

Abstract
Type I interferons (IFNs), including IFN-α, can enhance antigen presentation and promote the expansion, survival and effector function of CD8+ cytotoxic T lymphocytes (CTL) during viral infection. Since these are ideal characteristics for a vaccine adjuvant we examined the efficacy and mechanism of exogenous IFN-α as an adjuvant for anti-melanoma peptide vaccination. We studied the expansion of pmel-1 transgenic CD8+ T cells specific for the gp100 melanocyte differentiation antigen after vaccination of wild-type C57BL/6 or transgenic/knockout mice with gp10025-33 peptide in incomplete Freund's adjuvant. IFN-α synergized with peptide vaccination in a dose-dependent manner by boosting relative and absolute numbers of gp100-specific T cells that suppressed B16 melanoma growth. IFN-α dramatically increased the accumulation of gp100-specific, IFN-γ-secreting, CD8+ T cells in the tumor through reduced apoptosis and enhanced proliferation of antigen-specific CD8+ T cells. IFN-α treatment also greatly increased the long-term maintenance of pmel-1 CD8+ T-cells with an effector memory phenotype. IFN-α-enhanced persistence of pmel-1 T cells required expression of IFN-α receptor on the T cells and IL-15 in the host. These results demonstrate the efficacy of Type I IFN as an adjuvant for peptide vaccination, give insight into its mechanism of action, and provide a rationale for clinical trials in which vaccination is combined with standard-of-care IFN-α therapy for melanoma.
Keywords: T cells, Vaccination, Tumor immunity, Interleukin-15, Cytokines, Type I Interferon
Introduction
While vaccination against tumor-expressed antigens is a rational approach to cancer therapy, the efficacy of cancer vaccines in human trials against melanoma and other cancers has been disappointing (1). A major factor hampering the development of anti-melanoma vaccination is the paucity of strong, innate immune-activating vaccine adjuvants available for use in human clinical trials. One critical difference between an anti-viral and an anti-tumor immune response is the presence in the former of molecules that alert the immune system to pathogen invasion, such as ligands for the family of Toll-like receptors (TLRs). The activation of innate immunity also stimulates a robust adaptive immune response (2). Since tumors, unlike infectious organisms, do not appear to strongly induce innate immunity, anti-tumor vaccines require adjuvants which activate the innate immune system.
One such candidate adjuvant is Interferon-alpha (IFN-α). IFN-α is commonly expressed by many cell types in response to TLR triggering (3, 4) and is produced in great quantities by plasmacytoid dendritic cells (pDCs) in response to viral and other infections (5). IFN-α connects the innate and acquired immune systems because it promotes the activation, expansion, survival, and memory differentiation of T lymphocytes through a variety of mechanisms including enhanced antigen presentation, costimulation (6, 7) and cross-priming (8, 9) by antigen expressing cells. Subsequently, IFN-α has direct effects on proliferation and survival of T lymphocytes (10-13) and the formation and persistence of memory T cells (11, 14). An important potential mechanism by which IFN- α may promote memory formation is by upregulating IL-15 and IL-15Rα expression by myeloid cells (15, 16). IL-15 is a homeostatic cytokine whose primary role is to support the survival and maintenance of memory CD8+ T cells. While it shares both the β and γ subunits with the IL-2 receptor, the IL-15 receptor has a unique α subunit which allows trans-presentation of the IL-15/IL-15Rα complex to T cells with far greater efficiency than IL-15 alone(17, 18). IFN-α has been shown to upregulate both IL-15 and IL-15Rα in mDCs, which may then act to stimulate CD8+ T cells via trans-presentation (16).
There is evidence that Type I IFNs can act as adjuvants for a variety of experimental vaccines. Type I IFN, when used in conjunction with an influenza vaccine, boosted Th1-type humoral immune responses, improved protection against viral challenge (19), and enhanced CTL induction after immunization with an influenza-derived peptide (20). In tumor models, low endogenous levels of IFN-α promoted anti-tumor activity of CD8+ T cells (21), while exogenous IFN-α enhanced the efficacy of GM-CSF-secreting whole-cell melanoma vaccine (22), and IFN-α-secreting colorectal tumor cells promoted expansion and survival of tumor-specific CTL (23). While IFN-α is believed to have a direct cytostatic/cytocidal effect on human melanoma(24-26), there is strong evidence that it also acts through an immune-mediated mechanism. A recent large study showed that development of autoimmune phenomena in melanoma patients after IFN-α treatment strongly correlated with improved survival (27). However, the utility and mechanism of IFN-α as a melanoma vaccine adjuvant remains unclear. One clinical trial of gp100 peptide vaccination in which low dose IFN-α was co-administered with the vaccine lacked a vaccine-alone arm (28), precluding a conclusion regarding the impact, if any, of IFN-α on T cell induction. A second small trial of gp100 peptide vaccination with high-dose IFN-α (28, 29) showed that the vaccine plus IFN-α arm was no worse than vaccine alone, although there was a trend toward stronger T cell responses in patients receiving IFN-α.
Currently, the ability of type I IFNs to enhance tumor-specific CD8+ T cell responses to peptide vaccination is unknown, as is the potential mechanism or mechanisms by which this might occur. In the present study, we tested the vaccine adjuvant effect of IFN-α on the CD8+ T cell response to anti-tumor peptide vaccination targeting established B16 melanoma, and its mechanism. Our results demonstrate that IFN-α can serve as an effective adjuvant for anti-melanoma peptide vaccination, and may be a promising approach to boosting the efficacy of current melanoma vaccination strategies.
Materials and Methods
Mice and tumor cells
C57BL/6 mice were obtained from the National Cancer Institute. Pmel-1 TCR transgenic mice(30) on a pure C57BL/6 background (The Jackson Laboratory, Bar Harbor, ME) were bred and housed at the M.D. Anderson Cancer Center animal facilities and were used at 6–12 wk of age. Pmel-1 mice were crossed with IFNAR-/- mice(31) (kindly provided by Dr. Paul W. Dempsey, Department Of Microbiology and Molecular Genetics, UCLA and Dr. Tadatsugu Taniguchi, Department Of Immunology, Tokyo University, Japan) to generate pmel-1/IFNAR-/-mice. Direct flow cytometric analysis for IFNAR on PBMC from the backcrossed progeny was performed to confirm loss of both IFNAR alleles. IL-15Rα-/- mice(32) were generously provided by A. Ma (University of California at San Francisco) and backcrossed to C57BL/6 mice 15 generations. IL-15-/- mice(33) were obtained from Taconic (Hudson, NY). All animal experiments were reviewed and approved by the University of Texas MD Anderson Institutional Animal Care and Use Committee (IACUC). B16 is an H-2b+gp100+ spontaneous murine melanoma obtained from the National Cancer Institute tumor repository and maintained in culture medium containing RPMI 1640 with 10% heat-inactivated FBS, 0.03% L-glutamine, 100 μg/ml streptomycin, and 100 μg/ml penicillin (Invitrogen, Carlsbad, CA).
Peptides
All synthetic peptides were synthesized using regular Fmoc chemistry. The synthetic, H-2Db-restricted peptides, mouse mgp10025–33 (EGSRNQDWL), the high-affinity altered peptide ligand human hgp10025–33 (KVPRNQDWL) (34) and the H-2Kb-restricted chicken ovalbumin (OVA257-264) peptide (SIINFEKL) were synthesized by Multiple Peptide Systems (San Diego, CA) to a purity >95%.
Tumor treatment
C57BL/6 mice or knockout mice (n = 5/group unless indicated otherwise) were injected s.c. with 105 B16 melanoma cells and infused i.v. with 1 × 107 fresh TCR-transgenic pmel-1 or pmel-1/IFNAR-/- splenocytes (∼2 × 106 CD8+Vβ13+Thy1.1+ T cells). Mice were vaccinated with two separate s.c. injections to the flank with 100 μl of water/incomplete Freund's adjuvant (IFA) emulsion, each containing 100 μg of hgp10025–33 peptide or OVA peptide257-264. Hydrodynamic gene transfer (HGT) consisted of a single i.v. injection of indicated amount of endotoxin-free pORF plasmid encoding murine IFN-α (abbreviated to pIFN-α in the text for clarity) or pORF control plasmid DNA (InvivoGen, San Diego, CA) in 2 ml of saline on the day of vaccination as previously described (35). This plasmid contains the ampicillin resistance gene for antibiotic selection. Alternatively, we injected 100,000 IU recombinant Universal IFN-α protein (PBL Labs) i.p., once on the day of vaccination and twice a day on the 2 following days. Tumors were measured with calipers, and the products of perpendicular diameters were recorded. Animal experiments were approved by the institutional IACUC and mice were sacrificed when tumors reached 200 mm2 or became ulcerated or mice became moribund.
