

Abstract
AIM: To elucidate the possible crosstalk between angiogenesis, cytokeratin-18 (CK-18), and insulin resistance (IR) especially in patients with non-alcoholic steatohepatitis (NASH).
METHODS: Twenty-eight patients with NASH and 11 with simple fatty liver disease (FL) were enrolled in this study and underwent clinicopathological examination. The measures of angiogenesis, CK-18, and IR employed were CD34-immunopositive vessels, CK-18-immunopositive cells, and homeostasis model assessment of IR (HOMA-IR), respectively. The correlations of these factors with NASH were elucidated.
RESULTS: Significant development of hepatic neovascularization was observed only in NASH, whereas almost no neovascularization could be observed in FL and healthy liver. The degree of angiogenesis was almost parallel to liver fibrosis development, and both parameters were positively correlated. Similarly, CK-18 expression and HOMA-R were significantly increased in NASH as compared with FL and healthy liver. Furthermore, CK-18 and HOMA-IR were also positively correlated with the degree of neovascularization.
CONCLUSION: These results indicate that the crosstalk between angiogenesis, CK-18, and IR may play an important role in the onset and progression of NASH.
Keywords: Angiogenesis, Cytokeratin-18, Fatty liver, Insulin resistance, Non-alcoholic steatohepatitis, Liver fibrosis
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) ranges from simple steatosis to cirrhosis. Fatty liver (FL) has been recognized as a benign and non-progressive condition[1,2]. However, non-alcoholic steatohepatitis (NASH) is now widely known as a liver disease which may progress to liver cirrhosis and finally hepatocellular carcinoma (HCC). Fibrosis development is only seen in NASH but not in simple FL. The pathogenesis of NASH is not well understood, but it is unlikely that one of the recognized mechanisms explains all pathogenic processes of NASH. Thereby, NASH may develop as a consequence of a multifactorial process[2].
Angiogenesis i.e. formation of new vessels by sprouting from the pre-existing vasculature, is of central importance for embryonic development and organogenesis. Abnormally pathological angiogenesis is observed in rheumatoid arthritis, psoriasis, diabetic retinopathy, tumor growth, and even in fibrogenesis[3]. Although previous studies conducted to determine the molecular process associated with fibrosis and angiogenesis were performed independently, recent studies have revealed that both biological phenomena developed synergistically[4]. It was shown that neovascularization significantly increased during the development of liver fibrosis both in human and animal experimental studies[5,6]. In the NASH experimental model, we previously demonstrated that angiogenesis plays an important role in the progression of NASH[7]. We also reported that neovascularization developed in patients with NASH whereas no marked augmentation was observed in FL or the healthy liver[8].
Recent studies have suggested that increased hepatocyte apoptosis has an important role in progression from simple FL to NASH, and correlates with disease severity and hepatic fibrosis[9,10]. During apoptosis, some intracellular proteins are cleaved by caspase. A neoepitope in cytokeratin-18 (CK-18), the major intermediate filament protein in the liver, becomes available at an early caspase cleavage event during apoptosis and is not detectable in the viable or necrotic cells[11]. It has been reported that CK-18 fragments were markedly elevated in NAFLD patients as compared with healthy controls, and that the plasma levels of CK-18 correlated with the expression levels in the liver. A significant increase in CK-18 expression could be observed in patients with NASH as compared with the simple FL patients, and the expression level of CK-18 correlated with the presence of fibrosis[10]. The same group recently conducted a multicenter validation study which yielded similar results, and CK-18 was identified as an independent predictor of NASH[12].
NASH is well known as a liver disease that frequently complicates insulin resistance (IR) status[13]. Current evidence points to IR and subsequent hyperinsulinemia as the key pathogenic factors in NAFLD and progression from simple FL to NASH[13]. It has been reported that IR accelerated the progression in a NASH animal model, and that an insulin-sensitizing agent could reverse the underlying pathogenesis involved through improvement in IR[14]. We also have demonstrated that IR itself significantly promoted liver fibrosis development in diabetic rats[15]. Several studies have shown that insulin-sensitizing agents improved the metabolic and histologic parameters, most notably liver injury and fibrosis, not only in experimental models, but also in patients with NASH[16,17]. Collectively, these findings indicate that neovascularization, CK-18 expression, and IR status play pivotal roles in the progression of NASH. We previously demonstrated that angiogenesis and IR play important roles in animal NASH models[7,15]. However, no study has been conducted as yet to examine the interaction among these parameters in NASH using human cases.
