

Abstract
Johanson-Blizzard syndrome (JBS) is a rare autosomal recessive condition associated with exocrine pancreatic insufficiency, and is characterized by hypoplastic nasal alae, mental retardation, sensorineural hearing loss, short stature, scalp defects, dental abnormalities and abnormal hair patterns. Growth hormone deficiency, hypopituitarism, and impaired glucagon secretion response to insulin-induced hypoglycemia have been reported. Congenital heart defects have also been described in this condition. Mental retardation is typically moderate to severe in patients with JBS; however, normal intelligence can occur. In the pancreas, there is a selective defect of acinar tissue, whereas the islets of Langerhans and ducts are preserved. Diabetes has been reported in older children, suggesting the progressive nature of pancreatic disease. The molecular basis of JBS has recently been mapped to chromosome 15q15-q21 with identified mutations in the UBR1 gene. We report the case of a 7-year-old female with pancreatic insufficiency and mild phenotypic features, in whom the diagnosis of JBS was established using recently described molecular testing for the UBR1 gene.
Keywords: Johanson-Blizzard syndrome, Pancreatic insufficiency, Sensorineural hearing loss, UBR1 gene
INTRODUCTION
Johanson-Blizzard syndrome (JBS) is a rare autosomal recessive multisystem disorder in which the most characteristic feature is exocrine pancreatic insufficiency. Other common abnormalities include an abnormal facial appearance with a small beak-like nose, dental abnormalities, sensorineural hearing loss, midline scalp defects, hypothyroidism, genitourinary abnormalities, varying degrees of mental retardation, and growth failure[1,2]. In 2005 the disease-associated locus in individuals with this syndrome was mapped to chromosome 15q15-21 with identified mutations in the gene UBR1 encoding a ubiquitin ligase of the N-end rule pathway[3]. We report the case of a 7-year-old patient recently diagnosed with JBS, confirmed by genetic testing, who has been followed for longstanding pancreatic insufficiency of unknown etiology, but with only mild phenotypic features of JBS, mild sensorineural hearing loss, and who is of normal intelligence.
CASE REPORT
The patient is a 7-year-old girl who was initially evaluated at 18 mo of age for a history of growth failure and increased stool frequency. She was a term infant, birth weight 3230 g, born to non-consanguineous parents. She was initially breast-fed and transitioned to a soy-based formula at 3 mo of age and lactose-free milk at 1 year. She tolerated the introduction of solid foods at 4 mo of age. She had a history of 3-5 large bulky stools per day that contained partially undigested food and were described as being occasionally oily. Her growth was below the 3rd percentile for weight and height. She was otherwise healthy, with the exception of having five ear infections between the ages of 6 and 18 mo, which ultimately required pressure-equalizing tube placement. Her developmental history was normal, other than starting to walk independently at 18 mo of age. Family history was negative for cystic fibrosis, celiac disease, chronic diarrhea and growth failure. Her physical examination was remarkable for a weight of 9.3 kg (7% ile) and a height of 77.6 cm (< 5% ile). Her initial laboratory evaluation is shown in Table 1. She had evidence of significant fat malabsorption; however, testing was negative for celiac disease, cystic fibrosis, and Shwachman-Diamond syndrome. Based on the presumed diagnosis of pancreatic insufficiency, she was started on pancreatic enzyme replacement and fat soluble vitamin supplementation. She gained weight and grew along the 5%-10% ile for height and weight on this regimen. She was otherwise healthy and did not have recurrent or frequent infections. Her development was normal.
Table 1.
Initial evaluation
| Complete blood count |
| Hemoglobin 11.5 g/dL (normal, 10.5-13.5) |
| WBC 7420/μL (normal, 5.5-15.5) |
| ANC 1460/μL |
| Otherwise unremarkable |
| Complete metabolic panel |
| Total protein 6.6 g/dL (normal, 6-8.4) |
| Albumin 4.1 g/dL (normal, 3.5-5) |
| Alk phos 369 U/L (normal, 80-340) |
| Otherwise unremarkable |
| Lipase 8 U/L (normal, 12-70) |
| Amylase 49 U/L (normal, 0-137) |
| PT 11.6 s |
| Vitamin A and D levels were normal |
| Vitamin E 0.1 mg/dL (normal, 0.5-2) |
| Celiac antibody testing negative |
| Serum trypsinogen < 1.2 ng/mL (normal, 10-57) |
| TSH and free T4 were normal |
| Stool ova and parasites negative |
| Seventy-two fecal fat |
| Fecal fat quant 10.6 g/24 h (normal, < 7) |
| Coefficient of fat absorption 83% |
| Stool chymotrypsin < 3 U/10 g (normal, > 9) |
Sweat test: 45 mmol/L (normal, 8-45); CFTR gene mutation analysis: No known deleterious mutations; Shwachman-Diamond gene analysis: Negative.
