

Abstract
Clinical studies have indicated that the stent-eluting drugs sirolimus and paclitaxel impact restenosis; however, it is still elusive how these drugs affect the vascular endothelium at the molecular and cellular levels. The purpose of this study was to determine whether sirolimus and paclitaxel induce molecular and cellular alterations in the vascular endothelium. Endothelial regrowth was assessed in human aortic endothelial cells and rat aortic endothelium. Molecular and cellular alterations were analyzed in human aortic endothelial cells by Western blot analysis, transmission electron microscopy, and immunofluorescence staining. Green fluorescent protein-LC3 mice were used to analyze autophagic endothelium. Here, we show that sirolimus and paclitaxel differentially induce self-digesting autophagy in vascular endothelial cells with changes in expression of LC3B, p53, and Bcl-2, considerably suppressing re-endothelialization and revascularization. These results suggest that phenotypic alteration in the endothelium by sirolimus or paclitaxel might affect the rates of late stent thrombosis, myocardial infarction, and mortality.
Sirolimus and paclitaxel, which were originally used for immunosuppression or cancer treatment, have been shown to have a new therapeutic application in preventing restenosis after angioplasty.1,2 Angioplasty induces target vascular injury that often accelerates mitogen-mediated vascular smooth muscle proliferation and formation of neointimal hyperplasia. In this situation, both sirolimus and paclitaxel have been shown to inhibit cell cycle proliferation and migration of vascular smooth muscle cells.3 On the basis of these findings, drug-eluting stents (DES), including a sirolimus-eluting stent, and a paclitaxel-eluting stent, have been implanted in millions of patients with coronary artery disease undergoing percutaneous coronary intervention, and their efficacy and safety relative to bare-metal stents have been investigated in randomized clinical trials worldwide.4
While enthusiasm for DES use with this technology has been increasing, recent reports have sounded an alarm that patients with DES may encounter life threatening complications such as late stent thrombosis, in-stent thrombosis, and myocardial infarction.5,6 These clinical data, together with the effects of sirolimus and paclitaxel on cell cycle inhibition,3 indicate that endothelial dysfunction may exist. However, presently, little information is available regarding the direct effects of sirolimus and paclitaxel on the vascular endothelium. Here, we hypothesized that sirolimus and paclitaxel would have undefined effects on the vascular endothelium, which correlate with unfavorable outcomes after DES use.
Materials and Methods
Reagents
We purchased sirolimus (rapamycin), paclitaxel, and 3-methyladenine (3-MA) from Sigma (St. Louis, MO). Both sirolimus and paclitaxel were prepared as a stock solution in ethanol, aliquoted, and stored at −70°C. In each experiment, we adjusted final concentrations of ethanol in culture medium as 0.1% in all groups. First, we estimated the appropriate concentrations of sirolimus and paclitaxel in vitro by calculating local concentrations of these compounds in vessels with stents by our previous report.1
Cell Culture
Human aortic endothelial cells (HAECs) were obtained from Applied Cell Biology Research Institute (Kirkland, WA). We cultured HAECs in Modified MCDB131 medium supplemented with 2% fetal bovine serum and an endothelial growth supplement kit (Clonetics; Lonza, Basel, Switzerland), so-called endothelial growth medium (EGM) as previously described,7 and used them for experiments between passages 3 to 6. We used cells that had reached 80% confluence for the following experiments. Human aortic smooth muscle cells (HASMCs) were used for enzyme-linked immunosorbent (ELISA) assay as described below.
Proliferation Assay
HAECs cultured on 12-well culture plates were pre-incubated in medium with 0.5% fetal bovine serum overnight to synchronization of cells, and then incubated them in EGM with sirolimus or paclitaxel for 48 hours. The cell number in each group was manually counted with a hemocytometer. Proliferative activity of endothelial cells (ECs) was also analyzed by 5-bromo-2′-deoxyuridine ELISA according to the manufacturer’s instruction (Roche, Basel, Switzerland).
Tube Formation Assay
HAECs cultured on 12-well culture plates coated with Matrigel basement membrane matrix (BD Bioscience, San Jose, CA) were treated with sirolimus or paclitaxel for 18 hours. Cells were then stained with 4′,6-diamidino-2-phenylindole (Molecular Probes Inc, Eugene, OR), photographed by BIOREVO immunofluorescence microscope (Keyence, Osaka, Japan), and their total lengths quantified with imaging software (Kurabo, Osaka, Japan). All groups were studied in at least four independent experiments.
Animal Studies
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the NIH (Publication No. 85–23, revised 1996) and is approved by the Animal Care Committee of Gifu University Graduate School of Medicine. We performed all procedures according to protocols approved by the Animal Care Committee of Gifu University Graduate School of Medicine.
