

Abstract
Cholesterol gallstone formation is a complex process mediated by genetic and environmental factors. Until recently, the role of the immune system in the pathogenesis of cholesterol gallstones was not considered a valid topic of research interest. This review collates and interprets an extensive body of basic literature, some of which is not customarily considered to be related to cholelithogenesis, describing the multiple facets of the immune system that appear to be involved in cholesterol cholelithogenesis. A thorough understanding of the immune interactions with biliary lipids and cholecystocytes should modify current views of the pathogenesis of cholesterol gallstones, promote further research on the pathways involved, and lead to novel diagnostic tools, treatments, and preventive measures.
Bile is a complex aqueous colloidal system that is essential for a wide range of physiologic functions, including the excretion of lipids from the organism and intestinal fat absorption.1,2 Bile is formed primarily in hepatic canaliculi, small (1–2 μm) spaces formed between the tight junctions of hepatocytes. It is composed of water, electrolytes, and a variety of lipid solutes dispersed in mixed micelles and vesicles including bile salts, phospholipids (>96% being mixed phosphatidylcholines), and cholesterol, as well as proteins and bilirubin conjugates.3 Phospholipids and bile salts are essential for the removal from the organism of otherwise insoluble cholesterol molecules in an aqueous environment by solubilizing the sterol in mixed micelles, composed of the catabolic product bile salts, and unilamellar vesicles.2–4 Bile is transported from the canaliculi along tubules of increasingly greater diameter until it egresses into the gut at the midduodenum. In health, about half the secreted bile is stored, concentrated, and slightly acidified in the gallbladder during the interdigestive interval. The gallbladder is connected to the biliary tree via the cystic duct, which functions simultaneously as a gallbladder filling and emptying conduit.3
Alterations in the relative or absolute proportions of cholesterol, phospholipids, and bile salts can lead to phase separation of cholesterol from solution in bile. Most frequently these changes result from excess secretion of cholesterol from the liver.1–3 As the absolute cholesterol concentration increases, the excess cholesterol phase separates, forming unilamellar vesicles with biliary phospholipids (Figure 1). Under suitable physicochemical conditions, these can aggregate to form multilamellar vesicles (lamellar liquid crystals), and eventually cholesterol monohydrate crystals can separate from these and aggregate in the gallbladder.2,4 These crystals can progress to form cholesterol gallstones by agglomeration within a gallbladder-secreted mucin gel. Formation of cholesterol gallstones invariably occurs adjacent to the gallbladder wall, where cholesterol monohydrate crystals nucleate in the gelled mucin glycoprotein scaffolding.2,4–6 The majority of cholesterol gallstones form in the gallbladder; however, in rare situations especially and perhaps uniquely from genetic deficiency of biliary phospholipid secretion,7 the stones can form anywhere along the biliary tree, even intrahepatically.1–3 Although a relative excess of cholesterol is necessary for formation of cholesterol gallstones, it is not sufficient. Biliary sludge, composed of agglomerated cholesterol crystals of an ultrasonically detectable size (∼50 μm) suspended in mucin gel, is frequently observed and can progress to form cholesterol gallstones.8–10 Lee et al found that in 18% of patients studied by ultrasonography for a mean of more than 3 years, biliary sludge disappeared completely; in 60% of the patients, the biliary sludge disappeared and reappeared.10
Figure 1.
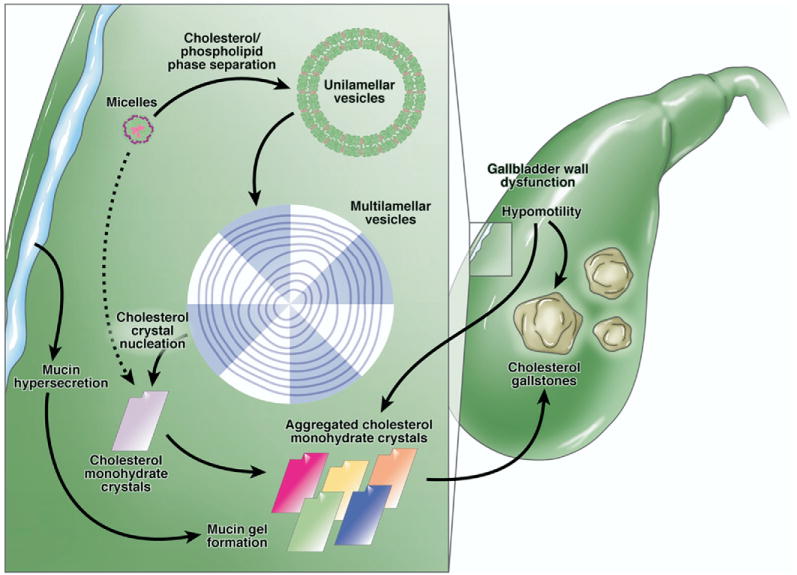
The physical-chemical processes involved in formation of cholesterol gallstones. The classic physical-chemical symbols for cholesterol (pink), phospholipid (green), and bile salt (purple) molecules are shown, along with the macromolecular structures they form. Cholesterol, phospholipids, and bile salts combine by hydrophobic interactions to form mixed micelles (micelles) and cholesterol and phospholipids form unilamellar vesicles. Normally the unilamellar vesicles would be ∼5–10 times larger than micelles (∼40 Å in radius), but for illustration purposes they are depicted here nonproportionately. As cholesterol concentration in gallbladder bile increases principally from hepatic hypersecretion of cholesterol, the true supersaturated state forms transiently. Supersaturated bile usually implies that phase separation of excess cholesterol from micelles has occurred, forming unilamellar vesicles with biliary phospholipids (mostly >95% phosphatidylcholine). In the most common nucleation sequence, unilamellar vesicles fuse to form multilamellar vesicles, or liquid crystals, which are visible by low-power polarizing microscopy. From these, plate-like cholesterol monohydrate crystals (solid cholesterol crystals) nucleate heterogeneously, usually in a mucin gel. The dotted arrow indicates how cholesterol can occasionally phase separate directly from supersaturated micelles. The solid resulting cholesterol monohydrate crystals are a polymorph of the classic cholesterol monohydrate plates into which they transform with passage of time. Once the nucleation sequence has occurred and solid cholesterol crystals have formed, the phase sequence is not repeated if bile remains continuously supersaturated. Gallbladder dysmotility and mucin gel formation also contribute to the aggregation of the plate-like cholesterol monohydrate crystals and contribute to their agglomeration and growth into macroscopic cholesterol gallstones.
The study of cholesterol gallstones relies on multiple methodologies, including physical-chemical studies of model bile and ex vivo bile systems, biliary lipid secretory studies, animal models with particular focus on cholesterol gallstone (Lith) genes, and epidemiologic studies analyzing susceptible and resistant human populations.2,11–14 These studies provide the basis for our understanding of the epidemiology and pathogenesis of cholesterol gallstones. Formation of cholesterol gallstones is principally a polygenic trait with significant contributions from the environment.3,15 Although these studies provide a strong basis for our understanding of the etiology and pathogenesis of cholesterol gallstones, many issues remain unresolved. For example, why do so few individuals with phase-separated and aggregated cholesterol monohydrate crystals (such as the biliary sludge that is frequently observed in pregnant women) proceed to gallstones?8–10 Additionally, why do genetically identical mouse models challenged with the same lithogenic diet but housed at different research institutions display such marked differences in their proclivity to form cholesterol gallstones?16
We proposed that the differences in the prevalence of gallstones in genetically identical mice might arise from differences in their colonization status with gastrointestinal microbes. Initially, we analyzed the ability of enterohepatic Helicobacter spp to contribute to lithogenicity and identified several enterohepatic Helicobacter spp that contributed to cholesterol gallstone nucleation and other Helicobacter strains that did not.16,17 The host immune system might be the primary mechanism whereby these organisms (and perhaps others) promote cholesterol gallstone formation as well as mucin gel and gallbladder inflammation. Subsequent studies showed that the response of the adaptive immune system was important, and probably essential, because Rag-deficient mice, which lack mature T and B cells, rarely (<10%) develop cholesterol gallstones.18 A number of previous studies showed invariably that gallbladder inflammation occurred concomitantly with cholesterol supersaturation and gallstone formation; none, however, conclusively showed that this process was a primary contributing factor rather than a secondary effect of the lithogenic process.19–23
This review focuses on the immune aspects of the pathogenesis of cholesterol gallstones and examines the role of infection, inflammation, and the response of the immune system during the formation of cholesterol gallstones. There is an extensive body of immunity literature related to cholesterol gallstones as well as a number of studies involving mouse models of infection, inflammation, and immunity. This review describes the critical contributions of the gallbladder epithelium and the immune system to modulation of cholesterol gallstone formation and progression, rather than the nonimmunologic factors (hypersecretion of cholesterol, alterations in cholesterol to bile salt and phospholipid ratios and concentrations, and so on) that are pivotal factors in the pathogenesis of cholesterol gallstones but have been reviewed elsewhere.3,24 Most of the immunologic factors described in this review occur in conjunction with the fundamental prolithogenic alterations in liver and bile; without these alterations, inflammation would not be sufficient to induce cholesterol supersaturation of bile and gallstone formation. The causes of biliary tree inflammation, as they relate to cholesterol gallstone disease, are far from being fully elucidated; nonetheless, the literature indicates that some noninfective inflammation occurs secondary to physical-chemical alterations of bile (cholesterol supersaturation and early stages in the nucleation and phase-separation sequences; Figure 1), whereas others occur independently of biliary lipid composition.
Biliary Epithelium and Immunity
Anatomically, the biliary tree is often divided into intrahepatic and extrahepatic portions that include the gallbladder epithelium. However, with respect to biliary tissue and disease, this distinction appears artificial when describing the interactions of the biliary epithelium with the immune system; it appears that biliary epithelial cells from either location have similar responses to immunogenic stimuli.25,26
Biliary epithelial cells participate in both innate and adaptive immunity.27–29 Briefly, the innate immune system induces no immunologic memory to an antigen but responds instead to a variety of evolutionarily conserved patterns present in foreign antigens. In addition to specific cellular subsets that include neutrophils, macrophages, eosinophils, basophils, mast cells, and natural killer cells, a variety of proteins also participate in innate immunity, including the complement cascade, cytokines, and proteins of the acute phase response.30–32 In contrast, the adaptive immune response induces “memory” to foreign antigens and is mediated by both B and T cells. Following activation, the adaptive immune response can also stimulate the production of a variety of cytokines and the recruitment of inflammatory cells.33–36 There is overlap in these 2 pathways; perturbation of either can alter the ability of the other to respond appropriately to a stimulus.36
Biliary epithelial cells express all known toll-like receptors (TLR), which mediate antigen-pattern recognition—a key response of the innate immune system.28,37–39 Additionally, myeloid differentiation protein 88 (MyD88), which is an important downstream effector of the TLR pathway, is also present in these cells.28 Biliary epithelial cells that express dominant-negative forms of MyD88 have increased susceptibility to infection with the intracellular biliary pathogen Cryptosporidium parvum, indicating that the TLR/MyD88 system functions in biliary epithelial cells.39 Additionally, C parvum infection up-regulates β-defensins in infected wild-type cells, but β-defensin production is ablated in cells with reduced levels of TLR2, TLR4, or nuclear factor κB, indicating an essential role for defensins in protection of cholangiocytes and, by implication, bile itself from infection.38
The relationship between biliary epithelial cells and the adaptive immune response requires a variety of receptor-ligand interactions that occur between epithelial cells, T cells, and “professional” antigen-presenting cells such as dendritic cells. Human biliary epithelium cell lines constitutively express HLA class I (major histocompatibility complex class I) antigens but only low levels of class II antigens (Figure 2).25,26 However, after stimulation with proinflammatory cytokines, the cells express a variety of HLA class II antigens, including HLA-DR, HLA-DP, and HLA-DQ (Figure 2).25 Essentially, this expression profile shows that biliary epithelial cells are likely to interact with both CD8+ T cells (cytotoxic T cells) and CD4+ T cells (helper T cells). In addition to these primary immune receptors, biliary epithelial cells also possess a variety of costimulatory molecules necessary for functional interaction with these immune cells, including CD40, intercellular adhesion molecule 1, lymphocyte function-associated antigen 3, and FAS (Figure 2).29,40–45 These protein expression profiles indicate that the biliary epithelium actively participates in adaptive immunity through interaction with T cells and local antigen-presenting cells.
Figure 2.
