

Abstract
Progressive axonal degeneration follows demyelination in many neurological diseases, including multiple sclerosis and inherited demyelinating neuropathies, such as Charcot-Marie-Tooth disease. One glial molecule, the myelin-associated glycoprotein (MAG), located in the adaxonal plasmalemma of myelin-producing cells, is known to signal to the axon and to modulate axonal caliber through phosphorylation of axonal neurofilament proteins. This report establishes for the first time that MAG also promotes resistance to axonal injury and prevents axonal degeneration both in cell culture and in vivo. This effect on axonal stability depends on the RGD domain around arginine 118 in the extracellular portion of MAG, but it is independent of Nogo signaling in the axon. Exploiting this pathway may lead to therapeutic strategies for neurological diseases characterized by axonal loss.
Keywords: myelin-associated glycoprotein, MAG, myelin, Schwann cells, axon–glia interaction, axonal protection
Introduction
A fundamental problem in neurology is the tendency for demyelinated nerve fibers to undergo degeneration and loss. In diseases of presumed immune pathogenesis, such as multiple sclerosis (MS) in the CNS and Guillain-Barré syndrome in the peripheral nervous system (PNS), local inflammation undoubtedly plays a role. There is now a wealth of evidence that demyelination in itself also can contribute to altered axonal transport, axonal degeneration, and loss (de Waegh et al., 1992; Trapp et al., 1998; Coleman and Perry, 2002; Ciccarelli et al., 2003; Oh et al., 2004). Examples include heritable demyelinating diseases of both the CNS and PNS. Many of the genetic defects in these diseases are abnormalities in genes encoding intrinsic myelin proteins and not expressed in axons. The clinical manifestations are often attributable to progressive, distally predominant axonal degeneration (Berciano et al., 2000; Krajewski et al., 2000).
The myelin-associated glycoprotein (MAG) is a component of all myelinated internodes, whether formed by oligodendrocytes in the CNS or by Schwann cells in the PNS. MAG is distinctively located in the adaxonal plasmalemma that apposes the axon as well as the paranodal loops, Schmidt-Lanterman incisures, and mesaxons (Trapp and Quarles, 1982). The normal role of MAG is poorly understood. MAG is not necessary for myelination, and myelin sheaths of MAG knock-out mice (MAG−/−) are largely normal (Yin et al., 1998). Its distribution has prompted the hypothesis that MAG prevents compaction of myelin membranes and contributes to the uniform intermembranous distance characteristic of the periaxonal space (Trapp and Quarles, 1982). MAG is known to signal to the axon, locally influencing the phosphorylation of axonal neurofilaments immediately beneath MAG-bearing membranes because of reduced interfilament spacing (Hsieh et al., 1994; Dashiell et al., 2002). Myelinated axons of MAG−/− mice have smaller diameters than normal as a result of hypophosphorylation of the neurofilament proteins NF-H and NF-M (Garcia et al., 2003; Rao et al., 2003).
Much of the published research on MAG has focused on its ability to inhibit axonal elongation during regeneration. Multiple axonal receptors have been proposed to mediate MAG-induced growth cone collapse. One is a multicomponent complex consisting of a ligand-binding Nogo-66 receptor (NgR) and two transmembrane coreceptors, LINGO-1 and either p75 or TROY (Fournier et al., 2003; Mi et al., 2004). A second is proposed to be gangliosides GD1a and GT1b. Interaction of MAG with axons involves at least two recognition sites: one around arginine 118 (R118) in Ig domain 1 and second in Ig domains 4 and 5 (Kelm et al., 1994; Tang et al., 1997; Cao et al., 2007). Whereas Ig domains 4 and 5 are believed to be important for the interaction with NgR, the R118 binding site is thought to involve in interactions with gangliosides. Other roles of MAG at the R118 binding site remain unclear.
MAG−/− mice develop axonal loss in the CNS and PNS (Yin et al., 1998; Pan et al., 2005), suggesting that MAG may influence axonal maintenance. This prompted us to hypothesize that, in addition to MAG's well described inhibitory effects on axonal outgrowth, an additional role of MAG in the normal nervous system may be to promote stability and survival of myelinated axons. In this study we have asked whether MAG has protective effects on axons and if so, what region of the MAG molecule is involved and whether these effects depend on NgR.
Materials and Methods
Reagents.
The following were obtained commercially: acrylamide (Sigma-Aldrich); Neurobasal medium (Invitrogen); fetal bovine serum (FBS) (Hyclone); l-glutamine (Invitrogen); B27 serum-free supplement (Invitrogen); nerve growth factor (NGF) (Sigma-Aldrich); trypsin (Invitrogen); collagenase I (Worthington); β-III tubulin (Promega, G7121); anti-MAG (R&D Systems, AF538); anti-Fc (R&D Systems, AF2049); anti-tyrosinated tubulin (Sigma-Aldrich, T9028); anti-detyrosinated tubulin (Millipore Bioscience Research Reagents, AB3201); MAG-Fc (R&D Systems, 538-MG); Fc (R&D Systems, AF2049); recombinant active MMP-7 (EMD Biosciences, catalog #444270). Mutant proteins MAG-Fc and OMgp were provided by Dr. Guo-Li Ming.
Animals.
MAG knock-out founder mice, kindly provided by Dr. John Roder (University of Toronto, Toronto, Ontario, Canada), were constructed by disruption of exon 5 of the MAG gene as previously reported (Ng et al., 1996). The strain provided (identical to that available from The Jackson Laboratory) was a mixture of C57BL/6, 129 inbred strains, and the CD1 random-bred strain. To enhance comparisons between mutant strains, mutant mice were repeatedly back-crossed onto a C57BL/6 background to >99% strain purity (Pan et al., 2005).
NgR1 knock-out and wild-type littermate mice, kindly provided by Dr. Stephen Strittmatter (Yale University School of Medicine, New Haven, CT), were constructed by disruption of exon 2 of the NgR gene as previously reported (Kim et al., 2004). The strain provided was on a C57BL/6 background.
Morphological analysis.
Mice were anesthetized with chloral hydrate and perfused through the ascending aorta with freshly prepared 4% paraformaldehyde in 0.1 m sodium phosphate, pH 7.4. The C5 spinal cord, sciatic nerves, and distal tibial nerves were harvested from groups of mice at 6, 12, and 15 months of age. Five mice were used for each group. The tissues were further fixed in 4% paraformaldehyde/3% glutaraldehyde in Sorenson's buffer overnight at 4°C, postfixed in OsO4, and embedded in Epon-Araldite resin.
Cross sections (1 μm thick) were stained with toluidine blue for analysis under light microscopy with a 60× or 100× oil-immersion objective lenses using stereotactic imaging software (Stereo Investigator version 5). Every myelinated fiber in easily defined regions of the CNS and PNS was counted (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The resulting data were free of counting biases that could affect both random and systematic (stereologic) schemes of number of axon counting. Actively degenerating fibers were defined as fibers at different stages of Wallerian-like degeneration, such as myelin figures and ovoids. Results from each studied group were compared by the two-tailed Student's t test. p < 0.05 was considered significant.
