E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 45.Mouton R. E., Venable M. E. Ceramide induces expression of the senescence histochemical marker, betagalactosidase, in human fibroblasts. Mech. Ageing Dev. 2000;113:169–181. doi: 10.1016/S0047-6374(99)00105-0. [DOI] [PubMed] [Google Scholar]
- 46.Tyner S. D., Venkatachalam S., Choi J., Jones S., Ghebranious N., Igelmann H., Lu X., Soron G., Cooper B., Brayton C., Hee Park S., Thompson T., Karsenty G., Bradley A., Donehower L. A. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 47.Jacobs J. J., de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr. Biol. 2004;14:2302–2308. doi: 10.1016/j.cub.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 48.Davis T., Singhrao S. K., Wyllie F. S., Haughton M. F., Smith P. J., Wiltshire M., Wynford-Thomas D., Jones C. J., Faragher R. G., Kipling D. Telomere-based proliferative lifespan barriers in Werner-syndrome fibroblasts involve both p53-dependent and p53-independent mechanisms. J. Cell Sci. 2003;116:1349–1357. doi: 10.1242/jcs.00331. [DOI] [PubMed] [Google Scholar]
- 49.Dickson M. A., Hahn W. C., Ino Y., Ronfard V., Wu J. Y., Weinberg R. A., Louis D. N., Li F. P., Rheinwald J. G. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/MCB.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.El-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K.W., Vogelstein B. WAF-1, a potential mediator of p53 tumour suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 51.Harper J. W., Adami G. R., Wei N., Keyomarsi K., Elledge S. J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-G. [DOI] [PubMed] [Google Scholar]
- 52.Noda A., Ning Y., Venable S. F., Pereira Smith O. M., Smith J. R. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp. Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 53.Evans R. J., Wyllie F. S., Wynford-Thomas D., Kipling D., Jones C. J. AP53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res. 2003;63:4854–4861. [PubMed] [Google Scholar]
- 54.Sharpless N. E., DePinho R. A. Cancer biology: gone but not forgotten. Nature. 2007;445:606–607. doi: 10.1038/nature05567. [DOI] [PubMed] [Google Scholar]
- 55.Rudolph K. L., Chang S., Lee H.W., Blasco M., Gottlieb G. J., Greider C., DePinho R. A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/S0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 56.Chin L., Artandi S. E., Shen Q., Tam A., Lee S. L., Gottlieb G. J., Greider C. W., DePinho R. A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/S0092-8674(00)80762-X. [DOI] [PubMed] [Google Scholar]
- 57.Choudhury A. R., Ju Z., Djojosubroto M.W., Schienke A., Lechel A., Schaetzlein S., Jiang H., Stepczynska A., Wang C., Buer J., Lee H.W., von Zglinicki T., Ganser A., Schirmacher P., Nakauchi H., Rudolph K. L. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- 58.Werner C. W.O. Über Katarakt in Verbindung mit Sklerodermie. Kiel: University of Kiel. Schmidt and Klaunig; 1904. [Google Scholar]
- 59.Goto M. Clinical characteristics of Werner syndrome and other premature aging syndromes: pattern of aging in progeroid syndromes. In: Goto M. and., Miller R. W., editors. From Premature Gray Hair to Helicase-Werner Syndrome: Implications for Aging and Cancer. Tokyo: Japan Scientific Societies Press; 2001. pp. 27–39. [Google Scholar]
- 60.Anderson N. E., Haas L. F. Neurological complications of Werner's syndrome. J. Neurol. 2003;250:1174–1178. doi: 10.1007/s00415-003-0168-3. [DOI] [PubMed] [Google Scholar]
- 61.Goto M. Hierarchical deterioration of body systems in Werner's syndrome: implications for normal ageing. Mech. Ageing Dev. 1997;98:239–254. doi: 10.1016/S0047-6374(97)00111-5. [DOI] [PubMed] [Google Scholar]
- 62.Epstein C. J., Martin G. M., Schultz A. L., Motulsky A. G. Werner's syndrome: a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 63.Payao S. L., de Labio R.W., Gatti L. L., Rigolin V. O., Bertolucci P. H., Smith Mde A. Werner helicase polymorphism is not associated with Alzheimer's disease. J. Alzheimers Dis. 2004;6:591–594. doi: 10.3233/jad-2004-6603. [DOI] [PubMed] [Google Scholar]
- 64.Goto M., Miller R.W., editors. From Premature Gray Hair to Helicase-Werner Syndrome: Implications for Aging and Cancer. Tokyo: Japan Scientific Societies Press; 2001. p. 178. [Google Scholar]
- 65.Huang S., Lee L., Hanson N. B., Lenaerts C., Hoehn H., Poot M., Rubin C. D., Chen D. F., Yang C. C., Juch H., Dorn T., Spiegel R., et al. The spectrum of WRN mutations in Werner syndrome patients. Hum. Mutat. 2006;27:558–567. doi: 10.1002/humu.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu C. E., Oshima J., Goddard K. A., Miki T., Nakura J., Ogihara T., Poot M., Hoehn H., Fraccaro M., Piussan C., Martin G. M., Schellenberg G.D., Wijsman E. M. Linkage disequilibrium and haplotype studies of chromosome 8p 11.1–21.1 markers and Werner syndrome. Am. J. Hum. Genet. 1994;55:356–364. [PMC free article] [PubMed] [Google Scholar]
- 67.Imbert A., Chaffanet M., Essioux L., Noguchi T., Adelaide J., Kerangueven F., Le Paslier D., Bonaiti-Pellie C., Sobol H., Birnbaum D., Pebusque M. J. Integrated map of the chromosome 8p12-p21 region, a region involved in human cancers and Werner syndrome. Genomics. 1996;32:29–38. doi: 10.1006/geno.1996.0073. [DOI] [PubMed] [Google Scholar]
