

Abstract
Presenilin-1 (PS1) and -2 (PS2), which when mutated cause familial Alzheimer disease, have been localized to numerous compartments of the cell, including the endoplasmic reticulum, Golgi, nuclear envelope, endosomes, lysosomes, the plasma membrane, and mitochondria. Using three complementary approaches, subcellular fractionation, γ-secretase activity assays, and immunocytochemistry, we show that presenilins are highly enriched in a subcompartment of the endoplasmic reticulum that is associated with mitochondria and that forms a physical bridge between the two organelles, called endoplasmic reticulum-mitochondria-associated membranes. A localization of PS1 and PS2 in mitochondria-associated membranes may help reconcile the disparate hypotheses regarding the pathogenesis of Alzheimer disease and may explain many seemingly unrelated features of this devastating neurodegenerative disorder.
Alzheimer disease (AD) is a late onset neurodegenerative disorder characterized by progressive neuronal loss, especially in the cortex and the hippocampus.1 The two main histopathological hallmarks of AD are the accumulation of extracellular neuritic plaques, consisting predominantly of β-amyloid (Aβ), and of neurofibrillary tangles, consisting mainly of hyperphosphorylated forms of the microtubule-associated protein tau.1
The vast majority of AD is sporadic, but mutations in amyloid precursor protein (APP), presenilin-1 (PS1), and presenilin-2 (PS2) have been identified in the rarer familial form, which is similar to sporadic AD but has an earlier age of onset.
PS1 and PS2 are aspartyl proteases that cleave their substrates within transmembrane regions. The active forms of PS1 and PS2 are N- and C-terminal fragments, which are produced by cleavage of full-length presenilin in its “loop” domain.2 PS1 and PS2 are components of the γ-secretase complex that processes a number of plasma-membrane proteins, including Notch, Jagged, E-cadherin, and, most relevant to AD, APP. The γ-secretase complex also contains three other structural subunits: APH1, nicastrin (also called APH2), and presenilin enhancer protein 2.2
Following cleavage of APP by β-secretase, γ-secretase cleaves the ∼100-aa C-terminal “β-stub” to release small amyloidogenic fragments, 40- and 42-aa in length (Aβ40 and Aβ42), that have been implicated in the pathogenesis of AD, as well as a ∼60-aa APP intracellular domain.1 Whereas the components of the γ-secretase complex are localized predominantly intracellularly,3,4 its substrates, including APP, are located mainly in the plasma membrane (PM).5 This discrepancy forms the basis of what has been called the “spatial paradox.”6
PS1 has been localized to numerous compartments of the cell, including the endoplasmic reticulum (ER),7 Golgi,7 the nuclear envelope,8 endosomes,9 lysosomes,10 mitochondria,11 kinetochores and centrosomes,12 and the plasma membrane, where it is especially enriched at intercellular contacts known as adherens junctions.13
The ER and mitochondria are linked, not only biochemically but also physically,14 via ER-mitochondria-associated membranes (ER-MAM, or MAM).14,15 MAM was described almost 20 years ago as a specific compartment involved in the synthesis and transfer of phospholipids between the ER and mitochondria.16 More than two dozen proteins are concentrated in MAM (see Supplemental Table S1 at http://ajp.amjpathol.org),15,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34 including proteins involved in calcium homeostasis (eg, inositol triphosphate receptor isoform 3), in lipid metabolism (eg, fatty acid co-A ligase 4 [FACL4]), in intermediate metabolism (eg, glucose-6-phosphatase), in cholesterol metabolism (eg, acyl-coenzyme A:cholesterol acyltransferase 1), and in the transfer of lipids between the ER and mitochondria. A few nonenzymatic proteins are also concentrated in MAM (see Supplemental Table S1 at http://ajp.amjpathol.org),15,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34 suggesting that it is a domain of the ER with specialized functions. Contacts between the two organelles are maintained by MAM-associated proteins, such as phosphofurin acidic cluster sorting protein 2, which controls the apposition of mitochondria with the ER and which appears to stabilize and regulate the interaction of ER and mitochondria.34
Using a combination of biochemical and morphological approaches, we show here that PS1 and PS2 are highly enriched in MAM. The discovery that presenilins are not distributed homogeneously in the ER, but rather are enriched in the MAM subcompartment, has potentially significant implications regarding the pathogenesis of familial AD in particular and AD in general.