Acrylamide treatment.
Groups of five 6-week-old male mice were treated with acrylamide by adding acrylamide to the drinking water at 400 ppm. Control mice drank regular water. Five mice were housed in each plastic cage throughout the experimental period. Experimental procedures followed the Society of Toxicology Guiding Principles in the Use of Animals in Toxicology and National Institutes of Health (NIH) guidelines (Guide for the Care and Use of Laboratory Animals, NIH Publication No. 86-23, 1985).
Rotarod studies.
Groups of five mice were trained 1 week before acrylamide treatment on 3 consecutive days by performing on the rotarod at a constant speed of 4 rotations/min (rpm) on day 1 and 8 rpm on days 2 and 3 for 3 min (pretraining), followed by an accelerating protocol in which the speed of rotation was initially set at 4 rpm and accelerated an additional 4 rpm every 30 s to a maximum of 40 rpm. Three trials were performed on each mouse on each of 3 consecutive days with ∼30 min rest between trials. After initiation of acrylamide treatment, the mice were further trained for another 3 consecutive days. Two weeks after acrylamide intoxication, mice were again pretrained and tested for 3 d, 3 trials/d. Unlike untreated mice, most MAG−/− mice fell off the rod even during pretraining (mice were not returned to the rod more than twice during pretraining).
To analyze data, we recorded the speed and duration during which the mouse stayed on the rod. Results of all three trials on day 3 of testing were pooled to generate a mean and SEM (n = 15). Results were compared by the two-tailed Student's t test. Values of p < 0.05 were considered significant. The tester was blinded to the treatment. No mice or values were excluded.
Electrophysiological studies.
Nerve conduction studies were performed on five wild-type and five MAG knock-out mice at 6–8 weeks of age. The mice were anesthetized with isoflurane using a nose cone. Body temperature was maintained on a warm blanket to keep the surface temperature between 32 and 38°C before the taking of measurements. All compound motor action potential (CMAP) measurements were performed with a PowerLab signal acquisition setup (ADInstruments). The CMAPs were measured by stimulation with subdermal needle electrodes placed near the sciatic nerve at the sciatic notch. Recording electrodes were placed in the tibial nerve-innervated intrinsic foot muscles in the plantar surface. Recordings were made with supramaximal stimulation. We determined the latencies, negative peak amplitudes, and durations of the sciatic compound muscle action potentials as well as the F-wave latencies and durations. The distance between stimulation sites, determined by calipers, was also used to calculate conduction velocities. CMAP measurements were obtained at the initiation of acrylamide intoxication and 2.5 weeks after intoxication.
DRG explant culture.
Postnatal day 4 (P4)–P5 dorsal root ganglia (DRGs) were removed from Sprague Dawley rat pups and maintained in Neurobasal medium containing 2 m l-glutamine, 2% B27 serum-free supplement, and 50–100 ng/ml NGF. Explant cultures were allowed to grow to a mature and maintenance state for 5 d, creating a lush outgrowth of neurites. This method of allowing neuritic outgrowth extension to proceed before addition of MAG and/or a neurotoxin tests their effect on established neurites as opposed to their effect on primary neuritic outgrowth. The neurons were examined under phase-contrast microscopy, and the average neurite length was quantified by using ImageJ, a public domain image-processing program (http://rsb.info.nih.gov/ij/). The axonal lengths of all axons were measured from the explant's border to the tips of intact axons. An average of all axonal lengths for each explant was then calculated. At least 8–10 explants were used for each test conditions.
In experiments involving MAG-CHO cells, DRG explants were allowed to grow to a mature and maintenance state for 5 d. MAG-CHO cells were then added to the culture and allowed to grow for additional 48 h before the effect of MAG-CHO cells on axonal protection was examined.
A tetracycline-repressible MAG system was generated in Chinese hamster ovary (CHO) cells as described previously (Milward et al., 2008). Several clones were derived and simultaneously assessed for expression of MAG and DsRed2, tetracycline repressibility of MAG, and membrane-bound expression of MAG. Addition of the tetracycline analog doxycycline at 1 μg/ml caused complete inhibition of MAG expression. In the present studies, MAG was allowed to be expressed throughout the experiments.
Results from each studied group were compared by the two-tailed Student's t test. p < 0.05 was considered significant.
Dissociated P4–5 DRG and cerebrocortical neurons.
P4–P5 DRGs and cortical neurons were removed from four animals and incubated with 0.25% trypsin. The digestion solution for postnatal DRG neurons also included 0.3% collagenase I. After 30 min, digestion was stopped with 10% FBS containing L-15 medium, and cells were plated at 10,000/cells per well. Cultures were maintained in Neurobasal medium containing 1% FBS, 2 m l-glutamine, 2% B27 serum-free supplement, and 50–100 ng/ml NGF. Neuronal cultures were allowed to grow to a mature and maintenance state for 5–7 d, creating a lush outgrowth of neurites. The average neurite lengths of 50 neurons immunostained for class III β-tubulin were quantified under fluorescent microscopy using ImageLab. Each experiment was performed in triplicate. Results from each studied group were compared by the two-tailed Student's t test. p < 0.05 was considered significant.
Campenot chamber.
Dissociated DRGs and cortical neurons were plated onto collagen-coated tissue culture dishes in the middle of a three-compartment chamber. Compartmentalized cultures were prepared as previously described (Campenot, 1982). Cells were maintained for the initial 2–7 d in growth medium containing cytosine arabinoside (10 μm) to eliminate Schwann cells and non-neuronal, dividing cells. After growing for 7 d, axons from the central chamber extended into the side chambers. The central compartment contained the cell bodies and proximal axons, and the side compartments contained the distal axonal processes and axon terminals. Treatment mediums were added to the side chambers, allowing selective delivery of the stimuli to axons. The average axonal lengths of 50 neurons immunostained for class III β-tubulin were quantified under fluorescent microscopy using ImageLab. Axonal length was measured from the edge of the chamber to the intact axonal tip. Each experiment was performed in 5–8 culture plates. Results from each studied group were compared by Student's t test. Values of p < 0.05 were considered significant.
Mutant MAG peptide with KGE domain.
Mutation on MAG (from RGD to KGE) was generated by site-directed mutagenesis (Stratagene) as described by the manufacturer. Mutant MAG-Fc with KGE domain was then transfected into 293 EBNA cells. The proteins were collected from the media and affinity purified using protein A Sepharose. Wild-type MAG-Fc with RGD domain also were expressed and purified in a similar manner.
Inhibitory substrata.