- 68.Yu C. E., Oshima J., Fu Y. H., Wijsman E. M., Hisama F., Alisch R., Matthews S., Nakura J., Miki T., Ouais S., Martin G. M., Mulligan J., Schellenberg G. D. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 69.Bachrati C. Z., Hickson I. D. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem. J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cordin O., Banroques J., Tanner N. K., Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 71.Gray M. D., Shen J. C., Kamath-Loeb A. S., Blank A., Sopher B. L., Martin G. M., Oshima J., Loeb L. A. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- 72.Huang S., Li B., Gray M. D., Oshima J., Mian I. S., Campisi J. The premature ageing syndrome protein, WRN, is a 3′-5′ exonuclease. Nat. Genet. 1998;20:114–116. doi: 10.1038/2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsumoto T., Imamura O., Goto M., Furuichi Y. Characterization of the nuclear localization signal in the DNA helicase involved inWerner's syndrome. Int. J. Mol. Med. 1998;1:71–76. doi: 10.3892/ijmm.1.1.71. [DOI] [PubMed] [Google Scholar]
- 74.von Kobbe C., Bohr V. A. A nucleolar targeting sequence in the Werner syndrome protein resides within residues 949–1092. J. Cell Sci. 2002;115:3901–3907. doi: 10.1242/jcs.00076. [DOI] [PubMed] [Google Scholar]
- 75.Suzuki T., Shiratori M., Furuichi Y., Matsumoto T. Diverged nuclear localization of Werner helicase in human and mouse cells. Oncogene. 2001;20:2551–2558. doi: 10.1038/sj.onc.1204344. [DOI] [PubMed] [Google Scholar]
- 76.Marciniak R. A., Lombard D. B., Johnson F. B., Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proc. Natl. Acad. Sci. USA. 1998;95:6887–6892. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bohr V. A. Deficient DNA repair in the human progeroid disorder, Werner syndrome. Mutat. Res. 2005;577:252–259. doi: 10.1016/j.mrfmmm.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 78.Moser M. J., Oshima J., Monnat R. J., Jr. WRN mutations in Werner syndrome. Hum. Mutat. 1999;13:271–279. doi: 10.1002/(SICI)1098-1004(1999)13:4<271::AID-HUMU2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 79.Kashino G., Kodama S., Suzuki K., Oshimura M., Watanabe M. Preferential expression of an intact WRN gene in Werner syndrome cell lines in which a normal chromosome 8 has been introduced. Biochem. Biophys. Res. Commun. 2001;289:111–115. doi: 10.1006/bbrc.2001.5933. [DOI] [PubMed] [Google Scholar]
- 80.Moser M. J., Kamath-Loeb A. S., Jacob J. E., Bennett S. E., Oshima J., Monnat R. J., Jr. WRN helicase expression in Werner syndrome cell lines. Nucleic Acids Res. 2000;28:648–654. doi: 10.1093/nar/28.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Killoran M. P., Keck J. L. Sit down, relax and unwind: structural insights into RecQ helicase mechanisms. Nucleic Acids Res. 2006;34:4098–4105. doi: 10.1093/nar/gkl538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamath-Loeb A. S., Welcsh P., Waite M., Adman E. T., Loeb L. A. The enzymatic activities of the Werner syndrome protein are disabled by the amino acid polymorphism R834C. J. Biol. Chem. 2004;279:55499–55505. doi: 10.1074/jbc.M407128200. [DOI] [PubMed] [Google Scholar]
- 83.Castro E., Edland S. D., Lee L., Ogburn C. E., Deeb S. S., Brown G., Panduro A., Riestra R., Tilvis R., Louhija J., Penttinen R., Erkkola R., Wang L., Martin G. M., Oshima J. Polymorphisms at the Werner locus. II. 1074Leu/Phe, 1367Cys/Arg, longevity, and atherosclerosis. Am. J. Med. Genet. 2000;95:374–380. doi: 10.1002/1096-8628(20001211)95:4<374::AID-AJMG14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 84.Passarino G., Shen P., Van Kirk J. B., Lin A. A., De Benedictis G., Cavalli Sforza L. L., Oefner P. J., Underhill P. A. The Werner syndrome gene and global sequence variation. Genomics. 2001;71:118–122. doi: 10.1006/geno.2000.6405. [DOI] [PubMed] [Google Scholar]
- 85.Smith M. A., Silva M. D., Araujo L. Q., Ramos L. R., Labio R.W., Burbano R. R., Peres C. A., Andreoli S. B., Payao S. L., Cendoroglo M. S. Frequency of Werner helicase 1367 polymorphism and age-related morbidity in an elderly Brazilian population. Braz. J. Med. Biol. Res. 2005;38:1053–1059. doi: 10.1590/S0100-879X2005000700008. [DOI] [PubMed] [Google Scholar]
- 86.Castro E., Ogburn C. E., Hunt K. E., Tilvis R., Louhija J., Penttinen R., Erkkola R., Panduro A., Riestra R., Piussan C., Deeb S. S., Wang L., et al. Polymorphisms at the Werner locus: I. Newly identified polymorphisms, ethnic variability of 1367Cy/Arg, and its stability in a population of Finnish centenarians. Am. J. Med. Genet. 1999;82:399–403. doi: 10.1002/(SICI)1096-8628(19990219)82:5<399::AID-AJMG8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 87.Ye L., Miki T., Nakura J., Oshima J., Kamino K., Rakugi H., Ikegami H., Higaki J., Edland S. D., Martin G. M., Ogihara T. Association of a polymorphic variant of the Werner helicase gene with myocardial infarction in a Japanese population [published erratum appears in Am. J. Med. Genet. 1997 May 2;70(1),103] Am. J. Med. Genet. 1997;68:494–498. doi: 10.1002/(SICI)1096-8628(19970211)68:4<494::AID-AJMG30>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 88.Bohr V. A., Metter E. J., Harrigan J. A., von Kobbe C., Liu J. L., Gray M. D., Majumdar A., Wilson D. M., 3rd, Seidman M. M. Werner syndrome protein 1367 variants and disposition towards coronary artery disease in Caucasian patients. Mech. Ageing Dev. 2004;125:491–496. doi: 10.1016/j.mad.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 89.Brosh R. M., Jr., Waheed J., Sommers J. A. Biochemical characterization of the DNA substrate specificity of Werner syndrome helicase. J. Biol. Chem. 2002;277:23236–23245. doi: 10.1074/jbc.M111446200. [DOI] [PubMed] [Google Scholar]
- 90.Salk D., Au K., Hoehn H., Martin G. M. Cytogenetics of Werner's syndrome cultured skin fibroblasts: variegated translocation mosaicism. Cytogenet. Cell. Genet. 1981;30:92–107. doi: 10.1159/000131596. [DOI] [PubMed] [Google Scholar]
- 91.Scappaticci S., Cerimele D., Fraccaro M. Clonal structural chromosomal rearrangements in primary fibroblast cultures and in lymphocytes of patients with Werner's Syndrome. Hum. Genet. 1982;62:16–24. doi: 10.1007/BF00295599. [DOI] [PubMed] [Google Scholar]