Ex Vivo Re-Endothelialization Assay
Ten-week-old male Sprague-Dawley rats were anesthetized with 5% inhaled isoflurane. The thoracic aorta was dissected, placed in ice-cold culture medium, cleaned of peripheral fat under a dissecting microscope (Leica, Solms, Deutschland), and cross-sectioned into 10-mm lengths of aorta. The aortic sections were horizontally cut, en face, fixed on culture dishes with pins, and then 3-mm spreads of the endothelial layers on the scratched wound were created. These sections were culture in EGM with 10 nM/L of sirolimus or paclitaxel for 7 days, and re-endothelialization was evaluated by the endothelial uptake of dioctadecyl indocarbocyanine (Dil)-labeled acetylated low-density lipoprotein (Biomedical Technologies, Stoughton, MA). These sections were photographed as gray scale images, and the re-endothelialized area was calculated using NIH image software. Sections of a scratched wound on day 0 were used as the negative control for 0% of re-endothelialization, and sections without a scratched wound as the positive control for 100% of re-endothelialization.
Aortic Ring Assay
The thoracic aorta was dissected from age-matched 10-week-old male Sprague-Dawley rats, placed in ice-cold Dulbecco’s Modified Eagle Medium (DMEM), cleaned of fibroadipose tissue around the adventitia, and sectioned into aortic rings (1-mm thickness) within 2 hours. These rings were placed in two layers of pre-prepared Matrigel (BD Bioscience) as shown in Supplementary Figure 1 at http://ajp.amjpathol.org, and cultured with EGM containing sirolimus (10 nmol/L) or paclitaxel (10 nmol/L). Capillary sprouts from each ring (six aortic rings from each animal) were monitored everyday under a microscope up to 6 days. Sprouting length was measured in 12 areas around the rings (each 30-degree). By using Dispase (BD Bioscience), a solution for isolating cells from Matrigel layers, we collected sprouting ECs in Matrigel on day 3, and confirmed that about 95% of cells represented endothelial specific uptake of DiI-labeled acetylated low-density lipoprotein.
Figure 1.

Sirolimus and paclitaxel suppress the capacity of the endothelium for re-endothelialization. A: HAEC cell number after 48 hours of sirolimus or paclitaxel treatment. Values are expressed as mean ± SEM. n = 9 per group (HAECs with three different passages from three donors). *P < 0.05 and **P < 0.01 vs. control group without stimulation. B: 5-Bromo-2′-deoxyuridine incorporation assay for endothelial proliferation after 48 hours of sirolimus or paclitaxel treatment. C: Representative immunoblot images of p27Kip1 and p21Cip1 after 24 hours of sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) treatment. n = 3 per group. D: Schematic indication of ex vivo re-endothelialization assay. A scratched wound was created on rat aortic endothelium on day 0. E and F: Effects of sirolimus and paclitaxel on re-endothelialization. Representative images of DiI-labeled acetylated low-density lipoprotein stained rat aortic endothelium 7 days after making a scratched wound in the sirolimus (10 nmol/L) group or paclitaxel (10 nmol/L) group (E) and quantification of % re-endothelialization on scratched area relative to unscratched vessels. n = 8 per group (F).
Detection of Apoptosis
HAECs cultured on 12-well culture plates were treated with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 12 hours, and then fixed with 1% paraformaldehyde in 0.1 M/L PBS (pH 7.4) for 15 minutes. Cells were then stained with antibody against cleaved caspase-3 (Cell Signaling, Beverly, MA) and a species-specific Alexa Fluor secondary antibody (Molecular Probes Inc). The number of apoptotic cells was counted and normalized with reference to the total number of cells in each fields.
Detection of Autophagy
HAECs were cultured with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 18 hours. They were then fixed with 2% paraformaldehyde/2% glutaraldehyde in 0.1 M/L-PBS (pH 7.4), and postfixed with 1% osmium tetroxide. Cell sections in epon were counterstained with uranyl acetate and lead citrate and examined with a Hitachi H7600 transmission electron microscopy (Hitach, Tokyo, Japan). Note that at least six cell sections from each group were prepared for transmission electron microscopy analysis. Autophagic HAECs were also visualized by microtubule-associated protein light-chain3 (LC3) staining of transduced green fluorescent protein (GFP)-LC3 construct as described previously, with some modifications.8 Briefly, HAECs were cultured in 4-well glass chamber slides and transfected with enhanced GFP plasmid or GFP-LC3 plasmid using TransIT-LT1 reagent (Mirus Corporation, Madison, WI) according to the manufacture’s instructions. Twenty-four hours after transfection, cells were treated with sirolimus or paclitaxel for 18 hours, and were fixed in 1% paraformaldehyde in PBS for 20 minutes and examined by epifluorescence Axiovert 200M microscope and AxioVision imaging software (Zeiss, Oberkochen, Deutschland). Cells with at least five enhanced-LC spots were counted as autophagic cells.
Ex Vivo Imaging of Autophagic Endothelium
Eight-week-old male GFP-LC3 transgenic mice were used as in vivo marker of autophagy induction.9 The thoracic aorta was dissected, placed in ice-cold EGM and cross-sectioned into 5-mm lengths of aorta. The aortic sections were horizontally cut, en face and were culture in EGM with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 24 hours. Autophagy induction in aortic endothelium was monitored as an enhanced GFP/LC3 expression by BIOREVO immunofluorescence microscope (Keyence). For quantitative analysis of GFP-LC3 dots, florescence intensity was analyzed on inverted gray scale images using NIH image software.