In the current study, we elucidated the possible correlation between angiogenesis, CK-18, and IR especially in fibrosis development, which is one of the specific characteristic features of NASH compared with simple FL.
MATERIALS AND METHODS
Patients and methods
We recruited a total of 39 patients with NAFLD between 2001 and 2007 at Nara Medical University, and three healthy volunteers were enrolled as a control group. Twenty-eight patients with NASH (17 males and 11 females) and 11 patients with simple FL (seven males and four females), diagnosed by histological examination, were enrolled in this study. First, all patients were re-evaluated clinically for evidence of diseases including diabetes mellitus and hypertension. Alcohol-induced hepatitis was excluded according to each patient’s self-report and was confirmed by the family. The height and weight were measured, and the body mass index (BMI) was calculated. Hepatitis B virus surface antigen and hepatitis C virus antibody were negative in all patients. The standard liver function tests were performed for all patients. The fasting blood levels of glucose and insulin were assessed, and the homeostasis model assessment parameter of IR (HOMA-IR) was calculated. The serum fibrosis markers, namely, hyaluronic acid, type IV collagen 7S (7S-collagen), and amino-terminal peptide of type-III pro-collagen, were measured by latex agglutination, enzyme immunoassay, and radioimmunoassay, respectively, using routine laboratory methods. The serum levels of leptin and adiponectin were measured by ELISA kit (R&D systems, Tokyo, Japan) according to the manufacturer’s instructions as described previously[18,19].
Histology and immunohistochemistry
Liver biopsies obtained for diagnostic purposes were histologically examined. The liver biopsy specimens exceeded 1.5-2 cm in length and 1.4 cm in width, and contained more than five portal areas in each sample that was sufficient to perform the immunohistochemical analysis. In all samples, serial sections were used for analysis. The first section was routinely stained with hematoxylin and eosin for histological examination. Another section was stained with Sirius Red to detect fibrosis development. The fibrosis stages was scored in an F0 to F4 scale, where F0 means absence of fibrosis; F1, portal fibrosis with few septa; F2, few septa; F3, abundant septa with cirrhosis; F4, cirrhosis, as described elsewhere[20]. Regarding the 28 patients with NASH, 10 had low-grade fibrosis (F0 and F1), and 18 had high-grade fibrosis (F2 to F4). For determination of neovascularization, we employed immunohistochemical detection of CD34, which is widely used as a marker of neovascularization, using paraffin-embedded sections as described previously[21]. We also performed immunohistochemical staining of CK-18 in the liver. Liver tissue samples were fixed in 4% formaldehyde solution and embedded in paraffin. Then, 5 μm tissue sections were incubated in 0.1% hydrogen peroxide in 70% methanol for 30 min to inhibit the endogenous peroxidase activity. Microwave antigen retrieval was performed at 500 W for 15 min with pH 9 antigen retrieval solution (Nichirei Bioscience Inc., Tokyo, Japan). Then, 10% fetal bovine serum with 0.3% Triton X was used to prevent nonspecific staining. The slides were subsequently incubated overnight at 4°C in humidified chambers with primary rabbit polyclonal anti-CD34 antibody at a dilution of 1:100 and primary rabbit polyclonal anti-CK-18 antibody (DAKO, Kyoto, Japan) at a dilution of 1:100. The sections were rinsed three times in a phosphate-buffered solution and further incubated with a biotinylated secondary antibody for 30 min at room temperature. Antigen-antibody complexes were detected by the avidin-biotin-peroxide method, using diaminobenzidine as a chromogenic substrate (DAKO, Carpentaria, CA, USA). Finally, the slides were counter-stained with hematoxylin and then examined microscopically. Immunopositive quantitation of CD34 and Sirius Red-positive liver fibrosis areas were evaluated with Adobe Photoshop software and NIH image software as described previously[22]. The intensity of CK-18 staining was scored from 0 to 4 as previously described[8] with minor modification; 0: no staining; 1: mild (punctured labeling); 2: mild to moderate (dense labeling in a few lesions); 3: moderate; 4: strong (dense and homogeneous labeling in large areas).