- 92.Fukuchi K., Martin G. M., Monnat R. J., Jr. Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc. Natl. Acad. Sci. USA. 1989;86:5893–5897. doi: 10.1073/pnas.86.15.5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Agrelo R., Cheng W. H., Setien F., Ropero S., Espada J., Fraga M. F., Herranz M., Paz M. F., Sanchez-Cespedes M., Artiga M. J., Guerrero D., Castells A., von Kobbe C., Bohr V. A., Esteller M. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc. Natl. Acad. Sci. USA. 2006;103:8822–8827. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lebel M., Spillare E. A., Harris C. C., Leder P. TheWerner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J. Biol. Chem. 1999;274:37795–37799. doi: 10.1074/jbc.274.53.37795. [DOI] [PubMed] [Google Scholar]
- 95.Cox L. S. Who binds wins: PCNA rings out the cell cycle changes. Trends in Cell Biol. 1997;7:493–498. doi: 10.1016/S0962-8924(97)01170-7. [DOI] [PubMed] [Google Scholar]
- 96.Warbrick E. PCNA binding through a conserved motif. Bioessays. 1998;20:195–199. doi: 10.1002/(SICI)1521-1878(199803)20:3<195::AID-BIES2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 97.Rodriguez-Lopez A. M., Jackson D. A., Nehlin J. O., Iborra F., Warren A. V., Cox L. S. Characterisation of the interaction between WRN, the helicase/exonuclease defective in progeroid Werner's syndrome, and an essential replication factor, PCNA. Mech. Ageing Dev. 2003;124:167–174. doi: 10.1016/S0047-6374(02)00131-8. [DOI] [PubMed] [Google Scholar]
- 98.Brosh R. M., Jr., von Kobbe C., Sommers J. A., Karmakar P., Opresko P. L., Piotrowski J., Dianova I., Dianov G. L., Bohr V. A. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sharma S., Otterlei M., Sommers J. A., Driscoll H. C., Dianov G. L., Kao H. I., Bambara R. A., Brosh R. M., Jr. WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol. Biol. Cell. 2004;15:734–750. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kao H. I., Bambara R. A. The protein components and mechanism of eukaryotic Okazaki fragment maturation. Crit. Rev. Biochem. Mol. Biol. 2003;38:433–452. doi: 10.1080/10409230390259382. [DOI] [PubMed] [Google Scholar]
- 101.Brosh R. M., Jr., Driscoll H. C., Dianov G. L., Sommers J. A. Biochemical characterization of the WRNFEN- 1 functional interaction. Biochemistry. 2002;41:12204–12216. doi: 10.1021/bi026031j. [DOI] [PubMed] [Google Scholar]
- 102.Zheng L., Zhou M., Chai Q., Parrish J., Xue D., Patrick S. M., Turchi J. J., Yannone S. M., Chen D., Shen B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005;6:83–89. doi: 10.1038/sj.embor.7400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pichierri P., Franchitto A., Mosesso P., Palitti F. Werner's syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol. Biol. Cell. 2001;12:2412–2421. doi: 10.1091/mbc.12.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rodriguez-Lopez A. M., Jackson D. A., Iborra F., Cox L. S. Asymmetry of DNA replication fork progression in Werner's syndrome. Aging Cell. 2002;1:30–39. doi: 10.1046/j.1474-9728.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 105.Rodriguez-Lopez A. M., Whitby M. C., Borer C. M., Bachler M. A., Cox L. S. Correction of proliferation and drug sensitivity defects in the progeroid Werner's syndrome by Holliday junction resolution. Rejuvenation Res. 2007;10:27–40. doi: 10.1089/rej.2006.0503. [DOI] [PubMed] [Google Scholar]
- 106.Sharma S., Sommers J. A., Gary R. K., Friedrich-Heineken E., Hubscher U., Brosh R. M., Jr. The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33:6769–6781. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Constantinou A., Tarsounas M., Karow J. K., Brosh R. M., Bohr V. A., Hickson I. D., West S. C. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takeuchi F., Hanaoka F., Goto M., Yamada M., Miyamoto T. Prolongation of S phase and whole cell cycle in Werner's syndrome fibroblasts. Exp. Gerontol. 1982;17:473–480. doi: 10.1016/S0531-5565(82)80009-0. [DOI] [PubMed] [Google Scholar]
- 109.Poot M., Hoehn H., Runger T. M., Martin G. M. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp. Cell Res. 1992;202:267–273. doi: 10.1016/0014-4827(92)90074-I. [DOI] [PubMed] [Google Scholar]
- 110.Griffiths T. D. S-phase transit times as a function of age in human diploid fibroblasts. Mech. Ageing Dev. 1984;24:273–282. doi: 10.1016/0047-6374(84)90113-1. [DOI] [PubMed] [Google Scholar]
- 111.Fujiwara Y., Higashikawa T., Tatsumi M. A retarded rate of DNA replication and normal level of DNA repair in Werner's syndrome fibroblasts in culture. J. Cell Physiol. 1977;92:365–374. doi: 10.1002/jcp.1040920305. [DOI] [PubMed] [Google Scholar]
- 112.Takeuchi F., Hanaoka F., Goto M., Akaoka I., Hori T., Yamada M., Miyamoto T. Altered frequency of initiation sites of DNA replication inWerner's syndrome cells. Hum. Genet. 1982;60:365–368. doi: 10.1007/BF00569220. [DOI] [PubMed] [Google Scholar]
- 113.Machwe A., Xiao L., Groden J., Orren D. K. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- 114.Pichierri P., Franchitto A., Mosesso P., Proietti de Santis L., Balajee A. S., Palitti F. Werner's syndrome lymphoblastoid cells are hypersensitive to topoisomerase II inhibitors in the G2 phase of the cell cycle. Mutat. Res. 2000;459:123–133. doi: 10.1016/s0921-8777(99)00065-8. [DOI] [PubMed] [Google Scholar]
- 115.Poot M., Gollahon K. A., Rabinovitch P. S. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum. Genet. 1999;104:10–14. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- 116.Lowe J., Sheerin A., Jennert-Burston K., Burton D., Ostler E. L., Bird J., Green M. H., Faragher R. G. Camptothecin sensitivity in Werner syndrome fibroblasts as assessed by the COMET technique. Ann. NY Acad. Sci. 2004;1019:256–259. doi: 10.1196/annals.1297.042. [DOI] [PubMed] [Google Scholar]