Western Immunoblot Analysis
HAECs at 80% confluence were treated with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for up to 24 hours. Whole-cell protein extracts (40 μg to 100 μg) were separated on 10% to 12.5% sodium dodecylsulfate polyacrylamide gels, and transferred them to nitrocellulose membrane (Amersham, Tokyo, Japan). The blotted membrane was proved with antibodies against LC3B (Abgent, San Diego, CA), p53 (Oncogene, Merk, Deutschland), Bcl-2 (Santa Cruz, San Diego, CA), Bax (Upstate Biotechnology, Lake Placid, New York), Akt, phosphorylated Akt (pAkt: Ser473), extracellular-signal-related-kinase (ERK), phosphorylated ERK (pERK), p27, p21 (all from Cell Signaling), and actin (Sigma). Densitometric results were calculated as mean ± SEM.
Real-Time Quantitative Reverse Transcription-PCR Analysis
Transcriptional regulation was determined by real-time quantitative reverse transcription-PCR technology using Primer Express 2.0, SYBR Green Reagents and ABI 7900 sequence detection system (Applied Biosystems, Foster City, CA). Primer pairs for the kinase insert domain receptor (KDR) (sense, 5′-TTTGACGGGCTGAGTCTATCCA-3′; antisense, 5′-AATGCATTTGCAGGCTCCAGTA-3′), endothelial nitric oxide synthase (eNOS) (sense, 5′-CGATGCTCCCAACTTGACCA-3′; antisense, 5′-CCTCAGGATGTCCTGCACGTAG-3′), Tie-2 (sense, 5′-AGGCCATCATTTGCCCAGATA-3′; antisense, 5′-ATGTTCTGTCCTAGGCCGCTTC-3′) and glyceraldehyde-3-phosphate dehydrogenase (sense, 5′-GCACCGTCAAGGCTGAGAAC-3′; antisense, 5′-ATGGTGGTGAAGACGCCAGT-3′) were used to detect target gene transcripts. Relative expression values were calculated from the cycle threshold (Ct) using the formula 2−Ct. Before this formulation, the eNOS, KDR, and Tie-2 Ct values from each sample were normalized by glyceraldehyde-3-phosphate dehydrogenase Ct value.
Nitric Oxide Release
HAECs cultured on 12-well culture plates were treated with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 24 hours. Total nitric oxide (NO) in conditioned medium was assayed as nitrites (NO2−) and nitrates (NO3−) using Griess reaction-based NO2/NO3 colorimetric assay kit (DOJINDO, Japan).
ELISA Assay
HASMCs were seeded on 12-well cell culture plates, and cultured in MCDB131 medium supplemented with 10% fetal bovine serum. Cells that reached 80% confluence were serum-starved for 24 hours, and treated with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 48 hours. Subsequently, vascular endothelial growth factor (VEGF) concentration was measured in conditioned medium from HASMCs using a specific ELISA kits (R&D systems, Minneapolis, MN).
Statistical Analysis
Each result was obtained in at least four separate experiments. All values are expressed as mean ± SEM. We analyzed values using a one-way analysis of variance, and then determine the significance of differences in multiple comparisons using Scheffé’s multiple comparison test. P < 0.05 was considered statistically significant.
Results
To test whether sirolimus and paclitaxel have effects on vascular endothelium, we first examined in vitro and ex vivo model of endothelial regrowth. We found that proliferative activity of HAECs was dose-dependently impaired by treatment with sirolimus or paclitaxel (Figure 1A), whose concentrations in culture medium were also enough to inhibit proliferation of HASMCs (data not shown). We also obtained consistent results that both compounds impaired endothelial proliferation by using 5-bromo-2′-deoxyuridine incorporation assay (Figure 1B). In addition, sirolimus and paclitaxel showed cell cycle inhibition in HAECs via regulation of p27 and p21, respectively (Figure 1C). Notably, the ex vivo model of re-endothelialization (Figure 1D) revealed that both sirolimus and paclitaxel significantly impaired re-endothelialization after 7 days of scratched wound in rat aortic endothelium, which was visualized by endothelial uptake of fluorescence (DiI)-labeled acetylated low-density lipoprotein (Figure 1E). In fact, 60% to 65% of the scratched lesion was re-endothelialized in control vessels without these compounds, while less than 10% of scratched lesions was re-endothelialized in vessels treated with sirolimus or paclitaxel (Figure 1F). Moreover, paclitaxel not only inhibited re-endothelialization of scratched endothelium, but also seemed to induce endothelial damage at untreated areas (Figure 1, E and F). These results indicate that both sirolimus and paclitaxel induce delay in the healing process of vascular endothelium.
As the capacity of ECs for arterial revascularization correlate with collateral growth and its protective nature against myocardial infarction, we also investigated whether sirolimus or paclitaxel affects arterial revascularization. As shown in Figure 2A, both sirolimus and paclitaxel inhibited tube formation of HAECs after 18 hours in an in vitro angiogenesis assay. In addition, we obtained similar results that both sirolimus and paclitaxel markedly inhibited vessels sprouting from aortic rings after 3 days of ex vivo culture (Figure 2, B and C). These data suggest the possibility that sirolimus and paclitaxel impair initial steps for revascularization, presumably leading to reduction in the number of well-grown collateral vessels in the heart.
Figure 2.