At 5 years of age, she failed her routine kindergarten hearing screen. Her parents had previously not noted any problem with her hearing or speech. An audiology evaluation was abnormal demonstrating a mild-to-moderate bilateral asymmetric sensorineural hearing loss, greater on the left than the right. She was referred for genetic evaluation given the known association between JBS and sensorineural hearing loss with her history of pancreatic insufficiency. At that time, mild phenotypic features of JBS were identified, including an abnormal hair pattern, hypoplasia of the nasal alae, small teeth and a narrow upper lip (Figure 1). A computed tomography (CT) scan of her abdomen demonstrated complete fatty replacement of the pancreas with no visualized gland residing in the pancreatic bed (Figure 2). Renal ultrasound to evaluate genitourinary abnormalities was negative.
Figure 1.
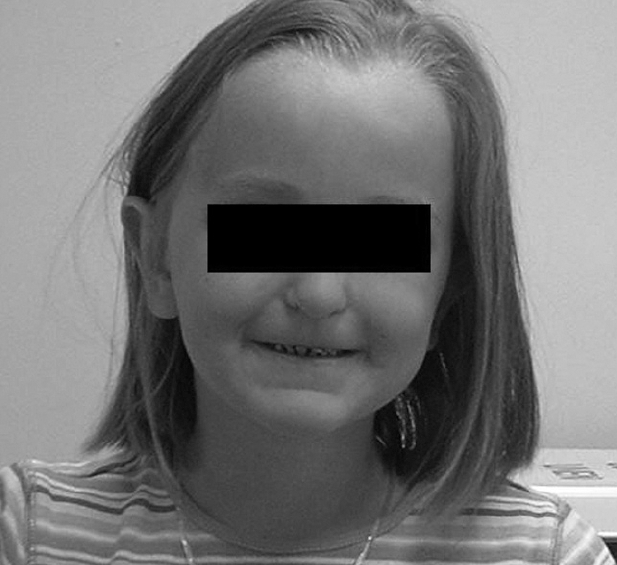
Phenotypic features of JBS in our patient: abnormal hair pattern, nasal alae hypoplasia, small teeth and narrow upper lip (with permission from parents).
Figure 2.
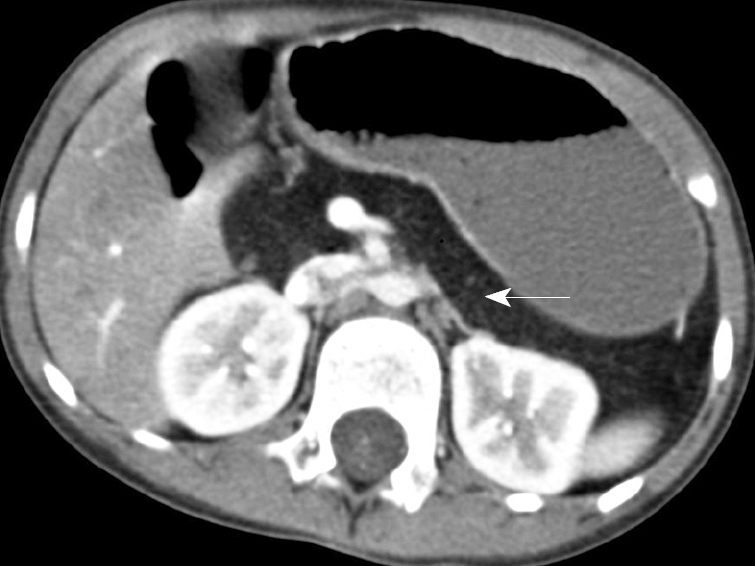
CT scan of the abdomen demonstrating fatty replacement of the pancreas (arrow).
Research testing for mutations in UBR1 revealed two novel mutations that molecularly confirmed the diagnosis of JBS, IVS1+5G>C (c.81+G>C) a splice site mutation (paternally inherited, father had a mosaic mutation present in only a subset of cells) and exon 17, c.1979_1981delTTG (p.V660del, which is a deletion of a highly conserved valine that was maternally inherited (Figure 3). While abnormal splicing at the splice donor site of exon 1 is predicted to lead to no expression of a functional protein (functional null allele), the maternally inherited deletion may represent a hypomorphic mutation, conferring partial residual function. Indeed, analysis of mRNA from lymphoblastoid cells from the patient by RT-PCR and sequencing indicated that mRNA from the allele with the splice site mutation underwent early degradation (Figure 3). However, we cannot exclude the possibility that some production of a functional UBR1 protein may also result from a low level of normal splicing despite the splice donor mutation at position +5. Our patient has continued to do well on pancreatic enzyme replacement and is being followed by otolaryngology for her hearing loss. She was referred to dental services for her tooth abnormalities.
