

Abstract.
Hsp70 proteins are central components of the cellular network of molecular chaperones and folding catalysts. They assist a large variety of protein folding processes in the cell by transient association of their substrate binding domain with short hydrophobic peptide segments within their substrate proteins. The substrate binding and release cycle is driven by the switching of Hsp70 between the low-affinity ATP bound state and the high-affinity ADP bound state. Thus, ATP binding and hydrolysis are essential in vitro and in vivo for the chaperone activity of Hsp70 proteins. This ATPase cycle is controlled by co-chaperones of the family of J-domain proteins, which target Hsp70s to their substrates, and by nucleotide exchange factors, which determine the lifetime of the Hsp70-substrate complex. Additional co-chaperones fine-tune this chaperone cycle. For specific tasks the Hsp70 cycle is coupled to the action of other chaperones, such as Hsp90 and Hsp100.
Key words. Hsp70, DnaK, DnaJ, heat shock proteins, chaperones, protein folding, quality control
Introduction
70-kDa heat shock proteins (Hsp70s) assist a wide range of folding processes, including the folding and assembly of newly synthesized proteins, refolding of misfolded and aggregated proteins, membrane translocation of organellar and secretory proteins, and control of the activity of regulatory proteins [1–7]. Hsp70s have thus housekeeping functions in the cell in which they are built-in components of folding and signal transduction pathways, and quality control functions in which they proofread the structure of proteins and repair misfolded conformers. All of these activities appear to be based on the property of Hsp70 to interact with hydrophobic peptide segments of proteins in an ATP-controlled fashion. The broad spectrum of cellular functions of Hsp70 proteins is achieved through (i) the amplification and diversification of hsp70 genes in evolution, which has generated specialized Hsp70 chaperones, (ii) co-chaperones which are selectively recruited by Hsp70 chaperones to fulfill specific cellular functions and (iii) cooperation of Hsp70s with other chaperone systems to broaden their activity spectrum. Hsp70 proteins with their co-chaperones and cooperating chaperones thus constitute a complex network of folding machines.
Protein folding processes assisted by Hsp70
The role of Hsp70s in the folding of non-native proteins can be divided into three related activities: prevention of aggregation, promotion of folding to the native state, and solubilization and refolding of aggregated proteins. In the cellular milieu, Hsp70s exert these activities in the quality control of misfolded proteins and the co- and posttranslational folding of newly synthesized proteins. Mechanistically related but less understood is the role of Hsp70s in the disassembly of protein complexes such as clathrin coats, viral capsids and the nucleoprotein complex, which initiates the replication of bacteriophage λ DNA. A more complex folding situation exists for the Hsp70-dependent control of regulatory proteins since several steps in the folding and activation process of these substrates are assisted by multiple chaperones.
Hsp70 proteins together with their co-chaperones of the J-domain protein (JDP) family prevent the aggregation of non-native proteins through association with hydrophobic patches of substrate molecules, which shields them from intermolecular interactions (‘holder’ activity). Some JDPs such as Escherichia coli DnaJ and Saccharomyces cerevisiae Ydj1 can prevent aggregation by themselves through ATP-independent transient and rapid association with the substrates. Only members of the Hsp70 family with general chaperone functions have such general holder activity.
Hsp70 chaperone systems assist non-native folding intermediates to fold to the native state (‘folder’ activity). The mechanism by which Hsp70-chaperones assist the folding of non-native substrates is still unclear. Hsp70-dependent protein folding in vitro occurs typically on the time scale of minutes or longer. Substrates cycle between chaperone-bound and free states until the ensemble of molecules has reached the native state. There are at least two alternative modes of action. In the first mechanism Hsp70s play a rather passive role. Through repetitive substrate binding and release cycles they keep the free concentration of the substrate sufficiently low to prevent aggregation, while allowing free molecules to fold to the native state (‘kinetic partitioning’). In the second mechanism, the binding and release cycles induce local unfolding in the substrate, e.g. the untangling of a misfolded β-sheet, which helps to overcome kinetic barriers for folding to the native state (‘local unfolding’) [8–11]. The energy of ATP may be used to induce such conformational changes or alternatively to drive the ATPase cycle in the right direction.
Protein aggregates can be solubilized by some Hsp70 homologs in cooperation with chaperones of the Hsp100 family (Hsp104/ClpB) and subsequently refolded into the native state [12–14]. In case of the E. coli Hsp70 and Hsp100 homologs, DnaK and ClpB, the requirement for ClpB is alleviated below a threshold size of the aggregates [15]. Small aggregates (2- to 3-mers) of glucose-6-phosphate dehydrogenase were efficiently solubilized and subsequently refolded by the DnaK system alone. Larger aggregates (up to 10-mers) could be solubilized only partially by the DnaK system, which then was necessary in a high molar excess. In the presence of ClpB these larger aggregates were disaggregated more efficiently even at stoichiometric concentrations of DnaK. Based on the dependence of the DnaK:substrate stoichiometry optimal for disaggregation on the aggregate size, Goloubinoff and co-workers proposed the ‘Brownian power stroke’ model for disaggregation. This model assumes that the relatively small Hsp70 molecule retains, when it is bound to the large protein aggregate, a higher thermal mobility as compared to the aggregate. This difference in thermal mobility creates a force that leads to the solubilization of the aggregated protein [16].
An increasing number of regulatory proteins of eukaryotes are known to be controlled in their biological activity through transient association with Hsp70. These proteins include nuclear receptors (steroid hormone receptors), kinases (Raf, eIF2α-kinase, CyclinB1/Cdk1) and transcription factors (HSF, c-Myc, pRb). Several features emerge that are common to many of these chaperone-substrate complexes. First, formation of active complexes depends on the interaction with Hsp70 already during the de novo synthesis of the substrate. Second, the association of the chaperone is sensitive to substrate-specific ligands (e.g. steroid, heme) or substrate phosphorylation. Third, the complexes involve not only the Hsp70 system but also the Hsp90 system, co-chaperones such as Cdc37 and p23, and additional proteins including immunophilins [17]. Through such interactions Hsp70 chaperones are involved in signal transduction, cell cycle regulation, differentiation and programmed cell death. It is therefore not surprising that Hsp70 chaperones are important factors in developmental and pathological processes such as oncogenesis, neurodegenerative and autoimmune diseases, viral infections and aging [18–24].
Hsp70 in cellular physiology and pathophysiology
Two Hsp70 functions are especially interesting, de novo folding of nascent polypeptides and interaction with signal transduction proteins, and therefore some aspects of these functions shall be discussed below in more detail. Hsp70 chaperones were estimated to assist the de novo folding of 10–20% of all bacterial proteins whereby the dependence on Hsp70 for efficient folding correlated with the size of the protein [1, 2]. Since the average protein size in eukaryotic cells is increased (52 kDa in humans) as compared to bacteria (35 kDa in E. coli) [25], it is to be expected that an even larger percentage of eukaryotic proteins will be in need of Hsp70 during de novo folding. This reliance on Hsp70 chaperones increases even more under stress conditions. Interestingly, mutated proteins [for example mutant p53, cystis fibrosis transmembrane regulator (CFTR) variant ΔF508, mutant superoxid dismutase (SOD) 1] seem to require more attention by the Hsp70 chaperones than the corresponding wild-type protein [26–29]. As a consequence of this interaction the function of the mutant protein can be preserved. Thereby Hsp70 functions as a capacitor, buffering destabilizing mutations [30], a function demonstrated earlier for Hsp90 [31, 32]. Such mutations are only uncovered when the overall need for Hsp70 action exceeds the chaperone capacity of the Hsp70 proteins, for example during stress conditions [30], at certain stages in development or during aging, when the magnitude of stress-induced increase in Hsp70 levels declines [33, 34]. Alternatively, the mutant protein can be targeted by Hsp70 and its co-chaperones to degradation as shown e.g. for CFTRΔF508 and some of the SOD1 mutant proteins [35, 36]. Deleterious mutant proteins may then only accumulate when Hsp70 proteins are overwhelmed by other, stress-denatured proteins. Both mechanisms may contribute to pathological processes such as oncogenesis (mutant p53) and neurodegenerative diseases, including amyotrophic, lateral sclerosis (SOD1 mutations), Parkinsonism (α-synuclein mutations), Huntington's chorea (huntingtin with polyglutamin expansions) and spinocerebellar ataxias (proteins with polyglutamin expansions).
De novo folding is not necessarily accelerated by Hsp70 chaperones. In some cases folding is delayed for different reasons. First, folding of certain proteins can only proceed productively after synthesis of the polypeptide is completed as shown, e.g. for the reovirus lollipop-shaped protein sigma 1 [37]. Second, proteins destined for posttranslational insertion into organellar membranes are prevented from aggregation and transported to the translocation pore [38]. Third, in the case of the caspase-activated DNase (CAD), the active protein is dangerous for the cell and therefore can only complete folding in the presence of its specific inhibitor (ICAD). Hsp70 binds CAD cotranslationally and mediates folding only to an intermediate state. Folding is completed after addition of ICAD, which is assembled into a complex with CAD in an Hsp70-dependent manner [39]. Similar folding pathways may exist also for other potentially dangerous proteins.
As mentioned above Hsp70 interacts with key regulators of many signal transduction pathways controlling cell homeostasis, proliferation, differentiation and cell death. The interaction of Hsp70 with these regulatory proteins continues in activation cycles that also involve Hsp90 and a number of co-chaperones. The regulatory proteins, called clients, are thereby kept in an inactive state from which they are rapidly activated by the appropriate signals. Hsp70 and Hsp90 thus repress regulators in the absence of the upstream signal and guarantee full activation after the signal transduction pathway is switched on [6]. Hsp70 can be titrated away from these clients by other misfolded proteins that may arise from internal or external stresses. Consequently, through Hsp70 disturbances of the cellular system induced by environmental, developmental or pathological processes act on these signal transduction pathways.