Flow cytometry, intracellular IFN-γ and apoptosis assays
To obtain appropriate lymphocyte samples, mice were either tail-bled on indicated days after vaccination or sacrificed for organ isolation. Erythrocytes were removed by hypotonic lysis or Ficoll gradient separation, and cells were stained with the indicated dilutions of mAbs against CD8α-APC (1:200, clone 53-6.7), Vβ13-FITC (1:200, clone MR12-3) and Thy1.1-PE (1:800, clone OX-7), all from BD Biosciences. Propidium iodide was used to exclude nonviable cells from analysis. Intracellular IFN-γ assay was performed using the Cytofix/Cytoperm kit (BD Biosciences, San Jose, CA) according to the manufacturer's recommendations after 4 h of stimulation with 1 μM mgp10025-33 peptide and using a 1:800 dilution of anti-IFN-γ-PE mAb (clone XMG1.2). Annexin staining was performed on unfixed cells with an annexin V-FITC staining kit according to the manufacturer's instructions (BD Biosciences). Goat anti-IL-15Rα-biotin and goat anti-IL-15-biotin (R&D Systems) was detected by streptavidin-APC (Jackson ImmunoResearch) after preincubation with Fc block (BD). A biotinylayed goat Ig control (Jackson ImmunoResearch) was used to determine background staining. The following mAbs were purchased from BD Biosciences : CD44, CD62L, CD8α, and CD127. Samples were analyzed using a FACScalibur or LSRII flow cytometer (BD Biosciences) and CELLquest (BD) or FloJo (Tree Star, Ashland, OR) software.
IFN-α ELISA and multiplex cytokine analysis (Luminex)
IFN-α ELISA was performed on serum using the Mu-IFN-α ELISA kit (PBL Biomedical Laboratories, Piscataway, NJ) according to manufacturer's recommendations. For Luminex (Luminex, Austin, TX) assays, fresh spleens were harvested and splenocytes obtained by mechanical disruption, mesh filtration, and lysis of red blood cells with ACK lysis buffer (BD bioscience). Splenocytes were cultured for 24 hours in the presence of 30 IU/ml of recombinant human IL-2 and 1 μM mgp10025-33 peptide. Supernatants were harvested, clarified by centrifugation and stored at -70 degrees until the time of assay. Luminex assay was performed according to the manufacturer's instructions.
CFSE labeling
Prior to adoptive transfer of Thy1.1+ donor pmel-1 splenocytes to Thy1.2+ C57BL/6 mice, red blood cells were lysed with ACK lysis buffer, and cells were labeled with 5 μM CFSE at 37°C for 10 minutes. Cells were then washed, counted, and 2 × 107 donor splenocytes transferred intravenously as described above.
Generation of Bone Marrow-Derived DCs and pmel-1 T cell lines
For generation of bone marrow-derived DCs, bone marrow cells from normal C57BL/6 (WT), IL-15Rα-/-, and IL-15-/- mice were isolated by flushing femurs with RPMI containing 2% FBS followed by lysis of RBCs. The cells were filtered through a 70-μm filter before resuspension in RPMI containing 2.5 mM HEPES, 5.5 × 10−5 M 2-ME, 100 U/ml penicillin, 100 μg/ml streptomycin, 5 mM glutamine, and 10% FBS (complete medium, CM). Cells were seeded at 1 × 106/ml in complete medium supplemented with 10 ng/ml GM-CSF and cultured at 37°C with 5% CO2 for 6 days total with a 1:2 passage with fresh medium and GM-CSF after 3 days. Loosely adherent cells (GM-DC) were then collected on day 6, analyzed for expression of CD11c, CD11b, CD86, MHC class II by flow cytometry and used to set up co-cultures with CD8+ T cell lines. DC cultures were at least 90% CD11c+CD11b+,CD86int, MHC classIIint. Pmel-1 T cell lines were generated from pmel-1 splenocytes cultured with GM-DCs prepulsed with the cognate peptide (splenocyte/APC ratio 10/1; peptide concentration 10 μg/ml) for at least 8-10 days in presence of 20 IU/ml of recombinant murine IL-2 (R&D). T cell lines were composed of at least 95% pure CD8+, tetramer+, CD44hi cells.
T cell/DC Co-cultures
Pmel-1 T cell lines were first resuspended in RPMI at 1×107 cells/ml and stained with CSFE (0.3μg/ml) for 10 minutes at 37 C. Cold RPMI containing 20% FCS was added to stop the reaction and cells were washed, resuspended in CM and cultured with or without soluble murine recombinant IL-15 (R&D) or with GM-DCs at a 1:1 ratio in CM (final CD8+ T cells concentration in the assay was 5×105/ml). DCs were either untreated or treated for 24 hours with recombinant Universal IFN-α at 1000 unit/ml (PBL Biomedical Lab) and then washed before co-culture with T cells. Biological activity of IFN-α was verified by flow cytometric analysis of upregulation of MHC I and CD86 molecules (data not shown). Aliquots were taken at different time points from each culture condition and cells were stained for CD8 and Thy1.1 expression to detect pmel-1 T cells in the samples. CSFE dilution for CD8+ Thy1.1+ T cells and percentages of CD8+ T cells in the co-cultures were analyzed by flow cytometry. Total CD8 T cells were counted by flow cytometry by acquiring events for a fixed time period of 100 seconds.
Statistics
Statistical analyses to compare T cell numbers and tumor sizes between treatment and control groups were determined by student's T test, with p values <= 0.05 considered significant. Where appropriate, Welch's correction for unequal variances was applied. All data shown is representative of at least two independent experiments with similar results. All statistical analyses were performed using GraphPad Prism version 5.01 for Windows, GraphPad Software, San Diego, CA.
Results
IFN-α enhances vaccine-induced CD8+ T cell responses and suppression of established B16 melanoma
To assess the effect of Type I IFN administration on the CD8+ T cell response to peptide vaccination we utilized hydrodynamic gene transfer of murine IFN-α expression vector (pIFN-α) or empty vector control (pORF). A single HGT with pIFN-α resulted in serum IFN-α levels which peaked at day 2 post-injection, and then persisted at slowly declining levels for at least 37 days (Fig 1a). These peak levels are comparable to those obtained during an acute viral infection (36, 37). Importantly, HGT with pORF control plasmid did not induce detectable levels of serum IFN-α. Mice were immunized with ovalbumin (OVA) peptide in IFA followed by HGT with pIFN-α or pORF control plasmid. Vaccination in combination with control plasmid did not lead to measurable induction of OVA tetramer-positive CD8+ T cells in PBL (Fig 1b). When vaccination was followed by HGT with IFN-α-expressing plasmid, a rise in OVA-specific CD8+ T cells was observed which peaked around day 20. This demonstrates that IFN-α enhances the response of the endogenous CD8+ T cell repertoire to peptide vaccination.
Figure 1. IFN-α enhances vaccine-induced CD8+ T cell response and tumor suppression.