- 117.Shao R. G., Cao C. X., Zhang H., Kohn K.W., Wold M. S., Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Laine J. P., Opresko P. L., Indig F. E., Harrigan J. A., von Kobbe C., Bohr V. A. Werner protein stimulates topoisomerase I DNA relaxation activity. Cancer Res. 2003;63:7136–7146. [PubMed] [Google Scholar]
- 119.Gerber J. K., Gogel E., Berger C., Wallisch M., Muller F., Grummt I., Grummt F. Termination of mammalian rDNA replication: polar arrest of replication fork movement by transcription termination factor TTF-I. Cell. 1997;90:559–567. doi: 10.1016/S0092-8674(00)80515-2. [DOI] [PubMed] [Google Scholar]
- 120.Caburet S., Conti C., Schurra C., Lebofsky R., Edelstein S. J., Bensimon A. Human ribosomal RNA gene arrays display a broad range of palindromic structures. Genome Res. 2005;15:1079–1085. doi: 10.1101/gr.3970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sinclair D. A., Mills K., Guarente L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science. 1997;277:1313–1316. doi: 10.1126/science.277.5330.1313. [DOI] [PubMed] [Google Scholar]
- 122.Sinclair D. A., Guarente L. Extrachromosomal rDNA circles-a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/S0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 123.Kamath-Loeb A. S., Loeb L. A., Johansson E., Burgers P. M., Fry M. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J. Biol. Chem. 2001;276:16439–16446. doi: 10.1074/jbc.M100253200. [DOI] [PubMed] [Google Scholar]
- 124.Orren D. K., Theodore S., Machwe A. The Werner syndrome helicase/exonuclease (WRN) disrupts and degrades D-loops in vitro. Biochemistry. 2002;41:13483–13488. doi: 10.1021/bi0266986. [DOI] [PubMed] [Google Scholar]
- 125.Machwe A., Xiao L., Orren D. K. TRF2 recruits the Werner syndrome (WRN) exonuclease for processing of telomeric DNA. Oncogene. 2004;23:149–156. doi: 10.1038/sj.onc.1206906. [DOI] [PubMed] [Google Scholar]
- 126.Opresko P. L., von Kobbe C., Laine J. P., Harrigan J., Hickson I. D., Bohr V. A. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J. Biol. Chem. 2002;277:41110–41119. doi: 10.1074/jbc.M205396200. [DOI] [PubMed] [Google Scholar]
- 127.Opresko P. L., Otterlei M., Graakjaer J., Bruheim P., Dawut L., Kolvraa S., May A., Seidman M. M., Bohr V. A. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell. 2004;14:763–774. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 128.Crabbe L., Verdun R. E., Haggblom C. I., Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 129.Schulz V. P., Zakian V. A., Ogburn C. E., McKay J., Jarzebowicz A. A., Edland S. D., Martin G. M. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum. Genet. 1996;97:750–754. doi: 10.1007/BF02346184. [DOI] [PubMed] [Google Scholar]
- 130.Tahara H., Tokutake Y., Maeda S., Kataoka H., Watanabe T., Satoh M., Matsumoto T., Sugawara M., Ide T., Goto M., Furuichi Y., Sugimoto M. Abnormal telomere dynamics of B-lymphoblastoid cell strains from Werner's syndrome patients transformed by Epstein-Barr virus. Oncogene. 1997;15:1911–1920. doi: 10.1038/sj.onc.1201377. [DOI] [PubMed] [Google Scholar]
- 131.Baird D. M., Davis T., Rowson J., Jones C. J., Kipling D. Normal telomere erosion rates at the single cell level in Werner syndrome fibroblast cells. Hum. Mol. Genet. 2004;13:1515–1524. doi: 10.1093/hmg/ddh159. [DOI] [PubMed] [Google Scholar]
- 132.Wyllie F. S., Jones C. J., Skinner J.W., Haughton M. F., Wallis C., Wynford-Thomas D., Faragher R. G., Kipling D. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat. Genet. 2000;24:16–17. doi: 10.1038/71630. [DOI] [PubMed] [Google Scholar]
- 133.Ostler E. L., Wallis C. V., Sheerin A. N., Faragher R. G. A model for the phenotypic presentation of Werner's syndrome. Exp. Gerontol. 2002;37:285–292. doi: 10.1016/S0531-5565(01)00194-2. [DOI] [PubMed] [Google Scholar]
- 134.Bird J., Ostler E. L., Faragher R. G. Can we say that senescent cells cause ageing? Exp. Gerontol. 2003;38:1319–1326. doi: 10.1016/j.exger.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 135.Ariyoshi, K., Kodama, S., and Watanabe, M. (in press) Increased chromosome instability and accumulation of DNA double-strand breaks in Werner syndrome cells. J. Radiat. Res. [DOI] [PubMed]
- 136.Crabbe L., Jauch A., Naeger C. M., Holtgreve-Grez H., Karlseder J. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc. Natl. Acad. Sci. USA. 2007;104:2205–2210. doi: 10.1073/pnas.0609410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Saintigny Y., Makienko K., Swanson C., Emond M. J., Monnat R. J., Jr. Homologous recombination resolution defect in werner syndrome. Mol. Cell Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Baynton K., Otterlei M., Bjoras M., von Kobbe C., Bohr V. A., Seeberg E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 2003;278:36476–36486. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- 139.Prince P. R., Emond M. J., Monnat R. J., Jr. Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001;15:933–938. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yamagata K., Kato J., Shimamoto A., Goto M., Furuichi Y., Ikeda H. Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc. Natl. Acad. Sci. USA. 1998;95:8733–8738. doi: 10.1073/pnas.95.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stewart E., Chapman C. R., Al-Khodairy F., Carr A. M., Enoch T. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 1997;16:2682–2692. doi: 10.1093/emboj/16.10.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cheng W. H., von Kobbe C., Opresko P. L., Arthur L. M., Komatsu K., Seidman M. M., Carney J. P., Bohr V. A. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J. Biol. Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- 143.Cheng W. H., Sakamoto S., Fox J. T., Komatsu K., Carney J., Bohr V. A. Werner syndrome protein associates with gamma H2AX in a manner that depends upon Nbs1. FEBS Lett. 2005;579:1350–1356. doi: 10.1016/j.febslet.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 144.Stravardi E. S., Halazonetis T. D. Nbs1 moving up in the world. Nat. Cell Biol. 2005;7:648–650. doi: 10.1038/ncb0705-648. [DOI] [PubMed] [Google Scholar]