In this way stress response and apoptosis are linked to each other. Hsp70 inhibits apoptosis acting on the caspase-dependent pathway at several steps both upstream and downstream of caspase activation and on the caspase-independent pathway. Overproduction of Hsp70 leads to increased resistance against apoptosis-inducing agents such as tumor necrosis factor-α (TNFα), staurosporin and doxorubicin, while downregulation of Hsp70 levels by antisense technology leads to increased sensitivity towards these agents [18, 40]. This observation relates to many pathological processes, such as oncogenesis, neurodegeneration and senescence. In many tumor cells increased Hsp70 levels are observed and correlate with increased malignancy and resistance to therapy. Downregulation of the Hsp70 levels in cancer cells induce differentiation and cell death [41]. Neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s corea and spinocerebellar ataxias are characterized by excessive apoptosis. In several different model systems overexpression of Hsp70 or one of its co-chaperones could overcome the neurodegenerative symptoms induced by expression of a disease-related gene (huntingtin, α-synuclein or ataxin) [20, 42]. Senescence in cell culture as well as aging in vivo is correlated with a continuous decline in the ability to mount a stress response [34, 43]. Age-related symptoms and diseases reflect this decreased ability to cope with cellular stresses. Interestingly, centenarians seem to be an exception to the rule, as they show a significant induction of Hsp70 production after heat shock challenge [44].
Mechanism of action
Hsp70 homologs consist of an N-terminal ATPase domain of 45 kDa and a C-terminal substrate binding domain of ca. 25 kDa which is further subdivided into a β-sandwich subdomain of 15 kDa and a C-terminal α-helical subdomain (fig. 1). Sequence alignment of Hsp70 protein sequences reveals a number of differences that may translate into functional differences, some of which will be described below. The ATPase cycle of Hsp70 consists of an alternation between the ATP state with low affinity and fast exchange rates for substrates, and the ADP state with high affinity and low exchange rates for substrates (fig. 2). The molecular mechanism of the ATPase and substrate binding/release cycles has been analyzed in detail only for a few Hsp70 homologs, including DnaK and HscA (Hsc66) of E. coli, DnaK of Thermus thermophilus, Ssa1 of S. cerevisiae, Hsc70 of bovine, and BiP of hamster and S. cerevisiae. These proteins show differences in their cycles, which have implications for their chaperone activities as outlined below.
Figure 1.

Structural aspects of Hsp70 chaperones and their interaction with co-chaperones and substrates. (a) ATPase domain of bovine Hsc70. Secondary structure representation of the ATPase domain of bovine Hsc70 in complex with ADP, inorganic phosphate and two potassium ions. To emphasized the flexibility and shearing motion of the ATPase domain an overlay of the x-ray structure (cyan; PDB entry code 1BUP; [90]) and a model derived by NMR using the method of residual dipolar coupling (dark blue; [46]) is shown. The two structures were aligned in the program WeblabViewer (Accelrys, San Diego) using tethers in subdomain IA only. (b) Compex of the ATPase domain of DnaK with a dimer of GrpE X-ray structure of the complex of the DnaK ATPase domain (blue) and the GrpE dimer (orange and yellow; PDB entry code 1DKG; [60]). In ball-and-stick and transparent surface representation are indicated the interacting residues. The ATPase domain is rotated by about 90° as compared to the representation shown in (a) as indicated at the bottom of panel (a). (c) Compex of the ATPase domain of Hsc70 with the Bag-domain fragment of Bag-1. X-ray structure of the complex of the human Hsc70 ATPase domain (green) and a fragment of Bag-1 (magenta; PDB entry code 1HX1; [61]). The interacting residues and the potential salt bridge (K56-E268) are indicated. The ATPase domain is rotated by about 90° as compared to the representation shown in (a) as indicated at the bottom of panel (a). (d) Model of the structure of the substrate binding domain. Ribbon representation of the structure of the substrate binding domain of E. coli DnaK (PDB entry code 1DKX; [74]) with bound peptide substrate (as stick model). Indicated are residues that contact the substrate in space-filling representation and residues that form the so-called latch of H-bonds and a salt bridge in ball-and-stick representation. (e) Interaction of E. coli DnaK with a peptide substrate. X-ray structure of the β-sheet domain of E. coli DnaK (PDB entry code 1DKX; [74]) in ribbon representation in complex with the peptide NRLLLTG in stick representation. Orientation as indicated in the inset with a-helices cut away along the dashed line and rotated by 90°. H-bonds to the backbone of the substrate are indicated as dotted lines. (f) Interaction of E. coli HscA with a peptide substrate. X-ray structure of the β-sheet domain of E. coli HscA (PDB entry code 1U00; [78] in ribbon representation in complex with the peptide ELPPVKIHC in stick representation. Orientation as indicated in the inset of panel (e) with α-helices cut away along the dashed line and rotated by 90°. H-bonds to the backbone of the substrate are indicated as dotted lines. Note the inverse orientation as compared to the peptide in (e).
Figure 2.
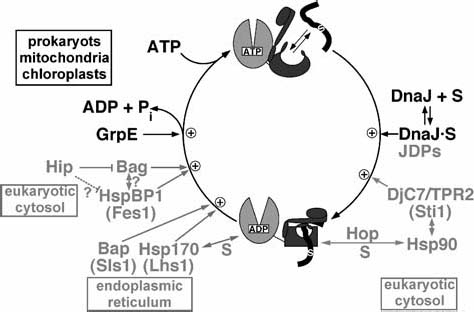
Model of the chaperone cycle of DnaK. In black is shown the basic cycle as elucidated for the DnaK system in E. coli. In gray are shown modifications found in the eukaryotic cytosol and endoplasmic reticulum
ATPase domain and ATPase cycle
The ATPase domain consists of two large globular subdomains (I and II), each further divided into two small subdomains (A and B; fig. 1a). The subdomains are separated by a deep cleft at the bottom of which the nucleotide binds in complex with one Mg2+ and two K+ ions contacting all four subdomains (IA, IB, IIA, IIB) [45]. The X-ray structures of the bovine Hsc70 ATPase domain complexed with several adenosine nucleotides (ADP, AMPPNP, ATP to mutant proteins) revealed that the adenosine nucleotide is positioned in the active site by interactions with two β- and γ-phosphate-binding loops and a hydrophobic adenosine binding pocket [45]. More recent nuclear magnetic resonance (NMR) investigations demonstrate a high flexibility of the ATPase domain with a shearing and tilting motion of the different subdomains towards each other, leading to an opening and closing of the nucleotide binding cleft (fig. 1a) [46]. A continuous opening and closing of the nucleotide binding cleft was proposed earlier to depend on the nucleotide present in the nucleotide binding site with the opening frequency being largest in the nucleotide-free state and decreasing in the order nucleotide-free > ADP > ADP+inorganic phosphate > ATP [47]. These observations explain the different dissociation rates for the different nucleotides and the influence of inorganic phosphate on these dissociation rates (see below).
Genetic and biochemical evidence clearly demonstrates that ATP hydrolysis is essential for the chaperone activity of all Hsp70 proteins tested so far. ATP hydrolysis is the ratelimiting step in the ATPase cycle of most Hsp70 proteins investigated except E. coli HscA and T. thermophilus DnaK. The rates of hydrolysis are very low in the ground state, ranging between 3·10−4 to 1.6·10−2 s−1, respectively [48]. The hydrolysis of ATP triggers the closing of the substrate binding cavity and the locking-in of associated substrates. In a thermodynamically coupled process, substrates stimulate the hydrolysis of ATP by typically 2–10-fold. This stimulation by substrates is too low to drive the functional cycle of Hsp70 chaperones. Instead, the activity of co-chaperones of the family of JDPs is required for productive tight coupling of ATP hydrolysis with substrate association [49–51]. J-domain co-chaperones are a heterogeneous class of multidomain proteins which share a conserved stretch of approx. 70 residues often located at the N-terminus (fig. 3b). This so-called J-domain is essential for the interaction of JDPs with their Hsp70 partner proteins. The mechanism of JDPs is best understood for the E. coli member, DnaJ.
Figure 3.

DnaJ and Bag-1 protein families. (a) Domain structure of the 3 DnaJ subfamilies. The different domains are marked in the following way: J, J-domain; G/F, Gly-Phe rich region; Zn, Zn2+ binding domain, C, C-terminal domain of homology. (b) Secondary structure representation of the J domain. NMR structure of the J-domain of E. coli DnaJ (PDB entry code 1XBL; [127], marked is the conserved HPD motif as ball-and-stick model. (c) Secondary structure representation of the Zn-binding and the C-terminal domain of S. cerevisiae Type I JDP Ydj1. X-ray structure of a fragment of yeast Ydj1 in complex with a peptide (red) (PDB entry code 1NLT; [128]. (d) Domain structure of the Bag family of proteins. Domains indicated: NLS, nuclear localization sequence; TRSEEX, Thr-Arg-Ser-Glu-Glu-Xaa repeat motif; Ub, ubiquitin-like domain; Bag, Bag homology region; WW, Trp-Trp-domain.
E. coli DnaJ and protein substrates synergistically stimulate the rate of ATP hydrolysis by >1000-fold [49, 51, 52]. The action of DnaJ requires both binding of protein substrates to the central hydrophobic pocket of DnaK’s substrate binding cavity and structural coupling between DnaK’s ATPase and substrate binding domains, which transmits the substrate binding event to the catalytic center [51]. Furthermore, DnaJ's coupling activity requires the ability to interact with DnaK through its J-domain [49, 53] and with substrates through its C-terminal substrate binding domain. DnaJ interacts with substrates in a rapid and transient fashion [54].