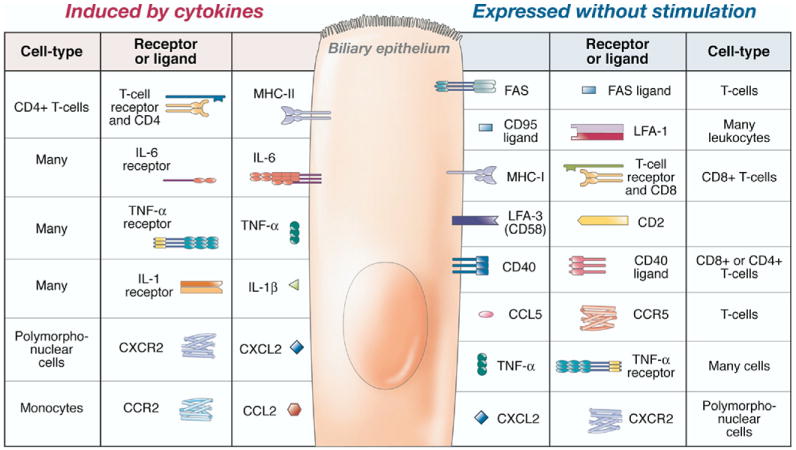
Receptors and ligands expressed by biliary epithelial cells. The receptors (displayed on the basolateral surface of the cell) and ligands (displayed extracellularly) expressed by biliary epithelial cells are shown in the unstimulated (right panel) and stimulated (left panel) state. The receptors, ligands, and cells that interact with these biliary expressed proteins are also shown. CCL, chemokine CC motif ligand; CCR, chemokine CC motif receptor; CXCL, chemokine CXC ligand; CXCR, chemokine CXC receptor; LFA, lymphocyte function-associated antigen; MHC, major histocompatibility complex.
The second aspect of adaptive immunity involves the B-cell response, mediated through immunoglobulins (Igs). All Ig classes, including IgA, IgG, and IgM, are present in low concentrations in bile.29 However, biliary epithelial cells have only minimal participation in Ig secretion. In humans, IgA is synthesized by plasma cells that line the bile ducts and then is bound to the polymeric Ig receptor at the basolateral surface of cholangiocytes.46–48 IgA is then transcytosed to the apical surface of the cells and secreted into bile. This intracellular transport system has an important role in protecting the biliary tree from intracellular pathogens by binding these antigens in transit.49 Similar to IgA secretion, IgM is transported via the polymeric Ig receptor, but in contrast, IgG is secreted using the FcRn.49–51 Igs that reach intraluminal bile aid in protecting bile from colonization with gastrointestinal pathogens.52,53
Aside from expressing the necessary receptors and downstream activators for participation in the immune response, biliary epithelial cells also secrete a variety of cytokines and undergo phenotypic changes in response to these inflammatory mediators. Cultured murine cholecystocytes (gallbladder epithelial cells) express Tnf-α, Ccl5 (RANTES), and Mip-2 (CXCL2) (Figure 2).54 Following treatment with lipopolysaccharide (LPS), these cells increase expression of Mip-2 and Tnf-α. Additionally, expression of Il-1β, Il-6, and Mcp1 is induced following exposure to LPS (Figure 2).54 Interestingly IL-10, the Th2 and T-regulatory cytokine, is not expressed in resting or LPS-stimulated gallbladder epithelium.54 It has been established that the biliary epithelium possess receptors for some of these cytokines and chemokines (including tumor necrosis factor [TNF]-α), indicating that these secreted cytokines can act in both an autocrine and a paracrine manner (Figure 2).29,55 The addition of exogenous cytokines to cultured biliary epithelial cells induces phenotypic changes in these cells, supporting an autocrine role for some of these cytokines (Figure 2). For example, the addition of TNF-α, LPS, interleukin (IL)-1α, and prostaglandin E2 to cultured human cholecystocytes alters the transport of sodium and chloride and diminishes the absorptive function of the gallbladder epithelium.56 Studies in guinea pigs (Cavia porcellus) with ligated bile ducts support these in vitro findings.57 Specifically, the instillation of IL-1 or LPS into the gallbladder induces gallbladder wall inflammation and stimulates the release of myeloperoxidase, prostaglandin E2, and water into the gallbladder lumen.57 Systemic administration of cytokines during chemotherapeutic treatment is also associated with changes in the biliary epithelium. For example, IL-2 administration to humans causes acalculous cholecystitis.58–60 The administration of transforming growth factor (TGF)-β to cultured rabbit (Oryctolagus cuniculi) gallbladder epithelium induces a mesenchymal, fibrotic phenotype.61 This in vitro phenomenon appears to have an in vivo correlate because TGF-β expression is associated with fibrosis and inflammation during formation of cholesterol gallstones in humans.62 Finally, histamines released during mast cell degranulation induce gallbladder smooth muscle contraction.63
There are several disease states associated with immune-mediated destruction of the biliary epithelium: primary biliary cirrhosis, primary sclerosing cholangitis, and graft-versus-host disease.40,42,64–67 The biliary epithelium clearly participates in cellular and humoral immunity through antigen presentation, cytokine and chemokine production, and Ig transport. Moreover, biliary epithelial cells possess all of the necessary cellular components required to participate in the innate immune system; perturbations of innate immunity appear to predispose to biliary infection.28,37–39
Role of Infection, Inflammation, and the Immune System in Formation of Cholesterol Gallstones
Igs
In vitro studies with model bile created with various concentrations of purified lipids have shown that biliary Igs, particularly IgM and IgG, promote the nucleation of solid cholesterol crystals.68,69 Interestingly, the source of the Igs appears to influence the ability of these proteins to nucleate solid cholesterol crystals from bile. IgM and IgA from commercial sources are incapable of nucleating supersaturated bile, whereas commercially available IgG has pronucleating activity.68,70 The anatomic source of the Igs also appears to influence the ability of these molecules to cause cholesterol crystal phase separation. Specifically, biliary Igs seem to promote solid cholesterol nucleation more than Igs isolated from blood.70 Of the biliary Igs, IgM most potently nucleates supersaturated bile, whereas IgG has slightly less facility and IgA has very little activity.68–70
Prospective studies utilizing animal models generally do not support a role for Igs in the pathogenesis of cholesterol gallstones. AKR or C57L mice fed a lithogenic diet undergo an initial decrease, followed by a temporary increase, in biliary IgM and IgA levels.22 In both strains of mice, the total amount of IgG remained relatively unchanged. Van Erpecum et al concluded that Igs are not likely to participate in formation of cholesterol gallstones because both gallstone-resistant (AKR) and gallstone-susceptible (C57L) mice displayed alterations in their Ig levels when placed on lithogenic diets.22 Actually, gallstone-resistant AKR mice had higher concentrations of biliary Igs than the gallstone-susceptible C57L mice.22 Although these changes appeared to negate any important role that Igs had in formation of cholesterol gallstones, data interpretation was complicated by pronounced differences in cholesterol saturation index (a quantifiable measurement of the relative amount of cholesterol in bile) values between C57L mice (∼2.0) and AKR mice (∼1.5).71 Therefore, the variable contributions of differing cholesterol saturation indexes of biliary lipids most likely confounded these results. Moreover, results of human studies indicated that Igs did not contribute to formation of cholesterol gallstones; in ex vivo bile samples from individuals with or without gallstones, there was no correlation between biliary Igs and cholesterol crystal detection times.72
One limitation of these studies is that the investigators focused primarily on the effect of Ig concentrations on solid cholesterol monohydrate crystal nucleation. Although Ig concentrations could be important, it is likely that other factors are relevant to this physical-chemical interaction. For example, the antigen specificity of the Igs may be crucial. Indeed, Fab fragments isolated from patients with cholesterol gallstones accelerated cholesterol crystal observation times in vitro, whereas commercially available Fab fragments did not.73 These data indicate that antigen-specific Fab fragments might contribute to cholesterol nucleation in vitro.73 Therefore, merely quantifying Ig concentrations in bile appears to provide limited functional information.
Recent studies have shown that mice that have only T cells (that lack B cells and Ig) develop gallstones at a greater frequency than wild-type mice. In contrast, mice possessing only B cells do not recapitulate the prevalence of gallstones observed in wild-type mice.18
Mucin Glycoproteins
Biliary mucin glycoproteins are the most thoroughly analyzed biliary proteins involved in the pathogenesis of cholesterol gallstones. Mucin genes that are expressed in human biliary epithelia include MUC1, MUC2, MUC3, MUC4, MUC5AC, MUC5B, and MUC6.74 Mucin gel accumulation appears to be important for formation of cholesterol gallstones in humans and in animal models.5,75–79 Moreover, these proteins promote solid cholesterol-phase nucleation in vitro.76,80–83 In vitro and in vivo data suggest that mucin gel accumulation precedes and promotes formation of cholesterol gallstones by nucleating cholesterol crystals from (liquid crystal) phase-separated bile.
In cultured cholangiocytes, many mucin genes are regulated by inflammatory mediators. Studies in 2 different cell culture systems have shown that mucin gel accumulation and mucin gene expression increased following exogenous addition of LPS and TNF-α. In cultured cholangiocytes, expression of Muc2 and Muc5ac increased following exposure to LPS or TNF-α (Figures 3 and 4).84 Utilizing the same cell culture system, the investigators showed that TNF-α stimulated protein kinase C, which appeared to signal increased production of Muc2 and Muc5ac (Figures 3 and 4).84 In the case of Muc3, its production was increased by LPS, but not TNF-α, possibly due to an interaction of these molecules with CD14 (Figure 5).84 In another study, LPS and other pathogen-associated molecular patterns were found to interact with TLR2 or TLR4 on the surface of biliary epithelium.85 This interaction induced expression of Cdx2, a gene whose product is associated with intestinal metaplasia of epithelial cells; CDX2 in turn induced expression of Muc2 (Figure 3).85 Furthermore, MUC2 protein expression is increased in the gallbladders of humans with cholesterol gallstones.86
Figure 3.
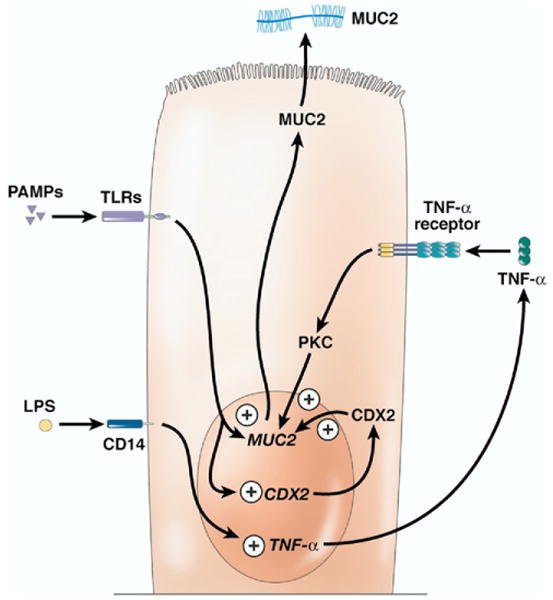
Immune system–stimulated production of MUC2. Various pathogen-associated molecular patterns (PAMPs), through their interaction with TLR molecules, increase the production of mucin 2 (MUC2). A portion of this increase seems to be due to increased production of caudal type homeobox transcription factor 2 (CDX2), which feeds back to stimulate MUC2 production. Further, LPS and TNF-α stimulate the production of MUC2. In the case of LPS, this stimulation seems to be mediated through the TNF-α pathway because inhibition of TNF-α blocks the effects of LPS treatment on MUC2 production.
Figure 4.
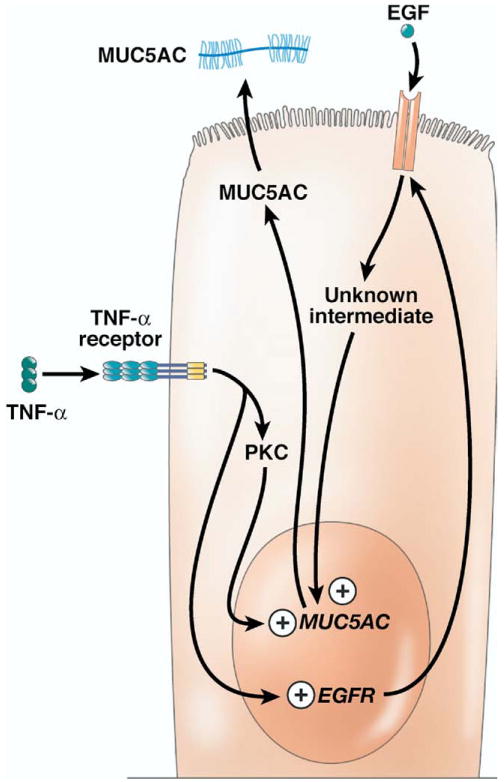
The points at which the immune system stimulates production of MUC5AC. As described for mucin 2, LPS stimulation leads to production of mucin 5AC (MUC5AC) in a TNF-α dependent manner (not shown). TNF-α acts via protein kinase C (PKC)-dependent and EGF-mediated pathways to up-regulate MUC5AC production. In the case of the EGF pathway, this up-regulation results from the production of apical EGF receptors and subsequent binding to apical EGF.