Materials and Methods
Cells and Reagents
Normal human fibroblasts (lines AE and TK), cultured primary rat cortical neurons, and mouse embryonic PS1-, PS2- and PS1/PS2-knockout fibroblasts, were kind gifts of Dr. Michio Hirano (Columbia University), Dr. David Sulzer (Columbia University), and Dr. Bart De Strooper (University of Leuven), respectively. Mutant FAD-A246E (AG06840) cells were obtained from the Coriell Institute for Medical Research (Camden, NJ). Mouse 3T3 cells and monkey COS-7 cells were available in the laboratory.
Antibodies
We used the antibodies to aa 31-46 (Sigma P4985), aa 450-467 (Sigma P7854), and aa 303-316 (Calbiochem PC267) of human PS1, and to aa 324-335 of human PS2 (Sigma P0482 and Cell Signaling 2192). We also used antibodies to APH-1 (ABR PA1-2010), APP (a kind gift of T.-W. Kim, Columbia University35), FACL4 (Abgent AP 2536b), Golgi matrix protein GM130/GOLGA2 (Monoclonal BD transduction #610822), inositol triphosphate receptor 3 (Millipore AB9076), Na,K-ATPase (Abcam ab7671), NDUFA9 (monoclonal; Molecular Probes A21344), nicastrin (Covance PRB-364P), presenilin enhancer protein 2 (Abcam ab62514), and SSRα (a generous gift of Howard Worman and Martin Wiedemann, Columbia University36). Mouse monoclonal anti-rabbit “bridge” antibodies were from Sigma (R1008; used at 1:2000).
For Western blotting, samples were resuspended in 2× Laemmli buffer, heated for 10 minutes at 60°C, electrophoresed, transferred to polyvinylidene difluoride, and probed with antibodies.
Immunofluorescence
To detect mitochondria, we labeled the cells with 1 nmol/L MitoTracker Red CMXRos (MT Red; Invitrogen) in Dulbecco’s modified essential medium for 20 minutes at 37°C. After washing, we fixed the cells in chilled methanol for 20 minutes at −20°C, blocked them in 2.5% normal goat serum, and 0.1% Tween-20 in 1× PBS, and incubated them with primary antibodies.
Cells were imaged in a single-plane by confocal microscopy with a Zeiss LSM510 microscope using a 63× and a 100× Plan-Neofluar, 1.25 NA objective lens. Percent co-localization of image signals was calculated using Image J (http://rsb.info.nih.gov/ij). Briefly, the area occupied by the signal for each marker (eg, MT Red, anti-PS1, anti-FACL4) was calculated from single confocal optical sections acquired with a 100×/1.4 objective. The thresholded image of the first marker signal (eg, MT Red) was used as a mask of the second marker signal (eg, anti-PS1). Percent colocalization was calculated as the area of the second marker signal within the mask divided by the total area of the second marker signal in the image. This analysis was performed on 5 to 6 images containing ∼5 to 10 cells per field, and the various colocalization data sets were compared using Manders’ overlap coefficient and Student’s t-test to measure statistical significance (P < 0.01).
Subcellular Fractionation
Purification of ER, MAM, and mitochondria was performed essentially as described.16,31 Cells and tissues were homogenized gently in isolation buffer (250 mmol/L mannitol, 5 mmol/L HEPES pH 7.4, and 0.5 mmol/L EGTA) with four strokes in a loose Potter-Elvehjem grinder (Kontes). The homogenate was centrifuged for 5 minutes at 600 × g to remove cells debris and nuclei. The supernatant was centrifuged for 15 minutes at 10,500 × g; the supernatant contained the ER/microsomal fraction and the pellet contained the crude mitochondrial fraction. The supernatant was centrifuged for 1 hour at 100,000 × g to pellet the ER/microsomal fraction. The crude mitochondrial fraction was layered on top of a 30% Percoll gradient and centrifuged for 30 minutes at 95,000 × g in a Beckman Coulter Ultracentrifuge. The upper band contained the MAM fraction and the lower band contained mitochondria free of ER. The upper band was diluted fivefold with isolation buffer and centrifuged at 6300 × g for 10 minutes, twice, to obtain the mitochondrial fraction in the pellet. The supernatant containing the MAM was centrifuged at 100,000 × g for 1 hour in a Beckman Ti70.1 rotor, and the resulting MAM pellet was resuspended in isolation buffer. The lower band was washed twice by centrifugation at 6300 × g for 10 minutes to remove the Percoll, after which the mitochondria were resuspended in isolation buffer and combined with the mitochondria derived from the upper band. All fractions were quantitated for total protein content using the Bradford system (BioRad).
