

Abstract
Background
Despite the widespread use of cardiac troponins for diagnosis of myocyte injury and risk stratification in acute cardiac disorders, little is known about the long term effects of the released troponins on cardiac function. Recently, we showed that an autoimmune response to cardiac troponin I induces severe inflammation and subsequent fibrosis in the myocardium. This autoimmune disorder predisposes in mice to heart failure and cardiac death.
Methods and Results
To investigate the role of cTnI-specific T-cells, T-cells were isolated from splenocytes of mice immunized with murine cardiac troponin I (mcTnI). WT mice receiving mcTnI-specific T-cells showed high mcTnI-specific antibody titers, increased production of pro-inflammatory cytokines IL-1β and TNF-α, severe inflammation and fibrosis in the myocardium, and reduced fractional shortening. To identify the antigenic determinants of troponin I responsible for the observed inflammation, fibrosis and heart failure, 16 overlapping 16-18mer peptides covering the entire amino acid sequence of mcTnI (211 residues) were synthesized. Only mice immunized with the residues 105-122 of mcTnI developed significant inflammation and fibrosis in the myocardium with increased expression of inflammatory chemokines RANTES, MCP-1, MIP-1α, MIP-1β, MIP-2, TCA-3, eotaxin and chemokine receptors CCR1, CCR2, CCR5. Mice immunized with the corresponding human cTnI residues 104-121 and the mcTnI residues 131-148 developed milder disease.
Conclusion
Transfer of troponin I-specific T-cells can induce inflammation and fibrosis in WT mice leading to deterioration of contractile function. Furthermore, two sequence motifs of cTnI that induce inflammation and fibrosis in the myocardium are characterized.
Keywords: inflammation, heart failure, myocarditis, immunology
Introduction
Heart failure has become an increasingly prevalent disorder with considerable morbidity and mortality. While many causal mechanisms such as inherited cardiomyopathies, ischemic cardiomyopathy or muscular overload are easily identified in clinical practice, the molecular mechanisms that determine the progression of heart failure or ventricular remodeling are largely unknown. There is compelling evidence that inflammatory mechanisms may contribute to progressive heart failure. Thus, myocardial infiltration of lymphocytes and mononuclear cells, increased expression of pro-inflammatory chemokines and cytokines and circulating autoantibodies are frequently observed in myocarditis and DCM (dilated cardiomyopathy). The antibodies identified in DCM patients are directed against various myocardial constituents1-3. Cardiac troponins in blood are the preferred biomarkers of myocardial injury. The fact that they are strictly intracellular proteins which are not found in the circulation of healthy individuals provides a high level of clinical sensitivity and specificity even when cardiac lesions are small. Thus, any significantly detectable troponin in circulation is considered a sign of acute myocardial cell damage4, 5.
Nishimura et al. reported that PD-1 receptor deficient mice developed autoantibodies against cardiac troponin I (cTnI) and -as a consequence-severe dilated cardiomyopathy6. They further found that administration of monoclonal anti-cTnI antibodies induced myocardial dysfunction highly likely by facilitating Ca2+ influx into cardiomyocytes7. We and others have shown, that autoantibodies to cTnI are present in patients with acute coronary syndrome3, 8. These antibodies may interfere with diagnostic assays leading to unpredictable results8 and may have an impact on the improvement of the left ventricular ejection fraction after myocardial infarction3. These findings indicate that induction of an autoimmune response to cTnI is not a rare event in patients. Recently, we showed that an autoimmune response to murine cardiac troponin I (mcTnI) induces severe inflammation in the myocardium followed by fibrosis and heart failure with increased mortality in mice9. Furthermore, we demonstrated that mice immunized with mcTnI prior to LAD ligation showed greater infarct size, more fibrosis, higher inflammation scores and reduced fractional shortening9.
In this study we investigated the role of cTnI-specific T-cells in this pro-inflammatory autoimmune response and identified the antigenic determinants of cTnI responsible.
Methods
Mice
Female A/J mice (5-6 weeks of age) obtained from Harlan Winkelmann GmbH (33176 Borchen, Germany), female SCID mice (5-6 weeks of age) obtained from Charles River (Sulzfeld, Germany) were maintained in the animal facility at the University of Heidelberg and used in all experiments. The animal work was approved by the Animal Care and Use Committee of the University of Heidelberg.
Preparation of recombinant murine cardiac troponin I
The murine cardiac troponin subunit mcTnI was expressed in E.coli and purified as previously described10. In addition to purification via ion exchange chromatography, mcTnI was applied to a cardiac troponin C affinity column as second purification step11. Isolated mcTnI-fractions were dialysed extensively against 1 mM HCl, then lyophilised and stored at -80 °C.
Cell sorting
CD90+, CD8+ and CD4+ T-cells were enriched to 90% purity from the spleen by magnetically activated cell sorting using anti-CD90, anti-CD8, anti-CD4- conjugated microbeads (Miltenyi-Biotec, Auburn, CA).