Sirolimus and paclitaxel suppress the capacity of the endothelium for arterial revascularization. A: Representative images of capillary tube formation of HAECs after 18 hours of treatment with sirolimus or paclitaxel (10 nmol/L or 100 nmol/L). n = 8 per group. Note that ECM (−) indicates the group without treatments of Matrigel and compounds. B: Representative images of vessel sprouting from rat aorta treated with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 3 days. Lower panels show high magnification views (×100) of vessel-sprouting lesions around rat aorta (Ao). C: Quantification of vessel-sprouting lengths from aortic rings after sirolimus or paclitaxel treatment. n = 8 per group. **P < 0.01 vs. control.
Consistent with the clinical interpretations that DES may induce endothelial dysfunction,5,6 we found that both sirolimus and paclitaxel significantly reduced transcriptional expression of eNOS, as well as NO production in HAECs (Figure 3A and B), which correlate with vascular remodeling and vascular formation. However, these compounds did not alter the mRNA expression of receptor for VEGF, KDR, in HAECs after 12 hours of treatment (Figure 3C). In addition, these compounds did not alter the secretion of VEGF from perivascular smooth muscle cells after 48 hours of treatment (Figure 3D), suggesting the possibility that both compounds induce molecular and cellular alterations in vascular endothelium before VEGF regulation.
Figure 3.
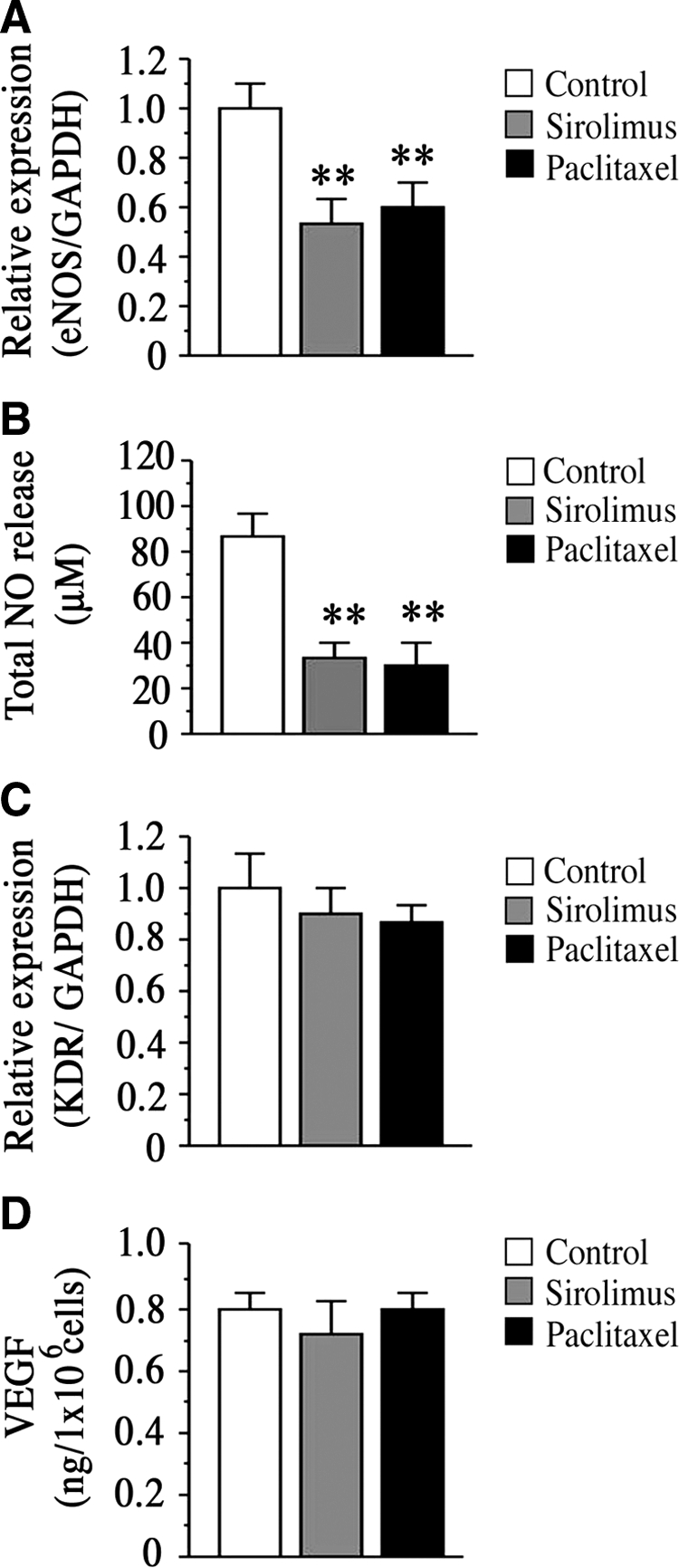
Effects of sirolimus and paclitaxel on expression of eNOS, KDR, and VEGF. A: Transcriptional expression of eNOS in HAECs after 12 hours of sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) treatment. B: Graph indicates total NO release from HAECs after 24 hours of sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) treatment. n = 8 per group. **P < 0.01 vs. control. C: Transcriptional expression of KDR in HAECs after 12 hours of sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) treatment. D: VEGF secretion from HASMCs after 24 hours of sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) treatment. All values are expressed as mean ± SEM. n = 5 per group. **P < 0.01 vs. control without sirolimus or paclitaxel treatment.