On the basis of the available data a model for the action of DnaJ has been proposed [49, 55]. Accordingly, DnaJ starts the functional cycle of the DnaK system by rapid and transient association with the substrate. The substrate can then be transferred onto DnaK in a two-step process involving the transient interaction of the J-domain with DnaK·ATP and the association of the substrate with the open substrate binding cavity of DnaK. Through an interdomain communication, the association of substrates and interaction with the J-domain lowers the activation energy for the hydrolysis of ATP by DnaK.
The synergistic stimulation of ATP hydrolysis by substrates and JDPs has also been observed in other Hsp70 systems, including the action of auxilin and clathrin on Hsc70, of Sec63 and substrates on BiP and of HscB and IscU on HscA [50, 56–58]. Thus, the coupling activity of DnaJ proteins appears to be a mechanism that is conserved at least in some Hsp70 chaperones.
The next step in the ATPase cycle, the release of ADP and Pi, allows the subsequent rapid binding of ATP and, consequently, the release of bound substrates and re-establishment of the starting point of the chaperone cycle. Under physiological conditions, i.e. high cytoplasmic ATP concentrations, nucleotide dissociation is rate limiting for substrate release. Nucleotide dissociation is thus a crucial step in the cycle, and it is therefore not surprising that this step is highly regulated and subject to strong evolutionary variation.
The dissociation of bound nucleotides requires an opening of the nucleotide binding cleft. Such movement has been observed for E. coli DnaK and bovine Hsc70 when their respective nucleotide exchange factors, GrpE and Bag-1, are bound. In the absence of such factors, the equilibrium between the closed and the opened states is probably the determining factor for the intrinsic rates of nucleotide dissociation. For DnaK, the ADP dissociation rates are very low, being 0.004–0.035 s−1 and 0.0004–0.0014 s−1 in the presence and absence of Pi, respectively [48, 59]. Although the ADP dissociation rate is 2–60-fold faster than the rate of hydrolysis in the unstimulated cycle, it nevertheless becomes rate limiting when the hydrolysis of ATP is stimulated by DnaJ and substrates. For bovine Hsc70, the dissociation rates for ADP±Pi are about 20-fold higher than those of DnaK [48,59]. Even more dramatic differences exist for HscA, which has dissociation rates for ADP±Pi that are 700-fold higher than those of DnaK [59].
What is the structural basis for these strong kinetic differences between Hsp70 homologs in nucleotide dissociation? Although the modeled ATPase domain structures of DnaK, HscA and Hsc70 are almost completely superimposable, they show subtle differences which allow classification of the entire Hsp70 family into three subfamilies with E. coli DnaK, E. coli HscA and human Hsc70 as prototypes [59]. First, variations exist in an exposed loop in subdomain IIB near the nucleotide binding cleft. DnaK subfamily members share a particularly long loop (A276-R302 in E. coli DnaK) with subfamily-specific sequence. Members of the Hsc70 subfamily share a loop with subfamily-specific sequence whose tip is 4 residues shorter, while members of the HscA subfamily share a loop that is 10 residues shorter and less conserved in sequence. Second, variations exist in the interface of the nucleotide binding cleft. DnaK proteins contain a hydrophobic patch (L257-V59 of DnaK) at the top of the cleft and two putative salt bridges (E264-R56, upper; E267-K55, lower) that are mainly responsible for the polarity of the interface. In contrast, Hsc70 proteins lack a DnaK-like hydrophobic patch and the upper salt bridge, while HscA proteins lack all three elements.
The loop and the salt bridges constitute a device that allows rapid association of ATP and slow dissociation of ATP and ADP±Pi. The salt bridges together with the hydrophobic contact probably function in a mouse-trap-like fashion to allow tight closure of the nucleotide binding cleft. The role of the loop is less obvious. Given the considerable size of its exposed part, it has been speculated that it may reach over to subdomain IB of the ATPase domain of DnaK, thereby acting as a latch [59]. The large variability within the Hsp70 family in nucleotide exchange rates is likely to contribute to the functional diversification of Hsp70 chaperone systems.
The differences between Hsp70 homologs in nucleotide dissociation go along with differences in the regulation of this step. DnaK homologs that have the described trap to prevent nucleotide dissociation require the nucleotide exchange factor, GrpE, for chaperone activities in vivo and in vitro. HscA homologs which lack this device and have high nucleotide dissociation rates neither interact with GrpE nor appear to have their own exchange factor [59]. Mammalian Hsc70 homologs that have intermediate dissociation rates possess chaperone activity without exchange factor. However, they can interact with a nucleotide exchange factor, Bag-1, perhaps to fulfill special chaperone activities. The two known nucleotide exchange factors for Hsp70 proteins, GrpE and Bag-1, have entirely different structures and mechanisms, although they generate the same conformational open state of their target chaperone [60–62]. These proteins may therefore have been generated by convergent functional evolution.
GrpE has a molecular weight of 22 kDa and forms a stable dimer in solution. The X-ray structure of GrpE in complex with the ATPase domain of DnaK was solved to 2.8 Å resolution [60]. The N-terminus of the truncated GrpE monomer constitutes an α-helix with the length of 100 Å. The N-terminal helix is connected over a rather unstructured loop to two additional short α-helices and a small β-sheet domain composed of six short β-strands. In addition to the N-terminal helix, the dimer interface is formed by the two short helices of each monomer which together build up a four-helix bundle. In the crystal structure GrpE forms an asymmetric and bent dimer whereby only one GrpE monomer (orange in fig. 1b) contacts the ATPase domain of DnaK through five major contact sites.
GrpE binds to the ADP bound and nucleotide-free states of DnaK with high affinity (Kd = 1 nM). Addition of ATP leads to the dissociation of the complex. As described above, the superposition of the structure of the nucleotidefree ATPase domain of DnaK in complex with GrpE with the structure of the ADP and Pi bound ATPase domain of Hsc70 [45] shows that subdomain IIB in DnaK is rotated outwards by 14°, thereby opening up the nucleotide binding cleft. This rotation displaces residues Ser274, Lys270 and Glu267 of DnaK by 2–3 Å. The corresponding amino acids in Hsc70 are involved in the coordination of the adenine and the ribose rings of the bound ADP. Furthermore, residues which constitute the hydrophobic patch and the upper salt bridge (E264-R56) interact with GrpE [60] (fig. 1b).
The mechanism by which GrpE accelerates the nucleotide release thus seems to rely on an active opening of the nucleotide binding cleft. GrpE furthermore stabilizes the open conformation of the nucleotide binding pocket, which facilitates the rapid binding of ATP to the nucleotide-free state of DnaK [47,59]. In the presence of ATP GrpE thereby regulates the lifetime of the DnaK-substrate complex [63].
Bag proteins form a heterogeneous family of multidomain proteins that share the Bag domain frequently located at their C-termini [64]. This domain is essential and sufficient for stimulation of nucleotide exchange by the mammalian homologs Hsc70 and Hsp70. The nucleotide exchange activity of Bag proteins was first described for the Bag-1 homolog by Höhfeld and co-workers [65]. Recent kinetic measurements using fluorescent labeled nucleotides showed that Bag-1 stimulates dissociation of ADP from Hsc70 and Hsp70 by up to 100-fold in the absence of inorganic phosphate and by 600-fold in its presence [47]. Interestingly, Bag-1 does not stimulate the dissociation of ATP and is strongly affected by Pi, which increased the apparent Kd by approx. 17-fold. This is in contrast to the interaction of GrpE with DnaK, which also stimulates ATP dissociation and is not affected by Pi. These observations suggest that the mechanisms by which GrpE and Bag-1 stimulate nucleotide release are different and that Bag-1 plays a more passive role than GrpE. Bag-1 may only bind and stabilize the open conformation of the ATPase domain of Hsc70 during the intrinsic fluctuations between the open and closed states.
The mammalian Bag domains form a three-helix bundle [61, 62, 66]. It associates as a monomer with the subdomains IB and IIB of the Hsc70 ATPase, through electrostatic interactions involving the conserved residues Glu212, Asp222, Arg237 and Gln245 in Bag-1 (fig. 1c). Both exchange factors, GrpE and Bag-1, cause the same 14° outwards rotation of subdomain IIB through a hinge.
In addition to GrpE and the Bag proteins other proteins have recently been found to act as nucleotide exchange factors for Hsp70 proteins. The yeast protein Sls1/Sil1, its mammalian homolog Bap and surprisingly the yeast Hsp70-related Hsp170 Lhs1 function as specific exchange factors for the endoplasmatic reticulum resident Hsp70 protein Bip [67–70]. Nucleotide exchange function for BiP seems to be essential since double deletion of Lhs1 and Sls1/Sil1 is lethal [71]. The yeast Fes1 and its mammalian homolog HspBP1 are nucleotide exchange factors for the cytosolic Hsc70 [72, 73]. In the mammalian cytosol, therefore, several exchange factors coexist, including Bag proteins with currently six distinct family members, one of which exists in four variants, and HspBP1. The physiological role of this diversity is not yet elucidated (see below).
Substrate binding
The substrate binding domain shows high sequence conservation within the Hsp70 family, although differences of unclear functional relevance exist. Most structural information on this domain exists for E. coli DnaK. The X-ray structure of the substrate binding domain was solved in complex with a heptameric peptide substrate at 2.0 Å resolution (fig. 1d, e) [74]. The peptide binding moiety (residues 389–607) forms a sandwich of 2 four-stranded β-sheets with four upwards protruding loops (two inner and two outer loops) and two helices A and B which are packed against the inner loops (L1,2 and L4,5). The substrate binding cavity is formed by the β-sheets 1 and 2 and the loops, L1,2 and L3,4. Helix B constitutes a lid which closes the cavity through a salt bridge and two hydrogen bonds to the outer loops L3,4 and L5,6. ATP binding to DnaK must at least open this lid to allow substrate release. The distal part of helix B, together with helices C, D, and E, forms a hydrophobic core of unknown function.