Transfer of T-cells
For the transfer experiments four groups of mice treated differently were used. Two groups of mice were first immunized with mcTnI on days 0 and 7. On day 21 purified T-cells from one group of mice were re-stimulated in vitro in the presence of dendritic cells and monocytes with 10μg/ml of mcTnI for 48h whereas T-cells from the second group were not re-stimulated with mcTnI. Additionally, two other groups of mice were immunized first with adjuvant alone on days 0 and 7. On day 21 purified T-cells from one group of mice were re-stimulated in vitro in the presence of dendritic cells and monocytes with 10μg/ml of mcTnI for 48 h whereas T-cells from the second group were not re-stimulated with mcTnI. Then 106-107 of stimulated T-cells were injected intraperitoneally (i.p.) to WT recipient mice irradiated with 600 rad or to non irradiated SCID mice. In order to survey the effect of the number of T-cells transferred, three additional groups of WT recipient mice irradiated with 600 rad were injected i.p. with either 106-107, 105-106 or 104-105 T-cells. Finally CD4+ and CD8+ subsets were isolated from the spleens of immunized mice and were re-stimulated in vitro in the presence of 10μg/ml of mcTnI for 48 h whereas CD8+ T-cells were re-stimulated in the presence of additional 50 IU/ml IL-2 (R&D Systems, 65205 Wiesbaden-Nordenstadt, Germany).
Determination of autoantibody titers
Antibody titers were essentially determined as described before12. In brief, to measure serum anti-peptide or troponin I titers, plates were coated either with 100μl/well of each peptide or cardiac troponin I (5μg/ml) in bicarbonate buffer (pH 9.6) and incubated overnight. Anti-mouse secondary antibody diluted to 1:5000 for IgG (Sigma) was used for detection. Serum samples from test mice were diluted to 1:100, 1:500, 1:2500, and 1:12500. Normal mouse serum was used as control. Optical densities were determined at 450nm. Antibody endpoint titers for each individual mouse were calculated as the greatest positive dilution of antibody yielding a positive signal.
Cardiac-troponin I dependent cytokine production by splenocytes
For in vitro cytokine production, the splenocytes were cultured at 5×106 per well in RPMI 1640 complete medium in the presence of 10μg/ml of either cTnI or medium alone for 48 h. Supernatant was collected, aliquoted and frozen at -20 °C. Cytokines (IL-1β, IL-2, IL-4, IL-6, IL-10, IL-13, IL-17, IFN-γ, and TNF-α) were measured by DuoSet ELISA Development Systems (R&D Systems, 65205 Wiesbaden-Nordenstadt, Germany), according to the manufacturer’s instructions.
Histopathological evaluation
For the histopathological evaluation of myocardium, mice were sacrificed on day 21 after transfer of T-cells and on day 28 after immunization with peptides respectively. Sections of 5μm thickness were cut at various depths in the myocardial tissue section and stained with haematoxylin and eosin to determine the level of inflammation and with Masson’s Trichrome to detect collagen deposition. Evidence of myocarditis and fibrosis was evaluated in a blinded manner by two independent investigators who used light microscopy, according to a scoring system: grade 0, no inflammation; grade 1, cardiac infiltration in up to 5% of the cardiac sections; grade 2, 6-10%; grade 3, 11-30%; grade 4, 31-50%; and grade 5, >50%.
RNA protection assay
The mCR-5 cytokine receptor multi-probe template set (BD Biosciences Pharmingen, Heidelberg, Germany) was used to measure mouse mRNAs encoding CCR1, CCR2, CCR1b, CCR3, CCR4, and CCR5. The mCK-5c multi-probe template set (BD Biosciences Pharmingen, Heidelberg, Germany) was used to measure mouse mRNAs encoding Ltn, RANTES, MIP-1b, MIP-1a, MIP-2, IP-10, MCP-1, TCA-3, and eotaxin. The measurement was done according to the manufacturer’s guidelines.
Echocardiography
Transthoracic echocardiography was performed as previously described9. The investigator who conducted the echocardiography was unaware of the treatment status.
Synthesis of peptides
Sixteen overlapping 16-18mer peptides covering the entire amino acid sequence of mcTnI (211 residues), the corresponding human cTnI residues 104-121 (VDKVDEERYDIEAKVTKN), the residues 73-90 (VEVVDEERYDIEAKCLHN) and 99-116 (LKVLDLRGKFKRPPLRRV) of slow skeletal troponin I, and the residues 98-115 (QKLFDLRGKFKRPPLRRV) of fast skeletal troponin I were synthesized and purified by HPLC (Peptide Specialty Laboratories, Heidelberg, Germany). The purity of the peptides was >90%.
Antigen preparation and administration
Each mouse was injected subcutaneously with an emulsion of 120μg of one of the peptides, or troponin I as positive control, or control buffer and adjuvant as negative control. The peptide/protein was supplemented with CFA containing 5 mg/ml of Mycobacterium tuberculosis H37Ra (Sigma, St. Louis, MO, USA) on days 0, 7 and 14. The mice were sacrificed on day 28 for histopathological evaluation, RNA protection assay analysis, and measurement of cytokines and antibody titers.
Statistical analysis
Results are expressed as mean ± SEM. Data were analyzed with the Kruskal-Wallis test followed by the Mann-Whitney U test to explore the significance between the treatment groups. P values of <0.05 were considered significant. The statistical software SPSS (ver 15.0) was used for all calculations.