Therefore, we next investigated whether sirolimus or paclitaxel induced programmed cell death in ECs as initial events, since a well-known form of programmed cell death, apoptosis, has been suggested to be a cellular response for the maintenance of vascular tissue homeostasis, including re-endothelialization and revascularization. As shown in Figure 4A, treatment of paclitaxel for 12 hours significantly increased the number of apoptotic HAECs (about 40% of total cells), as determined by the immunofluorescent staining with an antibody against activated-caspase-3 (green). In contrast, a low number of apoptotic ECs were observed in the sirolimus group and control group without these compounds (Figure 4, A and B).
Figure 4.

Sirolimus and Paclitaxel differentially induce endothelial autophagy. A: Representative images of HAECs stained with antibody against cleaved caspase-3 after 12 hours of sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) treatment. B: Quantification of activated-caspase3-positive cells treated with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) relative to control group. Values are expressed as mean ± SEM. n = 6 per group. **P < 0.01 vs. control. C: EM of HAECs treated with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 18 hours (upper panel: ×26, 000, lower panel: ×52, 000). Blue arrows indicate typical features of autophagy with formation of autophagosomes. n = 4 per group. D: Representative fluorescent images of HAECs, which were transfected with GFP-LC3 or GFP construct and cultured with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L) for 18 hours. Enhanced LC3 spots in the cytoplasm indicated autophagic cells. Graph shows quantification of autophagic cells with five or more LC3spots. n = 4 per group. *P < 0.05, **P < 0.01 vs. control.
Recent studies suggest that a new process of programmed cell death exists; the formation of autophagosomes and autolysosomes in which the content of the autophagosome is degraded by lysosomal hydrolases is called self-eating autophagy.10,11 Alternatively, it has also been suggested that rapamycin/sirolimus works as a mammalian target of rapamycin inhibitor and induce autophagy in several cell lines such as cancer cells and fibroblasts.12,13 Therefore, we next investigate whether sirolimus or paclitaxel induced endothelial autophagy. Strikingly, transmission electron microscopy of HAECs clearly revealed that sirolimus induced endothelial autophagy with formation of double-membrane vesicles containing cytoplasmic organelles after 18 hours of treatment (Figure 4C: middle panels), which was absent in most of the control cells without stimulation (Figure 4C: left panels). This formation was observed in about 45% of total cells in group with sirolimus, while in a low numbers of autophagic cells in control group (less than 5%). In contrast, paclitaxel not only induced endothelial autophagy (Figure 4C: right upper panels) but also induced advanced stages of apoptosis and necrosis (Figure 4C: right lower panels)(Supplementary Figure 2, A and C, at http://ajp.amjpathol.org). Notably, the rate of autophagy induction was lower in the paclitaxel group than that in the sirolimus group. To evaluate autophagy induction using another assay system, we transduced a GFP-LC3 construct into HAECs, and monitored live imaging of GFP-LC3 spots in cell cytosol. Fluorescence images showed consistent results that both sirolimus and paclitaxel induced endothelial autophagy with enhanced LC3 spots, but sirolimus rather than paclitaxel had strong effects on the induction of endothelial autophagy (45% vs. 20%, respectively) (Figure 4D). Together, these results indicate that human aortic ECs exposed to sirolimus or paclitaxel differentially undergo autophagy.
To further investigate whether sirolimus or paclitaxel induces endothelial autophagy in vascular tissue endothelium, we performed organ culture of murine vascular tissue form GFP-LC3 transgenic mice,9 which systemically expressing GFP fused to LC3. Notably, endothelial autophagy was clearly visualized in endothelial layers after 24 hours of sirolimus treatment (Figure 5: upper middle panels and graph). In contrast, however, paclitaxel showed weak effects on autophagy induction in vascular tissue endothelium (Figure 5: upper right panels and graph), suggesting the differential effects of sirolimus or paclitaxel on endothelial autophagy induction. These results indicate the possibility that sirolimus rather than paclitaxel have strong effects on endothelial autophagy induction in vivo situation.
Figure 5.

Imaging of autophagic endothelium. Representative fluorescent images of endothelial layers from GFP-LC3 trangenic mice or those from wild-type mice after 24 hours of tissue culture with sirolimus (10 nmol/L) or paclitaxel (10 nmol/L). Enhanced GFP/LC3 expression indicates autophagic endothelium. Graph shows quantification of autophagy. n = 8 per group. *P < 0.05, **P < 0.01 vs. control.
In seeking to define molecular alterations in ECs after treatment of sirolimus or paclitaxel, we found that these compounds regulated several critical factors in autophagy and/or apoptosis, including LC3B, p53, and Bcl-2.11,12,13,14,15 As shown in Figure 6 and Supplementary Figure 3 (at http://ajp.amjpathol.org), sirolimus markedly increased LC3B expression in HAECs just after 3 hours of treatment, indicating the induction of early stages of autophagy. Paclitaxel also increased LC3B expression after 24 hours of treatment. On the other hand, paclitaxel increased the expression levels of p53, a crucial mediator of cell death, in HAECs after 3 hours of treatment. In contrast, sirolimus did not affect p53 expression after 3 hours and 24 hours of treatment. We also measured the expression levels of Bcl-2 and Bax (a crucial mediators of cell survival and cell death, respectively) in HAECs, showing that both sirolimus and paclitaxel reduced the expression of Bcl-2 after 24 hours of treatment, while neither compound affected Bax expression. In addition, we found that sirolimus inhibited phosphorylation of mammalian target of rapamycin downstream of class1 phosphatidylinositol 3-kinase/Akt in HAECs (data not shown), but the compound did not affect Akt activation during our experimental time period. Moreover, paclitaxel, but not sirolimus, reduced activation of ERK, indicating the marked inhibition of cell proliferation and cell survival in HAECs. Together, these data suggest the possibility that induction of endothelial autophagy and/or apoptosis by these compounds is associated with the regulation of LC3B, p53, and Bcl-2.