MAG was extracted from purified myelin membranes using mild detergent and was adsorbed to culture surfaces as described (Mehta et al., 2007). Briefly, myelin was purified from brains freshly dissected from adult Sprague Dawley rats or adult wild-type or MAG-null mice and stored at −70°C before use. Myelin membranes were suspended at 1 mg of protein/ml in extraction buffer [0.2 m sodium phosphate buffer, pH 6.8, 0.1 m Na2SO4, 1 mm EDTA, 1 mm DTT, protease inhibitor mixture (Sigma), and 1% octylglucoside], incubated at 4°C for 16 h with gentle agitation, and then centrifuged at 100,000 × g for 1 h at 4°C. The supernatant was collected and diluted with an equal volume of detergent-free buffer, and an aliquot (50 μl) was added to each well of a poly-d-lysine (PDL)-coated 96-well plate. After 4 h at ambient temperature, the plate was washed with Dulbecco's PBS and then with the culture medium appropriate to the cell type before plating freshly prepared cells. DRGs and cortical neurons were grown on the substrate as described previously (Mehta et al., 2007).
After 48 h cultures were washed with PBS, fixed overnight with 2% paraformaldehyde in PBS, and then permeabilized using 0.1% Triton X-100 in PBS. DRG neurons were immunostained with anti-neuronal class III β-tubulin monoclonal antibody (TUJ1, 1:2000, Covance) followed by Cy3-conjugated anti-mouse IgG (1:200; Jackson ImmunoResearch Laboratories). After washing, multiple random fields were captured for image analysis using a Nikon TE300 epifluorescent microscope fitted with a Photometrics CoolSNAP HQ2 camera (Roper Scientific).
DRG neurons on control substrata extended long, thick neurites when grown in control conditions. In contrast, DRG neuronal cultures grown in the presence of vincristine (VIN; without myelin substrata) have neurites that progressively degenerate distally, where they become fragmented, comparatively thinner, and morphologically distinguishable from neurites in cultures without vincristine. To differentiate the integrity of neurites, we developed an image analysis protocol using NIS-Elements software (Nikon) to detect the intact, nonfragmented axons. The protocol selected neurites that were >150 μm long, and subtracted out the portion of neurites that are fasciculated. Neuritic lengths were summed and divided by the total number of DRG neuronal cell bodies to provide a single value for the presence of healthy unfasciculated neurites.
For each experimental condition, four to five random images from each of 5–10 independent wells from an average of three independent experiments were analyzed. Data are presented as the mean ± SEM. Results were compared using the two-tailed Student's t test. Values of p < 0.05 were considered significant.
Western blotting.
Cell monolayers were scraped and lysed in a detergent-containing buffer (10 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 2 mm sodium orthovanadate, 10 mm NaF, 10 mm sodium pyrophosphate, 1% IGEPAL, and 1 mm phenylmethylsulfonyl fluoride) with protease inhibitor mixture (Roche). Total protein (10 μg) was loaded on 4–12% Tris/glycine gradient gels and transferred to nitrocellulose membranes (Novex). Membranes were blocked in 10% bovine serum albumin (BSA) in Tris-buffered saline/0.05% Tween 20 for 1 h at room temperature and then incubated with the primary antibodies and their appropriate peroxidase-conjugated secondary antibodies for varying times. Blots were developed using enhanced chemiluminescence (NEN Life Science Products). Antibodies for specific proteins are listed above in Reagents.
Immunocytochemistry.
Skin tissue was fixed for 12–18 h in 2% paraformaldehyde/lysine/periodate fixative followed by cryoprotection. The tissue were sectioned with a sliding microtome into 50-mm-thick free-floating sections. Sections were stained with monoclonal antibodies to neurofilament (NF160 1:200, Millipore Bioscience Research Reagents) and developed with chromogens as previously described (Ebenezer et al., 2007).
Results
MAG contributes to normal axonal survival and maintenance in vivo
We confirmed that MAG−/− mice underwent progressive axonal degeneration within the CNS and PNS (Yin et al., 1998; Pan et al., 2005). The numbers of intact nerve fibers and those undergoing degeneration were enumerated in the rostral medial dorsal columns of the spinal cord at the C5 level (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). This site contains the preterminal regions of the central processes of neurons located in the sacral and lumbar dorsal root ganglia. These axons are thus among the longest axons in the body. In wild-type mice, there were very few actively degenerating fibers and no detectable axonal loss between 6 and 15 months of age (Fig. 1ai). In contrast, in the rostral medial dorsal columns of MAG knock-out mice, there was a nearly constant level of ongoing axonal degeneration involving 0.46% of fibers at any time between 6 and 15 months, resulting in progressive decrease in axonal numbers, cumulating in a 28% reduction by 15 months (Fig. 1aii). A similar progressive loss of axons was found in the sciatic and the tibial nerves of MAG−/− mice (Fig. 1b; supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 1.
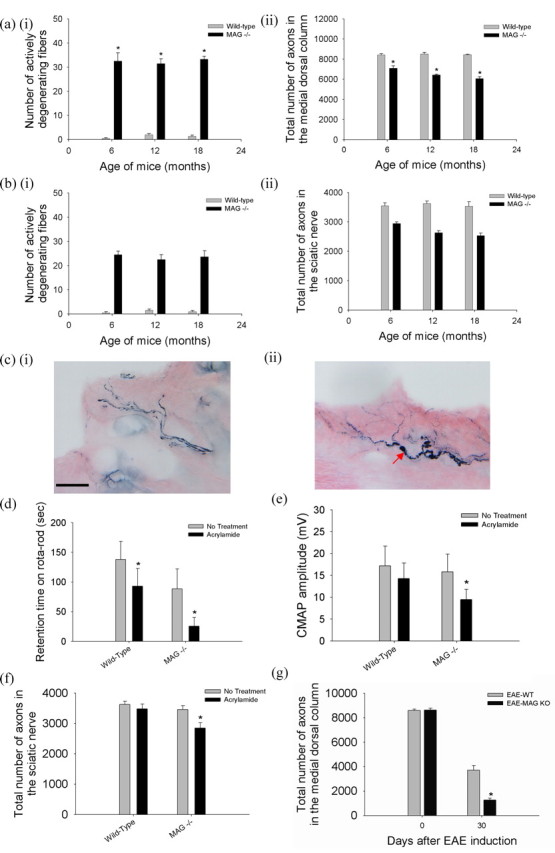
MAG promotes axonal stability in vivo. C5 spinal cords, sciatic nerves proximal to the bifurcation, and distal tibial nerves at the ankles were harvested from groups of five mice at 6, 12, and 15 months of age. Every myelinated fiber was counted in easily defined regions of the CNS and PNS as shown in supplemental Figure S1 (available at www.jneurosci.org as supplemental material). a, In the medial dorsal columns at C5 in wild-type mice, there were few degenerating fibers (ai), and axonal numbers remained constant between 6 and 15 months of age (aii). In contrast, MAG−/− mice had a high number of active Wallerian-like degeneration at all ages (ai), leading to progressive axonal loss reaching 28% at 15 months (aii) (*MAG−/− vs wild-type age matched mice, n = 5, p < 0.01). b, In the sciatic nerve, MAG−/− mice also exhibited large number of active Wallerian-like degeneration at all ages (bi), associated with progressive axonal loss reaching 28% at 15 months (bii) (*MAG−/− vs wild-type age matched mice, n = 5, p < 0.01). ci, Wild-type mice at 6-weeks of age exposed to 400 ppm acrylamide in the drinking water for 4 weeks showed normal appearing nerve fibers with minimal axonal swelling and segmentation consistent with mild cutaneous axonal degeneration and loss as shown by neurofilaments (NF160) immunostaining. Scale bar, 40 μm. cii, In contrast, MAG knock-out mice with equivalent age and exposure to acrylamide showed numerous axons with dramatic swelling, beading, and segmentation consistent with severe cutaneous axonal degeneration and loss (red arrow). d, Wild-type mice exposed to acrylamide as above showed modest decline in retention time on the rotarod, but the acrylamide-intoxicated MAG knock-out mice showed severe decrease in retention time. Most MAG knock-out mice fell off the rod even during pretraining. e, MAG knock-out mice exposed to acrylamide showed more decrease in CMAP amplitude in comparison that in wild-type controls. f, Six-week-old wild-type mice exposed to acrylamide as above showed no significant axonal loss in the sciatic nerve, but MAG−/−mice exposed to acrylamide showed ∼20% axonal loss in the sciatic nerve (*acrylamide treated vs untreated MAG−/− mice, n = 5, p < 0.01). g, EAE was induced by immunizing 6-week old wild-type and MAG knock-out mice with MOG peptide 35–55. MAG knock-out mice suffered >50% more axonal loss than wild-type mice at 4 weeks after MOG-EAE induction (*EAE-MAG−/− vs EAE wild-type mice, n = 5, p < 0.01).