The substrate peptide which has been co-crystallized with the substrate binding domain of DnaK is contacted over a stretch of five residues by two types of interactions [74]. Hydrogen bonds between the backbone of the cavity forming loops (L1,2 and L3,4) and the backbone of the substrate mediate the recognition of the extended peptide conformation. van der Waals interactions of hydrophobic side chains lining the substrate binding cavity with the substrate side chains explain DnaK’s preference for hydrophobic peptide segments. The central substrate residue, referred to as position 0, reaches down into a deep hydrophobic pocket of the substrate binding cavity which accommodates large hydrophobic amino acids, especially Leu [74]. This central substrate residue is completely buried from solvent. The adjacent residues of the bound substrate at positions −1 and +1 are only partially buried from solvent. They point upwards and contact Met404 and Ala429, which form a hydrophobic arch bridging over the substrate peptide backbone. The surface potential of the substrate binding domain surrounding the cavity is mainly negative which explains the contribution of positive charges in peptides to the affinity for DnaK.
The architectural constraints imposed by the substrate binding cavity agree well with the experimentally determined binding motif of DnaK [75]. By providing essential backbone contacts, the cavity allows DnaK to recognize only peptides composed of L-amino acids but not peptides composed of D-amino acids [76]. Furthermore, DnaK has different affinities for peptides with identical amino acid composition but authentic or inverse sequence [76]. This implies that a peptide exhibiting lower affinity for DnaK in the authentic sequence than a peptide in the inversed sequence cannot simply ‘turn around’ and bind to DnaK with the N-terminus on the other side. DnaKpeptide interactions therefore require a specific direction of the backbone (fig. 1e). Although the evolutionary conservation of the binding cavity suggests that this feature is conserved within the Hsp70 family, one Hsp70, HscA, was shown recently to bind a specific substrate peptide in the reverse orientation (fig. 1f) [77, 78].
Three structural elements of the cavity are of prime importance for substrate binding, the α-helical lid, the arch enclosing the substrate peptide and the central hydrophobic pocket. Removal of the α-helical lid by truncation of the C-terminus starting at a hinge point within helix B decreases the affinity of DnaK for peptide and protein substrates by 3–7-fold, primarily by increasing the dissociation rates [79–82].
Disturbing the arch by alteration of Met404 to Ala or Ala429 to Trp also decreases the affinity of DnaK for substrates by 2–4-fold in the ADP state with the major effects on the dissociation rates [79]. Alterations in the arch in addition confer changes in the specificity of DnaK for some peptide substrates [83]. This finding is interesting with regard to the fact that the arch-forming residues are the only substrate-contacting residues of the cavity that are subject of considerable evolutionary variation. It is therefore possible that the functional specialization of Hsp70 chaperones involves adaptive variation of the arch and the substrate specificity.
The main contribution to the binding affinity for substrates, however, comes from the central hydrophobic pocket. Introducing a steric hindrance in this pocket by alteration of Val436 to Phe increased the dissociation equilibrium constants of DnaK by 20- and 40-fold for protein and peptide substrates, respectively [79]. Interestingly, this decrease in affinity was solely caused by generally decreased association rates, while the dissociation rates in ADP and ATP states were identical to wild-type DnaK. Substrates may therefore bind to the mutant protein in a very similar way to wild-type DnaK, suggesting that the hydrophobic pocket has a considerable degree of flexibility and that substrates bind in an induced fit-like manner. Such a high degree of flexibility of the hydrophobic pocket was also suggested by the NMR structure of a DnaK substrate binding domain fragment [80, 84].
Nevertheless, despite the structural flexibility of the pocket, introducing a bulky side chain into the hydrophobic pocket is fatal for the chaperone activity in vitro and in vivo. Even high concentrations of the DnaK-V436F mutant could not refold chemically denatured luciferase or rescue a ΔdnaK-strain for growth at 40 °C [79].
The coupling mechanism: nucleotide-controlled opening and closing of the substrate binding cavity
All available structures of the substrate binding domain are believed to represent the high-affinity state of the Hsp70 chaperones. Since the architectural constraints of the binding cavity do not permit the dissociation of substrates through a lateral sliding mechanism, an opening of the lid, the arch and the cavity must occur to allow substrate release.
Association and dissociation of peptide and protein substrates is observed in the ADP state of Hsp70 chaperones [54, 56, 85–87]. This demonstrates that transitory opening of the substrate binding cavity also occurs, albeit slowly, in the absence of ATP. Christen and co-workers investigated the association of several related peptides to DnaK·ADP and found similar association rates for all peptides [88]. They concluded that substrate association to DnaK·ADP is limited by the opening rate of the substrate binding cavity. This opening rate in the ADP status of DnaK is limited by the lid and the arch as revealed by DnaK mutant analysis [79]. Both structural elements act independently in the control of substrate binding since the combination of lid truncation and arch alterations had only additive effects.
In the ATP state, the association of fluorescent peptide substrates to DnaK·ATP occurs at a rate of 105−106M−1 s−1 [79, 87, 88], and of a fluorescent peptide to Hsc70·ATP at a slower rate (700 M−1 s−1) [89]. Although these association rates are orders of magnitude higher than those in the ADP state, they are far from diffusion controlled (109 M−1 s−1).
The mechanism of the nucleotide controlled opening and closing of the substrate binding domain is still a matter of debate. Based on crystallographic evidence Hendrickson and co-workers suggested that ATP binding may induce an upwards movement of helix B starting at a hinge point (residues 536–538 of DnaK) located above the cavity [74]. This movement would disrupt the interactions of helix B with the outer loops and consequently destabilize the outer loops and open up the substrate binding cavity. However, recent structural and biochemical data indicate a more complicated mechanism. Deletion of the helical lid at the proposed hinge point increases the dissociation rates for bound substrates by only approx. 5-fold compared with the 400- to 2500-fold increase in the dissociation rate induced by ATP in case of wild-type DnaK [79, 87]. Furthermore, lidless mutant proteins of DnaK, BiP and Hsc70 have retained substantial activity as chaperones in vivo and in vitro and still respond to ATP binding with a release of bound substrates [56,79]. Even the isolated β-sheet domain lacking all helices A-E is not in an open conformation [80].
There is little known about the coupling mechanism that brings together all events which control the frequency of opening and closing of the substrate binding pocket: the DnaJ and substrate stimulated ATP hydrolysis, which stabilizes the closed state and the ATP induced stabilization of the open state. For discussion of the coupling mechanism, two aspects can be distinguished, the mechanics and the energetics of coupling. The mechanics of coupling considers the contact sites between the ATPase and substrate binding domains which mediate the nucleotide-dependent interdomain communication. The energetics of coupling considers the necessary energy transmissions. Binding of ATP frees energy which is necessary to surmount the activation barrier from the closed to the open conformation of the substrate binding domain, while binding of substrate and DnaJ deliver the energy to overcome the activation barrier for ATP hydrolysis. Several mutational alterations have been identified that affect the coupling mechanism. Some of them are clearly involved in ATP binding (Thr13 to Ser, Ala, or Val in Hsc70) or lower the activation barrier for the transition to the open substrate binding domain conformation (Glu543 to Lys in Hsc70) [90]. Other residues were found in sites where such an obvious effect on the energetics of coupling could not easily be implicated. These residues may therefore be necessary for the transmission of the signals.
The strongest coupling defects were detected in DnaK mutant proteins with alterations in the highly conserved segment, referred to as linker, which connects the ATPase domain with the substrate binding domain. The exchanges VLLL(389–392) to AAAA and LL(390,391) to DD completely abolish the interdomain communication in the sense that ATP fails to release bound substrates and substrate with DnaJ fail to stimulate ATP hydrolysis [51]. Interestingly, this linker was found in two different positions in the crystal structure of the substrate binding domain, in an extended position and in a position bound to a hydrophobic depression formed by the β-sheet domain proximal to helix A. The existence of these two structural variants led Hendrickson and co-workers to suggest that the linker may undergo an ATP-dependent movement in and out of this depression as part of a process that allows the ATPase domain to dock onto the substrate binding domain.
The targeting activity of co-chaperones
Since Hsp70s perform many different tasks within the same cellular compartment, regulation of the access to substrates seems to be essential. Many co-chaperones therefore act to target Hsp70 partner proteins to specific substrates or to mediate the targeted release of Hsp70 bound cargo. This regulatory activity relies on the ability of these co-chaperones to connect Hsp70 proteins with their target proteins, including substrates and other chaperones, through physical association with both, the chaperone and the targets.
J-domain proteins
The activity of JDPs to mediate the ATP hydrolysis-dependent locking of substrates into the binding cavity of Hsp70 proteins is essential for almost all chaperone activities of Hsp70 proteins [55, 91]. Cells usually encode a broad spectrum of JDPs which differ in their domain composition and cellular functions [92, 93]. Six JDPs exist in E. coli, 20 in Saccharomyces cerevisiae, 33 in Caenorhabditis elegans, 34 in Drosophila melanogaster and presumably 44 in human cells. The number of Hsp70 homologs in these organisms is smaller than the number of JDPs, e.g. 3 in E. coli and around 10 in the other organisms.