Figure 6.
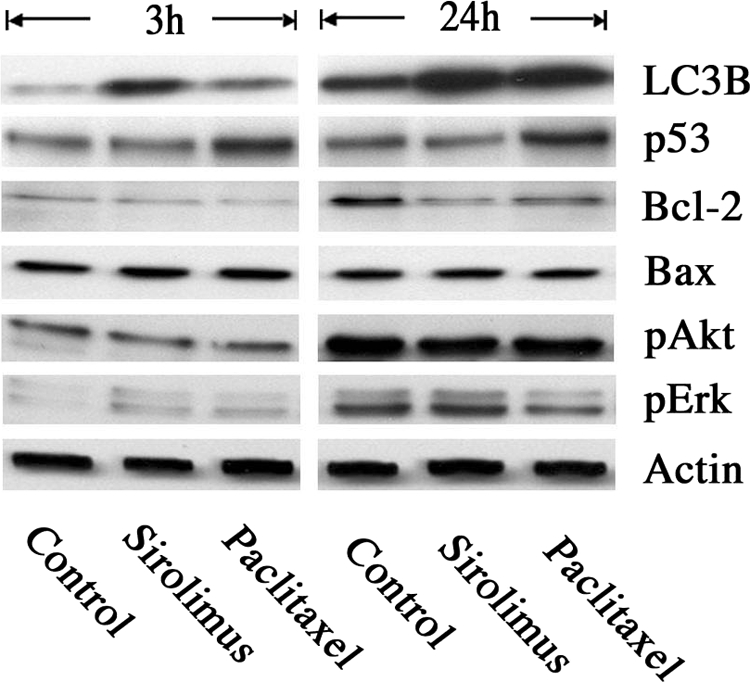
Molecular alterations in HAECs after treatment with Sirolimus or Paclitaxel. Immunoblot analysis of HAECs was performed after 3 or 24 hours of sirolimus or paclitaxel treatment. Antibodies against LC3B, p53, Bcl-2, Bax, pAkt, pERK, and Actin were used. n = 4 per group.
Finally, we addressed the question whether inhibition of autophagy can attenuate sirolimus- or paclitaxel-induced impairment of endothelial proliferation and re-endothelialization. Notably, 3-MA, a widely used inhibitor of autophagy as well as an inhibitor of class3 phosphatidylinositol 3-kinase,11,14 reduced the sirolimus-induced autophagy in HAECs (Figure 7A), and attenuated the impaired endothelial proliferation by sirolimus (Figure 7B). In contrast, however, 3-MA was not effective in attenuating paclitaxel-induced impairment of endothelial proliferation. Similarly, 3-MA attenuated sirolimus-induced impairment of re-endothelialization 7 days after making a scratched wound on rat aortic endothelium, while there was weak attenuation on re-endothelialization impairment by paclitaxel (Figure 7C). We also obtained consistent results that 3-MA attenuated sirolimus-induced impairment of vessel sprouting from aortic rings after 4 days of treatment, while was weak attenuation in the paclitaxel group (Figure 7D). These data suggest the possibility that selective inhibition of autophagy may be effective in restoring endothelial function in case of sirolimus-eluting stent use but less effective with paclitaxel-eluting stent use.
Figure 7.

Effects of autophagy inhibition on sirolimus- or paclitaxel-induced impairment of re-endothelialization and revascularization. A: EM of HAECs treated with sirolimus alone (upper panel) or ECs treated with both sirolimus (10 nmol/L) and 3-MA (1 mmol/L) (lower panel). B: HAECs cell number in the presence of sirolimus (10 nmol/L), paclitaxel (10 nmol/L) or 3-MA (1 mmol/L). n = 6 per group. **P < 0.01 vs. control group without treatment. *P < 0.05 vs. group with sirolimus. C: Inhibition of endothelial autophagy by 3-MA (1 mmol/L) attenuated sirolimus-induced impairment of re-endothelialization on day 7, whereas no attenuation of paclitaxel-induced impairment was observed. Values of % re-endothelialization below zero indicate damage of unscratched endothelium. n = 8 per group. **P < 0.01 vs. control group. *P < 0.05 vs. group with sirolimus. NS means no significant difference between two groups. D: Treatment with 3-MA attenuates sirolimus-induced impairment of aortic revascularization on day 4, while no attenuation of paclitaxel-induced impairment was observed. Values are expressed as mean ± SEM. n = 8 per group. **P < 0.01 vs. control. #P < 0.05 vs. sirolimus alone.