Figure 5.
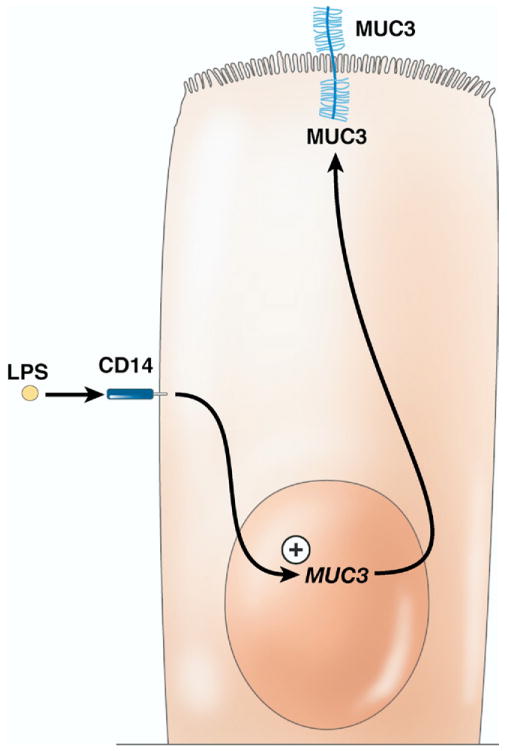
Stimulation of mucin 3 (MUC3) production by LPS via CD14 and likely TLR molecules. Stimulation of MUC3 by LPS is independent of TNF-α, in contrast to MUC2 and MUC5AC. It is currently unclear what downstream pathways are important in LPS stimulation of MUC3 production.
Choi et al87 obtained similar results utilizing cultured canine (Canis familiaris) gallbladder epithelial cells. The addition of LPS from any of 3 bacterial species stimulated mucin production in cultured dog cholecystocytes. There appeared to be differences in the ability of LPS from different bacterial species to induce mucin secretion. For example, LPS from Escherichia coli produced the greatest stimulation, whereas LPS from Pseudomonas aeruginosa stimulated the least mucin production. Furthermore, the addition of TNF-α to the culture media promoted mucin production, albeit to a lesser degree than LPS. The diminished response to TNF-α could have been due to the use of human rather than canine TNF-α. The investigators noted that the changes in mucin accumulation were not due to cytotoxicity of either LPS or TNF-α, because cell viability assays demonstrated survival of the cultured cells.87
Recent studies have shown that TNF-α regulates MUC5AC expression via an epidermal growth factor (EGF)-mediated pathway (Figure 4).88 Stimulation of gallbladder epithelium with TNF-α induced production of the EGF receptor (EGFR). EGF then binds to the EGFR and this interaction induces expression of Muc5ac, presumably through a second messenger protein88; inhibition of the EGF-EGFR pathway prevented production of MUC5AC.88
Findings from these in vitro and in vivo studies indicate that production and secretion of the mucin glycoproteins are influenced by inflammatory mediators. TNF-α, pathogen-associated molecular patterns, and their second messengers are likely to be only “the tip of the iceberg” with regard to inflammatory mediators that stimulate mucin production by the gallbladder. Perhaps a more important question to ask is not whether inflammatory mediators alter mucin production, but rather under what pathophysiological situations do inflammatory mediators exert their effects on the biliary epithelium?
Other Putative Pronucleating Biliary Proteins
Many other proteins nucleate cholesterol monohydrate crystals in vitro.72,89,90 Some of these putative pronucleating agents include haptoglobin, phospholipase C, α1-acid glycoprotein, albumin, and aminopeptidase N.72,89,90 In general, in vivo studies that analyzed the role of putative pronucleating proteins have been hampered by a number of factors: (1) large-scale proteomic analysis has only recently been undertaken to systematically analyze the protein complexity of gallbladder bile,91 (2) prior methods used to identify quantitative differences in these biliary proteins were relatively insensitive and might be inadequate to demonstrate significant differences in concentration, and (3) concentrations of individual proteins might not be important, but rather a cumulative or cooperative effect of multiple proteins could be required to promote phase separation of crystals or liquid crystals in bile and lead to formation of cholesterol gallstones. Moreover, there is little understanding of the physical-chemical interactions between these proteins. Large-scale, sensitive proteomic screens are becoming easier to perform and more widely used in pathophysiology and translational research; utilization of these techniques will likely increase our understanding of what role, if any, the myriad of secreted biliary proteins may play in formation of cholesterol gallstones.
Gallbladder Inflammation
In both animal models and humans, formation of cholesterol gallstones is preceded by histopathologic alterations in the gallbladder wall that indicate inflammation. These changes include edema, increased gallbladder wall thickness, the presence of inflammatory cells, and remodeling of the epithelium with concomitant increases in TGF-β production.19–21,62,92,93 These alterations are accompanied often by changes in gallbladder contractility and modifications in the ability of the gallbladder epithelium to transport a variety of substances.2,21,23,94,95 Furthermore, lithogenic bile can alter mechanisms associated with cytoprotection in the gallbladder muscle, demonstrated by the ability of the gallbladder muscle to respond to reactive oxygen species.96
A systematic histopathologic study23 showed that C57L mice, which are susceptible to gallstones, increase gallbladder wall thickness progressively when fed a lithogenic diet. In contrast, AKR mice, which are resistant to gallstones, display only a mild increase in thickness. Both strains accumulate subepithelial inflammatory cells and edema; however, C57L mice do so to a significantly greater degree. These changes coincide with decreases in the gallbladder expression of the aquaporin genes Aqp1 and Aqp8.23 These findings suggest that during formation of cholesterol gallstones, the gallbladder undergoes progressive changes that ultimately result in decreased motility, increased edema, and altered transport functions. The farnesoid X receptor (FXR) is a critical transcription factor that alters biliary lipid composition.92 In Fxr−/− mice, the gallbladder becomes thickened, edematous, and infiltrated with granulocytes following 1 week of lithogenic diet feeding; these changes were not observed in wild-type mice.92 Additionally, levels of potentially cytotoxic hydrophobic bile salts, such as deoxycholate conjugates, were significantly increased in Fxr−/− mice.92
It is likely that phase-separated cholesterol crystals and hydrophobic bile salts are responsible, in part, for the histopathologic changes noted in the murine gallbladder during formation of cholesterol gallstones. Ex vivo analysis of gallbladders from patients with cholesterol gallstones revealed smooth muscle cells that contained excess cholesterol in their plasma membranes.96 Furthermore, the isolated muscle tissues displayed less contractility and decreased levels of scavengers of reactive oxygen species.96 These changes appeared to be associated with dysfunction of the receptor for prostaglandin E2; prostaglandin E2 binding to its receptor usually has a cytoprotective effect. Others have reported correlations between human gallbladder muscle dysfunction, increases in the cholesterol saturation index of bile, and the histopathologic inflammation score of the gallbladder.97 Additionally, oxysterols are capable of inducing apoptosis in cultured gallbladder epithelial cells.98,99 The mechanism that mediates apoptosis appears to involve incorporation of oxysterols into the mitochondria of epithelial cells and subsequent cytochrome c activation.99
Based on these studies, it is not possible to determine whether the functional and histopathologic changes that occur in the gallbladder are due to cytotoxicity or inflammation; however, studies in the prairie dog model of cholelithogenesis indicated that inflammation is an essential element of cholesterol gallstone formation.100 Specifically, prairie dogs fed high doses of acetylsalicylic acid (aspirin) failed to form gallstones, whereas 100% of controls developed gallstones.100 Because both groups supersaturated their gallbladder biles, this study shows that cholesterol supersaturation is necessary but not sufficient for formation of cholesterol gallstones. Therefore, the proinflammatory effects of solid cholesterol itself, and likely other proinflammatory mediators, have an integral role in cholelithogenesis. However, subsequent studies failed to fully recapitulate these findings; it is difficult to make direct comparisons between these types of studies because they all used different doses of acetylsalicylic acid, dosing regimens, parameters of measured success, and lithogenic diets.101–105
Further evidence for the role of the immune system in the pathogenesis of cholesterol gallstones arose from studies with Rag-deficient mice, which do not have mature T or B cells (Rag mice). Both wild-type and Rag-deficient mice exhibited supersaturated gallbladder biles, evidenced by the phase separation of liquid crystals; however, only the wild-type mice had a high prevalence of cholesterol gallstones.18 Gallstone formation in this case appeared to be due in part to a potent Th1 immune response, which accompanied the formation of solid cholesterol monohydrate crystals in the gallbladder. These data indicate that most likely solid cholesterol crystals promote inflammation via the adaptive immune system, specifically via T-cell function.18
Hydrophobic bile salts promote inflammation and apoptosis in vitro and are likely to do so in vivo.106–108 In contrast, the hydrophilic bile acid ursodeoxycholic acid decreases mucosal inflammation and biliary protein concentrations.109 In C57L mice infected with Helicobacter spp, the relative concentration of hydrophobic bile salts (such as deoxycholate conjugates) increased in hepatic bile whereas that of hydrophilic bile salts (such as α- and β-muricholate conjugates) decreased compared with uninfected mice.110 It appears that one downstream effect of hydrophobic bile salts is the induction of proinflammatory cytokines.108 An increase in hydrophobic bile salts, principally deoxycholate conjugates, is a common finding in patients with cholesterol gallstone disease and in animal models; it is likely that bile salt–mediated inflammation is also important in cholelithogenesis.111–114
Bacterial Colonization of the Biliary Tract and Formation of Cholesterol Gallstones
Bacteria have historically been implicated in the pathogenesis of cholesterol gallstones; however, a definitive cause-and-effect relationship has never been established.115–117 A major factor contributing to this confusion is the chronic nature of cholecystolithiasis. The formation of cholesterol gallstones requires a long time, often decades. When gallstones do form, they do not disappear spontaneously except by migration into the duodenum, which is invariably followed by reformation.118–120 Furthermore, after cholesterol gallstones form in a patient, they do not always lead to symptoms; when they do, clinical expression can also take years.2,4,121 In prospective mouse models, organisms need not colonize the gallbladder to promote formation of cholesterol gallstones.16 Therefore, the identification of bacteria in bile or gallbladder tissue of patients with cholesterol gallstones does not mean that these organisms caused the gallstones. More likely, the bile, gallbladder mucosa, and biliary motility might have been altered to allow for subsequent bacterial colonization. Furthermore, bacteria that might have been present at the time of initial stone formation could have been cleared and therefore not detected by conventional culture techniques. Another issue is that some bacteria alter the composition of bile directly via β-glucuronidase, cholyl-glycyl hydrolase, phospholipase A1, or urease activities, thereby promoting calcium bilirubinate (brown pigment stone) formation.122
Despite these deficiencies, numerous studies have identified bacteria in bile, cholesterol stones, and gallbladder tissue from patients with cholesterol gallstones.123–127 Some propose that bacteria, through their β-glucuronidase activity, are responsible for nucleation and calcium salt deposition.128,129 Although this hypothesis is biologically plausible, no studies have shown conclusively that it occurs. In fact, in vitro studies utilizing E coli or calcium salts have failed to induce cholesterol crystal nucleation from supersaturated bile.130
Recently, there has been a great deal of interest in a possible connection between the gastric pathogen Helicobacter pylori and cholesterol gallstones.17,131–134 Interestingly, growth of this organism is inhibited by bile salts both in vivo and in vitro, and chemotactic assays show that bile salts actually repel the organism,17,135,136 findings that contradict the ability of H pylori to colonize a healthy gallbladder. Nonetheless, a number of groups claimed to have identified H pylori DNA in biliary tissue and gallstones.131,132,134,137 However, there are several problems with these studies. First, some samples were collected at endoscopic retrograde cholangiopancreatography134; because H pylori colonizes the gastric mucosa of more than half the world's population, the statistical chances for sample contamination by gastric H pylori are high. Additionally, 16S ribosomal RNA genus-specific primers are commonly used to identify these organisms in polymerase chain reaction (PCR) analysis, despite the fact that these primer sets can amplify other non–H pylori helicobacters.16,17 Furthermore, sequence evaluation of these primer products is generally inadequate to speciate H pylori properly, because of high homology with non–H pylori Helicobacter spp.138 For example, Avenaud et al139 identified H pylori–like organisms based on sequencing of the 16S ribosomal RNA gene from human liver samples; however, rigorous follow-up tests proved that these organisms were an unclassified Helicobacter spp.139 The development of species-specific primers to other regions of the genome may alleviate some of these concerns. Nonetheless, the efficacy of species-specific primers depends on their precise level of specificity. Finally, these organisms could invade the biliary tree following alterations in the biliary microenvironment and especially biliary dysmotility. Supporting this hypothesis is a recent study showing that H pylori DNA was present in gallbladder bile of patients with chronic cholecystitis, consistent with obstruction, but not in asymptomatic patients with cholesterol gallstones.131
In addition to the gastric pathogen H pylori, a variety of enterohepatic helicobacters (eg, Helicobacter bilis and Helicobacter hepaticus) exist.140,141 These reside in the small and large intestines and canalicular spaces of the liver of experimentally and naturally infected animals, including humans.140,141 DNA from these organisms was detected in nonwhite patients with chronic cholecystitis, gallstones, and malignant biliary tract diseases.142,143 Studies with experimental animal models indicate that these enterohepatic helicobacters promote formation of cholesterol gallstones in vivo.16 In contrast, in the same experimental model, H pylori did not promote formation of cholesterol gallstones.17 Interestingly, a follow-up study showed that urease-positive Helicobacter spp can precipitate calcium salts in vitro.144 The investigators of this study proposed that this activity might contribute, at least in part, to the ability of these organisms to promote cholesterol gallstones by inducing the formation of a calcium salt nidus, because monoinfection with urease-positive helicobacters or coinfection with at least one urease-positive helicobacter is necessary to induce cholesterol gallstones.16,144