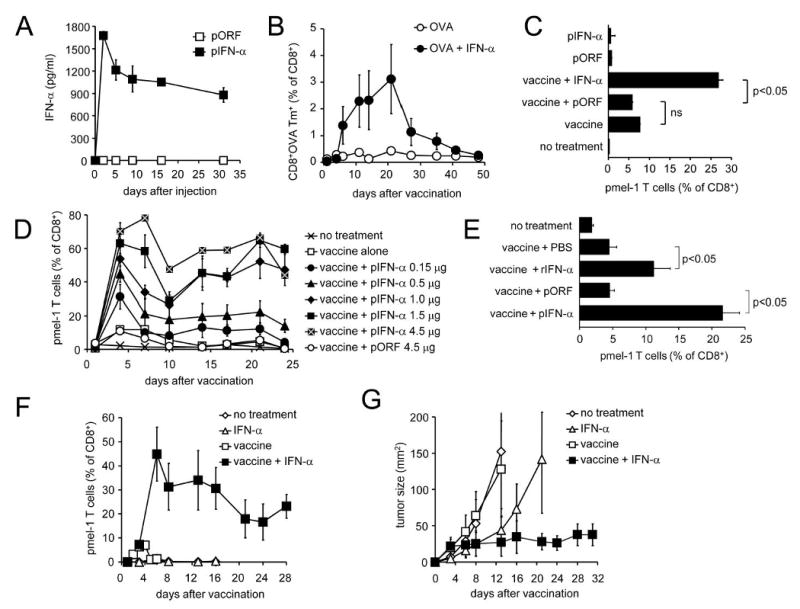
(a) C57BL/6 mice received one i.v. injection of 2 μg pIFN-α or control pORF plasmid and serum samples were tested for IFN-α by sandwich ELISA on indicated days. Results are the average of three mice +/- SEM. Differences are significant (p <0.01) on days 2-31. (b) C57BL/6 mice were vaccinated with OVA257-264 peptide in IFA followed by HGT with indicated doses of pIFN-α or pORF plasmid. CD8+ OVA-specific (tetramer-positive) T cells were detected by flow cytometry of peripheral blood on the indicated days. Difference is significant (p <0.05) on day 21. (c) Thy1.2+ C57BL/6 mice received either no treatment or 2×107 Thy1.1+ pmel-1 splenocytes. Where indicated, mice received gp100/IFA vaccination and/or HGT with 1 μg pIFN-α or pORF plasmid. On day 6, pmel-1 T cells were detected by flow cytometric analysis of peripheral blood for Thy1.1+Vβ13+CD8+ cells. (d) After adoptive transfer of pmel-1 T cells, Mice received pmel-1 T cells as in (c) followed by either no further treatment, or vaccination with gp100 peptide and HGT with the indicated plasmid. Pmel-1 T cells in peripheral blood were detected at the indicated times, as described above. Note that 4.5 μg pIFN-α was a lethal dose (4/5 mice dead on day 7). (e) Mice received pmel-1 splenocytes, vaccine and HGT with 1 μg pIFN-α or pORF plasmid as in (c); an additional group of mice received pmel-1 splenocytes and vaccine followed by 100,000 IU purified recombinant Universal IFN-α protein (rIFN-α) i.p. on the day of vaccination (day 0) and twice daily on days 1, 2 and 3. Pmel-1 T cells in PBMC were quantified by flow cytometry on day 4 post vaccination. (f, g) Tumor-bearing Thy 1.2+ C57BL/6 mice received either no treatment or adoptive transfer of Thy1.1+ pmel-1 T cells with or without gp100/IFA vaccine as described above. Where indicated, mice were also injected with 2 μg pIFN-α or pORF plasmid on the day of vaccination. Pmel-1 T cells in blood (f) were detected by FACS; the differences between pIFN-α and pIFN-α + vaccine groups were significant (p<0.05) on days 6, 13, and 16. Tumor growth (g) is plotted as the product of perpendicular diameters.
In order to follow the CD8+ T cell response to peptide vaccination against a melanoma-expressed self antigen, and examine the ability of type I IFN to enhance vaccine-induced tumor regression, we utilized the pmel-1 model of anti-gp100 vaccination (30). In this model, which has been extensively used by our laboratory and others to examine vaccine-specific CD8+ T cell responses in vivo, immunocompetent wild-type C57BL/6 mice receive anti-gp100 CD8+ T cells from pmel-1 mice which are transgenic for a Vα1/Vβ13 TCR which recognizes the mgp10025-33 peptide in the context of H-2Db. A Thy1.1/Thy1.2 mismatch between donor and host allows tracking of the expansion of Thy1.1+ gp100-specific donor CD8+ T cells in the Thy1.2+ recipient mouse over time. After adoptive transfer of gp100-specific pmel-1 T cells, vaccination with gp10025-33 peptide in IFA resulted in a measurable expansion of pmel-1 (Thy1.1+, Vβ13+) CD8+ (Fig 1c), which was significantly enhanced by HGT with IFN-α-expressing plasmid, but not empty vector control (pORF). IFN-α did not obviate the requirement for vaccination for the proliferation of pmel-1 T cells, but strongly increased the number of vaccine-induced pmel-1 T cells in a dose-dependent manner, up to 80% of circulating CD8+ T cells (Fig 1d). The maximum tolerated dose of pIFN-α was 3 μg (data not shown), while 4.5 μg killed 4/5 mice by day 7. To avoid possible confounding factors introduced by our HGT method and to more closely reflect clinical situations in which IFN-α is administered as a recombinant protein, we injected mice receiving pmel-1 T cells and gp100 peptide vaccine with 100,000 IU purified, recombinant Universal IFN-α (rIFN-α) twice a day for 3 days and analyzed pmel-1 T cell levels on day 4 (Fig 1e). As expected, results with rIFN-α were similar to those obtained with the IFN-α HGT method.
We and others have used the pmel-1 model to examine the ability of immunomodulatory agents to enhance vaccine-mediated anti-tumor activity, and have shown that in this model suppression of melanoma growth is strictly dependent upon CD8+ cytotoxic T cells(30, 34). Mice bearing established 7-day B16 melanoma responded to vaccination with hgp100 peptide in IFA with a small and transient rise in antigen-specific CD8+ T cells (Fig 1f) which failed to suppress tumor growth (Fig 1g). Addition of IFN-α to peptide vaccination produced a marked increase in vaccine-induced CD8+ T cells which strongly inhibited the growth of B16 melanoma (Fig 1f, g). Treatment with IFN-α alone in the absence of vaccination failed to induce a gp100-specific CD8+ T cell response (Fig 1f), although it did transiently inhibit tumor growth for approximately 2 weeks, possibly due to the known direct tumoristatic effect of IFN-α on melanoma cells or through activation of other immune cells (Fig 1g). Nevertheless, the full anti-tumor effect of IFN-α was dependent on a concurrent strong, vaccine-induced CD8+ T cell response. IFN-β, another type I IFN which signals through the same heterodimeric IFNAR, had similar effects on both vaccine-induced CD8+ T cell expansion and inhibition of tumor growth (data not shown), suggesting that the ability to serve as a vaccine adjuvant may be a common property of type I IFNs.
IFN-α enhances the accumulation of activated antigen-specific CD8+ T lymphocytes in tumor
To determine whether the highly elevated levels of vaccine-induced T cells seen in the circulation of IFN-α-treated mice were representative of pmel-1 accumulation in tumor and lymphoid tissues, we examined the absolute number of gp-100-specific pmel-1 T cells and other leukocytes in the blood, spleen, and tumor after adoptive transfer of pmel-1 T cells and vaccination. On day 7 after vaccination and transfer, absolute numbers of pmel-1 T cells were increased 6-15 fold in all assessed tissues in mice receiving pIFN-α compared to pORF control (2a-c). The increase in absolute number of total infiltrating cells after vaccination and IFN-α administration was limited to antigen-specific pmel-1 T cells, with no significant change seen in the numbers of NK cells, CD4+ T cells, and bystander (non-pmel-1) CD8+ T cells. While an increase in the total number of CD8+ T cells was seen in tumor from IFN-α-treated mice (2c), this was entirely accounted for by the increase in pmel-1 CD8+ T cells into tumor, rather than enhanced accumulation of nonspecific/bystander CD8+ T cells. IFN-α-induced accumulation of antigen-specific pmel-1 T cells tended to be stronger in tumor (15-fold increase) than in spleen (11-fold) and blood (6-fold).
We examined the activation status of FACS-sorted splenic pmel-1 T cells on day 7 after vaccination by assaying their mgp10025-33 peptide-induced production of inflammatory cytokines and chemokines associated with T cell activation using Luminex multiplex cytokine analysis (Fig 2d). Pmel-1 T cells isolated from IFN-α-treated mice secreted greater quantities of the effector cytokines TNF-α (2-fold) and IFN-γ (5.4-fold) as well as the T cell chemotactic factor MIP-1α (6-fold). Staining of ex-vivo stimulated pmel-1 T cells for intracellular IFN-γ likewise demonstrated that IFN-α treatment induced a significant increase in the number of gp100-specific CD8+ T cells producing IFN-γ in blood, spleen and tumor (Fig 3 a-d). In the case of tumor-derived pmel-1 T cells, a substantial proportion was highly activated at baseline, possibly through in situ recognition of tumor-derived gp100, and secreted IFN-γ even without peptide stimulation (6%); this proportion was increased (to 12%) in IFN-α-treated mice (data not shown), resulting in higher absolute number of IFN-γ producing pmel-1 T cells per gram of tumor (Fig 3d). Similarly, the percentage of pmel-1 T cells that produced IFN-γ after antigenic restimulation ex vivo was much higher in tumor than in spleen both without (21 vs. 4%) and with (47 vs. 9.6%) IFN-α treatment (data not shown and Fig 4d), suggesting that increased accumulation of highly-activated anti-tumor CD8+ T cells occurs through selectively enhanced infiltration of activated gp100-specific CD8+ T cells and/or preferential activation of gp100-specific CD8+ T cells within the tumor.
Figure 2. IFN-α promotes the specific accumulation of activated pmel-1 T cells.