Statistical analysis
All data are expressed as mean ± SD. The statistical analysis was performed using the χ2 test for independence, the two-tailed Student’s t-test, and simple regression analysis. Correlation between two parameters was tested by the Spearman rank correlation matrix.
RESULTS
Clinical features
The clinical features of both groups are shown in Table 1. Most of the clinical features in the FL and NASH patients were not significantly different except BMI, HOMA-IR, serum aspartate aminotransferase (AST), hyaluronic acid, leptin, and adiponectin. The NASH patients had significantly higher BMI (28.8 ± 4.33 vs 25.6 ± 1.20), HOMA-IR (5.79 ± 4.15 vs 2.26 ± 0.96), AST (59.4 ± 27.0 vs 37.5 ± 10.0), hyaluronic acid (62.0 ± 58.6 vs 15.0 ± 9.06), and leptin (7.79 ± 4.47 vs 2.58 ± 0.43), than the FL patients. On the other hand, significantly lower serum adiponectin levels were observed in the NASH patients compared with the FL patients (5.31 ± 1.70 vs 8.25 ± 0.95).
Table 1.
Characteristic features of patients with FL and NASH (mean ± SD)
| NASH (n = 28) | FL (n = 11) | P-value | |
| Age (yr) | 45.4 ± 14.7 | 43.6 ± 14.1 | 0.587 |
| Sex (M/F) | 17/11 | 7/4 | 0.660 |
| BMI (kg/m2) | 28.5 ± 6.91 | 25.8 ± 1.16 | 0.042 |
| Total cholesterol (mg/dL) | 218.6 ± 43.3 | 203.4 ± 48.3 | 0.350 |
| Triglycerides (mg/dL) | 186.1 ± 69.3 | 237.1 ± 98.2 | 0.075 |
| FFA (mmol/L) | 0.532 ± 0.207 | 0.897 ± 0.804 | 0.079 |
| Fasting plasma glucose (mg/dL) | 113.3 ± 30.0 | 98.8 ± 19.2 | 0.116 |
| Fasting plasma insulin (μU/mL) | 20.8 ± 11.5 | 10.0 ± 3.91 | 0.018 |
| HOMA-IR | 5.79 ± 4.15 | 2.26 ± 0.96 | 0.038 |
| HbA1c (%) | 6.09 ± 1.29 | 5.25 ± 0.53 | 0.130 |
| Serum albumin (g/dL) | 4.57 ± 0.33 | 4.54 ± 0.39 | 0.774 |
| AST (IU/L) | 63.4 ± 27.0 | 37.5 ± 10.0 | 0.012 |
| ALT (IU/L) | 105.5 ± 65.7 | 69.8 ± 31.2 | 0.035 |
| γ-GTP (IU/L) | 68.4 ± 37.1 | 64.2 ± 47.8 | 0.770 |
| ZTT (kU) | 7.87 ± 4.60 | 5.89 ± 2.39 | 0.224 |
| Total bilirubin (mg/dL) | 0.80 ± 0.25 | 1.00 ± 0.98 | 0.305 |
| ICG R15 (%) | 10.9 ± 4.28 | 9.00 ± 2.61 | 0.264 |
| Procollagen-III-peptide (U/mL) | 0.83 ± 0.31 | 0.96 ± 0.23 | 0.454 |
| Collagen IV-7S (ng/mL) | 4.46 ± 1.83 | 4.40 ± 1.52 | 0.762 |
| Hyaluronic acid (ng/mL) | 60.4 ± 57.6 | 15.0 ± 9.06 | 0.036 |
| Platelets (104/μL ) | 21.2 ± 8.83 | 23.2 ± 4.85 | 0.470 |
| Leptin (ng/mL) | 7.23 ± 3.98 | 2.58 ± 0.43 | 0.040 |
| Adiponectin (ng/mL) | 5.06 ± 1.90 | 8.25 ± 0.95 | 0.016 |
FL: Fatty liver; NASH: Non-alcoholic steatohepatitis; M: Male; F: Female.