The members of the JDP family have been subdivided according to their domain composition (fig. 3a) [94]. Class I proteins share all 4 domains characteristic for the prototype for all JDP E. coli DnaJ. DnaJ is composed of the N-terminal J-domain, followed by a glycine and phenylalanine-rich region (G/F-region), a Zn-binding domain and a C-terminal domain (fig. 3c). Class II proteins lack the Zn-binding domain. Class III proteins share with DnaJ only the J-domain. While the J-domain is always at the N-terminus of the mature protein in the first two classes, it may be at any place within the sequence of class III proteins. The class III family is the most diverse and contains proteins with additional distinct motifs or domains, e.g. transmembrane helices (E. coli DjlA, yeast Sec63, human DjC9/hSec63, yeast Mdj2), tetratricopeptide repeats (TPRs; mouse DjC2/Zrf1/Mida1, human DjC3/hp58 and DjC7/hTpr2), and cysteine-rich regions which are polypalmitoylated (cysteine string proteins). The question of how JDPs interact with substrates and mediate their transfer onto Hsp70 partner proteins is not answered for any of the three classes of JDPs. Some JDPs have broad substrate specificity, such as E. coli DnaJ and yeast Ydj-1, while others have more restricted substrate spectra. In particular the JDPs of class III seem either to bind a restricted number of substrates, such as clathrin-specific auxilin or kinesin light chain, or they may not bind substrates themselves but rather be positioned in close proximity to substrates, e.g. Sec63 at the translocation pore of the endoplasmic reticulum and Pam18/Tim14 at the translocase of the inner mitochondrial membrane. Stable substrate binding, however, does not seem to be an absolutely essential function of the JDPs. Point and deletion mutant proteins of DnaJ, which do not stably interact with substrates, can support in vitro refolding of a denatured model protein and the DNA replication of bacteriophage λ [53, 95]. In vivo such mutants are able to complement the temperature sensitivity phenotype of a δdnaJδcbpA strain and allow bacterophage λ plating on this strain [53, 96]. Nevertheless, direct interaction with the substrates increases the efficacy of the JDPs since in the in vitro and in vivo assays significantly higher concentrations of the defective DnaJ variants were necessary as compared with the wild-type proteins.
Bag proteins
The family of Bag proteins is composed in humans of the homologs Bag-1, Bag-2, Bag-3 (CAIR/BIS), Bag-4 (SODD), Bag-5 and Bag-6 (BAT3/Scythe) [64]. All homologs per definition contain one Bag domain except Bag-5, which stows five putative Bag domains. Except for Bag-5, a direct physical interaction with Hsp70 was shown for all members of the Bag domain protein family, and the Bag domain was responsible for this action [97–99]. The Bag proteins were therefore proposed to serve as targeting factors for the Hsp70 chaperone [64]. Outside the Bag domain the members of this family show a high degree of diversity in their domain composition, including known protein-protein interaction motifs such as the WW-domain, the PXXP-motif and the ubiquitin-like domain (fig. 3d). Therefore, Bag proteins differ from GrpE proteins by their ability to associate with ligands other than Hsp70 proteins. It is proposed that Bag proteins use this ability to connect the nucleotide dissociation-dependent delivery of Hsp70 bound cargo with cellular processes [47, 100], albeit the nucleotide exchange function of the Bag domain was so far only demonstrated for the Bag domain of Bag-1. The Bag domains of Bag-3, Bag-4 and Bag-5 are shorter by three to four turns of each of the helices which compose the three-helix bundle, but all of the Hsp70 interacting residues seems to be present in these shortened domains, and it is likely that they interact with both subdomains of the ATPase domain of Hsp70 in a similar way to the Bag domain of Bag-1 [66, 101].
The cellular processes in which the Bag-domain proteins are involved seem to be highly diverse since Bag homologs have been implicated in signal transduction processes related to cellular stress responses, including apoptosis, proliferation and development; in transcriptional regulation; and in degradation through the proteasome [64, 102]. Several of the Bag-domain proteins seem to be involved in tumor growth and apoptosis [103].
Whatever the functions of Bag proteins are in the above cellular processes, it is clear from the work of several laboratories that Bag proteins are not strictly essential for the chaperone activity of Hsp70 proteins in folding of non-native proteins [47]. This is in sharp contrast to the essential role for GrpE in the chaperone cycle of DnaK. The Bag domain rather seems to serve as a targeting domain to ensure a proper localization of Hsp70 chaperone action.
The biochemically and cell biologically best investigated member of the family is Bag-1. Bag-1 is produced in the cell as four translation initiation variants, Bag-1L (50 kDa), Bag-1M (46 kDa), Bag-1p33 (33 kDa) and Bag-1S (29 kDa) [64, 104, 105]. Bag-1M is identical to Rap46, a protein found in the cytosol of higher eukaryotes. The N-terminal segment which exists only in Bag-1L is rich in basic amino acids that have been implicated in DNA binding. Bag-1L is indeed found mainly in the nucleus [104]. The second region, present in Bag-1L and Bag-1M, contains a stretch of 6 TRSEEX repeats (threonine, arginine, serine, glutamic acid, glutamic acid, any amino acid; TRS is not identical in all repeats). Common to all four Bag-1 variants is an ubiquitin-like domain that was proposed to mediate the interaction of Bag-1 with the proteasome [106]. It is tempting to speculate that the proteasome associated Bag-1M by accelerating Hsp70’s nucleotide exchange dissociates substrates from a complex with Hsp70 for degradation by the proteasome. Such a role would directly link Hsp70 activity to proteolysis. Alternatively, Bag-1 may act as an anti-targeting factor that prevents Hsp70 from binding to substrates of the proteasome since this may interfere with the degradation process. A nucleotide exchange-independent function of Bag-1 was proposed for its regulation of Raf-1 [107]. From the fact that the binding site of Raf-1 in Bag-1 overlaps with the binding site of Hsp70, it was concluded that Raf-1 and Hsp70 compete with each other for binding to Bag-1 [107]. Mechanistically, however, this regulation of the Raf-1/Erk pathway is not clear.
Hip, Hop and CHIP
Hip has been identified in a yeast two-hybrid screen as a protein of 43 kDa which interacts with the ATPase domain of human Hsc70 [108]. Hip was claimed to stabilize the ADP state of Hsp70 and thereby to stimulate the chaperone activity of Hsp70 by preventing premature substrate release [65]. However, it is unclear how this may work, considering that Hip was used in a 75–250-fold excess (Hip-dimer) over the chaperone substrate. There are also reports that Hip does not affect Hsc70- and DjB-1/Hdj-1-mediated refolding activity [109]. Hip is currently considered to antagonize the substrate discharging function of Bag-1 by competing with Bag-1 for binding to the ATPase domain of Hsc70, thereby preventing Bag-1-stimulated nucleotide release [110, 111]. Indications for a Bag-independent function of Hip were found recently [112]. Hip overexpression stimulated activation of the glucocorticoid receptor but not of the mineralocorticoid receptor, estrogen receptor or progesterone receptor in the heterologous yeast system [113]. Interestingly, a TPR deletion variant of Hip and a point mutant with considerably reduced affinity to Hsp70 still had significant stimulatory effect on glucocorticoid receptor activation, arguing for an Hsp70-independent function of Hip in this process in the yeast system [112].
The 60-kDa protein Hop (yeast Sti1) has been identified in a genetic screen for proteins involved in the regulation of the heat shock response in yeast and was subsequently found to be a component of the progesterone receptor complex [114, 115]. Hop interacts with its three TPR domains with the C-terminal EEVD motifs of Hsp70 and Hsp90 proteins [116, 117]. In vitro reconstitution experiments showed that Hop enhances the maturation rate of the progesterone receptor [118]. The yeast Sti1 was recently found to stimulate the ATPase activity of the yeast Hsp70 Ssa1 [119], a function that could not be demonstrated for mammalian Hop [119]. However, in mammals there are other TPR domain-containing proteins, one of which contains in addition a J-domain which stimulates the ATPase activity of Hsp70 [120].
The 35-kDa protein CHIP was discovered in a search for TPR motif-containing co-chaperones of eukaryotic Hsp70s [121]. CHIP like Hop forms a dimer in solution and competes with Hop for binding to the C-terminus of Hsc70 and Hsp90 [122]. It acts as E3-ubiquitin ligase ubiquitinating Hsc70 substrates in vitro and in vivo and promoted their degradation by the proteasome [35, 123, 124]. CHIP therefore shares with Bag-1 the potential to connect the activity of Hsp70 chaperones with proteolysis. More recently evidence was found for a degradation-independent function of CHIP [125, 126].
Perspectives
The Hsp70 protein family and their co-chaperones constitute a complex network of folding machines which is utilized by cells in many ways. Despite considerable progress in the elucidation of the mechanistic basis of these folding machines, important aspects remain to be solved. With respect to the Hsp70 proteins it is still unclear whether their activity to assist protein folding relies on the ability to induce conformational changes in the bound substrates, how the coupling mechanism allows ATP to control substrate binding and to what extent sequence variations within the family translate into variations of the mechanism. With respect to the action of co-chaperones we lack a molecular understanding of the coupling function of JDPs and of how co-chaperones target their Hsp70 partner proteins to substrates. Furthermore, it can be expected that more cellular processes will be discovered that depend on the chaperone activity of Hsp70 chaperones.