Because PCR was used to identify Helicobacter spp and other bacteria in many of these studies, data interpretation can be challenging. PCR can detect relatively small quantities of bacterial DNA, a marker for colonization with organisms that might not be identified with standard culture methods. Unfortunately, the presence of bacterial DNA at the time of examination does not mean that it contributed to formation of gallstones. Theoretically, analyzing the core of stones might provide more reliable data on the inciting cause; however, due to the sensitive nature of PCR, the mere act of trying to isolate the core can contaminate it with nucleic acid from the external stone surface or from other sources. Additionally, no studies have analyzed the ability of cholesterol gallstones to absorb DNA.145 If stones are capable of imbibing DNA, then regardless of the analysis (be it core or external), the DNA detected might represent only the most recent DNA encountered. Furthermore, when multiple DNA fragments are present and nonspecific primers are used (designed to amplify 16S ribosomal RNA genes from multiple bacteria), there is the possibility that the DNA present at the highest concentration was preferentially amplified.146 Because PCR amplification is nonlinear, even a small bias in initial amplification can lead to logarithmic differences in the final DNA concentration. Unfortunately, the most prevalent bacteria are not necessarily the most important bacteria or might not be important at all. The use of more discriminatory PCR techniques such as those utilized to analyze complex environmental samples could circumvent this problem in the future.147,148
One of the classic bacterial pathogens of the biliary tree in humans is Salmonella enterica ser Typhi, which causes typhoid fever.149,150 This organism crosses the intestinal epithelial barrier, invades macrophages, and spreads systemically.151 After colonizing the liver, the organism can be shed into the gallbladder and chronically colonize the gallbladder wall (3%–5% of those infected).149,150 Although chronic colonization with S typhi is associated with gallstones and gallbladder cancer,149,150 it is not clear whether this organism contributes to stone formation or alternatively if the presence of gallstones promotes chronic colonization. Recent evidence indicates that S typhi forms bacterial biofilms on the surface of cholesterol gallstones.151 This biofilm formation would allow for chronic colonization and protect the organism during antibiotic treatment. Biofilm formation is dependent on the presence of bile because organisms cultured without bile do not readily form biofilms.151 This indicates that this organism is exquisitely adapted to survival in a gallbladder that contains cholesterol gallstones. Furthermore, biofilm formation is reduced when pebbles or glass beads are utilized instead of cholesterol gallstones, indicating that both bile and cholesterol gallstones are essential for biofilm formation. Moreover, colonization in the presence of cholesterol gallstones requires expression of several bacterial genetic components.151,152 These data argue that cholesterol gallstones promote chronic colonization with S typhi rather than S typhi–promoting gallstones. Regardless of whether gallstones promote colonization or vice versa, the concomitant presence of gallstones and S typhi markedly promotes the risk of gallbladder cancer, making the interaction between S typhi and cholesterol gallstones an important topic of clinical interest.149,151
It is not clear whether there is a causative role for bacteria in formation of cholesterol gallstones in humans. It is likely that some bacteria identified in humans with cholesterol gallstones are at least partially responsible for those gallstones due to their ability to promote inflammation and precipitate calcium salts. Unfortunately, most of these studies are primarily descriptive and fail to thoroughly analyze the organisms identified. For example, it is important to determine whether any of the organisms possess virulence factors that would allow for their invasion or colonization of a nondiseased biliary tree. Additional information could be garnered by culturing these organisms and utilizing them to prospectively infect mouse models. For example, when the C57L/J mouse model is acquired directly from The Jackson Laboratory (Bar Harbor, ME) and the mice are housed in microisolator cages under specific pathogen-free conditions and free of enzootic Helicobacter spp, they rarely (∼10%) develop cholesterol gallstones. However, purposeful infection with some enterohepatic strains of Helicobacter spp significantly increases the prevalence of gallstones to a high of 80%.16,17 It will prove valuable to test other putative lithogenic organisms utilizing this model system approach, bearing in mind the caveat that for the assay to be effective the organisms must be capable of colonizing the mouse strain being utilized.
Chronic Inflammatory Conditions and Gallstones
Chronic hepatitis C virus (HCV) infection is associated with cholesterol and pigment gallstone formation.153–156 Chronic hepatic damage might be responsible for this effect, leading to deranged liver function and cirrhosis.155,156 However, recent studies have shown that HCV infection, independent of liver cirrhosis, is associated with increased prevalence of cholesterol gallstones.153,154 HCV can replicate in the gallbladder epithelium,157,158 and damage of the biliary tree was noted in 22%–91% of biopsy specimens from patients with HCV.154 It is therefore possible that the association between noncirrhotic patients with HCV and gallstones depends on HCV replication in biliary epithelia, leading to local inflammation, alterations in gallbladder contraction, and inflammation-mediated alterations in mucin gene production. Interestingly, one such study reported a correlation between HCV and peaks in the prevalence of gallstones at 2 time points. The first peak occurred in patients between the ages of 31 and 40 years (7% gallstone prevalence) and the second peak occurred much later, at 61–70 years of age (16% gallstone prevalence).154 Perhaps the first peak represents the direct effect of HCV infection itself, whereas the second peak represents the effect of chronic liver dysfunction and hepatic cirrhosis. Future studies using biochemical and molecular techniques that focus on changes in the gallbladder itself could provide greater insight into the mechanistic relationships between HCV infection and formation of cholesterol gallstones.
Patients with Crohn's disease (CD) are at increased risk for the development of gallstones.159–162 Results vary regarding which factors correlate with pigment or cholesterol gallstone formation. Factors mentioned include age, female sex, site of disease, surgical resection, and duration of disease (independent of age).160,161,163 Some investigators noted that patients with CD with ileal disease or colonic resection supersaturated their bile with cholesterol, probably because of alterations in cholesterol and bile acid absorption from the small intestine.164,165 However, several studies have described a decrease in the cholesterol saturation in bile of patients with CD, especially when the ileum is involved.166,167 Furthermore, bile acid composition is altered in patients with CD, and these alterations are often characterized by a decrease in deoxycholate and an increase in ursodeoxycholate conjugates.164,166,167 These changes seem paradoxical, because ursodeoxycholate enrichment would seem to favor prevention of cholesterol gallstones.118,168,169 Unlike patients with CD, patients with ulcerative colitis are not at increased risk for cholesterol gallstones.159,161 Gallstone formation might vary between patients with ulcerative colitis and patients with CD because of differences in inflammatory changes.170–172 Although not true in all cases, CD is most commonly associated with a Th1 proinflammatory response whereas ulcerative colitis is more often associated with a Th2 response.170–172 No studies have described the gallbladder inflammatory profile of patients with either ulcerative colitis or CD; this would provide insight into the relationship of CD, inflammation, and gallstones.
Inflammation, Cholesterol Metabolism, and Gallstones
Inflammation, and more specifically the acute phase response, produces marked alterations in the metabolism of a variety of proteins and lipids; these changes are extensive and beyond the scope of this review (see Heller et al172 for a leading account). However, metabolic changes of the liver that alter cholesterol and bile acid metabolism can directly alter the composition of bile. Most studies of these phenomena were conducted using exogenous, acute administration of cytokines or LPS,173–177 which causes rapid (minutes to hours), relatively short-lived (24 hours or less) alterations in hepatic gene transcription and translation. Although these changes are significant, it is not clear if they are maintained for prolonged periods (days to weeks).176 In the case of cholesterol gallstone formation this could prove to be very important, because short-lived (hours to days) alterations in bile composition are less likely to promote disease than prolonged and sustained alterations.
With this proviso in mind, it is clear from these studies that acute inflammation does alter the hepatic metabolism of both cholesterol and bile salts. In rodents, administration of LPS or proinflammatory cytokines (IL-1, TNF) raises serum cholesterol levels and increases the production of HMG-CoA reductase at both the transcription and translational levels.173,175,178 This change augments de novo cholesterol synthesis. However, the increase in cholesterol production is rather modest because other enzymes involved in cholesterol biosynthesis, including squalene synthase and enzymes downstream of HMG-CoA reductase in the mevalonate pathway, are down-regulated.173,177–179 Some investigators propose that these changes maintain adequate cholesterol synthesis while redirecting mevalonate metabolites into non-sterol pathways.176 This hypothesis is supported by the observed increases in dolichol phosphate and glycosylated plasma proteins during the acute phase response.180,181 In contrast, primates decrease their serum cholesterol levels in response to a variety of proinflammatory mediators.182–184 Unfortunately, the mechanism underlying this change is less well understood but might arise from alterations in cholesterol secretion or ApoB synthesis.185,186
Profound alterations in bile salt production also occur under these conditions. Specifically, LPS down-regulates the classic and alternative pathways of bile salt synthesis by decreasing production of CYP7A1 (classic) and CYP7B1 or CYP27A1 (alternative) proteins.187,188 Additionally, exogenous administration of LPS decreases hepatocellular uptake and secretion of bile salts by down-regulating basolateral and apical bile salt transporters.189–193 Not surprisingly, because bile salts stimulate the biliary secretion of both phospholipids and cholesterol, administration of LPS also decreases expression of phospholipid (MDR2) and cholesterol transporters (ABCG5/ABCG8) on the canalicular (apical) membranes of the hepatocytes.190,193,194
These observations indicate that acute infections would decrease cholesterol secretion into bile due to intrahepatic cholestasis. Because formation of cholesterol gallstones requires only a relative excess of cholesterol, these changes may or may not be lithogenic. Additionally, the studies do not necessarily reflect the events following chronic immune and inflammatory stimulation. In fact, during chronic (10 weeks) infection with enterohepatic Helicobacter spp, hepatic bile salt secretion rate and bile flow increase.110 Interestingly, infection also alters the composition of the bile salt pool, rendering it more hydrophobic consistent with a proinflammatory response. These findings highlight the differences between acute studies involving exogenous administration of cytokines and LPS and those studies involving chronic infection. Furthermore, these data highlight the need for studies with pathogens that cause either or both chronic and acute infections, which could provide entirely different results than studies utilizing exogenous cytokine and LPS administration.
Lith (LITH) Genes
Large genetic screens have identified a variety of mouse Lith loci; products of genes at this loci promote cholesterol cholelithogenesis.3,93 Most of these alleles span large chromosomal regions, and the genes responsible for gallstone proclivity have not been identified (see Lyons et al93 for a detailed review of Lith alleles and their potential to encode inflammatory mediators). Unfortunately, the Lith loci identified to date occur on virtually every mouse chromosome and span relatively large regions encompassing numerous genes14; the proinflammatory genes are likely to be located in many, if not most, of these loci. Therefore, a more plausible and productive approach to study the role of inflammation in gallstone pathogenesis would be to utilize currently available transgenic and knockout mouse models, with alterations in genes important in inflammation and immune responses, and compare these with their wild-type counterparts. With the creation of these new models, the genetic loci involved in gallstone pathogenesis will become more refined and can be used to study the influence of potential roles of inflammation and immunity on formation of cholesterol gallstones.
Conclusions
The gallbladder, despite its simple structure and function, is a complex organ. Like most organs, when it becomes inflamed and damaged, it loses its ability to perform concentrative, pH modification, absorptive, and contractive functions. Unlike most other organs in the body, the gallbladder can be exposed to large concentrations of free (unesterified) cholesterol and potentially cytotoxic detergent-like bile salts. These molecules, as well as other proinflammatory molecules, can induce potent inflammatory responses under certain circumstances.23,106–108 Such inflammatory damage, through a variety of undetermined mediators, then induces gallbladder hypomotility and promotes the production of pronucleating agents.23,84,87 These effects appear to be due, at least in part, to T cells and a proinflammatory Th1 immune response.18 Furthermore, inflammation, at least in short-term studies, modifies hepatic lipid metabolism and secretion, which could promote cholelithogenesis by altering biliary cholesterol secretion and bile concentration.173,175,178 These findings raise the possibility that inflammatory mediators or their subsets are serologic biomarkers of cholesterol gallstone formation. If identified, these biomarkers could prove valuable for diagnosis and interventional therapy. Specifically, abatement of prolonged immune stimulation might prove useful in the prevention or treatment of cholesterol gallstones, either alone or in conjunction with other lipid-altering therapies.