Neovascularization
The typical features of CD34-immunopositive neovessels in the liver are shown in Figure 1A. Apparent CD34-positive vessels could be observed neither in the liver of the healthy subjects nor in the FL patients, whereas marked immunopositive vessels could be observed in NASH livers along with the development of liver fibrosis. In the liver of low-grade fibrosis, neovascularization could be observed around the central vein (zone III). These neovessels progressed to the portal area (zone I) and were also observed along the fibrotic septa in high-grade fibrosis. We next performed a semiquantitative analysis of the CD34-positive neovessels in conjunction with liver fibrosis development. There was no difference between the normal control liver and the FL (Figure 2A), which conformed with the immunohistochemical features. In NASH, a marked augmentation of neovascularization was found in the liver of NASH cases as compared with the FL. The magnitude of neovascularization in high-grade (F2 to F4) liver fibrosis was more than in low-grade (F0, F1) fibrosis. The increase in neovascularization was almost parallel to the development of liver fibrosis. As shown in Figure 2B, the degree of CD34-positive vessels positively correlated with fibrosis development (r2 = 0.6288).
Figure 1.
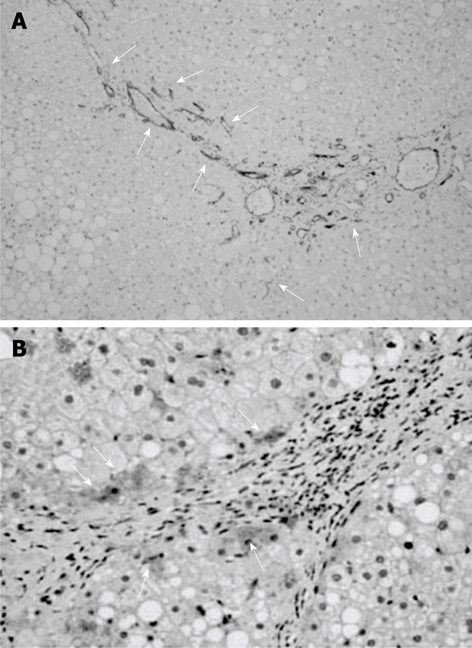
Representative microphotographs of CD34-positive neovessels and cytokeratin-18 (CK-18)-positive cells in the liver of patients. A: Marked CD34-positive immunopositive vessels could be found in NASH along with liver fibrosis development; B: The immunopositivity of CK-18 was mainly observed in the hepatocytes. The arrows indicate CD34 and CK-18-immunopositivity in A and B, respectively. The original magnification was × 100.
Figure 2.

Semi-quantitative analysis of CD34 and possible correlation with liver fibrosis development. A: Semi-quantitative analysis of the CD34-positive neovessels in the liver in non-alcoholic fatty liver disease (NAFLD). There was no difference between the control healthy liver and simple FL. In NASH, a marked augmentation of neovascularization was found in the liver of NASH as compared with FL. The magnitude of neovascularization in high-grade (F2 to F4) liver fibrosis was more than in low-grade (F0, F1) fibrosis. The proportional increase in neovascularization was almost parallel to the development of liver fibrosis. bP < 0.01 vs control group; dP < 0.01 vs low-grade fibrosis with NASH group. The number of patients in each group was as follows: Control (n = 3), FL (n = 11), low-grade fibrosis (F0 and F1: n = 10), and high-grade fibrosis (F2 to F4: n = 18). MV: Microvessel density; B: The relationship between the development of CD-34-positive vessels and fibrosis grade in NAFLD. The degree of angiogenesis correlated with the development of fibrosis. The equation was y = 509.82x - 6.7958. The correlation coefficient was 0.6288.
CK-18 and IR
We next examined CK-18 expression in the liver. The immunopositivity of CK-18 was mainly observed in the hepatocytes (Figure 1B). Similar to CD34, CK-18 expression was significantly increased in the NASH patients as compared with simple FL and healthy liver. Staining of CK-18 was barely visible in the healthy liver and FL, whereas most NASH cases showed positive staining. Among the NASH patients, those with high-grade fibrosis had greater hepatic expression of CK-18 than those with low-grade fibrosis (Figure 3A). We observed that the magnitude of CK-18 expression positively correlated with CD34-positive neovascularization (r2 = 0.6512, Figure 4A). We also examined the possible role of IR in progression of NASH, and observed that HOMA-IR was significantly higher in NASH patients than in simple FL and healthy liver. The HOMA-IR also increased with the progression of NASH, and there was a positive correlation between HOMA-IR and hepatic neovascularization (Figure 3B and Figure 4B, respectively).