To obtain the PM fraction, tissues were homogenized in STM 0.25 buffer (0.25 M/L sucrose, 10 mmol/L Tris·Cl pH 7.4, 1.0 mmol/L MgCl2; 4.5 ml/g tissue), using a loose-fitting Potter-Elvehjem grinder (Kontes) (10 strokes). Homogenates were centrifuged for 5 minutes at 260 × g and the supernatant was kept on ice. The pellet, containing nuclei and cell debris, was resuspended in half the volume of the same buffer and homogenized with three strokes on the same loose grinder and pelleted again for 5 minutes at 260 × g. Both supernatants were combined and centrifuged for 10 minutes at 1500 × g. The pellet, containing the PM, was resuspended in twice the volume of STM 0.25 used initially and was further homogenized by three strokes, but using a tight-fitting grinder (Kontes). The homogenate was diluted by adding an equal volume of STM 2 buffer (2 M/L sucrose, 10 mmol/L Tris·Cl pH 7.4, 1.0 mmol/L MgCl2), and centrifuged for 1 hour at 113,000 × g. The resultant low-density thin layer located near the top of the gradient, enriched in PM, was resuspended in 0.5 to 1 volume of STM 0.25 buffer.
γ-Secretase Activity Assays
Endogenous γ-secretase activity was determined by Western blotting to detect the amount of APP intracellular domain derived from the cleavage of endogenous APP, as described.35 We incubated 50 μg of protein from each fraction in reaction buffer (10 mmol/L Tris-HCl, 150 mmol/L NaCl, 5 mmol/L EDTA, pH 7.4) for 3 hours at 37°C, followed by Western blotting with anti-APP. As a control, the same samples were assayed in the presence of 2 μmol/L compound E ([(2S)−2-{[(3,5-difluorophenyl)acetyl]amino}-N-[(3S)−1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H−1,4-benzodiazepin- 3-yl]propanamide]; Alexis Biochemicals, ALX270-415-C250), a γ-secretase inhibitor.37 We also used a fluorescence based energy transfer-based γ-secretase activity assay to detect cleavage of an exogenously added secretase-specific peptide conjugated to two fluorescent reporter molecules (R&D Systems FP003) in serial dilutions of different subcellular fractions. As a control, the same samples were assayed in the presence of 2 μmol/L compound E.
In Vitro Import Assay
Human PS1 was transcribed and translated using a reticulocyte lysate system and imported into isolated mitochondria, as described.38
Results
We isolated PM, crude mitochondria, and ER from mouse brain, and fractionated crude mitochondria further by isopycnic centrifugation30 into a MAM fraction and a purified mitochondrial fraction. We evaluated each of these fractions by Western blot analysis, using antibodies to Na,K-ATPase as a marker for PM, to SSRα as a marker for ER, to Golgi matrix protein GM130 (GOLGA2) as a marker for Golgi, to inositol triphosphate receptor 3 as a marker for MAM, and to the subunit NDUFA9 as a marker for mitochondria (Figure 1A). All five markers were enriched in their respective compartments, but we note low levels of mitochondria NDUF9A were also present in the plasma membrane. Perhaps a number of mitochondrial proteins have been found in this compartment by others.39 The MAM fraction was enriched for inositol triphosphate receptor isoform 3, a known MAM marker,18 confirming our ability to separate MAM from bulk ER and mitochondria to a degree sufficient for further analysis. We quantitated the amount of protein recovered in each of the subcellular fractions analyzed from whole mouse brain. Of the total amount of protein recovered in the ER fraction, we estimate that ∼13% ± 0.3% (n = 6) was in the MAM subfraction. This value reflects the analysis of total mouse brain, and is likely to vary in different brain regions and in different tissues.
Figure 1.