It is important to note that many of the studies described in this review relied principally on observation using genetically homozygous mice (>99% identify among genomes). In contrast, even the most consanguineous of human populations are relatively heterogeneous. The role of inflammation in human cholesterol gallstone disease is therefore likely to be subtle and involve a wide variety of overlapping factors. Both environmental (exposure to pathogens and proinflammatory conditions) and genetic immune factors (polymorphisms in genes that promote the response to immune stimuli) are likely to contribute to pathogenesis. Clearly, much remains to be learned in this regard, and further in vitro, animal model, and human studies are likely to clarify many of these exciting possibilities.
Acknowledgments
K.J.M. was supported by grant K08-DK07728, J.G.F. was supported by grants P30-ES02109 and R01-CA067529, and M.C.C. was supported by grants R01-DK073687 and R37-DK036588.
Abbreviations used in this paper
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FXR
farnesoid X receptor
- IL
interleukin
- LPS
lipopolysaccharide
- MyD88
myeloid differentiation protein 88
- TGF
transforming growth factor
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- PCR
polymerase chain reaction
Footnotes
John P. Lynch and David C. Metz, Section Editors
The authors disclose no conflicts.
References
- 1.Carey MC, Lamont JT. Cholesterol gallstone formation. 1. Physical-chemistry of bile and biliary lipid secretion. Prog Liver Dis. 1992;10:139–163. [PubMed] [Google Scholar]
- 2.Carey MC. Pathogenesis of gallstones. Am J Surg. 1993;165:410–419. doi: 10.1016/s0002-9610(05)80932-8. [DOI] [PubMed] [Google Scholar]
- 3.Paigen B, Carey MC. Gallstones. In: King RA, Rotter JI, Motulsky AG, editors. Genetic basis of common diseases. 2nd. New York, NY: Oxford University Press; 2002. pp. 298–335. [Google Scholar]
- 4.Carey MC. Pathogenesis of gallstones. Recenti Prog Med. 1992;83:379–391. [PubMed] [Google Scholar]
- 5.Lee SP, LaMont JT, Carey MC. Role of gallbladder mucus hypersecretion in the evolution of cholesterol gallstones. J Clin Invest. 1981;67:1712–1723. doi: 10.1172/JCI110209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang DQ, Paigen B, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: physical-chemistry of gallbladder bile. J Lipid Res. 1997;38:1395–1411. [PubMed] [Google Scholar]
- 7.Rosmorduc O, Hermelin B, Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology. 2001;120:1459–1467. doi: 10.1053/gast.2001.23947. [DOI] [PubMed] [Google Scholar]
- 8.Carey MC, Cahalane MJ. Whither biliary sludge? Gastroenterology. 1988;95:508–523. doi: 10.1016/0016-5085(88)90513-6. [DOI] [PubMed] [Google Scholar]
- 9.Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit? Hepatology. 1994;20:523–525. [PubMed] [Google Scholar]
- 10.Lee SP, Maher K, Nicholls JF. Origin and fate of biliary sludge. Gastroenterology. 1988;94:170–176. doi: 10.1016/0016-5085(88)90626-9. [DOI] [PubMed] [Google Scholar]
- 11.Lammert F, Matern S. The genetic background of cholesterol gallstone formation: an inventory of human lithogenic genes. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:163–170. doi: 10.2174/1568008054064841. [DOI] [PubMed] [Google Scholar]
- 12.Portincasa P, Moschetta A, Palasciano G. Cholesterol gallstone disease. Lancet. 2006;368:230–239. doi: 10.1016/S0140-6736(06)69044-2. [DOI] [PubMed] [Google Scholar]
- 13.van Erpecum KJ. Biliary lipids, water and cholesterol gallstones. Biol Cell. 2005;97:815–822. doi: 10.1042/BC20040088. [DOI] [PubMed] [Google Scholar]
- 14.Wang DQ, Afdhal NH. Genetic analysis of cholesterol gallstone formation: searching for Lith (gallstone) genes. Curr Gastroenterol Rep. 2004;6:140–150. doi: 10.1007/s11894-004-0042-1. [DOI] [PubMed] [Google Scholar]
- 15.Katsika D, Grjibovski A, Einarsson C, et al. Genetic and environmental influences on symptomatic gallstone disease: a Swedish study of 43,141 twin pairs. Hepatology. 2005;41:1138–1143. doi: 10.1002/hep.20654. [DOI] [PubMed] [Google Scholar]
- 16.Maurer KJ, Ihrig MM, Rogers AB, et al. Identification of cholelithogenic enterohepatic helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology. 2005;128:1023–1033. doi: 10.1053/j.gastro.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 17.Maurer KJ, Rogers AB, Ge Z, et al. Helicobacter pylori and cholesterol gallstone formation in C57L/J mice: a prospective study. Am J Physiol Gastrointest Liver Physiol. 2006;290:G175–G182. doi: 10.1152/ajpgi.00272.2005. [DOI] [PubMed] [Google Scholar]
- 18.Maurer KJ, Rao VP, Ge Z, et al. T-cell function is critical for murine cholesterol gallstone formation. Gastroenterology. 2007;133:1304–1315. doi: 10.1053/j.gastro.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rege RV, Prystowsky JB. Inflammation and a thickened mucus layer in mice with cholesterol gallstones. J Surg Res. 1998;74:81–85. doi: 10.1006/jsre.1997.5213. [DOI] [PubMed] [Google Scholar]
- 20.Baig SJ, Biswas S, Das S, et al. Histopathological changes in gallbladder mucosa in cholelithiasis: correlation with chemical composition of gallstones. Trop Gastroenterol. 2002;23:25–27. [PubMed] [Google Scholar]
- 21.Hofmann AF. Pathogenesis of cholesterol gallstones. J Clin Gastroenterol. 1988;10(Suppl 2):S1–S11. [PubMed] [Google Scholar]
- 22.van Erpecum KJ, Wang DQ, Lammert F, et al. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: soluble pronucleating proteins in gallbladder and hepatic biles. J Hepatol. 2001;35:444–451. doi: 10.1016/s0168-8278(01)00173-8. [DOI] [PubMed] [Google Scholar]
- 23.van Erpecum KJ, Wang DQ, Moschetta A, et al. Gallbladder histopathology during murine gallstone formation: relation to motility and concentrating function. J Lipid Res. 2006;47:32–41. doi: 10.1194/jlr.M500180-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Strasberg SM, Harvey PR. Biliary cholesterol transport and precipitation: introduction and overview of conference. Hepatology. 1990;12:1S–5S. [PubMed] [Google Scholar]
- 25.Auth MK, Keitzer RA, Scholz M, et al. Establishment and immunological characterization of cultured human gallbladder epithelial cells. Hepatology. 1993;18:546–555. [PubMed] [Google Scholar]
- 26.Rumin S, Loreal O, Drenou B, et al. Patterns of intermediate filaments, VLA integrins and HLA antigens in a new human biliary epithelial cell line sensitive to interferon-gamma. J Hepatol. 1997;26:1287–1299. doi: 10.1016/s0168-8278(97)80464-3. [DOI] [PubMed] [Google Scholar]
- 27.Harada K, Ohba K, Ozaki S, et al. Peptide antibiotic human beta-defensin-1 and -2 contribute to antimicrobial defense of the intrahepatic biliary tree. Hepatology. 2004;40:925–932. doi: 10.1002/hep.20379. [DOI] [PubMed] [Google Scholar]
- 28.Harada K, Ohira S, Isse K, et al. Lipopolysaccharide activates nuclear factor-kappaB through toll-like receptors and related molecules in cultured biliary epithelial cells. Lab Invest. 2003;83:1657–1667. doi: 10.1097/01.lab.0000097190.56734.fe. [DOI] [PubMed] [Google Scholar]
- 29.Reynoso-Paz S, Coppel RL, Mackay IR, et al. The immunobiology of bile and biliary epithelium. Hepatology. 1999;30:351–357. doi: 10.1002/hep.510300218. [DOI] [PubMed] [Google Scholar]
- 30.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Fritz JH, Girardin SE. How Toll-like receptors and Nod-like receptors contribute to innate immunity in mammals. J Endotoxin Res. 2005;11:390–394. doi: 10.1179/096805105X76850. [DOI] [PubMed] [Google Scholar]
- 32.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 33.Banyer JL, Hamilton NH, Ramshaw IA, et al. Cytokines in innate and adaptive immunity. Rev Immunogenet. 2000;2:359–373. [PubMed] [Google Scholar]
- 34.Fagarasan S. Intestinal IgA synthesis: a primitive form of adaptive immunity that regulates microbial communities in the gut. Curr Top Microbiol Immunol. 2006;308:137–153. doi: 10.1007/3-540-30657-9_6. [DOI] [PubMed] [Google Scholar]
- 35.Firestein GS. The T cell cometh: interplay between adaptive immunity and cytokine networks in rheumatoid arthritis. J Clin Invest. 2004;114:471–474. doi: 10.1172/JCI22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flajnik MF, Du Pasquier L. Evolution of innate and adaptive immunity: can we draw a line? Trends Immunol. 2004;25:640–644. doi: 10.1016/j.it.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Harada K, Isse K, Sato Y, et al. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 2006;26:935–942. doi: 10.1111/j.1478-3231.2006.01325.x. [DOI] [PubMed] [Google Scholar]
- 38.Chen XM, O'Hara SP, Nelson JB, et al. Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappaB. J Immunol. 2005;175:7447–7456. doi: 10.4049/jimmunol.175.11.7447. [DOI] [PubMed] [Google Scholar]
- 39.Rogers KA, Rogers AB, Leav BA, et al. MyD88-dependent pathways mediate resistance to Cryptosporidium parvum infection in mice. Infect Immun. 2006;74:549–556. doi: 10.1128/IAI.74.1.549-556.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aoki CA, Bowlus CL, Gershwin ME. The immunobiology of primary sclerosing cholangitis. Autoimmun Rev. 2005;4:137–143. doi: 10.1016/j.autrev.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Adams DH, Afford SC. The role of cholangiocytes in the development of chronic inflammatory liver disease. Front Biosci. 2002;7:e276–e285. doi: 10.2741/a923. [DOI] [PubMed] [Google Scholar]
- 42.Adams DH, Afford SC. Effector mechanisms of nonsuppurative destructive cholangitis in graft-versus-host disease and allograft rejection. Semin Liver Dis. 2005;25:281–297. doi: 10.1055/s-2005-916320. [DOI] [PubMed] [Google Scholar]
- 43.Bloom S, Fleming K, Chapman R. Adhesion molecule expression in primary sclerosing cholangitis and primary biliary cirrhosis. Gut. 1995;36:604–609. doi: 10.1136/gut.36.4.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howell CD, Li J, Chen W. Role of intercellular adhesion molecule-1 and lymphocyte function-associated antigen-1 during nonsuppurative destructive cholangitis in a mouse graft-versus-host disease model. Hepatology. 1999;29:766–776. doi: 10.1002/hep.510290350. [DOI] [PubMed] [Google Scholar]
- 45.Korlipara LV, Leon MP, Rix DA, et al. Development of a flow cytometric assay to quantify lymphocyte adhesion to cytokine-stimulated human endothelial and biliary epithelial cells. J Immunol Methods. 1996;191:121–130. doi: 10.1016/0022-1759(96)00002-6. [DOI] [PubMed] [Google Scholar]
- 46.Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. 2005;206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x. [DOI] [PubMed] [Google Scholar]
- 47.Mostov KE. Transepithelial transport of immunoglobulins. Annu Rev Immunol. 1994;12:63–84. doi: 10.1146/annurev.iy.12.040194.000431. [DOI] [PubMed] [Google Scholar]
- 48.Norderhaug IN, Johansen FE, Schjerven H, et al. Regulation of the formation and external transport of secretory immunoglobulins. Crit Rev Immunol. 1999;19:481–508. [PubMed] [Google Scholar]