Figure 3.

Semi-quantitative analysis of the CK-18-positive cells in the liver (A) and HOMA-IR (B) in NAFLD. Similar to the CD34-positive neovascularization, there was no difference between the control healthy liver and FL. In NASH, a marked augmentation of CK-18 (A) and HOMA-IR (B) was found in the liver of NASH as compared with FL. The magnitude of neovascularization in high-grade (F2 to F4) liver fibrosis was more than in low-grade (F0, F1) fibrosis. The number of patients in each group was as follows; C (n = 3), FL (n = 11), low-grade fibrosis (F0 and F1: n = 10), and high-grade fibrosis (F2 to F4: n = 18). bP < 0.01 vs control group; dP < 0.01 vs low-grade fibrosis with NASH group.
Figure 4.

The relationship between angiogenesis, CK-18-positive vessels (A), and HOMA-IR (B). The degree of angiogenesis correlated with the CK-18-positive cells (A) and HOMA-IR (B). A: The magnitude of expression of CK-18 correlated positively with CD34-positive neovascularization. The equation was y = 500.63x + 4.6606. The correlation coefficient was 0.6512; B: Similarly, there was a positive correlation between HOMA-IR and hepatic neovascularization. The equation was y = 233.75x - 339.39. The correlation efficient was 0.6184.
DISCUSSION
Recent studies have revealed that angiogenesis plays an important role in many pathological events, including liver fibrosis development. Although previous studies conducted to determine the molecular processes associated with fibrosis and angiogenesis were performed independently, recent studies have revealed that both biological phenomena emerged concomitantly[4]. Angiogenesis in the liver is characterized by capillarization of the sinusoids[3]. It has been shown that capillarization and phenotypic changes of the hepatic sinusoidal endothelial cells (ECs) occur during liver fibrosis development[23]. It has been reported that CD34 is not expressed by healthy ECs, but when ECs alter their phenotype, they can express CD34[24]. Much attention is focused on a possible association with chronic liver diseases[25]. A recent study on chronic hepatitis C (CHC) has shown that the number of CD34-positive new vessels was significantly increased and positively correlated with the fibrosis stage[26]. As well as CHC, in the current study we found a significant development of hepatic CD34-positive neovascularization in NASH, whereas almost no development could be observed in FL. The degree of angiogenesis was almost parallel to the development of liver fibrosis in NASH. These results are consistent with our previous experimental finding that angiogenesis increased stepwise during hepatic fibrosis development in several fibrosis models, including the rodent dietary NASH model[5,7]. We also observed that HOMA-IR was significantly higher in NASH along with liver fibrosis development, and that it positively correlated with the development of neovascularization. We previously reported that the IR status, i.e. co-existence of high glucose and insulin, itself significantly promoted liver fibrosis development in rats along with augmentation of neovascularization. Furthermore, high glucose and insulin stimulated in vitro neovascularization, and the combination treatment with glucose and insulin significantly promoted the effect as compared with either agent alone[15]. Collectively, it is likely that IR-mediated neovascularization is involved in the development and progression of NASH.
Because NAFLD is a common manifestation of metabolic syndromes, various adipocytokines are involved in the progression of NASH[27,28]. Among them, adiponectin and leptin are well known to be involved in the pathogenesis of NASH[27,29,30]. Adiponectin administration alleviates NAFLD progression in mice, and liver fibrosis is accelerated in adiponectin knockout mice, indicating the protective effect of adiponectin against liver fibrosis development in NASH[31]. In the NASH patients, the circulating adiponectin is reportedly decreased[30], and we also observed that the serum level of adiponectin in NASH decreased significantly more than in FL. On the other hand, recent reports have revealed that leptin exerts pro-fibrogenic activity. Hepatic fibrogenesis is impaired in leptin- and leptin receptor-deficient animals[29,32]. Leptin also enhances proliferation of activated hepatic stellate cells (HSCs), which play a central role in the development of liver fibrosis[33]. In addition to these direct effects on HSC, recent studies have revealed that leptin possesses angiogenic activity[7,34]. We previously reported that leptin exerted a potent angiogenic effect, and that leptin-mediated neovascularization played an important role in the development of liver fibrosis in the rat NASH model[7]. In the current study, the serum leptin level was significantly higher in NASH than in FL. However, we only measured the serum level of leptin in the current study. The role of leptin in fibrosis development in NASH is still controversial. It has been reported that local leptin plays a more important role than serum leptin in the progression of NASH[35]. Further studies are required to elucidate the local leptin and leptin receptor interaction with in situ hybridization in the future.