- 145.Plank J. L., Wu J., Hsieh T. S. Topoisomerase IIIalpha and Bloom's helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc. Natl. Acad. Sci. USA. 2006;103:11118–11123. doi: 10.1073/pnas.0604873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Raynard S., Bussen W., Sung P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J. Biol. Chem. 2006;281(20):13861–13864. doi: 10.1074/jbc.C600051200. [DOI] [PubMed] [Google Scholar]
- 147.Wu L., Lung Chan K., Ralf C., Bernstein D. A., Garcia P. L., Bohr V. A., Vindigni A., Janscak P., Keck J. L., Hickson I. D. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 2005;24:2679–2687. doi: 10.1038/sj.emboj.7600740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Runger T. M., Bauer C., Dekant B., Moller K., Sobotta P., Czerny C., Poot M., Martin G. M. Hypermutable ligation of plasmid DNA ends in cells from patients with Werner syndrome. J. Invest. Dermatol. 1994;102:45–48. doi: 10.1111/1523-1747.ep12371730. [DOI] [PubMed] [Google Scholar]
- 149.Oshima J., Huang S., Pae C., Campisi J., Schiestl R. H. Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 2002;62:547–551. [PubMed] [Google Scholar]
- 150.Chen L., Huang S., Lee L., Davalos A., Schiestl R. H., Campisi J., Oshima J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;2:191–199. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 151.Li B., Navarro S., Kasahara N., Comai L. Identification and biochemical characterization of aWerner's syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1. J. Biol. Chem. 2004;279:13659–13667. doi: 10.1074/jbc.M311606200. [DOI] [PubMed] [Google Scholar]
- 152.Perry J. J., Yannone S. M., Holden L. G., Hitomi C., Asaithamby A., Han S., Cooper P. K., Chen D. J., Tainer J. A. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 153.Otsuki M., Seki M., Kawabe Y., Inoue E., Dong Y. P., Abe T., Kato G., Yoshimura A., Tada S., Enomoto T. WRN counteracts the NHEJ pathway upon camptothecin exposure. Biochem. Biophys. Res. Commun. 2007;355:477–482. doi: 10.1016/j.bbrc.2007.01.175. [DOI] [PubMed] [Google Scholar]
- 154.Prince P. R., Ogburn C. E., Moser M. J., Emond M. J., Martin G. M., Monnat R. J., Jr. Cell fusion corrects the 4-nitroquinoline 1-oxide sensitivity of Werner syndrome fibroblast cell lines. Hum. Genet. 1999;105:132–138. doi: 10.1007/s004390051075. [DOI] [PubMed] [Google Scholar]
- 155.Von Kobbe C., May A., Grandori C., Bohr V. A. Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. 2004;18:1970–1972. doi: 10.1096/fj.04-1895fje. [DOI] [PubMed] [Google Scholar]
- 156.Szekely A. M., Bleichert F., Numann A., Van Komen S., Manasanch E., Ben Nasr A., Canaan A., Weissman S. M. Werner protein protects nonproliferating cells from oxidative DNA damage. Mol. Cell. Biol. 2005;25:10492–10506. doi: 10.1128/MCB.25.23.10492-10506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.von Kobbe C., Harrigan J. A., May A., Opresko P. L., Dawut L., Cheng W. H., Bohr V. A. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol. Cell. Biol. 2003;23:8601–8613. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Blank A., Bobola M. S., Gold B., Varadarajan S., Kolstoe D.D., Meade E. H., Rabinovitch P. S., Loeb L. A., Silber J. R. The Werner syndrome protein confers resistance to the DNA lesions N3-methyladenine and O6-methylguanine: implications for WRN function. DNA Repair (Amst.) 2004;3:629–638. doi: 10.1016/j.dnarep.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 159.Harrigan J. A., Wilson D. M., 3rd, Prasad R., Opresko P. L., Beck G., May A., Wilson S. H., Bohr V. A. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006;34:745–754. doi: 10.1093/nar/gkj475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Harrigan J. A., Opresko P. L., von Kobbe C., Kedar P. S., Prasad R., Wilson S. H., Bohr V. A. The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activity. J. Biol. Chem. 2003;278:22686–22695. doi: 10.1074/jbc.M213103200. [DOI] [PubMed] [Google Scholar]
- 161.Ahn B., Harrigan J. A., Indig F. E., Wilson D. M., 3rd, Bohr V. A. Regulation of WRN helicase activity in human base excision repair. J. Biol. Chem. 2004;279:53465–53474. doi: 10.1074/jbc.M409624200. [DOI] [PubMed] [Google Scholar]
- 162.Frosina G., Fortini P., Rossi O., Carrozzino F., Raspaglio G., Cox L. S., Lane D. P., Abbondandalo A., Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 163.Brosh R. M., Jr., Orren D. K., Nehlin J. O., Ravn P. H., Kenny M. K., Machwe A., Bohr V. A. Functional and physical interaction between WRN helicase and human replication protein A. J. Biol. Chem. 1999;274:18341–18350. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- 164.Lebel M., Lavoie J., Gaudreault I., Bronsard M., Drouin R. Genetic cooperation between the Werner syndrome protein and poly(ADP-ribose) polymerase-1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am. J. Pathol. 2003;162:1559–1569. doi: 10.1016/S0002-9440(10)64290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.von Kobbe C., Harrigan J. A., Schreiber V., Stiegler P., Piotrowski J., Dawut L., Bohr V. A. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004;32:4003–4014. doi: 10.1093/nar/gkh721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gray M. D., Wang L., Youssoufian H., Martin G. M., Oshima J. Werner helicase is localized to transcriptionally active nucleoli of cycling cells. Exp. Cell Res. 1998;242:487–494. doi: 10.1006/excr.1998.4124. [DOI] [PubMed] [Google Scholar]