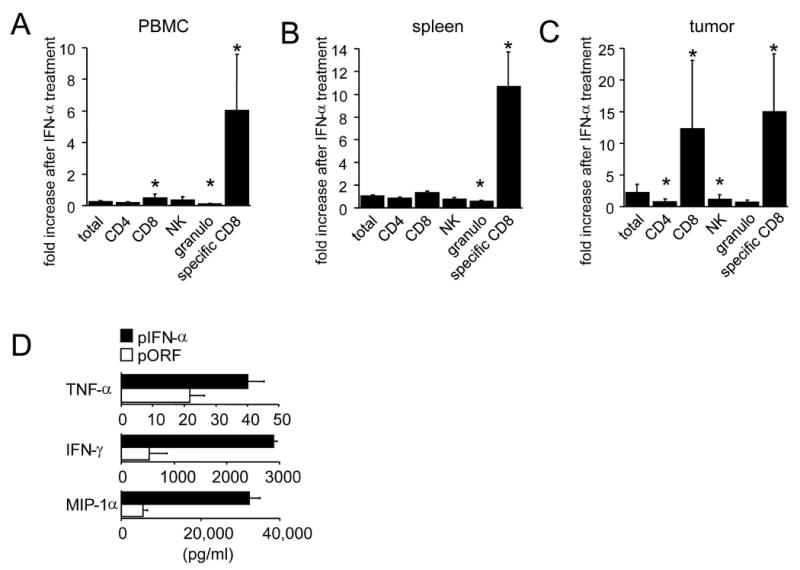
Thy1.2+ C57BL/6 mice bearing subcutaneous 7-day B16 melanoma received 2×107 Thy1.1+ pmel-1 splenocytes. Where indicated, mice received gp100/IFA vaccination and/or HGT with 2 μg pIFN-α or pORF plasmid. Seven days after vaccination organs and tumor were isolated and leukocytes were analyzed for indicated markers by flow cytometry. The IFN-α-induced increase in absolute numbers per organ for the indicated cell types is plotted for PBMC (a), spleen (b), and tumor (c). (d) Thy1.1+ pmel-1 CD8+ T cells were sorted from spleen homogenates obtained on day 7 after adoptive transfer, vaccination and treatment with pIFN-α or pORF plasmid as described above. Sorted pmel-1 T cells were cultured overnight in the presence of 30 IU/ml of IL-2 and 1 μM mgp10025-33 peptide and Luminex multiplex cytokine analysis was performed. Asterisk indicates significant difference (p<0.05).
Figure 3. IFN-α increases IFN-γ-production by pmel-1 T cells.
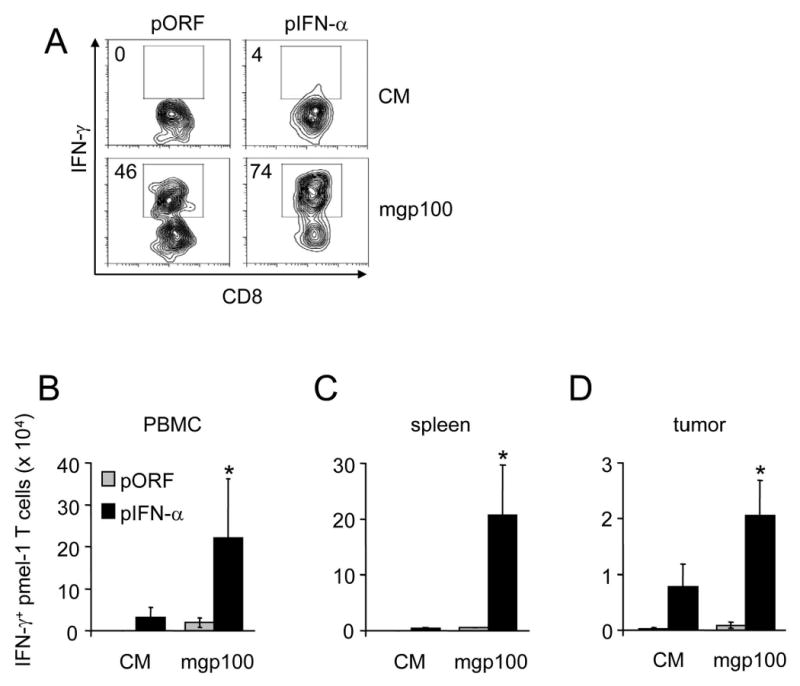
Thy1.2+ C57BL/6 mice bearing subcutaneous 7-day B16 melanoma received 2×107 Thy1.1+ pmel-1 splenocytes. Where indicated, mice received gp100/IFA vaccination and/or HGT with 2 μg pIFN-α or pORF plasmid. Seven days after vaccination, blood, spleens and tumors were isolated and total lymphocytes were isolated and incubated for 4 hours in the absence (CM) or presence of 1 μM mgp10025-33 peptide and in the presence of Brefeldin A prior to flow cytometry for CD8, Thy1.1 and intracellular IFN-γ. Representative scatter plots of IFN-γ production by CD8+Thy1.1+ pmel-1 T cells from PBMC are shown in (a); aggregated data representative of 5 mice per group are presented in (b-d). Asterisk indicates significant difference (p<0.05).
Figure 4. IFN-α prevents the death of pmel-1 T cells and enhances their proliferation.
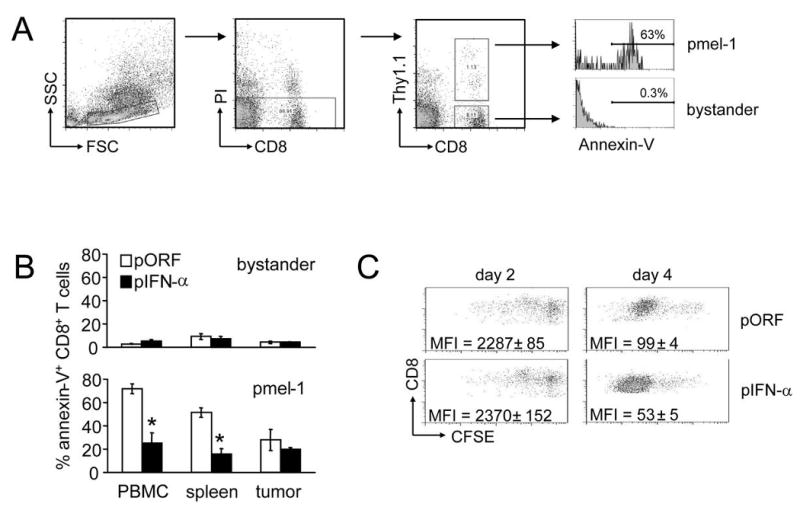
Thy1.2+ C57BL/6 mice bearing subcutaneous 7-day B16 melanoma received 2×107 Thy1.1+ pmel-1 splenocytes. Where indicated, mice received gp100/IFA vaccination and/or HGT with 1.25 μg pIFN-α or pORF plasmid. Organs and tumor were isolated on day 7 after vaccination and analyzed by flow cytometry for the presence of apoptotic (Annexin V+) pmel-1 (CD8+, Thy1.1+) T cells. A representative scatter plot is demonstrated in (a). The bar graphs (b) summarize the percentage of annexin V+ cells in the pmel-1 and bystander CD8+ T cell populations. Asterisk indicates significant (p<0.05) difference. (c) Thy1.1+ pmel-1 T cells were labeled with CFSE prior to intravenous transfer into C57BL/6 mice and gp100 vaccination and HGT with pIFN-α or control pORF plasmid. On day 2 and day 4 after vaccination splenocytes were harvested and CFSE dilution used to determine the rates of proliferation of pmel-1 T cells in vivo. Plots shown are gated on Thy1.1+ cells, and representative of results from 3 mice. Difference between pORF and pIFN-α groups on day 4 was statistically significant (p=0.002).
IFN-α protects vaccine-induced CD8+ T cells from activation-induced apoptosis and enhances their proliferation
The in vivo expansion of CD8+ T cells lacking the IFNAR during acute viral infection is impaired due to their greater rate of apoptosis (11, 12, 38). To determine whether IFN-α-mediated increase of tumor specific CD8+ is due to increased proliferation, increased T cell survival, or both mechanisms, we used annexin-V staining to examine the rates of apoptosis in vivo in both the endogenous and vaccine-induced CD8+ T cell compartments after vaccination and HGT with pIFN-α or control plasmid (Fig 4a-b). After using forward/side scatter profiles and propidium iodide exclusion to gate on the population of live lymphocytes, we stained for CD8+ and Thy1.1 to identify the bystander and antigen-specific pmel-1 T cell compartments (Fig. 4A). While low levels of apoptosis were observed in the bystander CD8+ T cell population (containing mostly unstimulated cells), a significantly higher fraction of apoptotic antigen-specific pmel-1 T cells was observed in PBL, spleen, and tumor from mice receiving pORF control plasmid (up to 75% annexin+ cells, vs. <10% in the bystander population). However, pmel-1 T cells from IFN-α-treated mice showed a dramatically lower rate of apoptosis in the PBL (28% vs. 75%) and spleen (15% vs. 52%). Antigen-specific pmel-1 T cells harvested from tumor demonstrated lower baseline rates of apoptosis than cells harvested from PBL or spleen and only a small, non-significant, decrease in apoptosis upon IFN-α treatment. This may be due to the availability of antigen or T cell survival factors present in the tumor milieu, or the result of preferential trafficking into tumor of T cells which have received survival signals.