As well as neovascularization, we observed that the hepatocyte apoptosis marker, CK-18, was also significantly increased in NASH as compared with simple FL. This finding was consistent with recent reports suggesting that the CK-18 expression can detect the presence of NASH[10,12,36]. Uncontrolled hepatocyte apoptosis proved to be an important event triggering liver fibrogenesis[9]. We also observed that the expression of CK-18 was increased along with liver fibrosis development in NASH. Moreover, there was a positive correlation between the CK-18 expression and hepatic neovascularization. These results suggested that there was some crosstalk between CK-18 and angiogenesis in the liver of NASH. As well as being a marker of apoptosis, CK-18 has been found to enhance the migratory and invasive potential of tumor cells[37]. Furthermore, it has been reported that CK-18 expression was significantly higher in HCC than in CHC[38]. We previously reported that hepatic neovascularization increased in a stepwise manner during hepatocarcinogenesis, and the increase in angiogenesis was mainly observed in the glutathione-S-transferase placental form (GST-P)-positive pre-neoplastic lesions as compared with the adjacent tissues[39,40]. A recent report has shown that CK-18 expression also significantly increased in GST-P-positive lesions[41]. These results indicate that, as well as being a marker of apoptosis, CK-18 may have a direct association with hepatic neovascularization.
It is important to elucidate whether the correlation among these factors is a cause or consequence of NASH. However, in this study, we could not identify what factors were the causes or consequences in the progression of NASH. Although the respective factors interact with each other, further sequential studies are required in the future to determine what factors developed prior to other factors during the progression of NASH. Also, the number of patients was not high enough in the current study. We are acquiring a larger number of patients’ files and when the sample of patients with NASH and simple FL is adequate, we will perform an advanced analysis of the current parameters.
In conclusion, we have shown that only the liver of NASH cases had marked neovascularization whereas simple FL and healthy livers did not. The hepatic neovascularization was proportional to the increase in grade of liver fibrosis. CK-18 expression and HOMA-IR were also significantly increased in NASH as compared with FL and healthy liver. Furthermore, CK-18 and HOMA-IR also positively correlated with the degree of neovascularization. These results indicate that the crosstalk between angiogenesis, CK-18, and IR may play an important role in the onset and progression of NASH.
COMMENTS
Background
It has been reported that angiogenesis, cytokeratin-18 (CK-18), and insulin resistance (IR) play important roles in the development of non-alcoholic steatohepatitis (NASH). The aim of the current study was to elucidate the possible crosstalk between angiogenesis, CK-18, and IR especially in patients with NASH.
Research frontiers
Significant development of hepatic neovascularization was observed only in NASH, and the degree of angiogenesis was almost parallel to liver fibrosis development. Similarly, CK-18 expression and IR were significantly increased in NASH. Furthermore, CK-18 and IR also positively correlated with the degree of neovascularization.
Innovations and breakthroughs
Only NASH was associated with marked neovascularization, which was proportional to the increase in grade of liver fibrosis. Moreover, the degree of neovascularization positively correlated with CK-18 and IR in NASH. These results emphasize the new findings that the crosstalk between angiogenesis, CK-18, and IR plays an important role in the onset and progression of NASH.
Applications
The novel findings may lead to a new alternative therapy for NASH in the near future.
Peer review
The manuscript is well written and the conclusions are appropriate for the results.