Statement of Responsibility
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Transfer of T-cells
Mice receiving T-cells from mcTnI immunized mice that have been re-stimulated in vitro with mcTnI showed high mcTnI-specific total IgG antibody titers (Figure 1a), severe inflammation (histoscore: 1.8±0.4) and fibrosis (histoscore: 1.9±0.4) in myocardium (Figure 1c), and increased mcTnI-induced production of the cytokines IL-1β, IL-4, IL-13, IL-17, and TNF-α (Figure 1b) compared to mice receiving T-cells from mcTnI immunized mice that have not been re-stimulated in vitro with mcTnI or receiving T-cells from mice given adjuvant alone irrespective of their in vitro re-stimulation. There was no significant difference in the production of the other cytokines (IL-2, IL-6, and IFN-γ). Transfer of T-cells from mcTnI immunized mice that have been re-stimulated in vitro with mcTnI led to enlarged hearts with significantly increased left ventricular end-diastolic (3.6±0.2mm) (Figure 1d) and end-systolic diameter (2.3±0.2mm) (Figure 1e). The fractional shortening was significantly reduced (35.6±3.1%) (Figure 1f). In contrast, mice receiving T-cells from mice given adjuvant alone irrespective their in vitro re-stimulation did not show any signs of inflammation, fibrosis, or alteration of heart function. One of six mice receiving T-cells from mcTnI immunized mice without in vitro re-stimulation showed slight inflammation and fibrosis but no significant alteration of heart function. To examine the expression of different chemokines and chemokine receptors, we measured the mRNA expression levels of the chemokines Ltn, RANTES, MIP-1β, MIP-1α, MIP-2, IP-10, MCP-1, TCA-3 and eotaxin as well as the levels of the chemokine receptors CCR1, CCR2, CCR1b, CCR3, CCR4 and CCR5 in the myocardium of the recipient mice. We were able to detect mRNA levels for RANTES, MIP-1β, MIP-1α, MIP-2, MCP-1, TCA-3 and eotaxin only in mice receiving mcTnI-specific T-cells unlike the otherwise treated mice (Figure 1g). In addition, we found that the mRNAs for CCR1, CCR2 and CCR5 were expressed only in the myocardium of mice receiving mcTnI-specific T-cells but not in the other groups of mice (Figure 1g).
Figure 1.



Transfer of T-cells into WT mice. Mice were immunized with mcTnI or adjuvant alone. On day 21 T-cells were isolated from splenocytes and re-stimulated in vitro in the presence of dendritic cells and monocytes with 10μg/ml of mcTnI (mcTnI/mcTnI or no/mcTnI) for 48h or were not re-stimulated with mcTnI (mcTnI/no or CFA/no). Then 106-107 T-cells per mouse were transferred to WT mice irradiated with 600 rad. Effects on production of mcTnI specific autoantibodies (total IgG (a), mean ± SEM) and cytokines ((b), mean ± SEM), on inflammation and fibrosis (c), on left ventricular end-diastolic ((d), mean ± SEM) and end-systolic diameter ((e), mean ± SEM) on fractional shortening ((f), mean ± SEM), and on expression of cytokines and cytokine receptors in the myocardium (g). Antibody endpoint titers for each individual mouse were calculated as the greatest positive dilution of antibody above normal mouse serum levels for day 21. *: p<0,05 (Kruskal-Wallis test and U test).
Further on, in order to study the effect of the number of T-cells transferred, three additional groups of WT recipient mice irradiated with 600 rad were injected i.p. with either 106-107, 105-106 or 104-105 T-cells. When we reduced the number of T-cells transferred the severity of disease decreased. Transfer of 104-105 or lower number of T-cells disclosed no significant inflammation in the recipient mice (Figure 2a). On the other hand, transfer of 106-107 T-cells in non-irradiated SCID mice induced in 10 out of 11 recipients an inflammation in the myocardium. Overall, the histoscores in SCID mice were lower compared to irradiated WT recipient mice (histoscore: 0.8±0.1, data not shown).
Figure 2.

WT recipient mice irradiated with 600 rad were injected i.p. with either 106-107, 105-106 or 104-105 T-cells (a). WT recipient mice irradiated with 600 rad were injected i.p. with either 106-107 CD90+ T-cells, 106-107 CD4+ or with 106-107 CD8+ T-cells (b). (Kruskal-Wallis test and U test).
In order to study the effect of T-cell subsets, CD4+ and CD8+ T-cells were isolated from the spleens of immunized mice and were re-stimulated in vitro with mcTnI or in the case of CD8+ T-cells were re-stimulated with mcTnI and supplemental IL-2. Transfer of whole T-cells (CD90+; histoscore: 2.1±0.5) and transfer of CD4+ subset of T-cells (histoscore: 2.4±0.4) induced moderate to severe disease, whereas transfer of CD8+ subset of T-cells alone did not induce any signs of inflammation in the recipient mice (Figure 2b).
Identity of peptides
To identify the antigenic sequences of murine cardiac troponin I triggering inflammation and fibrosis, sixteen overlapping 16-18mer peptides covering the entire amino acid sequence of murine troponin I (211 residues) were synthesized, followed by HPLC-purification (Figure 3).
Figure 3.

Sixteen overlapping 16-18 mer peptides covering the entire amino acid sequence of murine troponin I (211 residues) were synthesized followed by HPLC-purification.
Immunization and humoral immune response
The mice developed significant autoantibody titers against all injected peptides by day 28. However, total IgG autoantibody titers were significantly higher in mice immunized with the peptides 7, 8, 10 and 14 (data not shown).