To evaluate whether the absence of MAG results in increased vulnerability to axonal degeneration from additional stresses such as neurotoxins, we administered acrylamide, a well characterized toxin causing axonal “dying back” or degeneration without lymphocytic inflammatory response (Schaumburg et al., 1974; Ko et al., 2000) to wild-type and MAG−/− mice. When wild-type mice at 6 weeks of age were exposed to acrylamide for 3 weeks, there was mild axonal swellings and scattered, distally predominant axonal degeneration in the skin, sciatic nerve, and C5 spinal cord (Fig. 1ci; supplemental Fig. S3, available at www.jneurosci.org as supplemental material). On gross observation, there was only mild gait unsteadiness. When challenged with the rotarod test, acrylamide-intoxicated wild-type mice had mild difficulty in maintaining their balance on the rotating rods and had a modest decline in retention time on the rotarod in comparison with untreated mice (Fig. 1d). This was accompanied by a small decrease in amplitudes of CMAP on electrophysiological testing (Fig. 1e). Quantitative morphological analysis of the sciatic nerve and medial dorsal column of C5 spinal cord revealed a small loss in the total number of axons after acrylamide exposure (Fig. 1f). In contrast, MAG knock-out mice exposed to acrylamide showed markedly impaired gait with marked sensory ataxia and spreading of the toes of the hind limbs (supplemental Video 1, available at www.jneurosci.org as supplemental material). MAG knock-out mice had lower retention time on a rotarod than wild-type controls. After acrylamide exposure, MAG knock-out mice developed more rapid and severe motor impairment. Most MAG knock-out mice fell off the rod even during pretraining and had profound decline in the retention time on the rotarod (Fig. 1d). There was a 36% decrease in amplitudes of CMAP (Fig. 1e). Likewise, morphological analysis showed significant axonal degeneration with almost 20% loss in total number of axons in the sciatic nerve and medial dorsal column of C5 spinal cord (Fig. 1f; supplemental Fig. S3, available at www.jneurosci.org as supplemental material). This was accompanied by numerous axons in the skin with profound swelling, beading, and segmentation consistent with predegenerative changes of Aβ fibers (Fig. 1cii) (Schaumburg et al., 1974; Hsieh et al., 1996; Ebenezer et al., 2007).
To test whether the absence of MAG also results in increased vulnerability to axonal degeneration from CNS inflammation, we used a widely used animal model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). EAE was induced by immunizing wild-type and MAG knock-out mice with myelin oligodendrocyte glycoprotein (MOG) peptide 35–55. Wild-type mice induced with MOG-EAE exhibited severe axonal loss in the rostral medial column at 4 weeks. MAG−/− mice showed 52% more axonal loss at 4 weeks after MOG-EAE induction than wild-type mice at the same time point (Fig. 1g). Rotarod testing was not performed on mice in which EAE was induced; both EAE-MAG−/− and EAE-wild-type mice exhibited motor impairment sufficiently severe that rotarod testing was not informative.
Soluble MAG-Fc prevents axonal degeneration in cell culture
To further elucidate the effects of MAG on axons, we established postnatal (P5) primary rat dorsal root ganglion cultures. We confirmed that the addition of soluble MAG-Fc to the cultures resulted in phosphorylation of NF-H (data not shown) (Dashiell et al., 2002). We next asked whether MAG affected microtubule stability. Recently assembled and cold-labile microtubules contain greater amounts of tyrosinated tubulin than do older, more long-lived ones and cold-stable microtubules (Baas and Black, 1990). For this reason, the ratio of detyrosinated tubulin to tyrosinated tubulin is considered a marker of microtubule stability. After the addition of MAG-Fc to rat cerebrocortical cultures, we noted increased levels of detyrosinated tubulin and decreased levels of tyrosinated tubulin (Fig. 2a). These data suggest that MAG increases microtubule stability.
Figure 2.