Footnotes
Peer reviewers: Osman C Ozdogan, Associate Professor, Department of Gastroenterology, Liver Unit, Marmara University School of Medicine, Istanbul 34662, Turkey; Dr. Valentina Medici, University of California Davis, 4150 V Street, Suite 3500, Sacramento, 95814, United States
S- Editor Tian L L- Editor Cant MR E- Editor Zheng XM
References
- 1.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 2.Reid AE. Nonalcoholic steatohepatitis. Gastroenterology. 2001;121:710–723. doi: 10.1053/gast.2001.27126. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 4.Kalluri R, Sukhatme VP. Fibrosis and angiogenesis. Curr Opin Nephrol Hypertens. 2000;9:413–418. doi: 10.1097/00041552-200007000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Hicklin DJ, Wu Y, Yanase K, Namisaki T, Yamazaki M, Tsujinoue H, Imazu H, Masaki T, Fukui H. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut. 2003;52:1347–1354. doi: 10.1136/gut.52.9.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medina J, Caveda L, Sanz-Cameno P, Arroyo AG, Martín-Vílchez S, Majano PL, García-Buey L, Sánchez-Madrid F, Moreno-Otero R. Hepatocyte growth factor activates endothelial proangiogenic mechanisms relevant in chronic hepatitis C-associated neoangiogenesis. J Hepatol. 2003;38:660–667. doi: 10.1016/s0168-8278(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 7.Kitade M, Yoshiji H, Kojima H, Ikenaka Y, Noguchi R, Kaji K, Yoshii J, Yanase K, Namisaki T, Asada K, et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology. 2006;44:983–991. doi: 10.1002/hep.21338. [DOI] [PubMed] [Google Scholar]
- 8.Kitade M, Yoshiji H, Kojima H, Ikenaka Y, Noguchi R, Kaji K, Yoshii J, Yanase K, Namisaki T, Yamazaki M, et al. Neovascularization and oxidative stress in the progression of non-alcoholic steatohepatitis. Mol Med Report. 2008;1:543–548. [PubMed] [Google Scholar]
- 9.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 10.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 11.Ueno T, Toi M, Linder S. Detection of epithelial cell death in the body by cytokeratin 18 measurement. Biomed Pharmacother. 2005;59 Suppl 2:S359–S362. doi: 10.1016/s0753-3322(05)80078-2. [DOI] [PubMed] [Google Scholar]
- 12.Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, McCullough AJ. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. 2009;50:1072–1078. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polyzos SA, Kountouras J, Zavos C. Nonalcoholic fatty liver disease: the pathogenetic roles of insulin resistance and adipocytokines. Curr Mol Med. 2009;9:299–314. doi: 10.2174/156652409787847191. [DOI] [PubMed] [Google Scholar]
- 14.Ota T, Takamura T, Kurita S, Matsuzawa N, Kita Y, Uno M, Akahori H, Misu H, Sakurai M, Zen Y, et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology. 2007;132:282–293. doi: 10.1053/j.gastro.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Kaji K, Yoshiji H, Kitade M, Ikenaka Y, Noguchi R, Yoshii J, Yanase K, Namisaki T, Yamazaki M, Moriya K, et al. Impact of insulin resistance on the progression of chronic liver diseases. Int J Mol Med. 2008;22:801–808. [PubMed] [Google Scholar]
- 16.Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 17.Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, Podevin P, Lacorte JM, Bernhardt C, Bruckert E, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100–110. doi: 10.1053/j.gastro.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 19.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 20.Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. The French METAVIR Cooperative Study Group. Hepatology. 1994;20:15–20. [PubMed] [Google Scholar]
- 21.Di Carlo I, Fraggetta F, Lombardo R, Azzarello G, Vasquez E, Puleo S. CD 34 expression in chronic and neoplastic liver diseases. Panminerva Med. 2002;44:365–367. [PubMed] [Google Scholar]
- 22.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Hicklin DJ, Huber J, Nakatani T, Tsujinoue H, Yanase K, et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology. 2002;35:834–842. doi: 10.1053/jhep.2002.32541. [DOI] [PubMed] [Google Scholar]