- 49.Rojas R, Apodaca G. Immunoglobulin transport across polarized epithelial cells. Nat Rev Mol Cell Biol. 2002;3:944–955. doi: 10.1038/nrm972. [DOI] [PubMed] [Google Scholar]
- 50.Dickinson BL, Claypool SM, D'Angelo JA, et al. Ca2+-dependent calmodulin binding to FcRn affects immunoglobulin G transport in the transcytotic pathway. Mol Biol Cell. 2008;19:414–423. doi: 10.1091/mbc.E07-07-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blumberg RS, Koss T, Story CM, et al. A major histocompatibility complex class I-related Fc receptor for IgG on rat hepatocytes. J Clin Invest. 1995;95:2397–2402. doi: 10.1172/JCI117934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown WR, Kloppel TM. The liver and IgA: immunological, cell biological and clinical implications. Hepatology. 1989;9:763–784. doi: 10.1002/hep.1840090518. [DOI] [PubMed] [Google Scholar]
- 53.McQueen CE, Boedeker EC, Le M, et al. Mucosal immune response to RDEC-1 infection: study of lamina propria antibody-producing cells and biliary antibody. Infect Immun. 1992;60:206–212. doi: 10.1128/iai.60.1.206-212.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Savard CE, Blinman TA, Choi HS, et al. Expression of cytokine and chemokine mRNA and secretion of tumor necrosis factor-alpha by gallbladder epithelial cells: response to bacterial lipopolysaccharides. BMC Gastroenterol. 2002;2:23. doi: 10.1186/1471-230X-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi JS, Zhou LS, Han Y, et al. Expression of tumor necrosis factor and its receptor in gallstone and gallbladder carcinoma tissue. Hepatobiliary Pancreat Dis Int. 2004;3:448–452. [PubMed] [Google Scholar]
- 56.Rege RV. Inflammatory cytokines alter human gallbladder epithelial cell absorption/secretion. J Gastrointest Surg. 2000;4:185–192. doi: 10.1016/s1091-255x(00)80055-4. [DOI] [PubMed] [Google Scholar]
- 57.Prystowsky JB, Rege RV. Interleukin-1 mediates guinea pig gallbladder inflammation in vivo. J Surg Res. 1997;71:123–126. [PubMed] [Google Scholar]
- 58.Chung-Park M, Kim B, Marmolya G, et al. Acalculus lymphoeosinophilic cholecystitis associated with interleukin-2 and lymphokine-activated killer cell therapy. Arch Pathol Lab Med. 1990;114:1073–1075. [PubMed] [Google Scholar]
- 59.Dickey KW, Barth RA, Stewart JA. Recurrent transient gallbladder wall thickening associated with interleukin-2 chemotherapy. J Clin Ultrasound. 1993;21:58–61. doi: 10.1002/jcu.1870210114. [DOI] [PubMed] [Google Scholar]
- 60.Powell FC, Spooner KM, Shawker TH, et al. Symptomatic interleukin-2-induced cholecystopathy in patients with HIV infection. AJR Am J Roentgenol. 1994;163:117–121. doi: 10.2214/ajr.163.1.8010196. [DOI] [PubMed] [Google Scholar]
- 61.Mori M, Miyazaki K. Factors affecting morphogenesis of rabbit gallbladder epithelial cells cultured in collagen gels. Cell Tissue Res. 2000;300:331–344. doi: 10.1007/s004410000205. [DOI] [PubMed] [Google Scholar]
- 62.Koninger J, di Mola FF, Di Sebastiano P, et al. Transforming growth factor-beta pathway is activated in cholecystolithiasis. Langenbecks Arch Surg. 2005;390:21–28. doi: 10.1007/s00423-004-0517-4. [DOI] [PubMed] [Google Scholar]
- 63.Hemming JM, Guarraci FA, Firth TA, et al. Actions of histamine on muscle and ganglia of the guinea pig gallbladder. Am J Physiol Gastrointest Liver Physiol. 2000;279:G622–G630. doi: 10.1152/ajpgi.2000.279.3.G622. [DOI] [PubMed] [Google Scholar]
- 64.He XS, Ansari AA, Ridgway WM, et al. New insights to the immunopathology and autoimmune responses in primary biliary cirrhosis. Cell Immunol. 2006;239:1–13. doi: 10.1016/j.cellimm.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 65.Alba LM, Angulo P, Lindor KD. Primary sclerosing cholangitis. Minerva Gastroenterol Dietol. 2002;48:99–113. [PubMed] [Google Scholar]
- 66.Cullen S, Chapman R. Primary sclerosing cholangitis. Autoimmun Rev. 2003;2:305–312. doi: 10.1016/s1568-9972(03)00030-2. [DOI] [PubMed] [Google Scholar]
- 67.Koukoulis GK, Shen J, Karademir S, et al. Cholangiocytic apoptosis in chronic ductopenic rejection. Hum Pathol. 2001;32:823–827. doi: 10.1053/hupa.2001.26465. [DOI] [PubMed] [Google Scholar]
- 68.Harvey PR, Upadhya GA, Strasberg SM. Immunoglobulins as nucleating proteins in the gallbladder bile of patients with cholesterol gallstones. J Biol Chem. 1991;266:13996–14003. [PubMed] [Google Scholar]
- 69.Harvey PR, Upadhya GA. A rapid, simple high capacity cholesterol crystal growth assay. J Lipid Res. 1995;36:2054–2058. [PubMed] [Google Scholar]
- 70.Upadhya GA, Harvey PR, Strasberg SM. Effect of human biliary immunoglobulins on the nucleation of cholesterol. J Biol Chem. 1993;268:5193–5200. [PubMed] [Google Scholar]
- 71.Paigen B, Schork NJ, Svenson KL, et al. Quantitative trait loci mapping for cholesterol gallstones in AKR/J and C57L/J strains of mice. Physiol Genomics. 2000;4:59–65. doi: 10.1152/physiolgenomics.2000.4.1.59. [DOI] [PubMed] [Google Scholar]
- 72.Miquel JF, Nunez L, Amigo L, et al. Cholesterol saturation, not proteins or cholecystitis, is critical for crystal formation in human gallbladder bile. Gastroenterology. 1998;114:1016–1023. doi: 10.1016/s0016-5085(98)70322-1. [DOI] [PubMed] [Google Scholar]
- 73.Lipsett PA, Hildreth J, Kaufman HS, et al. Human gallstones contain pronucleating nonmucin glycoproteins that are immunoglobulins. Ann Surg. 1994;219:25–33. doi: 10.1097/00000658-199401000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andrianifahanana M, Moniaux N, Batra SK. Regulation of mucin expression: mechanistic aspects and implications for cancer and inflammatory diseases. Biochim Biophys Acta. 2006;1765:189–222. doi: 10.1016/j.bbcan.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 75.Lammert F, Wang DQ, Wittenburg H, et al. Lith genes control mucin accumulation, cholesterol crystallization, and gallstone formation in A/J and AKR/J inbred mice. Hepatology. 2002;36:1145–1154. doi: 10.1053/jhep.2002.36821. [DOI] [PubMed] [Google Scholar]
- 76.LaMont JT, Smith BF, Moore JR. Role of gallbladder mucin in pathophysiology of gallstones. Hepatology. 1984;4:51S–56S. doi: 10.1002/hep.1840040809. [DOI] [PubMed] [Google Scholar]
- 77.Lee KT, Liu TS. Mucin gene expression in gallbladder epithelium. J Formos Med Assoc. 2002;101:762–768. [PubMed] [Google Scholar]
- 78.Wang HH, Afdhal NH, Gendler SJ, et al. Targeted disruption of the murine mucin gene 1 decreases susceptibility to cholesterol gallstone formation. J Lipid Res. 2004;45:438–447. doi: 10.1194/jlr.M300468-JLR200. [DOI] [PubMed] [Google Scholar]
- 79.Wang HH, Afdhal NH, Gendler SJ, et al. Evidence that gallbladder epithelial mucin enhances cholesterol cholelithogenesis in MUC1 transgenic mice. Gastroenterology. 2006;131:210–222. doi: 10.1053/j.gastro.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 80.Afdhal NH, Niu N, Gantz D, et al. Bovine gallbladder mucin accelerates cholesterol monohydrate crystal growth in model bile. Gastroenterology. 1993;104:1515–1523. doi: 10.1016/0016-5085(93)90364-i. [DOI] [PubMed] [Google Scholar]
- 81.Levy PF, Smith BF, LaMont JT. Human gallbladder mucin accelerates nucleation of cholesterol in artificial bile. Gastroenterology. 1984;87:270–275. [PubMed] [Google Scholar]
- 82.Smith BF. Gallbladder mucin as a pronucleating agent for cholesterol monohydrate crystals in bile. Hepatology. 1990;12:183S–186S. 186S–188S. discussion. [PubMed] [Google Scholar]
- 83.Yamasaki T, Chijiiwa K, Endo M. Isolation of mucin from human hepatic bile and its induced effects on precipitation of cholesterol and calcium carbonate in vitro. Dig Dis Sci. 1993;38:909–915. doi: 10.1007/BF01295919. [DOI] [PubMed] [Google Scholar]
- 84.Zen Y, Harada K, Sasaki M, et al. Lipopolysaccharide induces overexpression of MUC2 and MUC5AC in cultured biliary epithelial cells: possible key phenomenon of hepatolithiasis. Am J Pathol. 2002;161:1475–1484. doi: 10.1016/S0002-9440(10)64423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ikeda H, Sasaki M, Ishikawa A, et al. Interaction of Toll-like receptors with bacterial components induces expression of CDX2 and MUC2 in rat biliary epithelium in vivo and in culture. Lab Invest. 2007;87:559–571. doi: 10.1038/labinvest.3700556. [DOI] [PubMed] [Google Scholar]
- 86.Sakamoto H, Mutoh H, Ido K, et al. A close relationship between intestinal metaplasia and Cdx2 expression in human gallbladders with cholelithiasis. Hum Pathol. 2007;38:66–71. doi: 10.1016/j.humpath.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 87.Choi J, Klinkspoor JH, Yoshida T, et al. Lipopolysaccharide from Escherichia coli stimulates mucin secretion by cultured dog gallbladder epithelial cells. Hepatology. 1999;29:1352–1357. doi: 10.1002/hep.510290515. [DOI] [PubMed] [Google Scholar]
- 88.Finzi L, Barbu V, Burgel PR, et al. MUC5AC, a gel-forming mucin accumulating in gallstone disease, is overproduced via an epidermal growth factor receptor pathway in the human gallbladder. Am J Pathol. 2006;169:2031–2041. doi: 10.2353/ajpath.2006.060146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Little TE, Madani H, Lee SP, et al. Lipid vesicle fusion induced by phospholipase C activity in model bile. J Lipid Res. 1993;34:211–217. [PubMed] [Google Scholar]
- 90.Offner GD, Gong D, Afdhal NH. Identification of a 130-kilodalton human biliary concanavalin A binding protein as aminopeptidase N. Gastroenterology. 1994;106:755–762. doi: 10.1016/0016-5085(94)90712-9. [DOI] [PubMed] [Google Scholar]
- 91.Zhou H, Chen B, Li RX, et al. Large-scale identification of human biliary proteins from a cholesterol stone patient using a proteomic approach. Rapid Commun Mass Spectrom. 2005;19:3569–3578. doi: 10.1002/rcm.2207. [DOI] [PubMed] [Google Scholar]
- 92.Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med. 2004;10:1352–1358. doi: 10.1038/nm1138. [DOI] [PubMed] [Google Scholar]
- 93.Lyons MA, Wittenburg H. Susceptibility to cholesterol gallstone formation: Evidence that LITH genes also encode immune-related factors. Biochim Biophys Acta. 2006;1761:1133–1147. doi: 10.1016/j.bbalip.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 94.Portincasa P, Di Ciaula A, Vendemiale G, et al. Gallbladder motility and cholesterol crystallization in bile from patients with pigment and cholesterol gallstones. Eur J Clin Invest. 2000;30:317–324. doi: 10.1046/j.1365-2362.2000.00639.x. [DOI] [PubMed] [Google Scholar]
- 95.Lee SP. Lessons from experimental cholelithiasis: gallbladder and mucosa, nonsteroidal antiinflammatory drugs, and gallstones. Gastroenterology. 1991;101:857–860. doi: 10.1016/0016-5085(91)90550-5. [DOI] [PubMed] [Google Scholar]
- 96.Xiao ZL, Amaral J, Biancani P, et al. Impaired cytoprotective function of muscle in human gallbladders with cholesterol stones. Am J Physiol Gastrointest Liver Physiol. 2005;288:G525–G532. doi: 10.1152/ajpgi.00261.2004. [DOI] [PubMed] [Google Scholar]