Soluble MAG-Fc and MAG expressed on CHO cells promote axonal stability. a, Cerebrocortical cultures treated with 25 μg/ml MAG-Fc for varying lengths of time were lysed and probed for detyrosinated and tyrosinated tubulin. MAG-Fc increased levels of detyrosinated tubulin and reduced levels of tyrosinated tubulin. b, MAG-Fc (25 μg/ml) application to the distal (axonal) chamber of a Campenot chamber inhibited longitudinal axonal growth over 48 h (*MAG vs control, n = 8 chambers/condition, p < 0.01). Application of 50 nm vincristine to the distal chamber resulted in axonal degeneration that was prevented by MAG coapplication (*VIN+MAG vs VIN, n = 5 chambers/condition, p < 0.01). Coadministration of MAG-Fc and vincristine shows similar protective effect when added to the center chamber. Note that the distal chamber contains exclusively axons but the center chamber contains both cell bodies and their proximal neurites. c, Panels 1–6 are representative images of DRG explants after exposure to 50 nm vincristine at 0, 36, and 48 h. Explants in panels 1, 2, and 3 were treated with 50 nm vincristine and 25 μg/ml MAG-Fc, whereas those in panels 4, 5, and 6 were treated with vincristine alone. Panel 6 shows almost complete axonal degeneration, but little axonal loss was seen in panel 3. d, Quantitative measurements of axonal length in the above DRG neurite degeneration in postnatal rat DRG cultures by vincristine (25 nm) at various time points over 72 h. Although vincristine caused marked axonal degeneration, the coadministration of MAG-Fc almost completely prevented the axonal “dying back” associated with vincristine. e, A dose–response curve of MAG-Fc axon-protective effect against vincristine. Coadministration with MAG-Fc prevented neurite degeneration in DRG neurons treated with vincristine, but the Fc-fragment alone did not provide protection. ED50 is ∼100 nm. (n = 10 chambers/condition). f, Administration of MAG-Fc prevented axonal degeneration in P5 DRG neurons by axonal toxic agents, including activated CD8 T cell supernatant (CD8 Sup), 1 nm granzyme B (GR-B), and 200 μm acrylamide (Acryl) (*CD8 Sup+MAG-Fc vs CD8 Sup, GR-B+MAG-Fc vs GR-B, Acryl+MAG-Fc vs Acryl, n = 6 chambers/condition, p values <0.01). g, Addition of MAG-CHO cells, or protease-released extracellular MAG domain from these cells (wash from MAG-CHO/MMP7), also prevented axonal degeneration in P5 DRG neurons. In contrast, the removal of the extracellular fragment of MAG with daily wash (MAG-CHO/MMP7+daily wash) did not provide protection against vincristine (*VIN+MAG-CHO vs VIN, n = 10 chambers/condition, p < 0.01; VIN+wash solution from MAG-CHO/MMP-7 vs VIN+MAG-CHO/MMP-7+daily wash to remove extracellular MAG fragment, n = 10 chambers/condition, p < 0.01).
To ensure that the effects were local effects on the axonal segments exposed to MAG, we used Campenot chambers (Campenot, 1992). These chambers use a grease seal to produce a physical, water-tight separation of distal axons from neuronal cell bodies. When soluble MAG (MAG-Fc) was added to the distal axonal compartment of these cultures, there was a decrease in subsequent axonal longitudinal growth (Fig. 2b), an observation consistent with the existing literature (DeBellard et al., 1996). To ask whether these stabilized axons were more resistant than untreated axons to injury, we added VIN, a chemotherapeutic agent with well characterized properties of axonal toxicity, to the distal chamber of postnatal DRG cultures. In cultures not treated with MAG-Fc, VIN produced a marked reduction in axonal length compared with baseline, and there was morphological evidence of axonal degeneration. When MAG-Fc was coadministered with vincristine to the distal axonal chamber, vincristine-induced axonal degeneration was largely prevented (Fig. 2b). Hence, this suggested that although MAG-Fc may discourage longitudinal axonal growth, it simultaneously promotes axonal stability and resistance to axonal degeneration.
MAG-Fc similarly conferred axonal protection from vincristine in postnatal (P5) DRG explant cultures (Fig. 2c,d) and dissociated embryonic day 15 cerebrocortical cultures (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). A dose–response curve of MAG-Fc axon-protective effect against vincristine is shown in Figure 2e. This axon-protective effect was not restricted to vincristine: MAG also prevented axonal degeneration in response to acrylamide, granzyme B, and supernatants from activated cytotoxic T cells (Fig. 2f).
MAG expressed on CHO cells prevents axonal degeneration in cell culture
To ensure that the effects were not limited to this soluble MAG-Fc fusion protein, the axonal protective ability of different forms of MAG was investigated. After allowing postnatal DRG explants to reach a mature and maintenance state, they were cocultured with MAG-CHO cells expressing a high level of membrane-associated MAG (Milward et al., 2008). In the presence of MAG-CHO cells, vincristine-induced axonal degeneration was largely prevented (Fig. 2g). To explore the functional domain of MAG's protection, MAG was cleaved at a juxtamembrane position on MAG-CHO cells by MMP-7 to release an intact extracellular fragment (Milward et al., 2008). When the extracellular MAG fragments were removed with regular interval washes, the ability of MAG-CHO cells to protect against vincristine was lost (Fig. 2g). When the medium containing the extracellular MAG fragments was placed on neurons grown without MAG-CHO cells, vincristine-induced axonal degeneration was now largely prevented (Fig. 2g). This suggests that like the growth-inhibitory effects, axonal protection depends on the extracellular Ig domains of MAG for the relevant functional domain for axonal protection (DeBellard et al., 1996).
MAG extract from native myelin prevents axonal degeneration in cell culture
Finally, postnatal DRG neurons were grown on poly-d-lysine matrix alone or with full-length native MAG detergent-extracted from adult rat brain myelin (Mehta et al., 2007). Because extension of axons of DRG neurons is inhibited in an NgR-mediated manner when grown on detergent-extracted myelin (Mehta et al., 2007), phosphatidylinositol-phospholipase C (PI-PLC) was added to the cultures to overcome MAG-mediated inhibition to study whether MAG could protect axons (Fournier et al., 2001; Liu et al., 2002; Mehta et al., 2007). When DRG neurons were grown on detergent extracts of myelin, the vincristine-induced axonal degeneration was largely abolished (Fig. 3a,c). Anti-MAG antibody reversed the protection against vincristine (Fig. 3b,c) (similar results in cortical neurons are not shown). Myelin extract from MAG−/− also failed to provide axonal protection (data not shown). Together, our in vitro and in vivo data thus suggest that the axonal protective effect of MAG is not restricted to a particular form of MAG, neuronal age, or neuronal cell type (CNS or PNS).
Figure 3.