- 23.Park YN, Yang CP, Fernandez GJ, Cubukcu O, Thung SN, Theise ND. Neoangiogenesis and sinusoidal "capillarization" in dysplastic nodules of the liver. Am J Surg Pathol. 1998;22:656–662. doi: 10.1097/00000478-199806000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Theuerkauf I, Zhou H, Fischer HP. Immunohistochemical patterns of human liver sinusoids under different conditions of pathologic perfusion. Virchows Arch. 2001;438:498–504. doi: 10.1007/s004280000364. [DOI] [PubMed] [Google Scholar]
- 25.Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604–620. doi: 10.1016/j.jhep.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Gabriel A, Kukla M, Wilk M, Liszka Ł, Petelenz M, Musialik J. Angiogenesis in chronic hepatitis C is associated with inflammatory activity grade and fibrosis stage. Pathol Res Pract. 2009;205:758–764. doi: 10.1016/j.prp.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Marra F, Aleffi S, Bertolani C, Petrai I, Vizzutti F. Adipokines and liver fibrosis. Eur Rev Med Pharmacol Sci. 2005;9:279–284. [PubMed] [Google Scholar]
- 28.Larter CZ, Farrell GC. Insulin resistance, adiponectin, cytokines in NASH: Which is the best target to treat? J Hepatol. 2006;44:253–261. doi: 10.1016/j.jhep.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 29.Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206–213. doi: 10.1016/s0168-8278(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 30.Musso G, Gambino R, Biroli G, Carello M, Fagà E, Pacini G, De Michieli F, Cassader M, Durazzo M, Rizzetto M, et al. Hypoadiponectinemia predicts the severity of hepatic fibrosis and pancreatic Beta-cell dysfunction in nondiabetic nonobese patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2005;100:2438–2446. doi: 10.1111/j.1572-0241.2005.00297.x. [DOI] [PubMed] [Google Scholar]
- 31.Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–1807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 32.Honda H, Ikejima K, Hirose M, Yoshikawa M, Lang T, Enomoto N, Kitamura T, Takei Y, Sato N. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology. 2002;36:12–21. doi: 10.1053/jhep.2002.33684. [DOI] [PubMed] [Google Scholar]
- 33.Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sierra-Honigmann MR, Nath AK, Murakami C, García-Cardeña G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 35.Ikejima K, Okumura K, Lang T, Honda H, Abe W, Yamashina S, Enomoto N, Takei Y, Sato N. The role of leptin in progression of non-alcoholic fatty liver disease. Hepatol Res. 2005;33:151–154. doi: 10.1016/j.hepres.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 36.Younossi ZM, Jarrar M, Nugent C, Randhawa M, Afendy M, Stepanova M, Rafiq N, Goodman Z, Chandhoke V, Baranova A. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH) Obes Surg. 2008;18:1430–1437. doi: 10.1007/s11695-008-9506-y. [DOI] [PubMed] [Google Scholar]
- 37.Chu YW, Runyan RB, Oshima RG, Hendrix MJ. Expression of complete keratin filaments in mouse L cells augments cell migration and invasion. Proc Natl Acad Sci USA. 1993;90:4261–4265. doi: 10.1073/pnas.90.9.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Athanassiadou P, Psyhoyiou H, Grapsa D, Gonidi M, Ketikoglou I, Patsouris E. Cytokeratin 8 and 18 expression in imprint smears of chronic viral hepatitis, autoimmune hepatitis and hepatocellular carcinoma. A preliminary study. Acta Cytol. 2007;51:61–65. doi: 10.1159/000325684. [DOI] [PubMed] [Google Scholar]
- 39.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Hicklin DJ, Wu Y, Yanase K, Namisaki T, Kitade M, et al. Halting the interaction between vascular endothelial growth factor and its receptors attenuates liver carcinogenesis in mice. Hepatology. 2004;39:1517–1524. doi: 10.1002/hep.20218. [DOI] [PubMed] [Google Scholar]
- 40.Yoshiji H, Noguchi R, Kitade M, Kaji K, Ikenaka Y, Namisaki T, Yoshii J, Yanase K, Yamazaki M, Tsujimoto T, et al. Branched-chain amino acids suppress insulin-resistance-based hepatocarcinogenesis in obese diabetic rats. J Gastroenterol. 2009;44:483–491. doi: 10.1007/s00535-009-0031-0. [DOI] [PubMed] [Google Scholar]
- 41.Kakehashi A, Inoue M, Wei M, Fukushima S, Wanibuchi H. Cytokeratin 8/18 overexpression and complex formation as an indicator of GST-P positive foci transformation into hepatocellular carcinomas. Toxicol Appl Pharmacol. 2009;238:71–79. doi: 10.1016/j.taap.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]