- 167.Shiratori M., Suzuki T., Itoh C., Goto M., Furuichi Y., Matsumoto T. WRN helicase accelerates the transcription of ribosomal RNA as a component of an RNA polymerase I-associated complex. Oncogene. 2002;21:2447–2454. doi: 10.1038/sj.onc.1205334. [DOI] [PubMed] [Google Scholar]
- 168.Balajee A. S., Machwe A., May A., Gray M. D., Oshima J., Martin G. M., Nehlin J. O., Brosh R., Orren D. K., Bohr V. A. The Werner syndrome protein is involved in RNA polymerase II transcription. Mol. Biol. Cell. 1999;10:2655–2668. doi: 10.1091/mbc.10.8.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Turaga, R. V.N., Massip, L., Chavz, A., Johnson, F. B., and Lebel, M. (in press) Werner syndrome protein prevents DNA breaks upon chromatin structure alteration. Aging Cell. [DOI] [PubMed]
- 170.Cogoni C., Macino G. Posttranscriptional gene silencing in Neurospora by a RecQ DNA helicase. Science. 1999;286:2342–2344. doi: 10.1126/science.286.5448.2342. [DOI] [PubMed] [Google Scholar]
- 171.Ketting R. F., Haverkamp T. H., van Luenen H. G., Plasterk R. H. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell. 1999;99:133–141. doi: 10.1016/S0092-8674(00)81645-1. [DOI] [PubMed] [Google Scholar]
- 172.Glazov E., Phillips K., Budziszewski G. J., Schob H., Meins F., Jr., Levin J. Z. A gene encoding an RNase D exonuclease-like protein is required for post-transcriptional silencing in Arabidopsis. Plant J. 2003;35:342–349. doi: 10.1046/j.1365-313X.2003.01810.x. [DOI] [PubMed] [Google Scholar]
- 173.Watt P. M., Louis E. J., Borts R. H., Hickson I. D. Sgs1: a eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell. 1995;81:253–260. doi: 10.1016/0092-8674(95)90335-6. [DOI] [PubMed] [Google Scholar]
- 174.Win T. Z., Mankouri H.W., Hickson I. D., Wang S. W. A role for the fission yeast Rqh1 helicase in chromosome segregation. J. Cell Sci. 2005;118:5777–5784. doi: 10.1242/jcs.02694. [DOI] [PubMed] [Google Scholar]
- 175.Uhlmann F., Nasmyth K. Cohesion between sister chromatids must be established during DNA replication. Curr. Biol. 1998;8:1095–1101. doi: 10.1016/S0960-9822(98)70463-4. [DOI] [PubMed] [Google Scholar]
- 176.Leng M., Chan D. W., Luo H., Zhu C., Qin J., Wang Y. MPS1-dependent mitotic BLM phosphorylation is important for chromosome stability. Proc. Natl. Acad. Sci. USA. 2006;103:11485–11490. doi: 10.1073/pnas.0601828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Salk D. Werner's syndrome: a review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Hum. Genet. 1982;62:1–15. doi: 10.1007/BF00295598. [DOI] [PubMed] [Google Scholar]
- 178.Faragher R. G., Kill I. R., Hunter J. A., Pope F. M., Tannock C., Shall S. The gene responsible for Werner syndrome may be a cell division “counting” gene. Proc. Natl. Acad. Sci. USA. 1993;90:12030–12034. doi: 10.1073/pnas.90.24.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Choi D., Whittier P. S., Oshima J., Funk W. D. Telomerase expression prevents replicative senescence but does not fully reset mRNA expression patterns in Werner syndrome cell strains. FASEB J. 2001;15:1014–1020. doi: 10.1096/fj.00-0104com. [DOI] [PubMed] [Google Scholar]
- 180.Ouellette M. M., McDaniel L. D., Wright W. E., Shay J.W., Schultz R. A. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 2000;9:403–411. doi: 10.1093/hmg/9.3.403. [DOI] [PubMed] [Google Scholar]
- 181.James S. E., Faragher R. G., Burke J. F., Shall S., Mayne L. V. Werner's syndrome T lymphocytes display a normal in vitro life-span. Mech. Ageing Dev. 2000;121:139–149. doi: 10.1016/S0047-6374(00)00205-0. [DOI] [PubMed] [Google Scholar]
- 182.Fukuchi K., Tanaka K., Kumahara Y., Marumo K., Pride M. B., Martin G. M., Monnat R. J., Jr. Increased frequency of 6-thioguanine-resistant peripheral blood lymphocytes in Werner syndrome patients. Hum.Genet. 1990;84:249–252. doi: 10.1007/BF00200569. [DOI] [PubMed] [Google Scholar]
- 183.Johnson F. B., Marciniak R. A., McVey M., Stewart S. A., Hahn W. C., Guarente L. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J. 2001;20:905–913. doi: 10.1093/emboj/20.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lebel M., Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular replicative capacity. Proc. Natl. Acad. Sci. USA. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wang L., Ogburn C. E., Ware C. B., Ladiges W. C., Youssoufian H., Martin G. M., Oshima J. Cellular Werner phenotypes in mice expressing a putative dominant-negative human WRN gene. Genetics. 2000;154:357–362. doi: 10.1093/genetics/154.1.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chang S., Multani A. S., Cabrera N. G., Naylor M. L., Laud P., Lombard D., Pathak S., Guarente L., DePinho R. A. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 187.Goto M., Miller R.W., Ishikawa Y., Sugano H. Excess of rare cancers in Werner syndrome (adult progeria) Cancer Epidemiol. Biomarkers Prev. 1996;5:239–246. [PubMed] [Google Scholar]
- 188.Yokote K., Hara K., Mori S., Kadowaki T., Saito Y., Goto M. Dysadipocytokinemia in werner syndrome and its recovery by treatment with pioglitazone. Diabetes Care. 2004;27:2562–2563. doi: 10.2337/diacare.27.10.2562. [DOI] [PubMed] [Google Scholar]