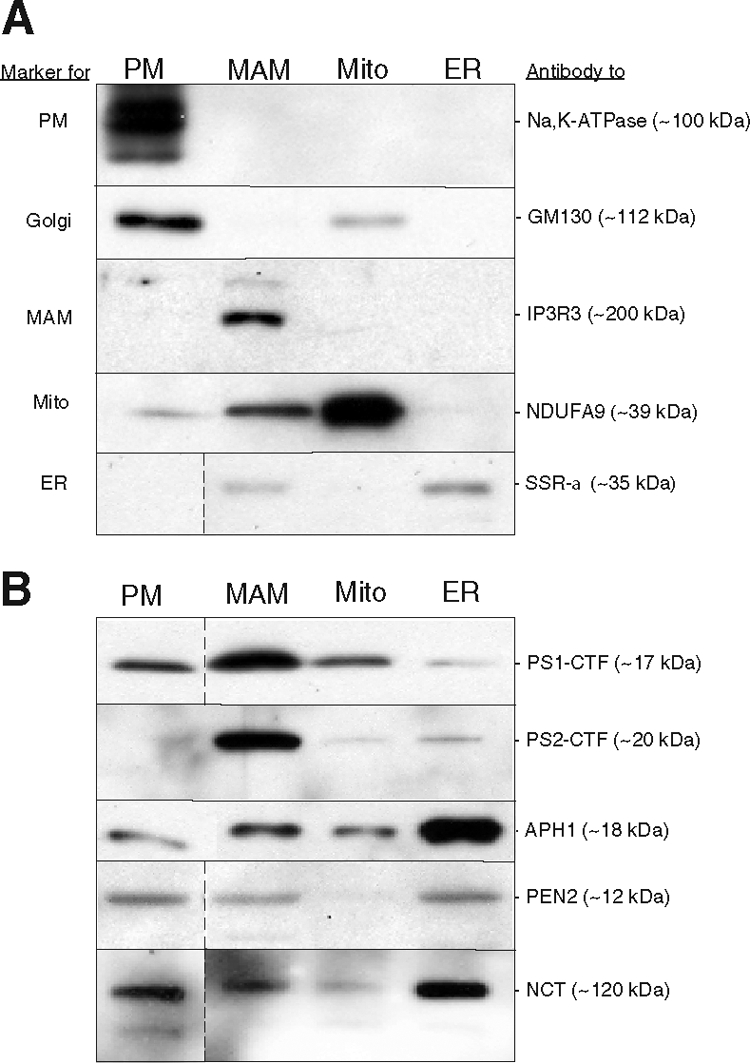
Western blot analysis of subcellular fractions of mouse brain. Thirty μg of total protein were loaded in each lane. A: Localization and predicted molecular masses of the indicated polypeptides were determined using the antibodies listed at right (see text). PM, plasma membrane. B: Fractions were probed using the indicated antibodies against PS1 (Calbiochem PC267) and PS2 (Cell Signaling 2192) and to other components of the γ-secretase complex. In the blots shown here, the intensity of both the PS1 and the PS2 signals in MAM was enriched ∼eightfold over that in the ER. In some blots, data represent nonadjacent lanes taken from a single blot; dividing lines indicate where lanes were pasted together.
We then performed Western blot analysis on these same fractions from mouse brain, using antibodies against PS1 and PS2 (Figure 1B). PS1 was found in the plasma membrane/Golgi fractions, as reported previously,3 but in our hands, PS1 is essentially an ER-resident protein (Figure 1B). However, within the ER, PS1 was not distributed homogeneously, but rather was enriched in ER membranes that are in close contact with mitochondria (ie, MAM) (Figure 1B). Like PS1, PS2 was also enriched in the MAM (Figure 1B). Analysis of the blots revealed that the amount of PS1 was enriched by 5- to 10-fold in MAM over that in “bulk” ER (n = 12).
We then assayed the various subcellular fractions of mouse brain for the presence and amount of γ-secretase activity, using two different assays (Figures 2, A and B).40 We detected most of the γ-secretase activity in MAM compared with the other fractions assayed, implying not only that PS1 and PS2 are enriched in this fraction, but that the other components of the γ-secretase complex—APH1, nicastrin, and presenilin enhancer protein 2—are present there as well.41 Using Western blotting, we found that those three polypeptides were enriched in the bulk ER, but were present in significant amounts in the MAM as well. We do not know why the amount of the various γ-secretase components are not distributed proportionally in the two compartments; perhaps this is a reflection of the different steps in the assembly pathway for the holoprotein.42 Moreover, APP itself was also present in high amounts in the MAM (Figure 2B). Thus, MAM contains both the enzymatic activity to cleave APP (ie, γ-secretase) and the APP substrate itself. The localization of γ-secretase activity in MAM could help explain the unexpected presence of Aβ in mitochondria.43
Figure 2.