- 97.van de Heijning BJ, van de Meeberg PC, Portincasa P, et al. Effects of ursodeoxycholic acid therapy on in vitro gallbladder contractility in patients with cholesterol gallstones. Dig Dis Sci. 1999;44:190–196. doi: 10.1023/a:1026635124115. [DOI] [PubMed] [Google Scholar]
- 98.Haigh WG, Lee SP. Identification of oxysterols in human bile and pigment gallstones. Gastroenterology. 2001;121:118–123. doi: 10.1053/gast.2001.25513. [DOI] [PubMed] [Google Scholar]
- 99.Seo DW, Choi HS, Lee SP, et al. Oxysterols from human bile induce apoptosis of canine gallbladder epithelial cells in monolayer culture. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1247–G1256. doi: 10.1152/ajpgi.00013.2004. [DOI] [PubMed] [Google Scholar]
- 100.Lee SP, Carey MC, LaMont JT. Aspirin prevention of cholesterol gallstone formation in prairie dogs. Science. 1981;211:1429–1431. doi: 10.1126/science.7466399. [DOI] [PubMed] [Google Scholar]
- 101.Adamek HE, Buttmann A, Weber J, et al. Can aspirin prevent gallstone recurrence after successful extracorporeal shockwave lithotripsy? Scand J Gastroenterol. 1994;29:355–359. doi: 10.3109/00365529409094849. [DOI] [PubMed] [Google Scholar]
- 102.Broomfield PH, Chopra R, Sheinbaum RC, et al. Effects of ursodeoxycholic acid and aspirin on the formation of lithogenic bile and gallstones during loss of weight. N Engl J Med. 1988;319:1567–1572. doi: 10.1056/NEJM198812153192403. [DOI] [PubMed] [Google Scholar]
- 103.Cohen BI, Mosbach EH, Ayyad N, et al. Aspirin does not inhibit cholesterol cholelithiasis in two established animal models. Gastroenterology. 1991;101:1109–1116. doi: 10.1016/0016-5085(91)90741-3. [DOI] [PubMed] [Google Scholar]
- 104.Kurata JH, Marks J, Abbey D. One gram of aspirin per day does not reduce risk of hospitalization for gallstone disease. Dig Dis Sci. 1991;36:1110–1115. doi: 10.1007/BF01297455. [DOI] [PubMed] [Google Scholar]
- 105.Li YF, Russell DH, Myers SI, et al. Gallbladder contractility in aspirin- and cholesterol-fed prairie dogs. Gastroenterology. 1994;106:1662–1667. doi: 10.1016/0016-5085(94)90424-3. [DOI] [PubMed] [Google Scholar]
- 106.Araki Y, Andoh A, Bamba H, et al. The cytotoxicity of hydrophobic bile acids is ameliorated by more hydrophilic bile acids in intestinal cell lines IEC-6 and Caco-2. Oncol Rep. 2003;10:1931–1936. [PubMed] [Google Scholar]
- 107.Yui S, Saeki T, Kanamoto R, et al. Characteristics of apoptosis in HCT116 colon cancer cells induced by deoxycholic acid. J Biochem (Tokyo) 2005;138:151–157. doi: 10.1093/jb/mvi106. [DOI] [PubMed] [Google Scholar]
- 108.Lamireau T, Zoltowska M, Levy E, et al. Effects of bile acids on biliary epithelial cells: proliferation, cytotoxicity, and cytokine secretion. Life Sci. 2003;72:1401–1411. doi: 10.1016/s0024-3205(02)02408-6. [DOI] [PubMed] [Google Scholar]
- 109.Kano M, Shoda J, Irimura T, et al. Effects of long-term ursodeoxycholate administration on expression levels of secretory low-molecular-weight phospholipases A2 and mucin genes in gallbladders and biliary composition in patients with multiple cholesterol stones. Hepatology. 1998;28:302–313. doi: 10.1002/hep.510280204. [DOI] [PubMed] [Google Scholar]
- 110.Maurer KJ. A systematic evaluation of the role of infection, immunity and inflammation in cholesterol gallstone pathogenesis [doctoral thesis] Cambridge, MA: Massachusetts Institute of Technology; 2007. p. 159. [Google Scholar]
- 111.Paumgartner G, Pauletzki J, Sackmann M. Ursodeoxycholic acid treatment of cholesterol gallstone disease. Scand J Gastroenterol Suppl. 1994;204:27–31. doi: 10.3109/00365529409103622. [DOI] [PubMed] [Google Scholar]
- 112.Paumgartner G, Sauerbruch T. Gallstones: pathogenesis. Lancet. 1991;338:1117–1121. doi: 10.1016/0140-6736(91)91972-w. [DOI] [PubMed] [Google Scholar]
- 113.Henkel A, Wei Z, Cohen DE, et al. Mice overexpressing hepatic Abcb11 rapidly develop cholesterol gallstones. Mamm Genome. 2005;16:903–908. doi: 10.1007/s00335-004-2465-2. [DOI] [PubMed] [Google Scholar]
- 114.Portincasa P, van de Meeberg P, van Erpecum KJ, et al. An update on the pathogenesis and treatment of cholesterol gallstones. Scand J Gastroenterol Suppl. 1997;223:60–69. [PubMed] [Google Scholar]
- 115.Merselis JG, Jr, Kaye D, Connolly CS, et al. The typhoid carrier state: quantitative bacteriology and preliminary observations on therapy. East Afr Med J. 1964;41:219–227. [PubMed] [Google Scholar]
- 116.Rains AJ, Barson GJ, Crawford N, et al. A chemical and bacteriological study of gallstones. The presence of an actinomycete. Lancet. 1960;2:614–618. doi: 10.1016/s0140-6736(60)91691-3. [DOI] [PubMed] [Google Scholar]
- 117.Scott AJ, Khan GA. Origin of bacteria in bileduct bile. Lancet. 1967;2:790–792. doi: 10.1016/s0140-6736(67)92231-3. [DOI] [PubMed] [Google Scholar]
- 118.Salen G. Gallstone dissolution therapy with ursodiol. Efficacy and safety. Dig Dis Sci. 1989;34:39S–43S. doi: 10.1007/BF01536661. [DOI] [PubMed] [Google Scholar]
- 119.Hellstern A, Leuschner U, Benjaminov A, et al. Dissolution of gallbladder stones with methyl tert-butyl ether and stone recurrence: a European survey. Dig Dis Sci. 1998;43:911–920. doi: 10.1023/a:1018811409538. [DOI] [PubMed] [Google Scholar]
- 120.Pauletzki J, Holl J, Sackmann M, et al. Gallstone recurrence after direct contact dissolution with methyl tert-butyl ether. Dig Dis Sci. 1995;40:1775–1781. doi: 10.1007/BF02212701. [DOI] [PubMed] [Google Scholar]
- 121.Lamont JT, Carey MC. Cholesterol gallstone formation. 2. Pathobiology and pathomechanics. Prog Liver Dis. 1992;10:165–191. [PubMed] [Google Scholar]
- 122.Cahalane MJ, Neubrand MW, Carey MC. Physical-chemical pathogenesis of pigment gallstones. Semin Liver Dis. 1988;8:317–328. doi: 10.1055/s-2008-1040553. [DOI] [PubMed] [Google Scholar]
- 123.Lee DK, Tarr PI, Haigh WG, et al. Bacterial DNA in mixed cholesterol gallstones. Am J Gastroenterol. 1999;94:3502–3506. doi: 10.1111/j.1572-0241.1999.01614.x. [DOI] [PubMed] [Google Scholar]
- 124.Swidsinski A, Lee SP. The role of bacteria in gallstone pathogenesis. Front Biosci. 2001;6:E93–E103. doi: 10.2741/swidsinski. [DOI] [PubMed] [Google Scholar]
- 125.Swidsinski A, Ludwig W, Pahlig H, et al. Molecular genetic evidence of bacterial colonization of cholesterol gallstones. Gastroenterology. 1995;108:860–864. doi: 10.1016/0016-5085(95)90461-1. [DOI] [PubMed] [Google Scholar]
- 126.Wu XT, Xiao LJ, Li XQ, et al. Detection of bacterial DNA from cholesterol gallstones by nested primers polymerase chain reaction. World J Gastroenterol. 1998;4:234–237. doi: 10.3748/wjg.v4.i3.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kawai M, Iwahashi M, Uchiyama K, et al. Gram-positive cocci are associated with the formation of completely pure cholesterol stones. Am J Gastroenterol. 2002;97:83–88. doi: 10.1111/j.1572-0241.2002.05425.x. [DOI] [PubMed] [Google Scholar]
- 128.Vitetta L, Sali A, Moritz V, et al. Bacteria and gallstone nucleation. Aust N Z J Surg. 1989;59:571–577. doi: 10.1111/j.1445-2197.1989.tb01633.x. [DOI] [PubMed] [Google Scholar]
- 129.Vitetta L, Best SP, Sali A. Single and multiple cholesterol gallstones and the influence of bacteria. Med Hypotheses. 2000;55:502–506. doi: 10.1054/mehy.2000.1101. [DOI] [PubMed] [Google Scholar]
- 130.Whiting MJ, Watts JM. Cholesterol gallstone pathogenesis: a study of potential nucleating agents for cholesterol crystal formation in bile. Clin Sci (Lond) 1985;68:589–596. doi: 10.1042/cs0680589. [DOI] [PubMed] [Google Scholar]
- 131.Farshad S, Alborzi A, Malek Hosseini SA, et al. Identification of Helicobacter pylori DNA in Iranian patients with gallstones. Epidemiol Infect. 2004;132:1185–1189. doi: 10.1017/s0950268804002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Monstein HJ, Jonsson Y, Zdolsek J, et al. Identification of Helicobacter pylori DNA in human cholesterol gallstones. Scand J Gastroenterol. 2002;37:112–119. doi: 10.1080/003655202753387455. [DOI] [PubMed] [Google Scholar]
- 133.Myung SJ, Kim MH, Shim KN, et al. Detection of Helicobacter pylori DNA in human biliary tree and its association with hepatolithiasis. Dig Dis Sci. 2000;45:1405–1412. doi: 10.1023/a:1005572507572. [DOI] [PubMed] [Google Scholar]
- 134.Silva CP, Pereira-Lima JC, Oliveira AG, et al. Association of the presence of Helicobacter in gallbladder tissue with cholelithiasis and cholecystitis. J Clin Microbiol. 2003;41:5615–5618. doi: 10.1128/JCM.41.12.5615-5618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mathai E, Arora A, Cafferkey M, et al. The effect of bile acids on the growth and adherence of Helicobacter pylori. Aliment Pharmacol Ther. 1991;5:653–658. doi: 10.1111/j.1365-2036.1991.tb00533.x. [DOI] [PubMed] [Google Scholar]
- 136.Worku ML, Karim QN, Spencer J, et al. Chemotactic response of Helicobacter pylori to human plasma and bile. J Med Microbiol. 2004;53:807–811. doi: 10.1099/jmm.0.45636-0. [DOI] [PubMed] [Google Scholar]
- 137.Kawaguchi M, Saito T, Ohno H, et al. Bacteria closely resembling Helicobacter pylori detected immunohistologically and genetically in resected gallbladder mucosa. J Gastroenterol. 1996;31:294–298. doi: 10.1007/BF02389534. [DOI] [PubMed] [Google Scholar]
- 138.O'Rourke JL, Solnick JV, Neilan BA, et al. Description of ‘Candidatus Helicobacter heilmannii’ based on DNA sequence analysis of 16S rRNA and urease genes. Int J Syst Evol Microbiol. 2004;54:2203–2211. doi: 10.1099/ijs.0.63117-0. [DOI] [PubMed] [Google Scholar]
- 139.Avenaud P, Marais A, Monteiro L, et al. Detection of Helicobacter species in the liver of patients with and without primary liver carcinoma. Cancer. 2000;89:1431–1439. [PubMed] [Google Scholar]
- 140.Fox J. Enterohepatic Helicobacters: natural and experimental models. Ital J Gastroenterol Hepatol. 1998;30(Suppl 3):S264–S269. [PubMed] [Google Scholar]
- 141.Solnick JV, Schauer DB. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin Microbiol Rev. 2001;14:59–97. doi: 10.1128/CMR.14.1.59-97.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fox JG, Dewhirst FE, Shen Z, et al. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology. 1998;114:755–763. doi: 10.1016/s0016-5085(98)70589-x. [DOI] [PubMed] [Google Scholar]
- 143.Matsukura N, Yokomuro S, Yamada S, et al. Association between Helicobacter bilis in bile and biliary tract malignancies: H. bilis in bile from Japanese and Thai patients with benign and malignant diseases in the biliary tract. Jpn J Cancer Res. 2002;93:842–847. doi: 10.1111/j.1349-7006.2002.tb01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Belzer C, Kusters JG, Kuipers EJ, et al. Urease induced calcium precipitation by Helicobacter species may initiate gallstone formation. Gut. 2006;55:1678–1679. doi: 10.1136/gut.2006.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sanabria JR, Upadhya GA, Harvey RP, et al. Diffusion of substances into human cholesterol gallstones. Gastroenterology. 1994;106:749–754. doi: 10.1016/0016-5085(94)90711-0. [DOI] [PubMed] [Google Scholar]