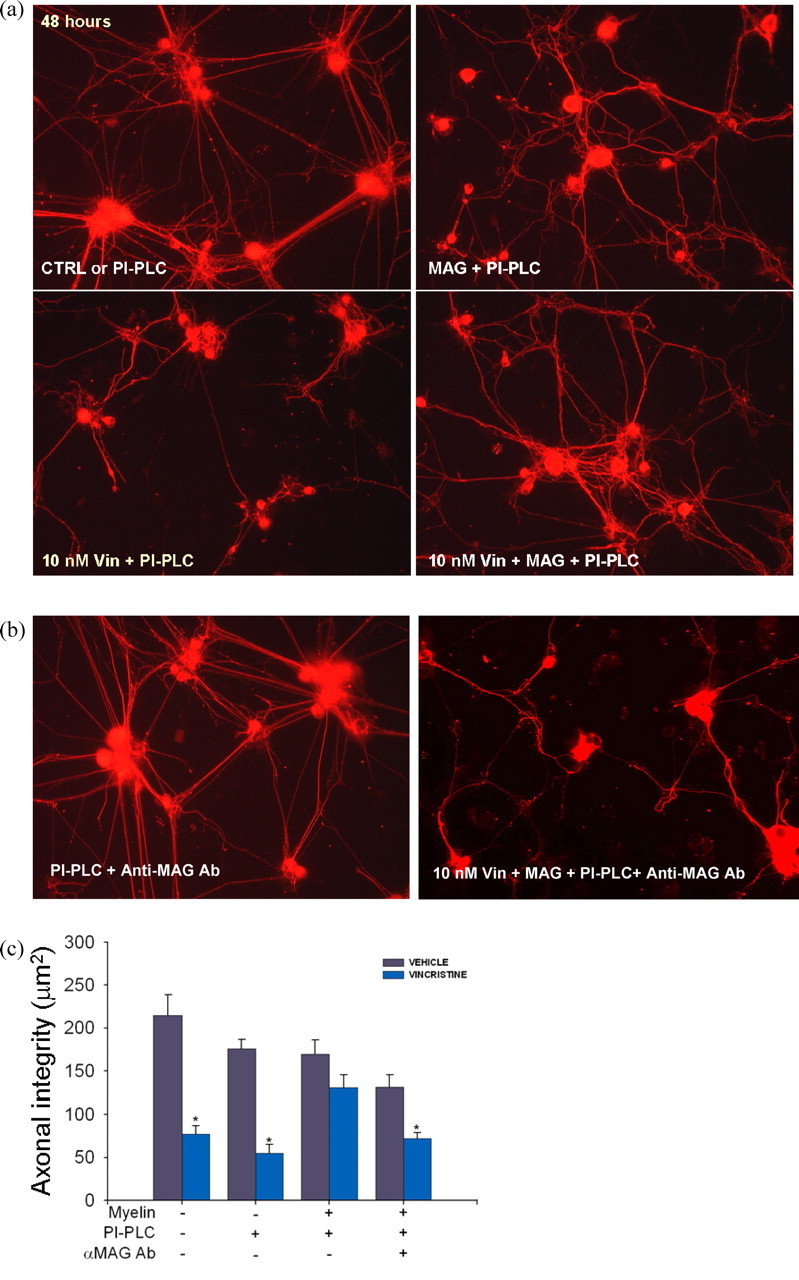
Native myelin extract containing MAG promotes axonal stability. a, Dissociated DRG neurons were grown for 48 h on poly-d-lysine matrix (PDL) alone (data not shown), PDL with soluble PI-PLC (PI-PLC), or with soluble PI-PLC plus native MAG detergent-extracted from adult rat brain myelin (“MAG”). At 48 h after plating, the application of 10 nm vincristine for an additional 48 h period caused marked axonal degeneration in cultures grown on poly-d-lysine matrix alone (data not shown). DRG axons grown on poly-d-lysine matrix with detergent-extracted myelin proteins and PI-PLC were not susceptible to axon degeneration induced by 10 nm vincristine (10 nm Vin+PI-PLC+MAG). b, In contrast, neurons grown on poly-d-lysine matrix, myelin extract, PI-PLC, and anti-MAG antibodies were highly susceptible to vincristine-induced axonal degeneration (right panel). Note that anti-MAG antibodies (PI-PLC+Anti-MAG Ab) did not cause axonal degeneration (left panel). c, Neurite integrity (mean ± SE) was quantified using image analysis (* vincristine vs vehicle solution, n = 4–10 plates/condition, p < 0.01). Detergent-extracted myelin proteins provided axonal protection against 10 nm vincristine. This effect was abolished by the addition of anti-MAG antibodies, suggesting that MAG was the myelin component providing axonal protection.
MAG-induced axonal protection is independent of Nogo signaling
We next asked whether a Nogo receptor (NgR), a member of a family of glycosylphosphatidylinositol (GPI)-linked receptors, was necessary for MAG-induced protection from axonal degeneration. In contrast to the axonal degeneration and loss seen in the MAG−/− mice, NgR1−/− mice exhibited no actively degenerating fibers in the rostral medial dorsal column of the spinal cord, in the sciatic nerves (Fig. 4a), or in the tibial nerves (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Moreover, in cerebrocortical and DRG neuronal cultures, removal of GPI-linked molecules like NgR from the cell surface by incubation with PI-PLC had no effect on the ability of MAG to prevent vincristine-induced neuritic degeneration (Fig. 4b) (Fournier et al., 2001; Domeniconi et al., 2002; Liu et al., 2002). Similarly, the activation of the Nogo signaling pathway with Nogo-66 or OMgp peptides, which nonselectively interact with various NgR receptor types, did not provide axonal protection against vincristine (Fig. 4b). These data suggest that an axonal receptor other than NgR mediates MAG-induced axonal protection.
Figure 4.

MAG mediated axonal stability via an NgR-independent pathway. a, NgR1 knock-out mice exhibited no evidence of Wallerian-like axonal degeneration (ai) or loss in axonal number (aii) within the medial dorsal column in C5 spinal cords or the sciatic nerve (harvested proximal to the bifurcation) at 12 months of age (n = 5 mice/group). b, MAG prevented axonal degeneration from vincristine in DRG neurons treated with PI-PLC. Coadministration of either Nogo or OMgp peptides, nonspecific NgR agonists, did not provide axonal protection against vincristine. Because MAG mediated axonal protection was unaltered by the inhibition of NgR with PI-PLC and activation of NgR by Nogo or OMgp did not confer axonal protection, our data suggest that MAG promotes axonal stability via an NgR-independent pathway. (*VIN+MAG-Fc vs VIN, VIN+MAG-Fc+PI-PLC vs VIN, Nogo vs VIN+ Nogo, Nogo+PI-PLC vs VIN+Nogo, n = 10 chambers/condition, p values <0.01). c, A MAG-Fc mutant with RGD altered to KGE (mtMAG) failed to prevent axonal degeneration from vincristine in primary DRG neuronal cultures (*VIN+wtMAG vs VIN, VIN+wtMAG vs VIN+mtMAG, n = 8 chambers/condition, p values <0.01). d, MAG did not promote axonal stability or prevent axonal degeneration in DRG cultures from B4galnt1 knock-out mice lacking complex gangliosides. (n = 8 chambers/condition).
The interaction of MAG with axons previously had been proposed to include a second binding site at arginine 118 (R118) in first Ig domain of MAG, which is not required for NgR-mediated inhibition of axonal elongation during regeneration (Kelm et al., 1994; Tang et al., 1997; Cao et al., 2007). In our system, an R118-mutated MAG failed to prevent axonal degeneration caused by vincristine (Fig. 4c). This suggests that MAG-induced axonal protection depends on the functional binding site around arginine 118.