γ-Secretase activity assays. A: Activity using a fluorescence based energy transfer-based assay, in the absence and presence of Compound E, a γ-secretase inhibitor.37 Serial dilutions of the indicated subcellular fractions from mouse brain were assayed for APP cleavage activity (in arbitrary units/μg protein). Bars = SD; *P < 0.05 in MAM compared with the other fractions n = 3 for all fractions. B: Αctivity using Western blotting to detect APP intracellular domain,40 in the absence and presence of Compound E. The identity of the lower bands in the first and third lanes is unknown, but may be cross-reaction to another APP-like polypeptide (eg, APLP1, APLP2). The specificity of the APP intracellular domain signal was confirmed in PS1/PS2 double-knockout mouse embryonic fibroblasts (not shown).
To further confirm that PS1 is a MAM-enriched protein, we compared the immunocytochemical localization of PS1 in human fibroblasts with that of FACL4, a known MAM-localized protein.24 We first stained cells with MT Red and then detected FACL4 by immunocytochemistry (Figure 3A). We found that FACL4 immunostain (green) “co-localized” with MT Red (red), but only partially: the “colocalization” was most predominant in the region around the nucleus (yellow arrowhead in Figure 3A), but not in the more distal regions of the cell (red arrowhead in Figure 3A). This result implies that the much of the yellow signal reflected the juxtaposition of MAM with mitochondria (see enlarged merge panel at right in Figure 3A). Like FACL4, PS1 partially colocalized with MT Red, and also predominantly in the perinuclear region (Figure 3B). The apparent colocalization of PS1 with MT Red in the perinuclear region was revealed to actually consist of small discrete regions of PS1 immunostain apposed to discrete MT Red-positive regions (enlarged merge panel at right in Figure 3B), a pattern highly similar to that observed with FACL4 (Figure 3A) and with the sigma-1 receptor, another MAM-resident protein.21 This result is also consistent with our finding that PS1 was not imported into mitochondria in an in vitro import assay (data not shown). Finally, when we double-stained cells for both PS1 and FACL4, the two proteins colocalized almost exactly, even at enlarged magnification (Figure 3C). These results imply that both PS1 and FACL4 reside in the same compartment, namely MAM. Quantitative analysis of the degree of colocalization confirmed these conclusions. In particular, the colocalization of PS1 with MT Red (as a decimal fraction) was 0.51 ± 0.08, which was not statistically different from the value of 0.47 ± 0.05 for the co-localization of FACL4, an authentic MAM protein, with MT Red. The quantitative data support the immunocytochemical results, namely, that PS1 is not a mitochondrial protein, but resides in a compartment adjacent to mitochondria, in a manner essentially identical to that of FACL4 (ie, MAM).
Figure 3.

Immunocytochemistry to detect FACL4 and presenilins in mammalian cells. A: Double staining of human fibroblasts with MT Red and anti-FACL4. FACL4 apparently colocalizes with MT Red, mainly in regions proximal to the nucleus (yellow arrowhead), with a lower degrees of co-localization in more distal mitochondria (red arrowhead). In an enlarged view of the perinuclear region from another merged field (rightmost panel), note discrete regions where the red and green signals (arrowheads) are in apposition and do not overlap. B: Double staining of human fibroblasts with MT Red and anti-PS1. Note the similarity of the colocalization pattern to that seen with FACL4. C: Double-staining of human fibroblasts with anti-FACL4 (red) and anti-PS1 (green). There is significant overlap between the red and green signals, even in the enlarged merged view of the perinuclear region, implying that both proteins are in the same compartment (ie, MAM). D: Double staining of mouse 3T3 cells with MT Red and anti-PS2. Note the similarity of the colocalization pattern to that seen in A and B. E: Double staining of confluent COS-7 cells with MT Red and anti-PS1, photographed in a plane of focus to reveal the localization of PS1 to adherens junctions (adherens junctions; arrowheads). The MT Red staining is fuzzy because almost all mitochondria are below the plane of focus. Note the absence of colocalization of PS1 with MT Red in adherens junctions. Immunostaining of anti-PS1 (Ab P7854) was suppressed in the presence of the peptide epitope used to generate the antibody, confirming its specificity (not shown).