Footnotes
Received 21 October 2004; received after revision 24 November 2004; accepted 6 December 2004
Contributor Information
M. P. Mayer, FAX: + 49 6221 54 5894, Email: M.Mayer@zmbh.uni-heidelberg.de
B. Bukau, Email: bukau@zmbh.uni-heidelberg.de
References
- 1.Bukau B., Deuerling E., Pfund C. and Craig E. A. (2000) Getting newly synthesized proteins into shape. Cell 101: 119–122 [DOI] [PubMed]
- 2.Hartl F. U. and Hayer-Hartl M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295: 1852–1858. [DOI] [PubMed]
- 3.Young J. C., Barral J. M. and Ulrich Hartl F. (2003) More than folding: localized functions of cytosolic chaperones. Trends. Biochem. Sci. 28: 541–547 [DOI] [PubMed]
- 4.Neupert W. and Brunner M. (2002) The protein import motor of mitochondria. Nat. Rev. Mol. Cell. Biol. 3: 555–565 [DOI] [PubMed]
- 5.Ryan M. T. and Pfanner N. (2002) Hsp70 proteins in protein translocation. Adv. Protein Chem. 59: 223–242 [DOI] [PubMed]
- 6.Pratt W. B. and Toft D. O. (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. (Maywood) 228: 111–133 [DOI] [PubMed]
- 7.Toft D. O. (1999) Control of hormone receptor function by molecular chaperones and folding catalysts. In: Molecular Chaperones and Folding Catalysts. Regulation, Cellular Function and Mechanism, pp. 313–327, Bukau B. (ed.), Harwood Academic Publishers, Amsterdam
- 8.Ben-Zvi A. P. and Goloubinoff P. (2001) Review: mechanisms of disaggregation and refolding of stable protein aggregates by molecular chaperones. J. Struct. Biol. 135: 84–93 [DOI] [PubMed]
- 9.Pierpaoli E. V., Sandmeier E., Baici A., Schönfeld H.-J., Gisler S. and Christen, P. (1997) The power stroke of the DnaK/DnaJ/GrpE molecular chaperone system. J. Mol. Biol. 269: 757–768 [DOI] [PubMed]
- 10.Mayer M. P., Rüdiger S. and Bukau B. (2000) Molecular basis for interactions of the DnaK chaperone with substrates. Biol. Chem. 381: 877–885 [DOI] [PubMed]
- 11.Slepenkov S. V. and Witt S. N. (2002) The unfolding story of the Escherichia coli Hsp70 DnaK: is DnaK a holdase or an unfoldase? Mol. Microbiol. 45: 1197–1206 [DOI] [PubMed]
- 12.Glover J. R. and Lindquist S. (1998) Hsp104, Hsp70 and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94: 73–82 [DOI] [PubMed]
- 13.Goloubinoff P., Mogk A., Peres Ben Zvi A., Tomoyasu T. and Bukau B. (1999) Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc. Natl. Acad. Sci. USA 96: 13732–13737 [DOI] [PMC free article] [PubMed]
- 14.Motohashi K., Watanabe Y., Yohda M. and Yoshida M. (1999) Heat-inactivated proteins are rescued by the DnaK.J-GrpE set and ClpB chaperones. Proc. Natl. Acad. Sci. USA 96: 7184–7189 [DOI] [PMC free article] [PubMed]
- 15.Diamant S., Peres Ben-Zvi A., Bukau B. and Goloubinoff P. (2000) Size-dependent disaggregation of stable protein aggregates by the DnaK chaperone machinery. J. Biol. Chem. 275: 21107–21113 [DOI] [PubMed]
- 16.Ben-Zvi A., De Los Rios P., Dietler G. and Goloubinoff P. (2004) Active solubilization and refolding of stable protein aggregates by cooperative unfolding action of individual Hsp70 chaperones. J. Biol. Chem. 279: 37298–37303 [DOI] [PubMed]
- 17.Pratt W. B. (1997) The role of the hsp90-based chaperone system in signal transduction by nuclear receptors and receptors signaling via MAP kinase. Annu. Rev. Pharmacol. Toxicol. 37: 297–326 [DOI] [PubMed]
- 18.Jäättelä M. (1999) Escaping cell death: survival proteins in cancer. Exp. Cell. Res. 248: 30–43 [DOI] [PubMed]
- 19.Jolly C. and Morimoto R. I. (2000) Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer. Inst. 92: 1564–1572 [DOI] [PubMed]
- 20.Bonini N. M. (2002) Chaperoning brain degeneration. Proc. Natl. Acad. Sci. USA 99Suppl. 4: 16407–16411 [DOI] [PMC free article] [PubMed]
- 21.Sakahira H., Breuer P., Hayer-Hartl M. K. and Hartl F. U. (2002) Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc. Natl. Acad. Sci. USA 99Suppl. 4: 16412–16418 [DOI] [PMC free article] [PubMed]
- 22.Millar D. G., Garza K. M., Odermatt B., Elford A. R., Ono N., Li, Z. et al. (2003) Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat. Med. 9: 1469–1476 [DOI] [PubMed]
- 23.Mayer M. P. (2004) Recruitment of Hsp70 chaperones: a crucial part of viral survival strategies. Rev. Physiol. Biochem. Pharmacol. 9 Jul. [Epub ahead of print] [DOI] [PubMed]
- 24.Kregel K. C. (2002) Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J. Appl. Physiol. 92: 2177–2186 [DOI] [PubMed]
- 25.Cagney G., Amiri S., Premawaradena T., Lindo M. and Emili A. (2003) In silico proteome analysis to facilitate proteomics experiments using mass spectrometry. Proteome Sci 1: 5 [DOI] [PMC free article] [PubMed]
- 26.Gaiddon C., Lokshin M., Ahn J., Zhang T. and Prives C. (2001) A subset of tumor-derived mutant forms of p53 downregulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 21: 1874–1887 [DOI] [PMC free article] [PubMed]
- 27.King F. W., Wawrzynow A., Hohfeld J. and Zylicz M. (2001) Co-chaperones Bag-1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild-type or mutant p53. EMBO J. 20: 6297–6305. [DOI] [PMC free article] [PubMed]
- 28.Meacham, G. C., Lu, Z., King, S., Sorscher, E., Tousson, A., and Cyr, D. M. (1999) The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 18: 1492–1505 [DOI] [PMC free article] [PubMed]
- 29.Shinder G. A., Lacourse M. C., Minotti S. and Durham, H. D. (2001) Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J. Biol. Chem. 276: 12791–12796 [DOI] [PubMed]
- 30.Roberts S. P. and Feder M. E. (1999) Natural hyperthermia and expression of the heat shock protein Hsp70 affect developmental abnormalities in Drosophila melanogaster. Oecologia 121: 323–329 [DOI] [PubMed]
- 31.Rutherford S. L. and Lindquist S. (1998) Hsp90 as a capacitor for morphological evolution. Nature 396: 336–342 [DOI] [PubMed]
- 32.Queitsch C., Sangster T. A. and Lindquist S. (2002) Hsp90 as a capacitor of phenotypic variation. Nature 417: 618–624 [DOI] [PubMed]
- 33.Rogue P. J., Ritz M. F. and Malviya A. N. (1993) Impaired gene transcription and nuclear protein kinase C activation in the brain and liver of aged rats. FEBS Lett. 334: 351–354 [DOI] [PubMed]
- 34.Njemini R., Abeele M. V., Demanet C., Lambert M., Vandebosch S. and Mets T. (2002) Age-related decrease in the inducibility of heat-shock protein 70 in human peripheral blood mononuclear cells. J. Clin. Immunol. 22: 195–205 [DOI] [PubMed]
- 35.Meacham G. C., Patterson C., Zhang W., Younger J. M. and Cyr D. M. (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3: 100–105 [DOI] [PubMed]
- 36.Urushitani M., Kurisu J., Tateno M., Hatakeyama S., Nakayama K., Kato S. et al. (2004) CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 90: 231–244 [DOI] [PubMed]
- 37.Leone G., Coffey M. C., Gilmore R., Duncan R., Maybaum L. and Lee P. W. (1996) C-terminal trimerization, but not Nterminal trimerization, of the reovirus cell attachment protein Is a posttranslational and Hsp70/ATP-dependent process. J. Biol. Chem. 271: 8466–8471 [DOI] [PubMed]
- 38.Young J. C., Hoogenraad N. J. and Hartl F. U. (2003) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112: 41–50 [DOI] [PubMed]
- 39.Sakahira H. and Nagata S. (2002) Co-translational folding of caspase-activated DNase with Hsp70, Hsp40 and inhibitor of caspase-activated DNase. J. Biol. Chem. 277: 3364–3370 [DOI] [PubMed]
- 40.Jäättelä M., Wissing D., Kokholm K., Kallunki T. and Egeblad M. (1998) Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 17: 6124–6134 [DOI] [PMC free article] [PubMed]
- 41.Nylandsted J., Rohde M., Brand K., Bastholm L., Elling F. and Jaattela M. (2000) Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 97: 7871–7876 [DOI] [PMC free article] [PubMed]
- 42.Klucken J., Shin Y., Masliah E., Hyman B. T. and McLean P. J. (2004) Hsp70 reduces alpha-synuclein aggregation and toxicity. J. Biol. Chem. 279: 25497–25502 [DOI] [PubMed]
- 43.Gutsmann-Conrad A., Heydari A. R., You S. and Richardson A. (1998) The expression of heat shock protein 70 decreases with cellular senescence in vitro and in cells derived from young and old human subjects. Exp. Cell. Res. 241: 404–413 [DOI] [PubMed]
- 44.Ambra R., Mocchegiani E., Giacconi R., Canali R., Rinna A., Malavolta M. et al. (2004) Characterization of the hsp70 response in lymphoblasts from aged and centenarian subjects and differential effects of in vitro zinc supplementation. Exp. Gerontol. 39: 1475–1484 [DOI] [PubMed]