Adoptive transfer studies with IFNAR-deficient CD8+ T cells undergoing expansion in response to acute viral infection have suggested that rates of antigen-induced proliferation in IFNAR-positive and -deficient T cells are similar, and that the impaired expansion of IFNAR-deficient CD8+ T cells is explained by increased susceptibility to apoptosis(11, 12). We used adoptive transfer of CFSE-labeled pmel-1 T cells to determine whether exogenously supplied IFN-α affected the vaccine-induced proliferation of CD8+ T cells (Fig 4c). On day 2 after gp100 vaccination, the majority of pmel-1 T cells had not yet divided in mice that had received either pIFN-α or control pORF plasmid (Fig 4c, left panels). By day 4 after vaccination (Fig 4c, right panels), pmel-1 T cells from both pIFN-α- and pORF-treated mice had undergone multiple rounds of division; however, pmel-1 T cells from IFN-α-treated mice displayed a slightly greater rate of proliferation (see representative scatter plot, as well as mean fluorescence intensity in Fig 4c, right panels). Thus, exogenously supplied IFN-α increases vaccine-induced CD8+ T cell numbers by both enhancing their rate of proliferation and protecting them from activation-induced apoptosis.
IFN-α promotes the generation and long-term persistence of CD8+ T cells with an effector memory phenotype
Anti-cancer vaccination has two related objectives: raising an acute response to destroy existing tumor bulk and establishing specific memory to protect against outgrowth of any remaining or newly arising tumor cells. We therefore studied the impact of IFN-α on the longevity of tumor-specific T cell responses after vaccination. We found that IFN-α maintained the downregulation of IL-7Rα expression on pmel-1 T cells on day 4 after vaccination (Fig 5A), consistent with activation of a greater proportion of antigen-specific CD8+ T cells in IFN-α-treated mice. To determine whether the antigen-specific CD8+ T cells induced in the presence of exogenous IFN-α can persist after initial exposure to antigen, we examined peripheral (lung), and lymphoid (spleen and lymph node) tissue at 90 days after vaccination. We found significantly higher absolute numbers of vaccine-induced anti-gp100 pmel-1 T cells in both non-lymphoid (lung) and lymphoid (spleen) tissues of IFN-α-treated mice (Fig 5B). Flow cytometric analysis of the CD44hi (antigen-experienced) population revealed that while pmel-1 T cells from control (pORF) mice displayed an IL-7Rαhi, and predominantly CD62Lhi phenotype consistent with the CD8+ T cells of a central memory phenotype, pmel-1 T cells from IFN-α treated mice displayed an IL-7Rαlo, CD62Llo phenotype consistent with persistently activated effector cells (Fig 5C, D). Since these mice were never re-vaccinated after induction of the primary response, the difference in phenotype (central memory vs. persistent activation) may be the result of either a developmental program initiated by high-dose IFN-α at the time of vaccination, or prolonged stimulation of pmel-1 T cells by persistent expression of IFN-α (see Fig 1a). In summary, IFN-α promotes the persistence and differentiation of anti-tumor memory CD8+ T cells which preferentially migrate to non-lymphoid tissues and resemble effector memory T cells.
Figure 5. IFN-α promotes IL-15Rα upregulation and long-term persistence of pmel-1 T cells.
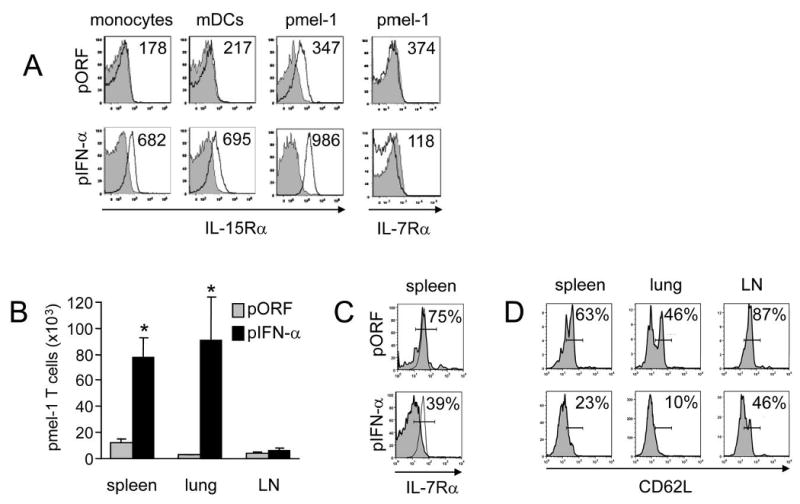
Thy1.2+ C57BL/6 mice received Thy1.1+ pmel-1 splenocytes, hgp10025-33 peptide vaccination, and HGT with 1 μg pIFN-α or pORF control. (a) Spleens were isolated on day 4 and stained to identify the monocyte, mDC, total CD8+ T cell and pmel-1 (CD8+Thy1.1+) populations; then stained with anti-IL-15Rα (solid lines) or isotype controls (shaded) and analyzed by flow cytometry. Alternatively, pmel-1 CD8+ T cells (solid lines) or bystander CD8+ T cells (shaded) were stained with anti-IL-7Rα. Numbers indicate average mean fluorescence intensity. (b-d) On day 90 post-vaccination, spleen, lungs and cervical, axial and mesenteric lymph nodes were isolated (b); asterisk indicates statistically significant difference (p<0.05). The total number of pmel-1 CD8+Thy1.1+CD44hi cells was determined by manual cell count and flow cytometry. (c) Staining and flow cytometry to determine the percentage of IL-7Rαhi cells was performed on CD44hiThy1.1+ pmel-1 T cells (shaded) using as a positive control CD44loThy1.1- naive CD8 T cells (solid line), both from vaccinated mice treated with pORF or pIFN-α. (d) Within the CD44hi population, percentage of CD62Lhi expression was determined by flow cytometry for pmel-1 T cells from vaccinated mice treated with pORF or pIFN-α; gating based on IgG control staining.
Increased expansion and persistence of antigen-specific CD8+ T cells by IFN-α requires IFNAR expression by the T cells and enhanced IL-15 transpresentation by host cells
To determine whether IFN-α acts directly on antigen-specific CD8+ T cells to support their proliferation and survival, we crossed pmel-1 mice with IFNAR knockout mice and compared the vaccine-induced expansion of wild type (IFNAR +/+) and IFNAR-/- pmel-1 T cells (Fig 6a). The expansion of IFNAR sufficient pmel-1 T cells after vaccination was greatly enhanced by the administration of IFN-α. In contrast, expansion of IFNAR-deficient pmel-1 T cells was comparable in pIFN-α and pORF-treated animals. This demonstrates that the adjuvant activity of IFN-α requires direct stimulation of antigen-specific CD8+ T cells through the IFNAR.
Figure 6. Both IFNAR expression on pmel-1 T cells and IL-15/IL-15Rα expression by antigen presenting cells are necessary for optimum expansion and persistence of antigen-specific CD8+ T cells.