The immunocytochemical results were confirmed numerous times in other cell types, including primary rat cortical neurons, mouse embryonic fibroblasts, mouse 3T3 cells, and monkey COS-7 cells (data not shown). Importantly, we obtained a similar result using immunocytochemistry to detect human PS2 in mouse cells (Figure 3D). Finally, besides the immunocytochemical localization to MAM, we also observed PS1 staining at adherens junctions in the plasma membrane in confluent COS-7 (Figure 3E) and in human 293T and mouse 3T3 cells (not shown), confirming the observations of others.13
Discussion
Taken together, the Western blotting, γ-secretase activity, and immunocytochemistry results imply that PS1 and PS2 are indeed MAM-enriched proteins, in both neuronal and non-neuronal cells. We believe that the discrepancy between our results and reports in which presenilins were found in fractions enriched in markers characteristic of other subcellular compartments, such as ER,7 Golgi,7 the trans-Golgi network,4 the ER-Golgi intermediate compartment,7 the nuclear envelope,8 endosomes,9 lysosomes,10 and mitochondria,11 is due mainly to technical issues. In some analyses of subcellular fractions, other organelles, including MAM, co-purified with ER,7 Golgi,7 or mitochondria.11 For example, after careful fractionation, sphingolipid-specific glycosyltransferase activity, which previously had been ascribed to the Golgi, was actually found to be in MAM27; in fact, MAM has been described as a pre-Golgi compartment for the secretory pathway.15 In other cases, the subcellular fractionation separated PS1 into a compartment that was almost certainly MAM, but in the absence of specific MAM markers was either not identified clearly or was identified in nonspecific terms as an ER-related subcompartment.44
We have shown here that presenilins residing in the MAM are functionally active, acting as the catalytic core of the γ-secretase complex. However, we cannot exclude the possibility that PS1 and/or PS2 are also involved in other functions in the MAM compartment. The finding that most of the γ-secretase activity is located in ER-mitochondria connections could help to explain the observation of mitochondrial oxidative damage associated with abnormal APP processing.45 Moreover, it could also help explain how Aβ accumulates in mitochondria,46 as well as provide the basis for the interaction between PS1 and a number of known mitochondrial proteins.
Numerous hypotheses have been proposed to explain the pathogenesis of AD, including altered APP processing and amyloid toxicity,46,47 tau hyperphosphorylation,48 altered lipid,49 cholesterol,50 and glucose51 metabolism, aberrant calcium homeostasis,52 glutamate excitotoxicity,53 inflammation,53 and mitochondrial dysfunction and oxidative stress.45 A localization of presenilin in MAM, a compartment intimately involved in lipid, glucose, cholesterol, and calcium homeostasis, may help reconcile these disparate hypotheses, and could explain many seemingly unrelated features of this devastating neurodegenerative disorder.
Supplementary Material
Acknowledgments
We thank Michio Hirano, Huang Yang, Vernice Jackson-Lewis, David Sulzer, and Bart De Strooper for providing cells and tissues; Theresa Swayne for expert assistance with the microscopy; and Wim Annaert, Tae-Wan Kim, Martin Wiedmann, Howard Worman, and Jean Vance for providing antibodies.
Footnotes
Address reprint requests to Eric A. Schon, Department of Neurology, Room 303A, Russ Berrie Medical Pavilion, 1150 St. Nicholas Ave., New York, NY 10032. E-mail: eas3@columbia.edu.
Supported by grants from the National Institutes of Health (HD83062 and NS11766 to E.A.S., GM45735, GM66037, and CA13696 to L.A.P., GM61721 to C.M.K., AG05136 to T.D.B., and a Pilot Grant to E.A.S. from the Columbia University Alzheimer Disease Research Center [AG08702 to Michael L. Shelanski]), the John Douglas French Alzheimer Foundation (to EAG), Veterans Administration Research Funds (to TDB), and the American Health Assistance Foundation, the Muscular Dystrophy Association, and the Marriott Foundation (to EAS).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Current address of A.J.C.deG.: Department of Cell Biology, Nijmegen Center for Molecular Life Sciences (NCMLS), Radboud University, Geert Grooteplein 28, 6525 GA Nijmegen, The Netherlands.