- 189.Hashimoto N., Hatanaka S., Yokote K., Kurosawa H., Yoshida T., Iwai R., Takahashi H., Yoshida K., Horie A., Sakurai K., Yagui K., Saito Y., Yoshida S. A patient with Werner syndrome and adiponectin gene mutation. Diabetes Res. Clin. Pract. 2007;75:27–29. doi: 10.1016/j.diabres.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 190.Yokote K., Honjo S., Kobayashi K., Fujimoto M., Kawamura H., Mori S., Saito Y. Metabolic improvement and abdominal fat redistribution in Werner syndrome by pioglitazone. J. Am. Geriatr. Soc. 2004;52:1582–1583. doi: 10.1111/j.1532-5415.2004.52430_4.x. [DOI] [PubMed] [Google Scholar]
- 191.Davis T., Baird D. M., Haughton M. F., Jones C. J., Kipling D. Prevention of accelerated cell aging in Werner syndrome using a p38 mitogen-activated protein kinase inhibitor. J. Gerontol. A Biol. Sci. Med. Sci. 2005;60:1386–1393. doi: 10.1093/gerona/60.11.1386. [DOI] [PubMed] [Google Scholar]
- 192.Hattori S., Kasai M., Namatame T., Hattori Y., Kasai K. Pioglitazone treatment of insulin resistance in a patient with Werner's syndrome. Diabetes Care. 2004;27:3021–3022. doi: 10.2337/diacare.27.12.3021. [DOI] [PubMed] [Google Scholar]
- 193.Fossel M. B. Cells, Aging and Human Disease. Oxford: Oxford University Press; 2004. p. 503. [Google Scholar]
- 194.De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C. L., Munnich A., LeMerrer M., Levy N. Lamin: a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 195.Eriksson M., Brown W. T., Gordon L. B., Glynn M.W., Singer J., Scott L., Erdos M. R., Robbins C. M., Moses T. Y., Berglund P., Dutra A., Pak E., Durkin S., Csoka A. B., Boehnke M., Glover T. W., Collins F. S. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Broers J. L., Machiels B. M., Kuijpers H. J., Smedts F., van den Kieboom R., Raymond Y., Ramaekers F. C. A- and B-type lamins are differentially expressed in normal human tissues. Histochem. Cell Biol. 1997;107:505–517. doi: 10.1007/s004180050138. [DOI] [PubMed] [Google Scholar]
- 197.Hutchison C. J., Worman H. J. A-type lamins: guardians of the soma? Nat. Cell Biol. 2004;6:1062–1067. doi: 10.1038/ncb1104-1062. [DOI] [PubMed] [Google Scholar]
- 198.Hutchison C. J., Bridger J. M., Cox L. S., Kill I. R. Weaving a pattern from disparate threads: lamin function in nuclear assembly and DNA replication. J. Cell Sci. 1994;107:3259–3269. doi: 10.1242/jcs.107.12.3259. [DOI] [PubMed] [Google Scholar]
- 199.Cox L. S., Hutchison C. J. Nuclear Envelope Assembly and Disassembly. In: Maddy A. H., Harris J. A., editors. Subcellular Biochemistry. New York: Plenum; 1994. pp. 263–325. [DOI] [PubMed] [Google Scholar]
- 200.Courvalin J. C., Segil N., Blobel G., Worman H. J. The lamin B receptor of the inner nuclear membrane undergoes mitosis-specific phosphorylation and is a substrate for p34cdc2-type protein kinase. J. Biol. Chem. 1992;267:19035–19038. [PubMed] [Google Scholar]
- 201.Heald R., McKeon F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell. 1990;61:579–589. doi: 10.1016/0092-8674(90)90470-Y. [DOI] [PubMed] [Google Scholar]
- 202.Goldman R. D., Gruenbaum Y., Moir R. D., Shumaker D. K., Spann T. P. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 2002;16:533–547. doi: 10.1101/gad.960502. [DOI] [PubMed] [Google Scholar]
- 203.Goldman R. D., Goldman A. E., Shumaker D. K. Nuclear lamins: building blocks of nuclear structure and function. Novartis Found Symp. 2005;264:3–16. doi: 10.1002/0470093765.ch2. [DOI] [PubMed] [Google Scholar]
- 204.Navarro C. L., Cau P., Levy N. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 2006;15(Suppl.2):R151–161. doi: 10.1093/hmg/ddl214. [DOI] [PubMed] [Google Scholar]
- 205.D'Apice M. R., Tenconi R., Mammi I., van den Ende J., Novelli G. Paternal origin of LMNA mutations in Hutchinson-Gilford progeria. Clin. Genet. 2004;65:52–54. doi: 10.1111/j..2004.00181.x. [DOI] [PubMed] [Google Scholar]
- 206.De Sandre-Giovannoli A., Levy N. Altered splicing in prelamin A-associated premature aging phenotypes. Prog. Mol. Subcell. Biol. 2006;44:199–232. doi: 10.1007/978-3-540-34449-0_9. [DOI] [PubMed] [Google Scholar]
- 207.Broers J. L., Hutchison C. J., Ramaekers F. C. Laminopathies. J. Pathol. 2004;204:478–488. doi: 10.1002/path.1655. [DOI] [PubMed] [Google Scholar]
- 208.Broers J. L., Ramaekers F. C., Bonne G., Yaou R. B., Hutchison C. J. Nuclear lamins: laminopathies and their role in premature ageing. Physiol. Rev. 2006;86:967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 209.Wuyts W., Biervliet M., Reyniers E., D'Apice M. R., Novelli G., Storm K. Somatic and gonadal mosaicism in Hutchinson-Gilford progeria. Am. J. Med. Genet. A. 2005;135:66–68. doi: 10.1002/ajmg.a.30663. [DOI] [PubMed] [Google Scholar]
- 210.Fong L. G., Ng J. K., Meta M., Cote N., Yang S. H., Stewart C. L., Sullivan T., Burghardt A., Majumdar S., Reue K., Bergo M. O., Young S. G. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc.Natl. Acad. Sci. USA. 2004;101:18111–18116. doi: 10.1073/pnas.0408558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Goldman R. D., Shumaker D. K., Erdos M. R., Eriksson M., Goldman A. E., Gordon L. B., Gruenbaum Y., Khuon S., Mendez M., Varga R., Collins F. S. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yang S. H., Bergo M. O., Toth J. I., Qiao X., Hu Y., Sandoval S., Meta M., Bendale P., Gelb M. H., Young S. G., Fong L. G. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Verstraeten V. L., Broers J. L., van Steensel M. A., Zinn- Justin S., Ramaekers F. C., Steijlen P. M., Kamps M., Kuijpers H. J., Merckx D., Smeets H. J., Hennekam R. C., Marcelis C. L., van den Wijngaard A. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum. Mol. Genet. 2006;15:2509–2522. doi: 10.1093/hmg/ddl172. [DOI] [PubMed] [Google Scholar]