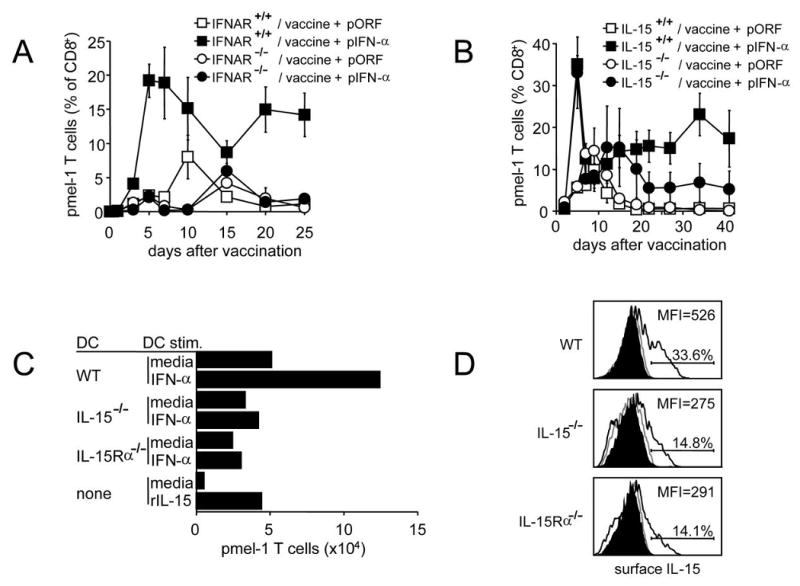
(a) C57BL/6 mice received 2×107 splenocytes from pmel-1/IFNAR+/+ or pmel-1/IFNAR-/- knockout mice, followed by hgp10025-33 peptide vaccination and HGT with 1 ug pIFN-α or pORF control. Hgp10025-33 tetramer-positive CD8+ pmel-1 T cells were detected by flow cytometry of peripheral blood on the indicated days. The difference between IFNAR+/+ + pIFN-α and IFNAR-/- + pIFN-α groups is significant (<0.05) on days 5, 7, 10, 20, and 25. (b) Thy1.2+ wild type or IL-15-/- KO C57BL/6 mice received 2×107 Thy1.1+ pmel-1 splenocytes, hgp10025-33 peptide vaccination, and HGT with 1 μg pIFN-α or pORF control. Thy1.1+ CD8+ pmel-1 T cells were detected by flow cytometry of peripheral blood on the indicated days. The difference between IL-15+/+ + pIFN-α and IL-15-/- + pIFN-α groups is significant (<0.05) on days 19-41. (c) Equivalent numbers of pmel-1 CD8+ T cells from pmel-1 CD8+ T cell lines were cultured in either media, media with soluble recombinant murine IL-15 (rIL-15) or with DCs derived from C57BL/6 (WT), IL-15-/-, or IL-15Rα-/- mice. DCs were either untreated or treated for 24 hours with murine IFN-α and washed prior to coculture. On day 19 of coculture, cells were stained for CD8 and Thy1.1 expression to detect pmel-1 CD8+ T cells by flow cytometry. Shown are absolute numbers of live pmel-1 T cells. (d) mDCs derived from C57BL/6 (WT), IL-15-/-, or IL-15Rα-/- mice were left untreated or treated for 24 hours with murine IFN-α. Surface expression of IL-15 was detected by flow cytometry. Shaded histograms represent isotype control, grey lines cells cultured in the absence of IFN-α, black lines cells cultured in the presence of IFN-α.
Since IL-15 is known to be important for the homeostatic maintenance of antigen-experienced memory CD8+ T cells and IFN-α induced in vivo upregulation of IL-15Ra, required for transpresentation of IL-15 to responding T cells (Fig. 5A), we compared the vaccination-induced expansion of adoptively transferred pmel-1 T cells in wild type and IL-15-/- knockout mice (Fig 6b). In the absence of IFN-α, pmel-1 T cells underwent a similar, modest, expansion in response to vaccination in both wild type and IL-15 KO mice. In contrast to the impairment of IFN-α enhanced proliferation seen with IFNAR-/- pmel-1, the early peak and decline in vaccine-induced pmel-1 numbers was nearly identical in wild type and IL-15 knockout mice. However, while IFN-α supported the continued persistence of pmel-1 T cells in wild type mice (15-25% of all CD8+ T cells for up to 40 days), in IL-15-/- knockout mice pmel-1 levels fell to a plateau of 5-6% of all CD8+ T cells. Thus IL-15 expression by host cells is required for the long-term maintenance of CD8+ T cells induced by vaccination and IFN-α.
To better define whether the observed IFN-α-induced, IL-15-dependent maintenance of pmel-1 T cells was mediated by a direct induction by IFN-α of IL-15 in mDCs, we stimulated purified mDCs from wild type, IL-15-/-, or IL-15Rα -/- mice with IFN-α for 1 day and co-cultured them with previously activated pmel-1 effector T cells (Fig 6c). Pmel-1 T cells cultured in medium alone failed to expand or persist. When co-cultured with bone marrow-derived mDCs from wild-type, IL-15-/-, or IL-15Rα -/- mice, or with soluble IL-15 in the absence of IFN-α, a modest increase in the number of persisting pmel-1 T cells was observed which was somewhat less in the co-cultures containing IL-15-/-, or IL-15Rα -/- mDCs. A one-day pre-stimulation of mDCs with IFN-α increased their ability to support pmel-1 T cell levels in an IL-15-dependent manner. We also observed that IFN-α treatment of mDCs upregulated IL-15Rα-dependent surface expression of IL-15 (Fig 6d), providing further evidence that IFN-α promotes enhanced trans-presentation of IL-15 to T cells. IFN-α also substantially upregulated expression of IL-15Rα on monocytes and mDCs in vivo (Fig 5A), suggesting that these cells may be able to more efficiently trans-present IL-15 to CD8+ T cells. These data suggest that exogenous IFN-α enhances persistence of vaccine-induced CD8+ T cells through the direct induction of increased IL-15Rα-dependent trans-presentation of IL-15 by host mDCs.
Discussion
The occasional and sometimes dramatic clinical responses in recent cancer vaccine trials suggest that vaccination against tumor-expressed antigens can induce clinically meaningful anti-tumor immunity, however currently these responses are only sporadic. One reason for the disappointing results of vaccine trials may be failure to induce adequate numbers of tumor-specific CD8+ T cells. While clinical vaccination trials demonstrate circulating antigen-specific CD8+ T cell responses on the order of 0.05-5% of total circulating CD8+ T cells (1, 39), levels achieved during viral infection can reach up to 20% or more of circulating CD8+ T cells (40) which may better suggest the magnitude of response necessary for efficacy against established tumors. Vaccine-elicited T cells may also become functionally impaired in the tumor-bearing host due to lack of innate immune activating signals such as TLR ligands, inhibition by regulatory T cells (41), or active immunosuppression by the tumor itself. Finally, tumor-specific CD8+ T cells may expand initially but fail to persist long enough to mediate total tumor destruction if they do not receive continued survival signals (such as common gamma-chain cytokines like IL-2, IL-7, and IL-15) (42) or are programmed improperly during priming and initial expansion (43).
Since the synthetic peptides that are widely used as vaccines are poorly immunogenic by themselves, adjuvants are required to induce significant T cell responses. Type I IFNs provide a critical link between the innate and acquired immune response during viral infection, suggesting that they may also be efficacious as vaccine adjuvants. Long-term systemic treatment with IFN-α is currently approved for the management of chronic viral infections such as with Hepatitis B and C viruses and several cancers, including melanoma. For our studies, we tested the vaccine adjuvant activity of IFN-α by inducing systemic IFN-α levels with the hydrodynamic gene transfer method. This method uses a brief hydrostatic pulse to transfect a microgram-amount of injected plasmid into normal hepatocytes, leading to long-term expression of the plasmid-encoded gene of choice. This method overcomes the significant obstacle of long-term daily injections of very costly recombinant IFN-α protein that has all but made impossible effective studies of the effects of prolonged, systemic in vivo treatment with IFN-α. Since even this small amount of plasmid DNA could conceivably contain sufficient CpG signals to trigger TLR9 on dendritic cells to enhance the vaccine adjuvant effect of IFN-α, we repeated the experiment with recombinant IFN-α and observed similar effects, effectively proving that the observed adjuvant activity does not depend on TLR9 triggering.
We found that IFN-α was capable of significantly boosting the antigen-specific (pmel-1) CD8+ T cell response to gp100 peptide vaccination. In order to achieve this effect, both interferon and vaccination were required; IFN-α alone did not produce a measurable rise in the pmel-1 CD8+ T cell response. IFN-α also boosted the response of the endogenous CD8+ T cell repertoire in response to ovalbumin peptide vaccination, demonstrating that Type I IFN can act as an adjuvant for vaccination of either endogenous or adoptively transferred T cells against both non-self and self antigens. However IFN-α adjuvant activity was limited to antigen-specific CD8+ T cells; the absolute number of non-specific bystander CD8+ T cells, and the absolute numbers of other leukocytes (CD4+ T cells, NK cells) were not significantly increased in the peripheral blood, spleen, or tumor of IFN-α-treated mice. The boost in pmel-1 T cell numbers required the expression of the IFNAR on the pmel-1 T cells, demonstrating that in addition to exerting indirect effects on T cells through antigen presenting cells, exogenous IFN-α also acts directly on activated antigen-specific CD8+ T cells.