References
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Wakabayashi T, De Strooper B. Presenilins: members of the γ-secretase quartets, but part-time soloists too. Physiology. 2008;23:194–204. doi: 10.1152/physiol.00009.2008. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, George-Hyslop PS, Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- Siman R, Velji J. Localization of presenilin-nicastrin complexes and γ-secretase activity to the trans-Golgi network. J Neurochem. 2003;84:1143–1153. doi: 10.1046/j.1471-4159.2003.01616.x. [DOI] [PubMed] [Google Scholar]
- Chyung JH, Raper DM, Selkoe DJ. γ-Secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J Biol Chem. 2005;280:4383–4392. doi: 10.1074/jbc.M409272200. [DOI] [PubMed] [Google Scholar]
- Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, De Strooper B, Annaert W. The discrepancy between presenilin subcellular localization and γ-secretase processing of amyloid precursor protein. J Cell Biol. 2001;154:731–740. doi: 10.1083/jcb.200104045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B. Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–294. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura N, Nakamura SI, Honda T, Takashima A, Nakayama H, Ono F, Sakakibara I, Doi K, Kawamura S, Yoshikawa Y. Age-related changes in the localization of presenilin-1 in cynomolgus monkey brain. Brain Res. 2001;922:30–41. doi: 10.1016/s0006-8993(01)03146-8. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ. Presenilin-1, nicastrin, amyloid precursor protein, and γ-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003;278:26687–26694. doi: 10.1074/jbc.m304009200. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Hultenby K. Presenilin-1 is located in rat mitochondria. Biochem Biophys Res Commun. 2002;295:766–770. doi: 10.1016/s0006-291x(02)00735-0. [DOI] [PubMed] [Google Scholar]
- Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinol AE, Cui Z, Chen MH, Vance JE. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem. 1994;269:27494–27502. [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G, Simmen T. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell. 2008;19:2777–2788. doi: 10.1091/mbc.E07-10-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem. 2005;280:40892–40900. doi: 10.1074/jbc.M506623200. [DOI] [PubMed] [Google Scholar]
- Kopach O, Kruglikov I, Pivneva T, Voitenko N, Fedirko N. Functional coupling between ryanodine receptors, mitochondria and Ca(2+) ATPases in rat submandibular acinar cells. Cell Calcium. 2007;43:469–481. doi: 10.1016/j.ceca.2007.08.001. [DOI] [PubMed] [Google Scholar]
- García-Pérez C, Hajnóczky G, Csordás G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. JBC. 2008;283:32771–32780. doi: 10.1074/jbc.M803385200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Man WC, Miyazaki M, Chu K, Ntambi J. Colocalization of SCD1 and DGAT2: implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J Lipid Res. 2006;47:1928–1939. doi: 10.1194/jlr.M600172-JLR200. [DOI] [PubMed] [Google Scholar]
- Jia Z, Moulson CL, Pei Z, Miner JH, Watkins PA. Fatty acid transport protein 4 is the principal very long chain fatty acyl-CoA synthetase in skin fibroblasts. J Biol Chem. 2007;282:20573–20583. doi: 10.1074/jbc.M700568200. [DOI] [PubMed] [Google Scholar]
- Lewin TM, Van Horn CG, Krisans SK, Coleman RA. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch Biochem Biophys. 2002;404:263–270. doi: 10.1016/s0003-9861(02)00247-3. [DOI] [PubMed] [Google Scholar]
- Bionda C, Portoukalian J, Schmitt D, Rodriguez-Lafrasse C, Ardail D. Subcellular compartmentalization of ceramide metabolism: mAM (mitochondria-associated membrane) and/or mitochondria? Biochem J. 2004;382:527–533. doi: 10.1042/BJ20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardail D, Popa I, Bodennec J, Louisot P, Schmitt D, Portoukalian J. The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem J. 2003;371:1013–1019. doi: 10.1042/BJ20021834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riekhof WR, Wu J, Jones JL, Voelker DR. Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2007;282:28344–28352. doi: 10.1074/jbc.M705256200. [DOI] [PubMed] [Google Scholar]