Figure 3.

Diagram of the UBR1 gene mutations in our patient and her parents. A: Exon 1-intron 1 transition showing a heterozygous nucleotide exchange at position +5 in the patient, IVS1+5G>C (c.81+G>C). Note the small peak in the father, indicating that he is mosaic for this mutation. B: Section of exon 17 showing a heterozygous 3 bp deletion in the patient and her mother, c.1978_1981delTTG (p.V660del), predicting the deletion of a highly conserved valine. In mRNA from lymphoblastoid cells from the patient the deletion is the predominant allele, indicating that mRNA from the allele with the splice site mutation is largely degradated.
DISCUSSION
JBS is a rare autosomal recessive multisystem disorder. The most prominent feature of this syndrome is exocrine pancreatic insufficiency. Other abnormalities include a characteristic facial appearance with a small beak-like nose (secondary to aplasia or hypoplasia of the nasal alae), long and narrow upper lip, small pointed chin, abnormalities of both deciduous and permanent teeth, sparse coarse hair/midline scalp defects, short stature in > 80%, hypothyroidism in 40%, sensorineural hearing loss in > 80%, mental retardation in 77%, imperforate anus in 39%, and genitourinary abnormalities in 38%[4]. Growth hormone deficiency, hypopituitarism, and impaired glucagon secretion response to insulin-induced hypoglycemia have been reported[5-7]. Congenital heart defects including atrial septal defect, ventricular septal defect, and dextrocardia with transposition of the great vessels have also been described in this condition[8]. Mental retardation is typically moderate to severe in patients with JBS, however, normal intelligence can occur[9]. Growth failure in patients with JBS typically begins in the intrauterine period and continues throughout childhood. Pancreatic hypoplasia with resultant exocrine insufficiency and malabsorption is thought to be responsible. In the pancreas of patients with this condition there is a selective defect of acinar tissue, whereas the islets of Langerhans and ducts are preserved[10,11]. This results in an almost complete absence of zymogens from duodenal juice, whereas bicarbonate secretion is much less impaired[10]. Diabetes has been reported in older children, suggesting the progressive nature of pancreatic disease[12,13].
The molecular basis of JBS has recently been mapped to chromosome 15q15-q21 with identified mutations in the UBR1 gene[3,4]. UBR1 expression is highest in predominantly skeletal muscle and pancreatic acinar cells. UBR1 encodes one of several E3 ubiquitin ligases of the N-end rule pathway, an ubiquitin-dependent proteolytic pathway. Ubiquitylation and subsequent degradation of proteins at the proteasome is the universal mechanism for regulated protein degradation and the control of many intracellular protein levels[14-16]. UBR1 is considered to play a critical role in the development and maintenance of acinar cells. In patients with JBS, destruction of acinar tissue which may begin in utero results in the development of exocrine pancreatic insufficiency and fatty replacement of the pancreas. Since the initial description of JBS in 1971, more than 60 cases have been reported[4]. The majority of these reports include children with significant pancreatic insufficiency, markedly abnormal facial features and moderate to severe mental retardation.
Our patient presented with pancreatic insufficiency and initially unrecognized mild phenotypic features of JBS. This diagnosis was only suspected when she failed a routine screening hearing test, without prior suspicion of hearing loss. In contrast to previous findings of biallelic UBR1 mutations predicting complete loss of function in the majority of patients with JBS[3], in our patient, the maternally inherited deletion is thought to be a hypomorphic mutation conferring partial residual function and explaining the more subtle phenotype. This is the first evidence for genotype-phenotype correlation in JBS. The purpose of this report is to highlight the broader spectrum of this syndrome which may have been previously unrecognized prior to the availability of specialized genetic testing. Once the diagnosis of JBS is established, patients with this condition need to be screened for renal anomalies, referred for dental evaluation, monitored for the development of hypothyroidism and diabetes and provided with appropriate genetic counseling.