When we measured T cell apoptosis and proliferation to determine their relative contribution to the IFN-induced accumulation of pmel-1 T cells, we found that exogenous IFN-α enhanced both proliferation and survival of vaccine-induced pmel-1 T cells. In the presence of IFN-α, antigen-specific pmel-1 T cells were protected from apoptosis in peripheral blood, spleen, and tumor, in a manner similar to the IFN-α-mediated protection of CD8+ T cells from apoptosis during viral infection. However, in contrast to the effect of endogenous IFN-α on CD8+ T cells during viral infection (11, 12), which seems limited to protecting them from apoptosis, we found a slight but consistent increase in the rate of antigen-specific CD8+ T cell proliferation after vaccination in the presence of exogenous IFN-α. This may be due to the continuous stimulation of the IFNAR on CD8+ T cells with high levels of IFN-α in our system compared to the more short-lived (1-5 days) systemic increase in IFN-α during acute viral infection in these reports(36, 37). Since these effects were seen over a range of non-toxic levels of IFN-α expression, our data suggest that adjuvant interferon at doses lower than required for anti-melanoma IFN-α monotherapy may increase both tumor-specific CD8+ T cell survival and proliferation in patients undergoing peptide vaccination.
Efficacy of the anti-tumor CD8+ T cell response depends not only on the induction of specific T cells but also on their effector function. We found increased numbers of vaccine-induced pmel-1 T cells producing IFN-γ in tumor, PBMC, and spleen, as well as higher mean IFN-γ release by pmel-1 T cells from IFN-α-treated mice. The observed increased production of TNF-α, MIP-1α, and IFN-γ by purified pmel-1 T cells from vaccinated, IFN-α-treated mice is consistent with enhanced development of an activated, effector CTL phenotype (44). We and others have shown that the T cell-mediated regression of B16 melanoma is partly dependent on tumor-specific T cell-derived IFN-γ (45). MIP-1α is a member of the group of CC chemokines, and plays a role in chemotaxis of T cells from the circulation into inflamed tissue (46). TNF-α is also produced by activated CTL, and has diverse pro-inflammatory and some direct anti-tumor activities (47). The coordinated upregulation of these cytokines in vaccinated mice treated with IFN-α is consistent with generation of a pro-inflammatory environment which supports the infiltration and anti-tumor activity of vaccine-induced CTL.
Prevention of tumor recurrence may depend on the induction of a stable and long-lasting population of specific CD8+ effector T cells. The increase in antigen-specific CD8+ T cell numbers and CTL activation correlated with inhibition of tumor growth in mice bearing established subcutaneous B16 melanoma, a fast-growing and poorly immunogenic tumor. While transient growth inhibition was seen in mice treated with IFN-α alone, uncontrolled tumor growth resumed within 2 weeks after treatment and rapidly killed these mice; however, tumor growth was suppressed for over 4 weeks in mice that received both vaccine and IFN-α. This correlates with the persistent increase in anti-tumor T cells in vaccinated mice treated with IFN-α. Even in vaccinated IFN-α-treated mice, tumor growth ultimately resumed, possibly due to the number of protective T cells dropping below a critical threshold, exhaustion of persistently-stimulated pmel-1 T cells, or outgrowth of resistant tumor clones. Further experiments will determine whether long-surviving tumor-specific cells are functional or have undergone exhaustion (48), and whether they depend on chronic IFN-α stimulation, persistent antigen stimulation, or both.
The phenotypic profile of IFN-α-induced CD8+ T cells may provide clues to their function and efficacy in vivo. IL-15 and IL-7 are constitutively produced common cytokine-receptor γ chain-family cytokines which play an important role in maintenance of antigen-specific CD8+ T cells, with IL-7 supporting the proliferation and survival of naïve cells, and IL-15 acting primarily on memory cells. IL-15 signals through a heterodimeric receptor composed of the IL-2/IL-15R β chain and the common γ chain which are expressed on CD8+ memory T cells. The IL-15Rα is expressed primarily on activated monocytes and dendritic cells where it acts to “trans-present” IL-15 to IL-15Rβ/γ-expressing target cells. IL-7Rα is expressed at high levels on naïve CD8+ T cells, and downregulated in response to signaling through the TCR. The phenotype of persistent IFN-α-induced pmel-1 cells (CD44hi, CD62Llo, IL-7Rαlo) is similar to that of effector memory cells, which would also be consistent with their residence in peripheral tissues (lung). One potential application for cancer vaccine therapy is in long-term control of minimal residual disease after debulking therapy. CD8+ T cells of effector memory phenotype may be suitable for this purpose, since they would allow prolonged maintenance of highly active tumor-specific T cells to allow for immune surveillance. Alternatively, these IL-7Ralo CD8+ T cells are reminiscent of a “short-lived effector” memory CD8+ T cells that have been recently identified (49). These cells have a longer survival than pure effector cells, but a shorter life span than memory T cells. Furthermore, these “short lived” memory CD8+ T cells have increased effector activities, and are induced by high levels of inflammation, yet still respond to a secondary stimulation.
The upregulation of IL-15Rα and requirement for host-derived IL-15 in supporting persistence of IFN-α-induced pmel-1 T cells suggests that at least some of the adjuvant activity of IFN-α is mediated by IL-15Rα-mediated transpresentation of IL-15. Our in vitro data demonstrating that abolishing either IL-15 or IL-15Rα expression by mDCs completely abrogates the IFN-α-mediated enhancement of pmel-1 T cell persistence suggests that expression of IL-15 and IL-15Rα by DCs may also be important in vivo. The upregulation of IL-15Rα we observed on IFN-α-stimulated mDCs in vitro and in vivo provides a straightforward mechanism by which Type I IFN may enhance CD8+ T cell persistence by increasing the efficiency of IL-15 trans-presentation by mDCs. Interestingly, we found that IFN-α also upregulated IL15Rα expression on total CD8+ T cells (data not shown) and pmel-1 T cells. The upregulation of IL-15Rα on antigen-specific CD8+ T cells has been previously demonstrated in a study of the ability of IFN-α to promote CD8+ T cell expansion through cross-priming, in which IL-15Rα expression on CD8+ T cells peaked on day 3 after immunization in the presence of IFN-α (9). There is also evidence that IL-15Rα expression can enhance the response of CD8+ T cells to limiting amounts of IL-15 (15). At present the role, if any, of enhanced IL-15Rα expression on CD8+ T cells after IFN-α treatment remains unclear.
Collectively, our data support a model in which IFN-α, when used as an adjuvant for anti-tumor peptide vaccination, acts on mDCs and vaccine-induced CD8+ T cells to increase the height, longevity and anti-tumor effect of the antigen-specific CD8+ T cell response. During the initial expansion and contraction of the antigen-specific CD8+ T cell compartment, in addition to the previously-described ability of Type I IFNs to enhance antigen presentation and costimulation by DCs, enhanced proliferation and survival requires direct stimulation of the IFNAR on the T cells themselves. Once IFN-α-enhanced CD8+ T cell levels decline to a plateau (in our studies, typically 50-80% of the peak response by day 12-15) their continued maintenance depends on the expression of IL-15 and probably IL-15Rα by host cells. Further studies will investigate whether the IFN-α-mediated increase in IL-15Rα on DCs and other cell types seen on day 4 post-vaccination continues to longer timepoints, as suggested by the ability of IFN-α to support IL-15-dependent long-term persistence of antigen-specific CD8+ T cells. Experiments are also ongoing to determine whether persistence of antigen-specific CD8+ T cells during the plateau phase requires continued delivery of exogenous IFN-α such as achieved during HGT with IFN-α expression plasmid. Alternatively, IFN-α and IL-15/IL-15Rα signaling in the early phase of the vaccine response may initiate a developmental program in antigen-specific CD8+ T cells which favors their persistence even after levels of exogenously-supplied IFN-α have waned.
In summary, these data demonstrate the activity of IFN-α as an adjuvant for anti-tumor peptide vaccination, and provide a model system for further dissecting the mechanisms of adjuvant activity and optimizing IFN-α-enhanced vaccination in pre-clinical studies. Since IFN-α is already an approved treatment for melanoma, our data point towards a potentially promising role as vaccine adjuvant in human clinical trials. In particular, patients with surgically resected stage III melanoma that are receiving IFN-α as part of their standard-of-care treatment might benefit from concurrently receiving a proven safe and non-toxic peptide vaccination to potentially reduce their risk of disease recurrence through the induction of tumor-specific T cell responses.
Acknowledgments
The authors wish to thank Drs. Yong-Jun Liu, Gregory Lizee and Laszlo Radvanyi for helpful discussion.