The ectodomain R118 of MAG has been reported to be necessary for its lectin activity and may suggest a possible association of MAG with sialic-acid residues on the axonal surface for the protective effects. We observed that MAG did not promote axonal stability or prevent axonal degeneration in DRG cultures from B4galnt1 knock-out mice lacking complex gangliosides (Fig. 4d). Although our data suggest that the axonal protective effect of MAG depends on the interaction with complex gangliosides, the nature of the interaction is beyond the scope of this study. Gangliosides could act as a coreceptor, binding receptor, or facilitator for clustering the full signaling complex assembly and cytoskeletal association within the membrane required for initiation of signal transduction.
Discussion
New data in this report support the conclusion that MAG, a constituent of the adaxonal membrane of myelin-forming cells, promotes axonal stability and survival in cell culture and in vivo. Although this effect on axonal stability is independent of Nogo signaling in the axon, it depends on arginine 118 in the RGD domain of the extracellular segment of the molecule. MAG signals to the axon to promote stability of axonal microtubules, and promotes axonal survival in the face of insults such as vincristine, acrylamide, and inflammatory mediators. This protective effect may be augmented by effects on phosphorylation of MAP proteins and tau (Dashiell et al., 2002; Zheng et al., 2007). These may also affect axonal transport, which is known to be affected locally by demyelination (de Waegh et al., 1992). Moreover, MAG is known to locally increase axonal diameter (Yin et al., 1998; Dashiell et al., 2002). Its role in limiting longitudinal growth is well known, and its presence in the internodal and paranodal adaxonal plasmalemma of the myelin-forming cell means that it is likely to inhibit collateral sprouting along the course of the myelinated fiber. In these ways MAG discourages plasticity of the mature myelinated axon (Griffin and Thompson, 2008). These data, together with previous studies, suggest that the biological roles of MAG include promoting axonal stability, survival, and maximal radial caliber, and discouraging either longitudinal or collateral growth. It thereby encourages stable and rapid point-to-point impulse transmission (Griffin and Thompson, 2008).
We speculate that the loss of axonal stabilizing and protective effects of MAG may contribute to the late axonal degeneration seen in demyelinating diseases. Although MAG is not a component of compact myelin, MAG is known to be lost from oligodendrocytes and Schwann cells that have undergone demyelination. There is a wealth of evidence that axons are liable to both prompt and progressive late Wallerian-like degeneration after demyelination (Dyck, 1975; Trapp et al., 1998; Scherer, 1999; Berciano et al., 2000; Krajewski et al., 2000; Bjartmar et al., 2003; Oh et al., 2004). In the PNS this is exemplified by demyelinating forms of Charcot-Marie-Tooth disease (CMT1), the most common forms of inherited peripheral neuropathy. In several demyelinating forms of CMT, the genetic defects are in components of the Schwann cells, such as PMP22 and P0 (Scherer, 1999). These defects lead to recurrent demyelination that can be detected by electrophysiologic changes early in childhood. However, the clinical manifestations often occur only years later and are attributable to progressive distal axonal loss in the peripheral nerves (Scherer, 1999). Distal axonal degeneration has been demonstrated in animal models of CMT, including P0, connexin 32, and PMP22 mutations in the PNS and PLP mutations in the CNS (Scherer, 1999). Importantly, axonal degeneration is now recognized to be a major component of the later progressive stage of the CNS inflammatory demyelinating disease, MS. Undoubtedly axonal injury can be produced by inflammatory mediators, but it is likely that demyelination renders the axon more vulnerable to axonal degeneration (Bjartmar et al., 2003). The loss of the axonal stabilizing and protective effects of adaxonal MAG may contribute to this type of “secondary” axonal loss.
An active field at the moment is developing therapies to overcome the effects of MAG that block axonal regeneration in the CNS. Such therapies will need to avoid destabilizing injured and uninjured axons, thus rendering them more vulnerable to axonal degeneration.
Footnotes
This work was supported by National Multiple Sclerosis Society Tissue Repair Grant R 3760-A-3, the Nancy Davis Center Without Walls, and the National Institutes of Health Grants K08 NS055135 and R37 NS037096. We thank Douglas Kerr and Harish Pant for technical help.
The authors declare no competing financial interests.
References
- Baas and Black, 1990.Baas PW, Black MM. Individual microtubules in the axon consist of domains that differ in both composition and stability. J Cell Biol. 1990;111:495–509. doi: 10.1083/jcb.111.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berciano et al., 2000.Berciano J, García A, Calleja J, Combarros O. Clinico-electrophysiological correlation of extensor digitorum brevis muscle atrophy in children with charcot-marie-tooth disease 1A duplication. Neuromuscul Disord. 2000;10:419–424. doi: 10.1016/s0960-8966(99)00114-5. [DOI] [PubMed] [Google Scholar]
- Bjartmar et al., 2003.Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Campenot, 1982.Campenot RB. Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev Biol. 1982;93:13–21. doi: 10.1016/0012-1606(82)90233-0. [DOI] [PubMed] [Google Scholar]
- Campenot, 1992.Campenot RB. Compartmented culture analysis of nerve growth. In: Stevenson BR, Gallin WJ, Hall DL, editors. Cell-cell interactions: a practical approach. New York: Oxford UP; 1992. pp. 275–298. [Google Scholar]
- Cao et al., 2007.Cao Z, Qiu J, Domeniconi M, Hou J, Bryson JB, Mellado W, Filbin MT. The inhibition site on myelin-associated glycoprotein is within Ig-domain 5 and is distinct from the sialic acid binding site. J Neurosci. 2007;27:9146–9154. doi: 10.1523/JNEUROSCI.2404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli et al., 2003.Ciccarelli O, Werring DJ, Barker GJ, Griffin CM, Wheeler-Kingshott CA, Miller DH, Thompson AJ. A study of the mechanisms of normal-appearing white matter damage in multiple sclerosis using diffusion tensor imaging—evidence of Wallerian degeneration. J Neurol. 2003;250:287–292. doi: 10.1007/s00415-003-0992-5. [DOI] [PubMed] [Google Scholar]
- Coleman and Perry, 2002.Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- Dashiell et al., 2002.Dashiell SM, Tanner SL, Pant HC, Quarles RH. Myelin-associated glycoprotein modulates expression and phosphorylation of neuronal cytoskeletal elements and their associated kinases. J Neurochem. 2002;81:1263–1272. doi: 10.1046/j.1471-4159.2002.00927.x. [DOI] [PubMed] [Google Scholar]