Immunization with the residues 105-122 (peptide 9) and 131-148 (peptide 11) of murine cTnI induces myocardial inflammation
Four of five mice immunized with the residues 105-122 of mcTnI (peptide 9: VDKVDEERYDVEAKVTKN) showed inflammation with a histoscore of ≥ 1 (Figures 3 and 4a-d). One out of five mice immunized with the residues 131-148 of mcTnI (peptide 11: QKIYDLRGKFKRPTLRRV) showed signs of inflammation (histoscore=1) (Figures 3 and 4e). No inflammation was observed in the myocardium of mice immunized with the other synthesized mcTnI residues or control buffer and adjuvant alone (Figures 3 and 4f).
Figure 4.

Histological examination of the hearts on day 28 (stain with haematoxylin and eosin (a-f) or Masson’s trichrome (g-i)). Representative heart section of the four mice immunized with the peptide 9 with the inflammation scores of 3 (a), 2 (b-c), and 1 (d). The heart section of the one mouse immunized with peptide 11 and a histoscore of 1 (e). Representative heart section of mice without significant inflammation (f). Representative heart sections of mice immunized with the peptide 9 and fibrosis scores of 1 (g), 3 (h), and 2 (i). Fibrosis was indicated by bright blue staining for collagen deposition. (a, b: 5x magnification; c-i: 20x magnification).
Immunization with the residues 105-122 (peptide 9) of murine cTnI causes myocardial fibrosis
Comparable to the observed inflammation four out of five mice immunized with the residues 105-122 of mcTnI (peptide 9) showed significant deposition of collagen in the myocardium with a fibrosis score of ≥ 1 (Figures 4g-i) whereas no fibrosis was detected in the mice immunized with the other peptides or control buffer and adjuvant alone. Myocardial fibrosis was indicated by bright blue staining for collagen deposition and was associated with myocardial inflammation.
Immunization with the residues 105-122 (peptide 9) of murine cTnI induces expression of chemokines and chemokine receptors in the myocardium
In order to investigate the expression of different chemokines and chemokine receptors we measured the mRNA-expression levels of the chemokines Ltn, RANTES, MIP-1β, MIP-1α, MIP-2, IP-10, MCP-1, TCA-3, eotaxin and of the chemokine receptors CCR1, CCR2, CCR1b, CCR3, CCR4 and CCR5 in the myocardium of the immunized mice. We were able to detect mRNA levels for RANTES, MIP-1β, MIP-1α, MIP-2, MCP-1, TCA-3, eotaxin only in mice immunized with the residues 105-122 of mcTnI (peptide 9) but not in mice immunized with the other peptides or control buffer and adjuvant alone (Figure 5a). In addition, we found that the mRNAs for the chemokine receptors CCR1, CCR2 and CCR5 were solely expressed in the myocardium of mice immunized with the residues 105-122 of mcTnI (peptide 9) (Figure 5b).
Figure 5.

RNA protection assay and antibody titers: mRNA expression levels of some tested chemokines (a) and mRNA expression levels of the chemokine receptors CCR1-5 (b) in the myocardium of mice immunized with the different peptides and mcTnI as a positive control on day 28. Antibody titers directed against the whole mcTnI protein and against the peptide 9 were measured in mice immunized with either mcTnI ((c), mean ± SEM) or peptide 9 ((d), mean ± SEM). Antibody endpoint titers for each individual mouse were calculated as the greatest positive dilution of antibody above normal mouse serum levels for day 21.
Immunization with the residues 105-122 (peptide 9) of murine cTnI induces production of antibodies reacting with the complete protein mcTnI
First we tested whether immunization with the complete protein mcTnI also induces also antibodies to the residues 105-122 of mcTnI (peptide 9). On day 28, mice immunized with mcTnI developed not only antibody against the whole mcTnI protein but also against the residues 105-122 of mcTnI (peptide 9) (Figure 5c). Conversely, immunization with the residues 105-122 of mcTnI (peptide 9) induced production of antibodies reacting with the complete protein mcTnI (Figure 5d).
hcTnI and other proteins with similar amino acid sequences as the antigenic murine amino acid sequences 105-122 and 131-148
When comparing the antigenic residues 105-122 and 131-148 of murine cTnI to corresponding amino acid sequences of hcTnI or other proteins with similar amino acid sequences, we found that the residues 104-121 and residues 130-147 of human cTnI (hcTnI) were different in only 1 amino acid to the major (residues 105-122) and minor epitope (residues 131-148) of mcTnI, respectively (Figure 6). The next protein with amino acid sequences similar to both mcTnI epitopes (105-122 and 131-148) was skeletal troponin I (residues 73-90 and 99-116 in slow skeletal troponin and residues 98-115 in fast skeletal troponin). Residues 73-90 of slow skeletal troponin were already different in 6 amino acids to the main epitope of mcTnI (105-122; peptide 9) (Figure 6). Residues 98-115 of fast skeletal troponin I were different in 3 amino acids and residues 99-116 of slow skeletal troponin I were different in 4 amino acids to the minor epitope of mcTnI (131-148; peptide 11).
Figure 6.

Corresponding proteins with similar sequences to the residues 105-122 and 131-148 of mcTnI.