- 146.Bruce IJ. Nucleic acid amplification mediated microbial identification. Sci Prog. 1993;77:183–206. [PubMed] [Google Scholar]
- 147.Davis RE, Jomantiene R, Kalvelyte A, et al. Differential amplification of sequence heterogeneous ribosomal RNA genes and classification of the ‘Fragaria multicipita’ phytoplasma. Microbiol Res. 2003;158:229–236. doi: 10.1078/0944-5013-00201. [DOI] [PubMed] [Google Scholar]
- 148.King S, McCord BR, Riefler RG. Capillary electrophoresis single-strand conformation polymorphism analysis for monitoring soil bacteria. J Microbiol Methods. 2005;60:83–92. doi: 10.1016/j.mimet.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 149.Dutta U, Garg PK, Kumar R, et al. Typhoid carriers among patients with gallstones are at increased risk for carcinoma of the gallbladder. Am J Gastroenterol. 2000;95:784–787. doi: 10.1111/j.1572-0241.2000.01860.x. [DOI] [PubMed] [Google Scholar]
- 150.Lai CW, Chan RC, Cheng AF, et al. Common bile duct stones: a cause of chronic salmonellosis. Am J Gastroenterol. 1992;87:1198–1199. [PubMed] [Google Scholar]
- 151.Prouty AM, Schwesinger WH, Gunn JS. Biofilm formation and interaction with the surfaces of gallstones by Salmonella spp. Infect Immun. 2002;70:2640–2649. doi: 10.1128/IAI.70.5.2640-2649.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Prouty AM, Gunn JS. Comparative analysis of Salmonella enterica serovar Typhimurium biofilm formation on gallstones and on glass. Infect Immun. 2003;71:7154–7158. doi: 10.1128/IAI.71.12.7154-7158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bini EJ, McGready J. Prevalence of gallbladder disease among persons with hepatitis C virus infection in the United States. Hepatology. 2005;41:1029–1036. doi: 10.1002/hep.20647. [DOI] [PubMed] [Google Scholar]
- 154.Chang TS, Lo SK, Shyr HY, et al. Hepatitis C virus infection facilitates gallstone formation. J Gastroenterol Hepatol. 2005;20:1416–1421. doi: 10.1111/j.1440-1746.2005.03915.x. [DOI] [PubMed] [Google Scholar]
- 155.Elzouki AN, Nilsson S, Nilsson P, et al. The prevalence of gallstones in chronic liver disease is related to degree of liver dysfunction. Hepatogastroenterology. 1999;46:2946–2950. [PubMed] [Google Scholar]
- 156.Sheen IS, Liaw YF. The prevalence and incidence of cholecystolithiasis in patients with chronic liver diseases: a prospective study. Hepatology. 1989;9:538–540. doi: 10.1002/hep.1840090405. [DOI] [PubMed] [Google Scholar]
- 157.Loriot MA, Bronowicki JP, Lagorce D, et al. Permissiveness of human biliary epithelial cells to infection by hepatitis C virus. Hepatology. 1999;29:1587–1595. doi: 10.1002/hep.510290527. [DOI] [PubMed] [Google Scholar]
- 158.Nouri Aria KT, Sallie R, Sangar D, et al. Detection of genomic and intermediate replicative strands of hepatitis C virus in liver tissue by in situ hybridization. J Clin Invest. 1993;91:2226–2234. doi: 10.1172/JCI116449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bargiggia S, Maconi G, Elli M, et al. Sonographic prevalence of liver steatosis and biliary tract stones in patients with inflammatory bowel disease: study of 511 subjects at a single center. J Clin Gastroenterol. 2003;36:417–420. doi: 10.1097/00004836-200305000-00012. [DOI] [PubMed] [Google Scholar]
- 160.Fraquelli M, Losco A, Visentin S, et al. Gallstone disease and related risk factors in patients with Crohn disease: analysis of 330 consecutive cases. Arch Intern Med. 2001;161:2201–2204. doi: 10.1001/archinte.161.18.2201. [DOI] [PubMed] [Google Scholar]
- 161.Kratzer W, Haenle MM, Mason RA, et al. Prevalence of cholelithiasis in patients with chronic inflammatory bowel disease. World J Gastroenterol. 2005;11:6170–6175. doi: 10.3748/wjg.v11.i39.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Whorwell PJ, Hawkins R, Dewbury K, et al. Ultrasound survey of gallstones and other hepatobiliary disorders in patients with Crohn's disease. Dig Dis Sci. 1984;29:930–933. doi: 10.1007/BF01312482. [DOI] [PubMed] [Google Scholar]
- 163.Hutchinson R, Tyrrell PN, Kumar D, et al. Pathogenesis of gall stones in Crohn's disease: an alternative explanation. Gut. 1994;35:94–97. doi: 10.1136/gut.35.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pereira SP, Bain IM, Kumar D, et al. Bile composition in inflammatory bowel disease: ileal disease and colectomy, but not colitis, induce lithogenic bile. Aliment Pharmacol Ther. 2003;17:923–933. doi: 10.1046/j.1365-2036.2003.01529.x. [DOI] [PubMed] [Google Scholar]
- 165.Freudenberg F, Carey MC. Hepatobiliary manifestations in inflammatory bowel disease. In: Blumberg RS, Gangl A, Manns MP, et al., editors. Gut-Liver Interactions: Basic and Clinical Concepts. Dordrecht, The Netherlands: Springer; 2005. pp. 165–176. [Google Scholar]
- 166.Lapidus A, Einarsson C. Bile composition in patients with ileal resection due to Crohn's disease. Inflamm Bowel Dis. 1998;4:89–94. doi: 10.1002/ibd.3780040204. [DOI] [PubMed] [Google Scholar]
- 167.Lapidus A, Einarsson K. Effects of ileal resection on biliary lipids and bile acid composition in patients with Crohn's disease. Gut. 1991;32:1488–1491. doi: 10.1136/gut.32.12.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hofmann AF. Medical dissolution of gallstones by oral bile acid therapy. Am J Surg. 1989;158:198–204. doi: 10.1016/0002-9610(89)90252-3. [DOI] [PubMed] [Google Scholar]
- 169.Jungst D, Brenner G, Pratschke E, et al. Low-dose ursodeoxycholic acid prolongs cholesterol nucleation time in gallbladder bile of patients with cholesterol gallstones. J Hepatol. 1989;8:1–6. doi: 10.1016/0168-8278(89)90154-2. [DOI] [PubMed] [Google Scholar]
- 170.Bamias G, Sugawara K, Pagnini C, et al. The Th1 immune pathway as a therapeutic target in Crohn's disease. Curr Opin Investig Drugs. 2003;4:1279–1286. [PubMed] [Google Scholar]
- 171.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 172.Heller F, Fuss IJ, Nieuwenhuis EE, et al. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17:629–638. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- 173.Feingold KR, Pollock AS, Moser AH, et al. Discordant regulation of proteins of cholesterol metabolism during the acute phase response. J Lipid Res. 1995;36:1474–1482. [PubMed] [Google Scholar]
- 174.Hardardottir I, Grunfeld C, Feingold KR. Effects of endotoxin on lipid metabolism. Biochem Soc Trans. 1995;23:1013–1018. doi: 10.1042/bst0231013. [DOI] [PubMed] [Google Scholar]
- 175.Hardardottir I, Moser AH, Memon R, et al. Effects of TNF, IL-1, and the combination of both cytokines on cholesterol metabolism in Syrian hamsters. Lymphokine Cytokine Res. 1994;13:161–166. [PubMed] [Google Scholar]
- 176.Khovidhunkit W, Kim MS, Memon RA, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 177.Memon RA, Shechter I, Moser AH, et al. Endotoxin, tumor necrosis factor, and interleukin-1 decrease hepatic squalene synthase activity, protein, and mRNA levels in Syrian hamsters. J Lipid Res. 1997;38:1620–1629. [PubMed] [Google Scholar]
- 178.Feingold KR, Hardardottir I, Memon R, et al. Effect of endotoxin on cholesterol biosynthesis and distribution in serum lipoproteins in Syrian hamsters. J Lipid Res. 1993;34:2147–2158. [PubMed] [Google Scholar]
- 179.Yoo JY, Desiderio S. Innate and acquired immunity intersect in a global view of the acute-phase response. Proc Natl Acad Sci U S A. 2003;100:1157–1162. doi: 10.1073/pnas.0336385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mookerjea S, Coolbear T, Sarkar ML. Key role of dolichol phosphate in glycoprotein biosynthesis. Can J Biochem Cell Biol. 1983;61:1032–1040. doi: 10.1139/o83-132. [DOI] [PubMed] [Google Scholar]
- 181.Sarkar M, Mookerjea S. Differential effect of inflammation and dexamethasone on dolichol and dolichol phosphate synthesis. Biochem Cell Biol. 1988;66:1265–1269. doi: 10.1139/o88-146. [DOI] [PubMed] [Google Scholar]
- 182.Auerbach BJ, Parks JS. Lipoprotein abnormalities associated with lipopolysaccharide-induced lecithin: cholesterol acyltransferase and lipase deficiency. J Biol Chem. 1989;264:10264–10270. [PubMed] [Google Scholar]
- 183.Ettinger WH, Miller LD, Albers JJ, et al. Lipopolysaccharide and tumor necrosis factor cause a fall in plasma concentration of lecithin: cholesterol acyltransferase in cynomolgus monkeys. J Lipid Res. 1990;31:1099–1107. [PubMed] [Google Scholar]
- 184.Sammalkorpi K, Valtonen V, Kerttula Y, et al. Changes in serum lipoprotein pattern induced by acute infections. Metabolism. 1988;37:859–865. doi: 10.1016/0026-0495(88)90120-5. [DOI] [PubMed] [Google Scholar]
- 185.Ettinger WH, Varma VK, Sorci-Thomas M, et al. Cytokines decrease apolipoprotein accumulation in medium from Hep G2 cells. Arterioscler Thromb. 1994;14:8–13. doi: 10.1161/01.atv.14.1.8. [DOI] [PubMed] [Google Scholar]
- 186.Schectman G, Kaul S, Mueller RA, et al. The effect of interferon on the metabolism of LDLs. Arterioscler Thromb. 1992;12:1053–1062. doi: 10.1161/01.atv.12.9.1053. [DOI] [PubMed] [Google Scholar]
- 187.Feingold KR, Spady DK, Pollock AS, et al. Endotoxin, TNF, and IL-1 decrease cholesterol 7 alpha-hydroxylase mRNA levels and activity. J Lipid Res. 1996;37:223–228. [PubMed] [Google Scholar]
- 188.Memon RA, Moser AH, Shigenaga JK, et al. In vivo and in vitro regulation of sterol 27-hydroxylase in the liver during the acute phase response. potential role of hepatocyte nuclear factor-1. J Biol Chem. 2001;276:30118–126. doi: 10.1074/jbc.M102516200. [DOI] [PubMed] [Google Scholar]
- 189.Green RM, Beier D, Gollan JL. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology. 1996;111:193–198. doi: 10.1053/gast.1996.v111.pm8698199. [DOI] [PubMed] [Google Scholar]
- 190.Hartmann G, Cheung AK, Piquette-Miller M. Inflammatory cytokines, but not bile acids, regulate expression of murine hepatic anion transporters in endotoxemia. J Pharmacol Exp Ther. 2002;303:273–281. doi: 10.1124/jpet.102.039404. [DOI] [PubMed] [Google Scholar]
- 191.Moseley RH, Wang W, Takeda H, et al. Effect of endotoxin on bile acid transport in rat liver: a potential model for sepsis-associated cholestasis. Am J Physiol. 1996;271:G137–G146. doi: 10.1152/ajpgi.1996.271.1.G137. [DOI] [PubMed] [Google Scholar]
- 192.Trauner M, Arrese M, Lee H, et al. Endotoxin downregulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J Clin Invest. 1998;101:2092–2100. doi: 10.1172/JCI1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Vos TA, Hooiveld GJ, Koning H, et al. Up-regulation of the multidrug resistance genes, Mrp1 and Mdr1b, and down-regulation of the organic anion transporter, Mrp2, and the bile salt transporter, Spgp, in endotoxemic rat liver. Hepatology. 1998;28:1637–1644. doi: 10.1002/hep.510280625. [DOI] [PubMed] [Google Scholar]
- 194.Tygstrup N, Bangert K, Ott P, et al. Messenger RNA profiles in liver injury and stress: a comparison of lethal and nonlethal rat models. Biochem Biophys Res Commun. 2002;290:518–525. doi: 10.1006/bbrc.2001.6216. [DOI] [PubMed] [Google Scholar]