- DeBellard et al., 1996.DeBellard ME, Tang S, Mukhopadhyay G, Shen YJ, Filbin MT. Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci. 1996;7:89–101. doi: 10.1006/mcne.1996.0007. [DOI] [PubMed] [Google Scholar]
- de Waegh et al., 1992.de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- Domeniconi et al., 2002.Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin M. Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron. 2002;35:283–290. doi: 10.1016/s0896-6273(02)00770-5. [DOI] [PubMed] [Google Scholar]
- Dyck, 1975.Dyck PJ. Philadelphia: Saunders; 1975. Inherited neuronal degeneration and atrophy affecting peripheral motor, sensory, and autonomic neurons. [Google Scholar]
- Ebenezer et al., 2007.Ebenezer GJ, McArthur JC, Thomas D, Murinson B, Hauer P, Polydefkis M, Griffin JW. Denervation of skin in neuropathies: the sequence of axonal and Schwann cell changes in skin biopsies. Brain. 2007;130:2703–2714. doi: 10.1093/brain/awm199. [DOI] [PubMed] [Google Scholar]
- Fournier et al., 2001.Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fournier et al., 2003.Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia et al., 2003.Garcia ML, Lobsiger CS, Shah SB, Deerinck TJ, Crum J, Young D, Ward CM, Crawford TO, Gotow T, Uchiyama Y, Ellisman MH, Calcutt NA, Cleveland DW. NF-M is an essential target for the myelin-directed “outside-in” signaling cascade that mediates radial axonal growth. J Cell Biol. 2003;163:1011–1020. doi: 10.1083/jcb.200308159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin and Thompson, 2008.Griffin JW, Thompson WJ. Biology and pathology of nonmyelinating Schwann cells. Glia. 2008;56:1518–1531. doi: 10.1002/glia.20778. [DOI] [PubMed] [Google Scholar]
- Hsieh et al., 1994.Hsieh ST, Kidd GJ, Crawford TO, Xu Z, Lin WM, Trapp BD, Cleveland DW, Griffin JW. Regional modulation of neurofilament organization by myelination in normal axons. J Neurosci. 1994;14:6392–6401. doi: 10.1523/JNEUROSCI.14-11-06392.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh et al., 1996.Hsieh ST, Choi S, Lin WM, Chang YC, McArthur JC, Griffin JW. Epidermal denervation and its effects on keratinocytes and Langerhans cells. J Neurocytol. 1996;25:513–524. doi: 10.1007/BF02284819. [DOI] [PubMed] [Google Scholar]
- Kelm et al., 1994.Kelm S, Pelz A, Schauer R, Filbin MT, Tang S, de Bellard ME, Schnaar RL, Mahoney JA, Hartnell A, Bradfield P, Crocker PR. Sialoadhesin, myelin-associated glycoprotein and CD22 define a new family of sialic acid-dependent adhesion molecules of the immunoglobulin superfamily. Curr Biol. 1994;4:965–972. doi: 10.1016/s0960-9822(00)00220-7. [DOI] [PubMed] [Google Scholar]
- Kim et al., 2004.Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Ko et al., 2000.Ko MH, Chen WP, Hsieh ST. Cutaneous nerve degeneration induced by acrylamide in mice. Neurosci Lett. 2000;293:195–198. doi: 10.1016/s0304-3940(00)01523-8. [DOI] [PubMed] [Google Scholar]
- Krajewski et al., 2000.Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, Kamholz J, Shy ME. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123:1516–1527. doi: 10.1093/brain/123.7.1516. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2002.Liu BP, Fournier A, GrandPré T, Strittmatter SM. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science. 2002;297:1190–1193. doi: 10.1126/science.1073031. [DOI] [PubMed] [Google Scholar]
- Mehta et al., 2007.Mehta NR, Lopez PH, Vyas AA, Schnaar RL. Gangliosides and Nogo receptors independently mediate myelin-associated glycoprotein inhibition of neurite outgrowth in different nerve cells. J Biol Chem. 2007;282:27875–27886. doi: 10.1074/jbc.M704055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi et al., 2004.Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Milward et al., 2008.Milward E, Kim KJ, Szklarczyk A, Nguyen T, Melli G, Nayak M, Deshpande D, Fitzsimmons C, Hoke A, Kerr D, Griffin JW, Calabresi PA, Conant K. Cleavage of myelin associated glycoprotein by matrix metalloproteinases. J Neuroimmunol. 2008;193:140–148. doi: 10.1016/j.jneuroim.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng et al., 1996.Ng WP, Cartel N, Li C, Roder J, Lozano A. Myelin from MAG-deficient mice is a strong inhibitor of neurite outgrowth. Neuroreport. 1996;7:861–864. doi: 10.1097/00001756-199603220-00005. [DOI] [PubMed] [Google Scholar]
- Oh et al., 2004.Oh J, Henry RG, Genain C, Nelson SJ, Pelletier D. Mechanisms of normal appearing corpus callosum injury related to pericallosal T1 lesions in multiple sclerosis using directional diffusion tensor and 1H MRS imaging. J Neurol Neurosurg Psychiatry. 2004;75:1281–1286. doi: 10.1136/jnnp.2004.039032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan et al., 2005.Pan B, Fromholt SE, Hess EJ, Crawford TO, Griffin JW, Sheikh KA, Schnaar RL. Myelin-associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: neuropathology and behavioral deficits in single- and double-null mice. Exp Neurol. 2005;195:208–217. doi: 10.1016/j.expneurol.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao et al., 2003.Rao MV, Campbell J, Yuan A, Kumar A, Gotow T, Uchiyama Y, Nixon RA. The neurofilament middle molecular mass subunit carboxyl-terminal tail domains is essential for the radial growth and cytoskeletal architecture of axons but not for regulating neurofilament transport rate. J Cell Biol. 2003;163:1021–1031. doi: 10.1083/jcb.200308076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaumburg et al., 1974.Schaumburg HH, Wiśniewski HM, Spencer PS. Ultrastructural studies of the dying-back process. I. Peripheral nerve terminal and axon degeneration in systemic acrylamide intoxication. J Neuropathol Exp Neurol. 1974;33:260–284. doi: 10.1097/00005072-197404000-00006. [DOI] [PubMed] [Google Scholar]
- Scherer, 1999.Scherer S. Axonal pathology in demyelinating diseases. Ann Neurol. 1999;45:6–7. doi: 10.1002/1531-8249(199901)45:1<6::aid-art3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Tang et al., 1997.Tang S, Shen YJ, DeBellard ME, Mukhopadhyay G, Salzer JL, Crocker PR, Filbin MT. Myelin-associated glycoprotein interacts with neurons via a sialic acid binding site at ARG118 and a distinct neurite inhibition site. J Cell Biol. 1997;138:1355–1366. doi: 10.1083/jcb.138.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp and Quarles, 1982.Trapp BD, Quarles RH. Presence of the myelin-associated glycoprotein correlates with alterations in the periodicity of peripheral myelin. J Cell Biol. 1982;92:877–882. doi: 10.1083/jcb.92.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp et al., 1998.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Yin et al., 1998.Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng et al., 2007.Zheng YL, Li BS, Kanungo J, Kesavapany S, Amin N, Grant P, Pant HC. Cdk5 Modulation of mitogen-activated protein kinase signaling regulates neuronal survival. Mol Biol Cell. 2007;18:404–413. doi: 10.1091/mbc.E06-09-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]