- 214.Plasilova M., Chattopadhyay C., Pal P., Schaub N. A., Buechner S. A., Mueller H., Miny P., Ghosh A., Heinimann K. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson- Gilford progeria syndrome. J. Med. Genet. 2004;41:609–614. doi: 10.1136/jmg.2004.019661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Navarro C. L., De Sandre-Giovannoli A., Bernard R., Boccaccio I., Boyer A., Genevieve D., Hadj-Rabia S., Gaudy-Marqueste C., Smitt H. S., Vabres P., Faivre L., Verloes A., Van Essen, et al. LaminAand ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 216.Navarro C. L., Cadinanos J., De Sandre-Giovannoli A., Bernard R., Courrier S., Boccaccio I., Boyer A., Kleijer W. J., Wagner A., Giuliano F., Beemer F. A., Freije J. M., et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 2005;14:1503–1513. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- 217.Moulson C. L., Go G., Gardner J. M., van der Wal A. C., Smitt J. H., van Hagen J. M., Miner J. H. Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J. Invest. Dermatol. 2005;125:913–919. doi: 10.1111/j.0022-202X.2005.23846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Agarwal A. K., Fryns J. P., Auchus R. J., Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 219.Bridger J. M., Kill I. R. Aging of Hutchinson-Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosis. Exp. Gerontol. 2004;39:717–724. doi: 10.1016/j.exger.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 220.Scaffidi P., Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson- Gilford progeria syndrome. Nat. Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Columbaro M., Capanni C., Mattioli E., Novelli G., Parnaik V. K., Squarzoni S., Maraldi N. M., Lattanzi G. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell. Mol. Life Sci. 2005;62:2669–2678. doi: 10.1007/s00018-005-5318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Meier J., Campbell K. H., Ford C. C., Stick R., Hutchison C. J. The role of lamin LIII in nuclear assembly and DNA replication, in cell-free extracts of Xenopus eggs. J. Cell Sci. 1991;98:271–279. doi: 10.1242/jcs.98.3.271. [DOI] [PubMed] [Google Scholar]
- 223.Moir R. D., Spann T. P., Herrmann H., Goldman R. D. Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J. Cell Biol. 2000;149:1179–1192. doi: 10.1083/jcb.149.6.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Spann T. P., Moir R. D., Goldman A. E., Stick R., Goldman R. D. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J. Cell Biol. 1997;136:1201–1212. doi: 10.1083/jcb.136.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Goldberg M., Jenkins H., Allen T., Whitfield W. G., Hutchison C. J. Xenopus lamin B3 has a direct role in the assembly of a replication competent nucleus: evidence from cell-free egg extracts. J Cell Sci. 1995;108:3451–3461. doi: 10.1242/jcs.108.11.3451. [DOI] [PubMed] [Google Scholar]
- 226.Spann T. P., Goldman A. E., Wang C., Huang S., Goldman R. D. Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J. Cell Biol. 2002;156:603–608. doi: 10.1083/jcb.200112047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Van Berlo J. H., Voncken J.W., Kubben N., Broers J. L., Duisters R., van Leeuwen R. E., Crijns H. J., Ramaekers F. C., Hutchison C. J., Pinto Y. M. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum. Mol. Genet. 2005;14:2839–2849. doi: 10.1093/hmg/ddi316. [DOI] [PubMed] [Google Scholar]
- 228.Liu B., Wang J., Chan K. M., Tjia W. M., Deng W., Guan X., Huang J. D., Li K. M., Chau P. Y., Chen D. J., Pei D., Pendas A. M., et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005;11:780–785. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- 229.Pendas A. M., Zhou Z., Cadinanos J., Freije J.M., Wang J., Hultenby K., Astudillo A., Wernerson A., Rodriguez F., Tryggvason K., Lopez-Otin C. prelaminA processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 230.Mounkes L. C., Kozlov S., Hernandez L., Sullivan T., Stewart C. L. progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003;423:298–301. doi: 10.1038/nature01631. [DOI] [PubMed] [Google Scholar]
- 231.Varela I., Cadinanos J., Pendas A. M., Gutierrez-Fernandez A., Folgueras A. R., Sanchez L. M., Zhou Z., Rodriguez F. J., Stewart C. L., Vega J. A., Tryggvason K., Freije J. M., Lopez-Otin C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 232.Capanni C., Mattioli E., Columbaro M., Lucarelli E., Parnaik V. K., Novelli G., Wehnert M., Cenni V., Maraldi N. M., Squarzoni S., Lattanzi G. Altered prelamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005;14:1489–1502. doi: 10.1093/hmg/ddi158. [DOI] [PubMed] [Google Scholar]
- 233.Kim J. B., Wright H. M., Wright M., Spiegelman B. M. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA. 1998;95:4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Krimm I., Ostlund C., Gilquin B., Couprie J., Hossenlopp P., Mornon J. P., Bonne G., Courvalin J. C., Worman H. J., Zinn-Justin S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002;10:811–823. doi: 10.1016/S0969-2126(02)00777-3. [DOI] [PubMed] [Google Scholar]
- 235.Huang S., Chen L., Libina N., Janes J., Martin G. M., Campisi J., Oshima J. Correction of cellular phenotypes of Hutchinson-Gilford Progeria cells by RNA interference. Hum. Genet. 2005;118:444–450. doi: 10.1007/s00439-005-0051-7. [DOI] [PubMed] [Google Scholar]
- 236.Mallampalli M. P., Huyer G., Bendale P., Gelb M. H., Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 2005;102:14416–14421. doi: 10.1073/pnas.0503712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yang S. H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., Majumdar S., Bergo M. O., Young S. G., Fong L. G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndromemutation. J. Clin. Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]