Residues 104-121 of human cTnI but not residues 73-90, 98-115 or 99-116 of skeletal troponin I induce myocardial inflammation and fibrosis
In order to study whether hcTnI or skeletal troponin I, different peptides with the most similarity in amino acid sequence to both pathogenic epitopes of mcTnI (105-122 and 131-148), have a pathogenic effect on the myocardium, mice were immunized with either hcTnI (104-121), or slow skeletal troponin I (residues 73-90 or 99-116), or fast skeletal troponin I (residues 98-115). Residues 105-122 (peptide 9) of mcTnI were used as a positive control (Figures 7a-g). All mice immunized with residues 105-122 of mcTnI developed mild to severe inflammation and fibrosis (Figures 7a-e). Four of eight mice immunized with the residues 104-121 of hcTnI showed mild inflammation and fibrosis (Figures 7a and 7f-g), some located perivascularly (Figure 7g). In contrast, none of the mice immunized with skeletal troponin I residues 73-90 (Figure 7a), residues 98-115, or residues 99-116 (data not shown) showed signs of inflammation.
Figure 7.

(a) Groups of mice were immunized with either mcTnI (residues 105-122), or hcTnI (residues 104-121), or skeletal troponin (residues 73-90). Effects on severity of inflammation and fibrosis on day 28 (Kruskal-Wallis test and U test). Histological examination of the hearts (stain with haematoxylin and eosin (b-g)). Representative heart section of mice immunized with mcTnI (105-121) (peptide 9) with the inflammation scores of 3,5 (b and d) and 2,5 (c and e). Representative heart section of mice immunized with hcTnI (104-121) and inflammation score of 1 (f) and with perivascular inflammation (g) on day 28. (b and c: 10x magnification; d-g: 20x magnification).
3-D structure of troponin I
The localisation of the peptide 9 sequences is marked with an arrow in an actual 3-D structure of troponin I in association to troponin T (with kind permission of ‘Nature Publishing Group’) (Figure 8)13.
Figure 8.
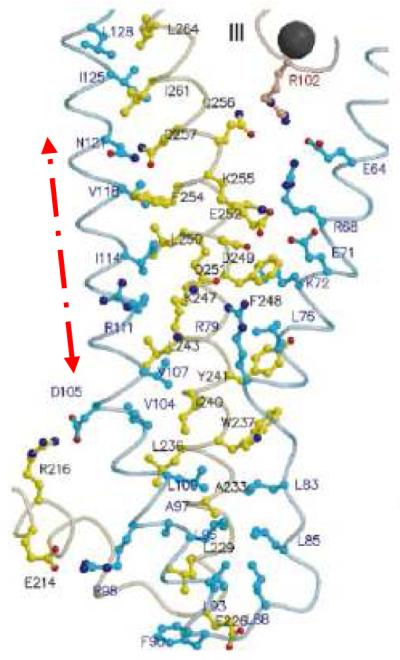
The localisation of the peptide 9 amino acid sequences is marked with an arrow in an actual 3-D structure of troponin I in association to troponin T (with kind permission of ‘Nature Publishing Group’) (Figure 7)13. TnC and TnT are coloured in red and yellow, respectively. TnI is coloured in cyan, except for the two stretches of amphiphilic helices (TnC-binding sites), which are dark blue.
Discussion
Here we showed for the first time that transfer of troponin I-specific T-cells led to severe inflammation and fibrosis in healthy recipient WT mice resulting in enlarged hearts, increased end-systolic and end-diastolic diameters, reduced fractional shortening, inflammation, fibrosis and heart failure. Furthermore, we identified two amino acid sequences of murine troponin I that led to heart failure by inducing inflammation and fibrosis. Hereby, the residues 105-122 (peptide 9) of mcTnI were the strongest inductor of inflammation and fibrosis in the myocardium which is accompanied by increased expression of inflammatory chemokines RANTES, IP-10, MCP-1, MIP-1α, MIP-1β, MIP-2, TCA-3 and eotaxin, and of the chemokine receptors CCR1, CCR2, CCR5. McTnI residues 131-148 (peptide 11) were a minor epitope inducing milder inflammation. While the corresponding human cTnI residues 104-121 which were different in one amino acid from the murine cTnI residues 105-122 also induced milder inflammation in mice, none of the mice immunized with skeletal troponin I showed significant signs of inflammation. Furthermore, we demonstrated that mice immunized with mcTnI also developed antibodies directed against the residues 105-122 of mcTnI (peptide 9). Conversely, immunization with the residues 105-122 of mcTnI (peptide 9) induced production of antibodies reacting with the complete protein mcTnI. Thus, the residues 105-122 of mcTnI must be immunogenic in the complex structure of the whole mcTnI protein as well. This epitope becomes more interesting, since it forms a parallel helix (residues 90-135 of TnI) with the helix of TnT (residues 226-271) and it has been suggested that this coiled coil between TnI and TnT has important physiological roles that are characteristic of troponin13.