JBS is a rare cause of pancreatic insufficiency, usually associated with typical phenotypic features. The genetic basis for this syndrome has been recently identified, and is related to UBR1 deficiency which leads to perturbation of pancreatic acinar cells as well as other organs. Gastroenterologists should be aware of the availability of genetic testing for JBS. Recognition of more subtle presentations of this syndrome may help to identify other patients with this autosomal recessive condition, previously thought to have idiopathic pancreatic insufficiency.
Footnotes
Supported by Grant from the German Research Foundation (DFG) to MZ
Peer reviewer: Dr. Venkata Muddana, Internal Medicine, University of Pittsburgh Medical Center, 404 Noble St, Pittsburgh 15232, United States
S- Editor Zhong XY L- Editor Webster JR E- Editor Lin YP
References
- 1.Johanson A, Blizzard R. A syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption. J Pediatr. 1971;79:982–987. doi: 10.1016/s0022-3476(71)80194-4. [DOI] [PubMed] [Google Scholar]
- 2.Hurst JA, Baraitser M. Johanson-Blizzard syndrome. J Med Genet. 1989;26:45–48. doi: 10.1136/jmg.26.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, Beier M, Hulskamp G, Guzman C, Rehder H, et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome) Nat Genet. 2005;37:1345–1350. doi: 10.1038/ng1681. [DOI] [PubMed] [Google Scholar]
- 4.Zenker M, Mayerle J, Reis A, Lerch MM. Genetic basis and pancreatic biology of Johanson-Blizzard syndrome. Endocrinol Metab Clin North Am. 2006;35:243–253, vii-viii. doi: 10.1016/j.ecl.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Sandhu BK, Brueton MJ. Concurrent pancreatic and growth hormone insufficiency in Johanson-Blizzard syndrome. J Pediatr Gastroenterol Nutr. 1989;9:535–538. doi: 10.1097/00005176-198911000-00026. [DOI] [PubMed] [Google Scholar]
- 6.Kristjansson K, Hoffman WH, Flannery DB, Cohen MJ. Johanson-Blizzard syndrome and hypopituitarism. J Pediatr. 1988;113:851–853. doi: 10.1016/s0022-3476(88)80015-5. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi T, Fujishima M, Tsuchida S, Enoki M, Takada G. Johanson-blizzard syndrome: loss of glucagon secretion response to insulin-induced hypoglycemia. J Pediatr Endocrinol Metab. 2004;17:1141–1144. doi: 10.1515/jpem.2004.17.8.1141. [DOI] [PubMed] [Google Scholar]
- 8.Alpay F, Gul D, Lenk MK, Ogur G. Severe intrauterine growth retardation, aged facial appearance, and congenital heart disease in a newborn with Johanson-Blizzard syndrome. Pediatr Cardiol. 2000;21:389–390. doi: 10.1007/s002460010089. [DOI] [PubMed] [Google Scholar]
- 9.Moeschler JB, Lubinsky MS. Johanson-Blizzard syndrome with normal intelligence. Am J Med Genet. 1985;22:69–73. doi: 10.1002/ajmg.1320220107. [DOI] [PubMed] [Google Scholar]
- 10.Jones NL, Hofley PM, Durie PR. Pathophysiology of the pancreatic defect in Johanson-Blizzard syndrome: a disorder of acinar development. J Pediatr. 1994;125:406–408. doi: 10.1016/s0022-3476(05)83286-x. [DOI] [PubMed] [Google Scholar]
- 11.Daentl DL, Frias JL, Gilbert EF, Opitz JM. The Johanson-Blizzard syndrome: case report and autopsy findings. Am J Med Genet. 1979;3:129–135. doi: 10.1002/ajmg.1320030203. [DOI] [PubMed] [Google Scholar]
- 12.Steinbach WJ, Hintz RL. Diabetes mellitus and profound insulin resistance in Johanson-Blizzard syndrome. J Pediatr Endocrinol Metab. 2000;13:1633–1636. doi: 10.1515/jpem.2000.13.9.1633. [DOI] [PubMed] [Google Scholar]
- 13.Trellis DR, Clouse RE. Johanson-Blizzard syndrome. Progression of pancreatic involvement in adulthood. Dig Dis Sci. 1991;36:365–369. doi: 10.1007/BF01318210. [DOI] [PubMed] [Google Scholar]
- 14.Ciechanover A, Schwartz AL. The ubiquitin system: pathogenesis of human diseases and drug targeting. Biochim Biophys Acta. 2004;1695:3–17. doi: 10.1016/j.bbamcr.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 15.Pickart CM. Back to the future with ubiquitin. Cell. 2004;116:181–190. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 16.Hershko A, Ciechanover A, Varshavsky A. Basic Medical Research Award. The ubiquitin system. Nat Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]