Discussion
Increasing evidence suggests that DES-associated endothelial dysfunction is likely to be essential for early/late stent thrombosis, in-stent restenosis, and myocardial infarction.5,6,16 However, little is known about the precise mechanisms behind the association between DES use and endothelial dysfunction. We demonstrate here that sirolimus and paclitaxel differentially induce endothelial autophagy and apoptosis with intracellular regulation of LC3B, Bcl-2, and p53. We also show that these phenotypic alterations of ECs by sirolimus or paclitaxel correlate with impaired endothelial regeneration.
A variety of clinical case reports and a limited number of autopsy studies suggest that DES-dependent thrombosis correlates with delayed arterial healing characterized by lack of endothelialization. However, it is not yet known when and how generalized the negative effect of DES on endothelium is. The appropriate antiplatelet therapy is generally used after DES implantation, while this may also put a mask on what happen in endothelium at early and late time periods. Here, we show that stent-eluting drugs, sirolimus and paclitaxel, induce phenotypic alterations of endothelium with induction of autophagy, and these alterations lead to impairment of re-endothelialization, which probably occur within 30 days after DES implantation. However, because of the slow drug-releasing kinetics of stent polymer, it is possible that these drugs continuously induce molecular and cellular alterations in ECs at the site of DES deployment. These might associate with DES-dependent early and late stent thrombosis. It is also possible that autophagy and other forms of programmed cell death may differentially affect the timing of DES-dependent endothelial dysfunction and stent thrombosis, since pathways toward cell death in autophagy often take longer time periods than those in apoptosis or necrosis. In addition, timing of late stent thrombosis might be affected by the patient related risk factors of stent thrombosis, such as diabetes, renal failure, heart failure, and withdraw of antiplatelet therapy.16
There exist several independent interpretations about the drug-mediated impairment of angiogenesis. Previous studies focused on the clinical benefit of sirolimus or paclitaxel-mediated impairment of target vessel angiogenesis, and suggested that these might reduce the incidences of in-stent remodeling and thrombosis. However, DES use seems to lack benefit against myocardial infarction and death.4 In addition, recent report suggest that DES-dependent endothelial dysfunction occurs not only inside the target vessels, but also beside the stent deployment.17 Since the capacity of ECs for angiogenesis correlates with tissue regeneration in diseased heart, collateral growth and its protective nature against myocardial infarction, our data that both drugs impaired angiogenesis would allow for the interpretation that drug-mediated impairment of angiogenesis involves DES-dependent myocardial infarction and death.
One of our crucial findings is that sirolimus and paclitaxel differentially induce cellular and molecular alterations in ECs. Notably, sirolimus-induced phenotypic alteration of ECs was mostly limited to autophagy. In this case, inhibition of autophagy by 3-MA seems to be effective in restoring endothelial function for regrowth. This implies that endothelium treated with sirolimus is still in reversed state from cell death in the short term, although autophagy induction may lead to cell death in the long term.10,11 The number of viable ECs in group with sirolimus also support this interpretation (Supplementary Figure 2B at http://ajp.amjpathol.org). In contrast, however, ECs treated with paclitaxel represent a mixed population of cells undergoing autophagy and apoptosis (20% vs. 40%, respectively). Unexpectedly, selective inhibition of autophagy in this case fails to restore endothelial function for regrowth, suggesting the possibility that endothelium treated with paclitaxel is in more serious condition, in which ECs cannot restore its function, as compared with ECs treated with sirolimus. In support of this, paclitaxel rather than sirolimus markedly reduces the number of viable cells (see Supplementary Figure 2B at http://ajp.amjpathol.org).
The signaling events toward autophagy and/or apoptosis may explain the differential effects of sirolimus or paclitaxel on vascular endothelium. For instance, sirolimus increases LC3B expression at an early time point, indicating the enhanced signaling events toward autophagy. In contrast, paclitaxel increases p53 expression at early time point before the regulation of LC3B, Bcl-2, and Erk. It is likely that increased expression of p53 regulates the induction of endothelial apoptosis by paclitaxel. However, p53 may also regulate induction of endothelial autophagy by paclitaxel because of the possible involvement of p53 in apoptosis and autophagy induction.12,13 As Bcl-2 can also modulate apoptosis and autophagy induction,14,15 ECs treated with paclitaxel seem to be in more complicated condition with mixed signaling pathways toward autophagy and apoptosis. In addition to these genetic events, there exists another possibility—that ECs treated with paclitaxel undergo necrosis without genetic modification. Further studies are required to determine whether induction of endothelial autophagy and apoptosis are separate events or parallel events during the use of DES, and whether endothelial autophagy contributes to induction of apoptosis or necrosis.
In case of cell cycle regulation, it seems that sirolimus and paclitaxel induce similar cell cycle inhibition in both ECs and smooth muscle cells (Figure 1C).3 However, the number of autophagosomes in cell cytosol after drug use was apparent lower in smooth muscle cells, as compared with that in ECs (data not shown). In addition, drug-mediated endothelial specific changes were observed in re-endothelialization, revascularization, and NO production, indicating the possibility that sirolimus and paclitaxel have both endothelial specific effects and nonspecific effects.