It has been reported that autoantibodies against cTnI induced heart failure by chronic stimulation of Ca2+-influx in cardiomyocytes6, 7. Recently, we reported that a humoral and cellular autoimmune response against mcTnI induced severe inflammation and fibrosis in the myocardium of mice with persistent, prominent inflammation and fibrosis over 270 days and reduced long-term survival9. We also demonstrated that mice pre-immunized with TnI prior to LAD ligation showed greater infarct size, more fibrosis, higher inflammation scores and reduced fractional shortening than mice without pre-immunization9. These results indicate that an autoimmune response against troponin I aggravates the outcome of acute cardiac damage and may have a significant influence on post-infarct remodeling. Now we demonstrate that the troponin induced heart failure is T-cell dependent. Furthermore, CD4+ T-cells are thereby necessary for successful transfer of disease. As opposed to CD8+ T-cells which seem not to play a significant role. We could neither transfer disease with CD8+ T-cells alone nor was the severity of disease higher when both CD4+ and CD8+ were transferred compared to transfer of CD4+ T-cells alone. In addition, we found significantly elevated levels of mc-TnI specific TNF-α, IL-1, IL-4, IL13, IL-17 production. This cytokine profile suggests that mcTnI induced myocarditis in A/J mice exhibits a Th2 (IL-4, IL13) and Th17 (IL-17) like phenotype. In the past, we demonstrated that EAM in A/J mice induced by cardiac myosin has a Th2 phenotype14. Others described an important role for Th1 subsets in EAM, so that the relative contributions of the CD4+ Th1 and Th2 subsets are still unclear14-16. Recently, Rangachari et al.17 demonstrated a significant role for another subset of CD4+ T-cells characterized by IL-17 production in EAM Balb/c mice induced by cardiac myosin peptide. We believe that the strain of mice used in the experiments is a crucial factor in deciding which subsets of CD4+ cells are involved in disease induction and progression.
Proinflammatory cytokines are important in the development of autoimmune myocarditis. We previously showed that administration of either IL-1 or TNF-α promoted virus- and myosin-induced myocarditis in genetically resistant B10.A mice18. Recently, we demonstrated that the presence of myocarditis is associated with increased levels of TNF-α from cardiac myosin-stimulated splenocytes in culture19. Furthermore, when A/J mice were infected with CB3 and treated with an IL-1 receptor antagonist, myocardial injury was diminished20. Thus, IL-1 and TNF-α are clearly critical in the pathogenesis of autoimmune myocarditis.
We have identified two pathogenic epitopes of the troponin I molecule responsible for the induction of inflammation, fibrosis and heart failure. All of the peptide sequences used to immunize mice led to an increase in total IgG autoantibody titers. Even though mice immunized with peptide sequences 7,8,10 and 14 showed higher total IgG antibody titers compared to the other peptides used in the experiments only mice immunized with peptide 9 and 11 showed inflammation and fibrosis in the myocardium. We conclude that in our model the initiation of the inflammatory process in the myocardium, followed by fibrosis and alteration of heart function is primarily T-cell dependent. This is supported by i) our findings that we can transfer disease to healthy WT mice with isolated T-cells alone; ii) the findings by Smith et al.21. They demonstrated that myosin induced myocarditis is a T-cell-mediated disease; iii) the fact that Okazaki, et al.7 and Nishimura, et al.6 described no inflammation in the myocardium in mice treated with antibodies against troponin. They demonstrated that antibodies to cTnI (humoral immune response only) induced heart dysfunction due to chronic stimulation of Ca2+ influx in cardiomyocytes by these antibodies. We also recognize the possibility that once the inflammatory process is induced, antibodies may have additional effects in the broadening this ongoing inflammatory process that is followed by fibrosis and alteration of heart function.
When mice were immunized with the whole troponin protein (mcTnI) we expect to induce an immune response (cellular and humoral) directed against different peptide sequences (epitopes) of this protein. By testing the sera of mice immunized with the whole troponin protein for presence of antibodies directed against the pathogenic peptide 9 we demonstrated that an immune response against peptide 9 can be induced not only by immunization with the synthesized peptide 9 alone but also when the whole troponin protein is used for immunization. This demonstrates that the peptide 9 sequences are immunodominant in the whole troponin protein. On the contrary, when mice were immunized with peptide 9 alone we would expect initially an immune response directed only against the amino acid sequences of peptide 9. These antibodies are able to bind to the whole protein TnI as well. This major finding suggests that these amino acid sequences can be bound by antibodies even in the complex structure of the whole protein. These results document that the peptide 9 sequence is not cryptic (i.e. buried in the core of the whole troponin protein complex). That is confirmed with the actual 3-D structure of troponin I. Furthermore, mice immunized with peptide 9 show at day 21 only antibodies binding itself (peptide 9) and the whole troponin protein TnI but do not have antibodies binding the other peptides used in our experiments. The utility of our peptide study could be to suggest the use of peptide 9 (+11) sequences instead of the whole troponin molecule in testing patient sera to identify patients at risk for myocardial damage induced by autoimmune inflammation. Thus the use of the peptide 9 (+11) sequences in tests would be more specific for identifying the risk of inflammation than using the whole troponin protein (TnI) since we demonstrated that immunization with the other peptide sequences of troponin I did not induce an inflammation or fibrosis in the myocardium. The use of whole troponin protein will identify all antibodies directed against different sequences of troponin including those that might have no pathogenic effect.
Chemokines, such as RANTES, MCP-1, MIP1-α and their major receptors, CCR2 and CCR5, play important roles in the pathogenesis of many inflammatory diseases22-24. MCP-1 mRNA expression has been shown in endomyocardial biopsy specimens from patients with dilated cardiomyopathy and suggested a significant role of this chemokine in the regulation of inflammatory cell infiltration into the myocardium24. Recently, we described an important role for MCP-1 and MIP1-α and their major receptors CCR2 and CCR5, in the initiation of autoimmune myocarditis23. In our experiments, only mice immunized with residues 105-122 of mcTnI (peptide 9) showed increased expression of these chemokines, which correlated with increased myocardial inflammation.