- 45.Flaherty K. M., Deluca-Flaherty C. and McKay D. B. (1990) Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 346: 623–628 [DOI] [PubMed]
- 46.Zhang Y. and Zuiderweg E. R. (2004) The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc. Natl. Acad. Sci. USA 101: 10272–10277 [DOI] [PMC free article] [PubMed]
- 47.Gässler C. S., Wiederkehr T., Brehmer D., Bukau B. and Mayer, M. P. (2001) Bag-1M accelerates nucleotide release for human Hsc70 and Hsp70 and can act concentration-dependent as positive and negative cofactor. J. Biol. Chem. 276: 32538–32544 [DOI] [PubMed]
- 48.Ha J.-H., Johnson E. R., McKay D. B., Sousa M. C., Takeda S. and Wilbanks S. M. (1999) Structure and mechanism of Hsp70 proteins. In: Molecular Chaperones and Folding Catalysts. Regulation, Cellular Function and Mechanism, pp. 573–607, Bukau B. (ed.), Harwood Academic Publishers, Amsterdam
- 49.Karzai A. W. and McMacken R. (1996) A bipartite signaling mechanism involved in DnaJ-mediated activation of the Escherichia coli DnaK protein. J. Biol. Chem. 271: 11236–11246 [DOI] [PubMed]
- 50.Barouch W., Prasad K., Greene L. and Eisenberg E. (1997) Auxilin-induced interaction of the molecular chaperone Hsc70 with clathrin baskets. Biochemistry 36: 4303–4308 [DOI] [PubMed]
- 51.Laufen T., Mayer M. P., Beisel C., Klostermeier D., Reinstein J. and Bukau B. (1999) Mechanism of regulation of Hsp70 chaperones by DnaJ co-chaperones. Proc. Natl. Acad. Sci. USA 96: 5452–5457 [DOI] [PMC free article] [PubMed]
- 52.Liberek K., Marszalek J., Ang D., Georgopoulos C. and Zylicz M. (1991) Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl. Acad. Sci. USA 88: 2874–2878 [DOI] [PMC free article] [PubMed]
- 53.Wall D., Zylicz M. and Georgopoulos C. (1994) The NH2-terminal 108 amino acids of the Escherichia coli DnaJ protein stimulate the ATPase activity of DnaK and are sufficient for λ replication. J. Biol. Chem. 269: 5446–5451 [PubMed]
- 54.Gamer J., Multhaup G., Tomoyasu T., McCarty J. S., Rudiger S., Schonfeld H. J. et al. (1996) A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 15: 607–617 [PMC free article] [PubMed]
- 55.Laufen T., Zuber U., Buchberger A. and Bukau B. (1998) DnaJ proteins. In: Molecular Chaperones in Proteins: Structure, Function and Mode of Action, pp. 241–274, Fink A. L. and Goto Y. (eds), Marcel Dekker, New York
- 56.Misselwitz B., Staeck O. and Rapoport T. A. (1998) J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol. Cell. 2: 593–603 [DOI] [PubMed]
- 57.Silberg J. J. and Vickery L. E. (2000) Kinetic characterization of the ATPase cycle of the molecular chaperone Hsc66 from Escherichia coli. J. Biol. Chem. 275: 7779–7786 [DOI] [PubMed]
- 58.Silberg J. J., Tapley T. L., Hoff K. G. and Vickery L. E. (2004) Regulation of the HscA ATPase reaction cycle by the cochaperone HscB and the iron-sulfur cluster assembly protein IscU. J. Biol. Chem. 279: 53924–53931 [DOI] [PubMed]
- 59.Brehmer D., Rüdiger S., Gässler C. S., Klostermeier D., Packschies L., Reinstein J. et al. (2001) Tuning of chaperone activity of Hsp70 proteins by modulation of nucleotide exchange. Nat. Struct. Biol. 8: 427–432 [DOI] [PubMed]
- 60.Harrison C. J., Hayer-Hartl M., Di Liberto M., Hartl F.-U. and Kuriyan J. (1997) Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276: 431–435 [DOI] [PubMed]
- 61.Sondermann H., Scheufler C., Schneider C., Hohfeld J., Hartl F. U. and Moarefi I. (2001) Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science 291: 1553–1557. [DOI] [PubMed]
- 62.Briknarova K., Takayama S., Brive L., Havert M. L., Knee D. A., Velasco J. et al. (2001) Structural analysis of BAG1 cochaperone and its interactions with Hsc70 heat shock protein. Nat. Struct. Biol. 8: 349–352 [DOI] [PubMed]
- 63.Brehmer D., Gassler C., Rist W., Mayer M. P. and Bukau B. (2004) Influence of GrpE on DnaK-substrate interactions. J. Biol. Chem. 279: 27957–27964 [DOI] [PubMed]
- 64.Takayama S. and Reed J. C. (2001) Molecular chaperone targeting and regulation by BAG family proteins. Nat. Cell. Biol. 3: E237–241 [DOI] [PubMed]
- 65.Höhfeld J. and Jentsch S. (1997) GrpE-like regulation of the Hsc70 chaperone by the anti-apoptotic protein BAG-1. EMBO J. 16: 6209–6216 [DOI] [PMC free article] [PubMed]
- 66.Briknarova K., Takayama S., Homma S., Baker K., Cabezas E., Hoyt D. W. et al. (2002) BAG4/SODD protein contains a short BAG domain. J. Biol. Chem. 277: 31172–31178 [DOI] [PubMed]
- 67.Kabani M., Beckerich J. M. and Gaillardin C. (2000) Sls1p stimulates Sec63p-mediated activation of Kar2p in a conformation-dependent manner in the yeast endoplasmic reticulum. Mol. Cell. Biol. 20: 6923–6934 [DOI] [PMC free article] [PubMed]
- 68.Chung K. T., Shen Y. and Hendershot L. M. (2002) BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 277: 47557–47563 [DOI] [PubMed]
- 69.Steel G. J., Fullerton D. M., Tyson J. R. and Stirling C. J. (2004) Coordinated activation of Hsp70 chaperones. Science 303: 98–101 [DOI] [PubMed]
- 70.Craven R. A., Tyson J. R. and Stirling C. J. (1997) A novel subfamily of Hsp70s in the endoplasmic reticulum. Trends Cell Biol. 7: 277–282 [DOI] [PubMed]
- 71.Tyson J. R. and Stirling C. J. (2000) LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J. 19: 6440–6452 [DOI] [PMC free article] [PubMed]
- 72.Kabani M., McLellan C., Raynes D. A., Guerriero V. and Brodsky J. L. (2002) HspBP1, a homologue of the yeast Fes1 and Sls1 proteins, is an Hsc70 nucleotide exchange factor. FEBS Lett. 531: 339–342 [DOI] [PubMed]
- 73.Kabani M., Beckerich J. M. and Brodsky J. L. (2002) Nucleotide exchange factor for the yeast Hsp70 molecular chaperone Ssa1p. Mol. Cell. Biol. 22: 4677–4689 [DOI] [PMC free article] [PubMed]
- 74.Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. et al. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272: 1606–1614 [DOI] [PMC free article] [PubMed]
- 75.Rüdiger S., Germeroth L., Schneider-Mergener J. and Bukau B. (1997) Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16: 1501–1507 [DOI] [PMC free article] [PubMed]
- 76.Rüdiger S., Schneider-Mergener J. and Bukau B. (2001) Its substrate specificity characterizes the DnaJ chaperone as scanning factor for the DnaK chaperone. EMBO J. 20: 1–9 [DOI] [PMC free article] [PubMed]
- 77.Tapley T. L. and Vickery L. E. (2004) Preferential substrate binding orientation by the molecular chaperone HscA. J. Biol. Chem. 279: 28435–28442 [DOI] [PubMed]
- 78.Cupp-Vickery J. R., Peterson J. C., Ta D. T. and Vickery L. E. (2004) Crystal structure of the molecular chaperone HscA substrate binding domain complexed with the IscU recognition peptide ELPPVKIHC. J. Mol. Biol. 342: 1265–1278 [DOI] [PubMed]
- 79.Mayer M. P., Schröder H., Rüdiger S., Paal K., Laufen T. and Bukau B. (2000) Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 7: 586–593 [DOI] [PubMed]
- 80.Pellecchia M., Montgomery D. L., Stevens S. Y., Vander Kooi C. W., Feng H., Gierasch L. M. et al. (2000) Structural insights into substrate binding by the molecular chaperone DnaK. Nat. Struct. Biol. 7: 298–303 [DOI] [PubMed]
- 81.Buczynski G., Slepenkov S. V., Sehorn M. G. and Witt S. N. (2001) Characterization of a lidless form of the molecular chaperone DnaK: deletion of the lid increases peptide on- and off-rate constants. J. Biol. Chem. 276: 27231–27236 [DOI] [PubMed]
- 82.Slepenkov S. V. and Witt S. N. (2002) Kinetic analysis of interdomain coupling in a lidless variant of the molecular chaperone DnaK: DnaK's lid inhibits transition to the low affinity state. Biochemistry 41: 12224–12235 [DOI] [PubMed]
- 83.Rüdiger S., Mayer M. P., Schneider-Mergener J. and Bukau B. (2000) Modulation of the specificity of the Hsp70 chaperone DnaK by altering a hydrophobic arch. J. Mol. Biol. 304: 245–251 [DOI] [PubMed]
- 84.Wang H., Kurochkin A. V., Pang Y., Hu W., Flynn G. C. and Zuiderweg E. R. P. (1998) NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: a preview of chaperone-protein interaction. Biochemistry 37: 7929–7940 [DOI] [PubMed]
- 85.Flynn G. C., Chappell T. G. and Rothman J. E. (1989) Peptide binding and release by proteins implicated as catalysts of protein assembly. Science 245: 385–390 [DOI] [PubMed]
- 86.Palleros D. R., Shi L., Reid K. L. and Fink A. L. (1994) Hsp70-protein complexes. J. Biol. Chem. 269: 13107–13114 [PubMed]