In any case, both sirolimus and paclitaxel delayed the tissue response for endothelial regrowth, and this involves drug-mediated induction of endothelial autophagy. Further studies will need to clarify the effects of these compounds on endothelial autophagy in clinical settings.
Overall, our results suggest that differential induction of endothelial autophagy is one of the potential mechanisms in the sirolimus- or paclitaxel-induced endothelial dysfunction. Given the critical function of vascular ECs for healing of endothelium, collateral formation, and protecting against thrombosis, these phenotypic alterations of endothelium may in part correlate with the recent crisis in the risk of late stent thrombosis, myocardial infarction, and mortality in patients with DES. Our findings further suggest that control of endothelial autophagy may be attractive therapeutic strategy for patients with DES and other vascular-related diseases, such as cancer and Alzheimer’s disease.
Supplementary Material
Acknowledgments
We thank Hiroko Omori (Research Institute for Microbial Diseases, Osaka University) for her advice on the GFP-LC3 construct, and Yasukazu Ohmoto (Research Institute in Otsuka Pharmaceutical Co., ltd.) for technical support with quantitative reverse transcription-PCR analysis. We also thank Noboru Mizushima for contributions to this work.
Footnotes
Address reprint requests to Shin-ichiro Hayashi, Department of Cell Signaling, Gifu University Graduate School of Medicine, 1-1 Yanagido, Gifu City, Gifu 501-1194, Japan. E-mail: Shin559182@aol.com.
Supported by a Dean’s grant from Gifu University Graduate School of Medicine to S-i.H.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Suzuki T, Kopia G, Hayashi S, Bailey LR, Llanos G, Wilensky R, Klugherz BD, Papandreou G, Narayan P, Leon MB, Yeung AC, Tio F, Tsao PS, Falotico R, Carter AJ. Stent-based delivery of sirolimus reduces neointimal formation in a porcine coronary model. Circulation. 2001;104:1188–1193. doi: 10.1161/hc3601.093987. [DOI] [PubMed] [Google Scholar]
- Axel DI, Kunert W, Göggelmann C, Oberhoff M, Herdeg C, Küttner A, Wild DH, Brehm BR, Riessen R, Köveker G, Karsch KR. Paclitaxel inhibits arterial smooth muscle cell proliferation and migration in vitro and in vivo using local drug delivery. Circulation. 1997;96:636–645. doi: 10.1161/01.cir.96.2.636. [DOI] [PubMed] [Google Scholar]
- Wessely R, Schömig A, Kastrati A. Sirolimus and paclitaxel on polymer-based drug eluting stents: similar but different. J Am Coll Cardiol. 2006;47:708–714. doi: 10.1016/j.jacc.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Stettler C, Wandel S, Allemann S, Kastrati A, Morice MC, Schömig A, Pfisterer ME, Stone GW, Leon MB, de Lezo JS, Goy JJ, Park SJ, Sabaté M, Suttorp MJ, Kelbaek H, Spaulding C, Menichelli M, Vermeersch P, Dirksen MT, Cervinka P, Petronio AS, Nordmann AJ, Diem P, Meier B, Zwahlen M, Reichenbach S, Trelle S, Windecker S, Jüni P. Outcomes associated with drug-eluting and bare-metal stents; a collaborative network meta-analysis. Lancet. 2007;370:937–948. doi: 10.1016/S0140-6736(07)61444-5. [DOI] [PubMed] [Google Scholar]
- McFadden EP, Stabile E, Regar E, Cheneau E, Ong AT, Kinnaird T, Suddath WO, Weissman NJ, Torguson R, Kent KM, Pichard AD, Satler LF, Waksman R, Serruys PW. Late thrombosis in drug-eluting coronary stents after discontinuation of antiplatelet therapy. Lancet. 2004;364:1519–1521. doi: 10.1016/S0140-6736(04)17275-9. [DOI] [PubMed] [Google Scholar]
- Meier P, Zbinden R, Togni M, Wenaweser P, Windecker S, Meier B, Seiler C. Coronary collateral function long after drug-eluting stent implantation. J Am Coll Cardiol. 2007;49:15–20. doi: 10.1016/j.jacc.2006.08.043. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Asahara T, Masuda H, Isner JM, Losordo DW. Functional ephrin-B2 expression for promoting interaction between arterial and venous vessels in postnatal neovascularization. Circulation. 2005;111:2210–2218. doi: 10.1161/01.CIR.0000163566.07427.73. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Cell Biol. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. Dram, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of bcl-2 family proteins in a non-apoptotic programmed cell death depending on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Finn AV, Nakazawa G, Joner M, Kolodgie FD, Mont EK, Gold HK, Virmani R. Vascular responses to drug eluting stents: importance of delayed healing. Arterioscler Thromb Vasc Biol. 2007;27:1500–1510. doi: 10.1161/ATVBAHA.107.144220. [DOI] [PubMed] [Google Scholar]
- Hamilos MI, Ostojic M, Beleslin B, Sagic D, Mangovski L, Stojkovic S, Nedeljkovic M, Orlic D, Milosavljevic B, Topic D, Karanovic N, Wijns W, NOBORI CORE Investigators Differential effects of drug-eluting stents on local endothelium-dependent coronary vasomotion. J Am Coll Cardiol. 2008;51:2123–2129. doi: 10.1016/j.jacc.2007.12.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.