Eriksson et al. have shown that troponin autoantibodies are present in patients and can produce false negative results by blocking the binding of troponin antibodies used in analytical assays to the target protein8. The report of the presence of autoantibodies against cTnI in patients with acute coronary syndrome8 points to an early induction of an autoimmune response to cTnI in these patients. In addition to the analytical interference in the assays, troponin I autoantibodies may have clinical consequences. Okazaki et al. showed that troponin autoantibodies induced dilation and cardiac dysfunction by the chronic stimulation of calcium influx in cardiomyocytes7. Recently, we showed that provoking an autoimmune response to cardiac troponin I induces severe inflammation in the myocardium followed by fibrosis and heart failure with increased mortality in mice9. These results suggest that some of the patients who develop an autoimmune response to released cTnI may have a higher risk of heart failure due to inflammation in the myocardium. These autoimmune responses may also explain the discrepancy observed in some patients with involvement of one or two coronary arteries while the entire heart is diffusely hypokinetic. Furthermore, the role of autoantibodies in heart failure has been supported by many clinical studies demonstrating that removing immunoglobulins by immunoadsorption can improve the ejection fraction and reduce the morbidity in patients with dilated cardiomyopathy25. Now we identified one major and one minor epitope of troponin I (peptide 9 and 11, respectively) that seem to be responsible for disease induction. Using these newly identified epitope sequences of troponin I instead of the whole troponin molecule; a more specific screening test of patients with high risk for progressive autoimmune inflammatory heart failure may be possible. Furthermore, we demonstrated the important role of troponin-specific T-cells in inducing the autoimmune inflammation. These findings may aid in developing new approaches to the early treatment of heart failure in some patients and in initiating further (clinical) studies to investigate the role of troponin I release, induction of an autoimmune response to released troponin I during acute cardiac damage, its role on post-infarct remodeling and heart failure.
Acknowledgments
The authors thank Özay Kaya, Theresa Tretter, Martin Andrassy and Simone Höger for critically reading the manuscript and thank Marc Bischof and Carmen Timke for their help in irradiating the mice.
Funding Sources:
This work was supported by the Deutsche Forschungsgemeinschaft, research grant KA 1797/4-1 (to Dr Kaya), by the Ernst und Berta Grimmke Stiftung (to Dr Kaya) and in part by National Institutes of Health research grant HL067290 and HL077611 (to Dr Rose).
Footnotes
Conflict of Interest Disclosures
Hugo A. Katus has developed the troponin T assay and holds a patent on this assay jointly with Roche Diagnostics. H.A.K. and Z.K. have applied for the patent entitled ‘measuring troponin antibodies to assess cardiovascular risk’ jointly with Roche Diagnostics. There is nothing else to disclose.
CLINICAL PERSPECTIVE
Despite the widespread use of cardiac troponin I (cTnI) for diagnosis of myocyte injury and risk stratification in acute cardiac disorders, little is known about the precise role of an autoimmune response to the troponins on cardiac function. Recently, investigators made the surprising discovery that mice treated with monoclonal anti-cTnI antibodies developed myocardial dysfunction. Shortly after, it has been reported that autoantibodies to cTnI are also present in patients with acute coronary syndrome (ACS). These findings indicate that induction of an autoimmune response to cTnI is not a rare event in patients. Lately, we demonstrated that the prevalence of cTnI antibodies in patients with ACS has an impact on the improvement of the left ventricular ejection fraction. Furthermore, the role of autoantibodies in heart failure has been supported by clinical studies demonstrating that removing immunoglobulins by immunoadsorption can improve the ejection fraction in patients with dilated cardiomyopathy. Recently, we showed that inducing an autoimmune response to cTnI leads to severe inflammation in the myocardium followed by fibrosis and heart failure with increased mortality in mice. Now we demonstrate that this disease is primarily a CD4+ T cell dependent disease with a Th2, Th17 phenotype and identified the antigenic determinants of cTnI responsible. We believe that these findings can change fundamentally the understanding of the pathophysiology of inflammatory cardiovascular diseases and post-infarct remodeling.