- 87.Schmid D., Baici A., Gehring H. and Christen P. (1994) Kinetics of molecular chaperone action. Science 263: 971–973 [DOI] [PubMed]
- 88.Pierpaoli E. V., Gisler S. M. and Christen P. (1998) Sequence-specific rates of interaction of target peptides with the molecular chaperones DnaK and DnaJ. Biochemistry 37: 16741–16748 [DOI] [PubMed]
- 89.Takeda S. and McKay D. B. (1996) Kinetics of peptide binding to the bovine 70 kDa heat shock cognate protein, a molecular chaperone. Biochemistry 35: 4636–4644 [DOI] [PubMed]
- 90.Sousa M. C. and McKay D. B. (1998) The hydroxyl of threonine 13 of the bovine 70-kDa heat shock cognate protein is essential for transducing the ATP-induced conformational change. Biochemistry 37: 15392–15399 [DOI] [PubMed]
- 91.Kelley W. L. (1999) Molecular chaperones: how J domains turn on Hsp70s. Curr. Biol. 9: R305–308 [DOI] [PubMed]
- 92.Kelley W. L. (1998) The J-domain family and the recruitment of chaperone power. Trends Biochem. Sci. 23: 222–227 [DOI] [PubMed]
- 93.Mayer M. and Bukau B. (1998) Hsp70 Chaperone systems: diversity of cellular functions and mechanism of action. Biol. Chem. 379: 261–268 [PubMed]
- 94.Cheetham M. l. E. and Caplan A. J. (1998) Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chap. 3: 28–36 [DOI] [PMC free article] [PubMed]
- 95.Linke K., Wolfram T., Bussemer J. and Jakob U. (2003) The roles of the two zinc binding sites in DnaJ. J. Biol. Chem. 278: 44457–44466 [DOI] [PubMed]
- 96.Kelley W. L. and Georgopoulos C. (1997) The T/t common exon of simian virus 40, JC and BK polyomavirus T antigens can functionally replace the J-domain of the Escherichia coli DnaJ molecular chaperone. Proc. Natl. Acad. Sci. USA 94: 3679–3684 [DOI] [PMC free article] [PubMed]
- 97.Takayama S., Xie Z. and Reed J. C. (1999) An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 274: 781–786 [DOI] [PubMed]
- 98.Thress K., Song J., Morimoto R. I. and Kornbluth S. (2001) Reversible inhibition of Hsp70 chaperone function by Scythe and Reaper. EMBO J. 20: 1033–1041 [DOI] [PMC free article] [PubMed]
- 99.Miki K. and Eddy E. M. (2002) Tumor necrosis factor receptor 1 is an ATPase regulated by silencer of death domain. Mol. Cell. Biol. 22: 2536–2543 [DOI] [PMC free article] [PubMed]
- 100.Alberti S., Esser C. and Hohfeld J. (2003) BAG-1 — a nucleotide exchange factor of Hsc70 with multiple cellular functions. Cell Stress Chap. 8: 225–231 [DOI] [PMC free article] [PubMed]
- 101.Brockmann C., Leitner D., Labudde D., Diehl A., Sievert V., Bussow K. et al. (2004) The solution structure of the SODD BAG domain reveals additional electrostatic interactions in the HSP70 complexes of SODD subfamily BAG domains. FEBS Lett. 558: 101–106 [DOI] [PubMed]
- 102.Gehring U. (2004) Biological activities of HAP46/BAG-1. The HAP46/BAG-1 protein: regulator of HSP70 chaperones, DNA-binding protein and stimulator of transcription. EMBO Rep. 5: 148–153 [DOI] [PMC free article] [PubMed]
- 103.Doong H., Vrailas A. and Kohn E. C. (2002) What's in the ‘BAG'? — a functional domain analysis of the BAG-family proteins. Cancer Lett. 1188: 25–32 [DOI] [PubMed]
- 104.Takayama S., Krajewski S., Krajewski M., Kitada S., Zapata J. M., Kochel K. et al. (1998) Expression and location of Hsp70/Hsc-binding anti-apoptotic protein BAG-1 and its variants in normal tissues and tumor cell lines. Cancer Res. 58: 3116–3131 [PubMed]
- 105.Lüders J., Demand J., Papp O. and Hohfeld J. (2000) Distinct isoforms of the cofactor BAG-1 differentially affect Hsc70 chaperone function. J. Biol. Chem. 275: 14817–14823 [DOI] [PubMed]
- 106.Lüders J., Demand J. and Hohfeld J. (2000) The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 275: 4613–4617 [DOI] [PubMed]
- 107.Song J., Takeda M. and Morimoto R. I. (2001) Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat. Cell Biol. 3: 276–282 [DOI] [PubMed]
- 108.Höhfeld J., Minami Y. and Hartl F. U. (1995) Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell 83: 589–598 [DOI] [PubMed]
- 109.Gebauer M., Zeiner M. and Gehring U. (1997) Proteins interacting with the molecular chaperone hsp70/hsc70: physical associations and effects on refolding activity. FEBS Lett. 417: 109–113 [DOI] [PubMed]
- 110.Kanelakis K. C., Murphy P. J., Galigniana M. D., Morishima Y., Takayama S., Reed J. C. et al. (2000) hsp70 interacting protein Hip does not affect glucocorticoid receptor folding by the hsp90-based chaperone machinery except to oppose the effect of BAG-1. Biochemistry 39: 14314–14321 [DOI] [PubMed]
- 111.Lambert C. and Prange R. (2003) Chaperone action in the posttranslational topological reorientation of the hepatitis B virus large envelope protein: Implications for translocational regulation. Proc. Natl. Acad. Sci. USA 100: 5199–5204 [DOI] [PMC free article] [PubMed]
- 112.Nelson G. M., Prapapanich V., Carrigan P. E., Roberts P. J., Riggs D. L. and Smith D. F. (2004) The heat shock protein 70 cochaperone hip enhances functional maturation of glucocorticoid receptor. Mol. Endocrinol. 18: 1620–1630 [DOI] [PubMed]
- 113.Picard D., Khursheed B., Garabedian M. J. Fortin M. G., Lindquist S. and Yamamoto K. R. (1990) Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature 348: 166–168 [DOI] [PubMed]
- 114.Nicolet C. and Craig E. (1989) Isolation and characterization of STI1, a stress-inducible gene from Saccharomyces cerevisiae. Mol. Cell. Biol. 9: 3638–3646 [DOI] [PMC free article] [PubMed]
- 115.Smith D. F., Sullivan W. P., Marion T. N., Zaitsu K., Madden B., McCormick, D. J. et al. (1993) Identification of a 60-Kilodalton Stress-Related Protein, p60, which interacts with hsp90 and hsp70. Mol. Cell. Biol. 13: 869–876 [DOI] [PMC free article] [PubMed]
- 116.Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H., et al. (2000) Struture of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell 101: 199–210 [DOI] [PubMed]
- 117.Odunuga O. O., Hornby J. A., Bies C., Zimmermann R., Pugh, D. J. and Blatch G. L. (2003) Tetratricopeptide repeat motif-mediated Hsc70-mSTI1 interaction. Molecular characterization of the critical contacts for successful binding and specificity. J. Biol. Chem. 278: 6896–6904 [DOI] [PubMed]
- 118.Morishima Y., Kanelakis K. C., Silverstein A. M., Dittmar K. D., Estrada L. and Pratt W. B. (2000) The Hsp organizer protein Hop enhances the rate of but is not essential for glucocorticoid receptor folding by the multiprotein Hsp90-based chaperone system. J. Biol. Chem. 275: 6894–6900 [DOI] [PubMed]
- 119.Wegele H., Haslbeck M., Reinstein J. and Buchner J. (2003) Sti1 is a novel activator of the Ssa proteins. J. Biol. Chem. 278: 25970–25976 [DOI] [PubMed]
- 120.Brychzy A., Rein T., Winklhofer K. F., Hartl F. U., Young J. C. and Obermann W. M. (2003) Cofactor Tpr2 combines two TPR domains and a J domain to regulate the Hsp70/Hsp90 chaperone system. EMBO J. 22: 3613–3623 [DOI] [PMC free article] [PubMed]
- 121.Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin, L. Y. et al. (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19: 4535–4545 [DOI] [PMC free article] [PubMed]
- 122.Nikolay R., Wiederkehr T., Rist W., Kramer G., Mayer M. P. and Bukau B. (2004) Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J. Biol. Chem. 279: 2673–2678 [DOI] [PubMed]
- 123.Höhfeld J., Cyr D. M. and Patterson C. (2001) From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2: 885–890 [DOI] [PMC free article] [PubMed]
- 124.Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Hohfeld J. et al. (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell. Biol. 3: 93–96 [DOI] [PubMed]
- 125.Jiang J., Cyr D., Babbitt R. W., Sessa W. C. and Patterson C. (2003) Chaperone-dependent regulation of endothelial nitricoxide synthase intracellular trafficking by the co-chaperone/ubiquitin ligase CHIP. J. Biol. Chem. 278: 49332–49341 [DOI] [PubMed]
- 126.Dai Q., Zhang C., Wu Y., McDonough H., Whaley R. A., Godfrey V. et al. (2003) CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 22: 5446–5458 [DOI] [PMC free article] [PubMed]
- 127.Pellecchia M., Szyperski T., Wall D., Georgopoulos C. and Wüthrich K. (1996) NMR structure of the J-domain and the Gly/Phe-rich region of the Escherichia coli DnaJ chaperone. J. Mol. Biol. 260: 236–250 [DOI] [PubMed]
- 128.Li J., Qian X. and Sha B. (2003) The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure 11: 1475–1483 [DOI] [PubMed]