- Pottekat A, Menon AK. Subcellular localization and targeting of N-acetylglucosaminyl phosphatidylinositol de-N-acetylase, the second enzyme in the glycosylphosphatidylinositol biosynthetic pathway. J Biol Chem. 2004;279:15743–15751. doi: 10.1074/jbc.M313537200. [DOI] [PubMed] [Google Scholar]
- Vance DE, Walkey CJ, Cui Z. Phosphatidylethanolamine N-methyltransferase from liver. Biochim Biophys Acta. 1997;1348:142–150. doi: 10.1016/s0005-2760(97)00108-2. [DOI] [PubMed] [Google Scholar]
- Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem. 2000;275:34534–35540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- Goetz JG, Nabi IR. Interaction of the smooth endoplasmic reticulum and mitochondria. Biochem Soc Trans. 2006;34:370–373. doi: 10.1042/BST0340370. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24:717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landman N, Jeong SY, Shin SY, Voronov SV, Serban G, Kang MS, Park MK, Di Paolo G, Chung S, Kim TW. Presenilin mutations linked to familial Alzheimer’s disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc NatlAcad Sci USA. 2006;103:19524–19529. doi: 10.1073/pnas.0604954103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio G, Nicchitta CV, Blobel G. The signal sequence receptor, unlike the signal recognition particle receptor, is not essential for protein translocation. J Cell Biol. 1992;117:15–25. doi: 10.1083/jcb.117.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M. Nicastrin, presenilin, APH-1, and PEN-2 form active γ-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654–51660. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- Leuenberger D, Bally NA, Schatz G, Koehler CM. Different import pathways through the mitochondrial intermembrane space for inner membrane proteins. EMBO J. 1999;18:4816–4822. doi: 10.1093/emboj/18.17.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae TJ, Kim MS, Kim JW, Kim BW, Choo HJ, Lee JW, Kim KB, Lee CS, Kim JH, Chang SY, Kang CY, Lee SW, Ko YG. Lipid raft proteome reveals ATP synthase complex in the cell surface. Proteomics. 2004;4:3536–3548. doi: 10.1002/pmic.200400952. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Jung KM, Huang YZ, Bennett LB, Lee JS, Mei L, Kim TW. Presenilin-dependent γ-secretase-like intramembrane cleavage of ErbB4. J Biol Chem. 2002;277:6318–6323. doi: 10.1074/jbc.M110371200. [DOI] [PubMed] [Google Scholar]
- Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, Wolfe MS. Active γ-secretase complexes contain only one of each component. J Biol Chem. 2007;282:33985–33993. doi: 10.1074/jbc.M705248200. [DOI] [PubMed] [Google Scholar]
- Spasic D, Annaert W. Building γ-secretase: the bits and pieces. J Cell Sci. 2008;121:413–420. doi: 10.1242/jcs.015255. [DOI] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lah JJ, Thinakaran G, Levey A, Sisodia SS. Subcellular localization of presenilins: association with a unique membrane pool in cultured cells. Neurobiol Dis. 2000;7:99–117. doi: 10.1006/nbdi.1999.0280. [DOI] [PubMed] [Google Scholar]
- Atamna H, Frey WH., 2nd Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion. 2007;7:297–310. doi: 10.1016/j.mito.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Small DH, Mok SS, Bornstein JC. Alzheimer’s disease and Aβ toxicity: from top to bottom. Nature Rev Neurosci. 2001;2:595–598. doi: 10.1038/35086072. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc Natl Acad Sci USA. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JK, Kim NH, Lee YJ, Kim YS, Choi EK, Kozlowski PB, Park MH, Kim HS, Min do S. Phospholipase D1 is up-regulated in the mitochondrial fraction from the brains of Alzheimer’s disease patients. Neurosci Lett. 2006;407:263–267. doi: 10.1016/j.neulet.2006.08.062. [DOI] [PubMed] [Google Scholar]
- Canevari L, Clark JB. Alzheimer’s disease and cholesterol: the fat connection. Neurochem Res. 2007;32:739–750. doi: 10.1007/s11064-006-9200-1. [DOI] [PubMed] [Google Scholar]
- Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimer’s Dis. 2006;9:1–12. doi: 10.3233/jad-2006-9101. [DOI] [PubMed] [Google Scholar]
- Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427–437. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Ringheim GE, Szczepanik AM. Brain inflammation, cholesterol, and glutamate as interconnected participants in the pathology of Alzheimer’s disease. Curr Pharm Des. 2006;12:719–738. doi: 10.2174/138161206775474215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.