Subject codes: [130]Animal_models_of_human_disease, [11]other_heart_failure, [105]contractile_function
References
- 1.Caforio AL, Grazzini M, Mann JM, Keeling PJ, Bottazzo GF, McKenna WJ, Schiaffino S. Identification of alpha- and beta-cardiac myosin heavy chain isoforms as major autoantigens in dilated cardiomyopathy. Circulation. 1992;85:1734–42. doi: 10.1161/01.cir.85.5.1734. [DOI] [PubMed] [Google Scholar]
- 2.SchulzMenger J, Maisch B, bdel-Aty H, Pankuweit S. Integrated biomarkers in cardiomyopathies: cardiovascular magnetic resonance imaging combined with molecular and immunologic markers--a stepwise approach for diagnosis and treatment. Herz. 2007;32:458–72. doi: 10.1007/s00059-007-3046-4. [DOI] [PubMed] [Google Scholar]
- 3.Leuschner F, Li J, Goser S, Reinhardt L, Ottl R, Bride P, Zehelein J, Pfitzer G, Remppis A, Giannitsis E, Katus HA, Kaya Z. Absence of auto-antibodies against cardiac troponin I predicts improvement of left ventricular function after acute myocardial infarction. Eur Heart J. 2008;29:1949–55. doi: 10.1093/eurheartj/ehn268. [DOI] [PubMed] [Google Scholar]
- 4.Horwich TB, Patel J, MacLellan WR, Fonarow GC. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation. 2003;108:833–8. doi: 10.1161/01.CIR.0000084543.79097.34. [DOI] [PubMed] [Google Scholar]
- 5.La VL, Mezzena G, Zanolla L, Paccanaro M, Varotto L, Bonanno C, Ometto R. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. J Heart Lung Transplant. 2000;19:644–52. doi: 10.1016/s1053-2498(00)00120-0. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 7.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, Ishida M, Hiai H, Matsumori A, Minato N, Honjo T. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9:1477–83. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson S, Hellman J, Pettersson K. Autoantibodies against cardiac troponins. N Engl J Med. 2005;352:98–100. doi: 10.1056/NEJM200501063520123. [DOI] [PubMed] [Google Scholar]
- 9.Goser S, Andrassy M, Buss SJ, Leuschner F, Volz CH, Ottl R, Zittrich S, Blaudeck N, Hardt SE, Pfitzer G, Rose NR, Katus HA, Kaya Z. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. 2006;114:1693–702. doi: 10.1161/CIRCULATIONAHA.106.635664. [DOI] [PubMed] [Google Scholar]
- 10.Kruger M, Pfitzer G, Stehle R. Expression and purification of human cardiac troponin subunits and their functional incorporation into isolated cardiac mouse myofibrils. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;786:287–96. doi: 10.1016/s1570-0232(02)00763-8. [DOI] [PubMed] [Google Scholar]
- 11.al-Hillawi E, Minchin SD, Trayer IP. Overexpression of human cardiac troponin-I and troponin-C in Escherichia coli and their purification and characterisation. Two point mutations allow high-level expression of troponin-I. Eur J Biochem. 1994;225:1195–201. doi: 10.1111/j.1432-1033.1994.1195b.x. [DOI] [PubMed] [Google Scholar]
- 12.Kaya Z, Afanasyeva M, Wang Y, Dohmen KM, Schlichting J, Tretter T, Fairweather D, Holers VM, Rose NR. Contribution of the innate immune system to autoimmune myocarditis: a role for complement. Nat Immunol. 2001;2:739–45. doi: 10.1038/90686. [DOI] [PubMed] [Google Scholar]
- 13.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 14.Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, Hill SL, Rose NR. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. 2001;159:193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Afanasyeva M, Wang Y, Kaya Z, Stafford EA, Dohmen KM, Sadighi Akha AA, Rose NR. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 2001;104:3145–51. doi: 10.1161/hc5001.100629. [DOI] [PubMed] [Google Scholar]
- 16.Cunningham MW. Cardiac myosin and the TH1/TH2 paradigm in autoimmune myocarditis. Am J Pathol. 2001;159:5–12. doi: 10.1016/S0002-9440(10)61665-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, Penninger JM, Eriksson U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–19. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane JR, Neumann DA, Lafond-Walker A, Herskowitz A, Rose NR. Interleukin 1 or tumor necrosis factor can promote Coxsackie B3-induced myocarditis in resistant B10.A mice. J Exp Med. 1992;175:1123–9. doi: 10.1084/jem.175.4.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Afanasyeva M, Hill SL, Rose NR. Characterization of murine autoimmune myocarditis induced by self and foreign cardiac myosin. Autoimmunity. 1999;31:151–62. doi: 10.3109/08916939908994060. [DOI] [PubMed] [Google Scholar]
- 20.Neumann DA, Lane JR, Allen GS, Herskowitz A, Rose NR. Viral myocarditis leading to cardiomyopathy: do cytokines contribute to pathogenesis? Clin Immunol Immunopathol. 1993;68:181–90. doi: 10.1006/clin.1993.1116. [DOI] [PubMed] [Google Scholar]
- 21.Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147:2141–7. [PubMed] [Google Scholar]
- 22.Rao AR, Quinones MP, Garavito E, Kalkonde Y, Jimenez F, Gibbons C, Perez J, Melby P, Kuziel W, Reddick RL, Ahuja SK, Ahuja SS. CC chemokine receptor 2 expression in donor cells serves an essential role in graft-versus-host-disease. J Immunol. 2003;171:4875–85. doi: 10.4049/jimmunol.171.9.4875. [DOI] [PubMed] [Google Scholar]
- 23.Goser S, Ottl R, Brodner A, Dengler TJ, Torzewski J, Egashira K, Rose NR, Katus HA, Kaya Z. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation. 2005;112:3400–7. doi: 10.1161/CIRCULATIONAHA.105.572396. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann MH, Kuhnert H, Muller S, Sigusch HH. Monocyte chemoattractant protein 1 (MCP-1) gene expression in dilated cardiomyopathy. Cytokine. 1998;10:739–46. doi: 10.1006/cyto.1998.0354. [DOI] [PubMed] [Google Scholar]
- 25.Dorffel WV, Wallukat G, Dorffel Y, Felix SB, Baumann G. Immunoadsorption in idiopathic dilated cardiomyopathy, a 3-year follow-up. Int J Cardiol. 2004;97:529–34. doi: 10.1016/j.ijcard.2004.03.001. [DOI] [PubMed] [Google Scholar]