Footnotes
This work was partially supported by VENI grant 916.046.014 from the Netherlands Organization for Scientific Research (NWO) to WWO, the MDACC SPORE in melanoma P50 CA093459 and by NIH grant AI070910 and the MD Anderson Trust Fellowship to KS
Conflict of Interest Disclosures: None.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 3.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 4.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 6.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 7.Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, Seya T, Taniguchi T. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A. 2003;100:10872–10877. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho HJ, Hayashi T, Datta SK, Takabayashi K, Van Uden JH, Horner A, Corr M, Raz E. IFN-alpha beta promote priming of antigen-specific CD8+ and CD4+ T lymphocytes by immunostimulatory DNA-based vaccines. J Immunol. 2002;168:4907–4913. doi: 10.4049/jimmunol.168.10.4907. [DOI] [PubMed] [Google Scholar]
- 9.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol. 2006;176:4682–4689. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 10.Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- 11.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- 13.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 14.Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol. 2006;177:1746–1754. doi: 10.4049/jimmunol.177.3.1746. [DOI] [PubMed] [Google Scholar]
- 15.Berard M, Brandt K, Bulfone-Paus S, Tough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol. 2003;170:5018–5026. doi: 10.4049/jimmunol.170.10.5018. [DOI] [PubMed] [Google Scholar]
- 16.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001;167:1179–1187. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 17.Schluns KS, Klonowski KD, Lefrancois L. Transregulation of memory CD8 T-cell proliferation by IL-15Ralpha+ bone marrow-derived cells. Blood. 2004;103:988–994. doi: 10.1182/blood-2003-08-2814. [DOI] [PubMed] [Google Scholar]
- 18.Schluns KS, Stoklasek T, Lefrancois L. The roles of interleukin-15 receptor alpha: trans-presentation, receptor component, or both? Int J Biochem Cell Biol. 2005;37:1567–1571. doi: 10.1016/j.biocel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 19.Bracci L, Canini I, Venditti M, Spada M, Puzelli S, Donatelli I, Belardelli F, Proietti E. Type I IFN as a vaccine adjuvant for both systemic and mucosal vaccination against influenza virus. Vaccine. 2006;24 2:S2–56. doi: 10.1016/j.vaccine.2005.01.121. [DOI] [PubMed] [Google Scholar]
- 20.Takasu H. Interferon-alpha: an effective adjuvant for peptide-based cytotoxic T-cell vaccines. Kurume Med J. 2001;48:171–174. doi: 10.2739/kurumemedj.48.171. [DOI] [PubMed] [Google Scholar]
- 21.Curtsinger JM, Gerner MY, Lins DC, Mescher MF. Signal 3 availability limits the CD8 T cell response to a solid tumor. J Immunol. 2007;178:6752–6760. doi: 10.4049/jimmunol.178.11.6752. [DOI] [PubMed] [Google Scholar]
- 22.Prell RA, Li B, Lin JM, VanRoey M, Jooss K. Administration of IFN-alpha enhances the efficacy of a granulocyte macrophage colony stimulating factor-secreting tumor cell vaccine. Cancer Res. 2005;65:2449–2456. doi: 10.1158/0008-5472.CAN-04-1975. [DOI] [PubMed] [Google Scholar]
- 23.Hiroishi K, Tuting T, Lotze MT. IFN-alpha-expressing tumor cells enhance generation and promote survival of tumor-specific CTLs. J Immunol. 2000;164:567–572. doi: 10.4049/jimmunol.164.2.567. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein D, Laszlo J. The role of interferon in cancer therapy: a current perspective. CA Cancer J Clin. 1988;38:258–277. doi: 10.3322/canjclin.38.5.258. [DOI] [PubMed] [Google Scholar]
- 25.Krasagakis K, Garbe C, Zouboulis CC, Orfanos CE. Growth control of melanoma cells and melanocytes by cytokines. Recent Results Cancer Res. 1995;139:169–182. doi: 10.1007/978-3-642-78771-3_12. [DOI] [PubMed] [Google Scholar]
- 26.Maellaro E, Pacenti L, Del Bello B, Valentini MA, Mangiavacchi P, De Felice C, Rubegni P, Luzi P, Miracco C. Different effects of interferon-alpha on melanoma cell lines: a study on telomerase reverse transcriptase, telomerase activity and apoptosis. Br J Dermatol. 2003;148:1115–1124. doi: 10.1046/j.1365-2133.2003.05301.x. [DOI] [PubMed] [Google Scholar]
- 27.Gogas H, Ioannovich J, Dafni U, Stavropoulou-Giokas C, Frangia K, Tsoutsos D, Panagiotou P, Polyzos A, Papadopoulos O, Stratigos A, Markopoulos C, Bafaloukos D, Pectasides D, Fountzilas G, Kirkwood JM. Prognostic significance of autoimmunity during treatment of melanoma with interferon. N Engl J Med. 2006;354:709–718. doi: 10.1056/NEJMoa053007. [DOI] [PubMed] [Google Scholar]
- 28.Di Pucchio T, Pilla L, Capone I, Ferrantini M, Montefiore E, Urbani F, Patuzzo R, Pennacchioli E, Santinami M, Cova A, Sovena G, Arienti F, Lombardo C, Lombardi A, Caporaso P, D'Atri S, Marchetti P, Bonmassar E, Parmiani G, Belardelli F, Rivoltini L. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gp100 peptides plus IFN-alpha results in the activation of specific CD8(+) T cells and monocyte/dendritic cell precursors. Cancer Res. 2006;66:4943–4951. doi: 10.1158/0008-5472.CAN-05-3396. [DOI] [PubMed] [Google Scholar]
- 29.Smith JW, 2nd, Walker EB, Fox BA, Haley D, Wisner KP, Doran T, Fisher B, Justice L, Wood W, Vetto J, Maecker H, Dols A, Meijer S, Hu HM, Romero P, Alvord WG, Urba WJ. Adjuvant immunization of HLA-A2-positive melanoma patients with a modified gp100 peptide induces peptide-specific CD8+ T-cell responses. J Clin Oncol. 2003;21:1562–1573. doi: 10.1200/JCO.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 30.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 32.Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, Charrier K, Sedger L, Willis CR, Brasel K, Morrissey PJ, Stocking K, Schuh JC, Joyce S, Peschon JJ. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. Journal of Experimental Medicine. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 36.Hughes GC, Thomas S, Li C, Kaja MK, Clark EA. Cutting edge: progesterone regulates IFN-alpha production by plasmacytoid dendritic cells. J Immunol. 2008;180:2029–2033. doi: 10.4049/jimmunol.180.4.2029. [DOI] [PubMed] [Google Scholar]
- 37.Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, Odermatt B, Conrad C, Ittner LM, Bauer S, Luther SA, Uematsu S, Akira S, Hengartner H, Zinkernagel RM. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–145. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 38.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189:521–530. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee KH, Wang E, Nielsen MB, Wunderlich J, Migueles S, Connors M, Steinberg SM, Rosenberg SA, Marincola FM. Increased vaccine-specific T cell frequency after peptide-based vaccination correlates with increased susceptibility to in vitro stimulation but does not lead to tumor regression. Journal of Immunology. 1999;163:6292–6300. [PubMed] [Google Scholar]
- 40.Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O'Callaghan CA, Steven N, McMichael AJ, Rickinson AB. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J Exp Med. 1998;187:1395–1402. doi: 10.1084/jem.187.9.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang RF. Regulatory T cells and innate immune regulation in tumor immunity. Springer Semin Immunopathol. 2006;28:17–23. doi: 10.1007/s00281-006-0022-7. [DOI] [PubMed] [Google Scholar]
- 42.Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 43.van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, Schoenberger SP. Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol. 2003;4:361–365. doi: 10.1038/ni912. [DOI] [PubMed] [Google Scholar]
- 44.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 45.Overwijk WW, de Visser KE, Tirion FH, de Jong LA, Pols TW, van der Velden YU, van den Boorn JG, Keller AM, Buurman WA, Theoret MR, Blom B, Restifo NP, Kruisbeek AM, Kastelein RA, Haanen JB. Immunological and antitumor effects of IL-23 as a cancer vaccine adjuvant. J Immunol. 2006;176:5213–5222. doi: 10.4049/jimmunol.176.9.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maurer M, von Stebut E. Macrophage inflammatory protein-1. Int J Biochem Cell Biol. 2004;36:1882–1886. doi: 10.1016/j.biocel.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 47.Smyth MJ, Norihisa Y, Ortaldo JR. Multiple cytolytic mechanisms displayed by activated human peripheral blood T cell subsets. J Immunol. 1992;148:55–62. [PubMed] [Google Scholar]
- 48.Wodarz D, Klenerman P, Nowak MA. Dynamics of cytotoxic T-lymphocyte exhaustion. Proc Biol Sci. 1998;265:191–203. doi: 10.1098/rspb.1998.0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]