

Abstract
Purpose of review
The application of mutation analysis is becoming an integral part of the complete evaluation of patients with primary immunodeficiencies, and as such, clinicians caring for these patients must develop a better understanding of the utility and challenges of this important laboratory technology.
Recent findings
Genomic DNA sequencing is currently the standard approach used to characterize a possible gene mutation causing a specific primary immunodeficiency. There are clinical situations in which this approach is revealing of a genetic defect and other circumstances in which this generates a false-positive or false-negative result. One case study is presented that reviews a straightforward analysis that clarifies the genetic basis of a primary immunodeficiency, and four cases are presented that required additional studies to clarify the underlying basis of the immunodeficiency. In the latter circumstances, the rationale for additional studies is outlined and the outcome of these is presented.
Summary
The identification of a gene mutation as the underlying basis of a primary immunodeficiency begins with the evaluation of the clinical presentation focusing on the infection history so as to develop a differential diagnosis including potential genetic causes. The next step is to obtain specific laboratory studies, including immunologic function evaluation, and, based on these findings, to proceed with DNA sequencing of one or several selected candidate genes. Genomic DNA sequencing has certain limitations, and alternative follow-up approaches may be necessary to establish the molecular basis of the primary immunodeficiency in a given patient.
Keywords: bioinformatics tools, DNA sequencing, immune function, mutation analysis, primary immunodeficiency
Introduction
In 1986, the automation of the Sanger method of DNA sequencing revolutionized modern molecular biology and facilitated clinical applications of this technology [1,2]. Although next generation DNA sequencing technologies have been developed, including pyrosequencing [3] and sequencing by hybridization [4], the well refined Sanger method continues to be the mainstay in DNA sequencing for clinical mutation analysis. As this is a rapidly developing field, alternative methods will likely move into the clinical laboratory for a comprehensive analysis of genomes; however, targeted Sanger sequencing will likely remain the technology of choice in the immediate future when one or several candidate disease genes are suspected based on clinical presentation and/or family history.
As few clinicians are directly involved in the sequencing necessary to screen candidate genes, we highlight a set of analyses for confirming a genetic diagnosis using case studies of referred patients with a suspected primary immunodeficiency disorder (PIDD). This review will present the initial data generated from Sanger sequencing, in which a disease-causing mutation is easily identified as well as situations in which first-pass genomic sequencing yields an apparent negative result. The latter represents cases evaluated in our institution and include the following circumstances: successful amplification of one allele and failure to amplify the second, noncoding mutations in intronic areas (e.g. splice sites) or regulatory elements [promoter regions and polyadenylation (polyA) signal site], and mutation analysis in the presence of a pseudogene. Each of these situations can result in a false-negative or false-positive result or misclassification when using standard genomic DNA sequencing. Another potential source for a false-negative result is the inability to detect a somatic mutation when the DNA used for sequencing contains primarily wild-type DNA from unaffected cells. In the following sections, discussion will be focused on the interpretation of mutation analysis in patients with a clinical phenotype strongly suggestive of a specific PIDD. The cases presented are not inclusive of all possible issues that can arise during evaluation for possible disease-causing mutations but represent select issues that have been encountered during the evaluation of individual patients referred for a probable primary immunodeficiency.
Case study 1
This case is that of a male patient with a history of recurrent sinopulmonary bacterial infections and Pneumocystis jerovicci pneumonia found to have elevated IgM levels with markedly decreased IgG, IgA, and IgE levels. The clinical and laboratory data strongly suggest a diagnosis of X-linked hyper-IgM syndrome [XHIGM; Online Mendelian Inheritance in Man (OMIM) 308230], and genomic DNA sequencing of CD40L using exon-specific primers was undertaken.
A hemizygous missense CD40L mutation (c.530A-G, p.Y170C) was detected in the patient’s sample, and the same mutation was detected in a heterozygous state in the mother (Fig. 1). Note in reviewing the mother’s sequencing data that, in addition to the presence of a mutant peak, there is also a distinct decrease in the wild-type peak height compared with that of the wild-type control. Comparing test sequencing data with wild-type control data is critical for discriminating a true heterozygous peak from potential baseline noise. As DNA polymerases may occasionally misincorporate a base during elongation in the PCR amplification, an independent PCR product should be sequenced to confirm any mutation detected. Further, when analyzing a relative for the presence of a previously identified family mutation, it is recommended to simultaneously test an affected individual or a known carrier to confirm that the correct region was sequenced in case of typographical or laboratory error, or nonstandard mutation notation.
Figure 1. Alignment of genomic DNA sequencing data for CD40L; wild-type control (top), male patient (middle), and female carrier (bottom).
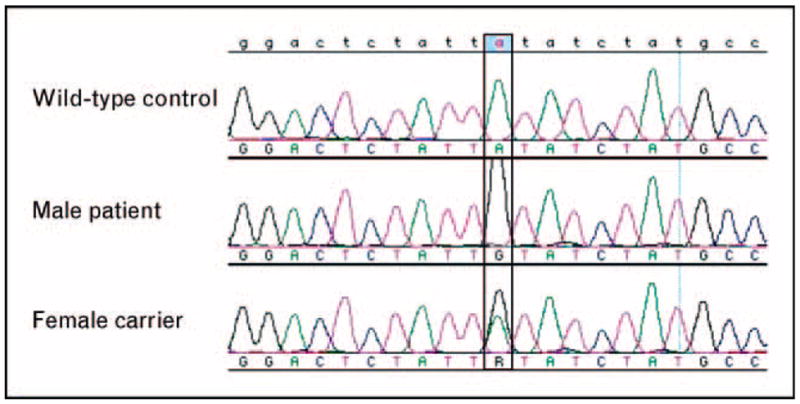
As the male patient has one X-chromosome, only the mutant allele appears in the chromatogram, whereas the female carrier with two X-chromosomes demonstrates both wild-type and mutant alleles.
The CD40L Y170C mutation has been previously described associated with the X-linked hyper-IgM syndrome in terms of its predicted location and effect [5,6]. Specifically, this mutation is located in the tumor necrosis factor (TNF) homology domain and is one of the two residues binding the three CD40L monomers into the functional, homotrimeric molecule [7]. The hydrophobic tyrosine residue contains an aromatic ring; substitution with cysteine, a small, neutral amino acid, is predicted by bioinformatic tools, such as those listed in Table 1, to be deleterious. This example illustrates how identification of a nucleotide change causing an amino acid substitution can be directly correlated with published data to confidently predict the genetic basis of disease in a patient. It also illustrates the use of bioinformatics that can help clarify the potential functional impact of a specific mutation. The following case studies focus on approaches used to expand the genetic evaluation in the setting of a negative or inconclusive initial mutation analysis to determine whether or not a gene mutation is actually present in a patient with a strongly suggestive clinical phenotype.
Table 1.
Online informatics tools and useful databases
| ConSeq | Sequence conservation | http://conseq.tau.ac.il/ |
| PolyPhen | Deleterious effects | http://coot.embl.de/PolyPhen/ |
| SIFT | Deleterious effects | http://sift.jcvi.org/ |
| OMIM | Database of human genetic disorders | http://www.ncbi.nlm.nih.gov/omim/ |
| dbSNP | Identified SNPs | http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp |
| GeneTests | Laboratories performing genetic testing | http://www.ncbi.nlm.nih.gov/sites/GeneTests/ |
| IL2RGbase | Published mutations in IL2RG | http://research.nhgri.nih.gov/scid/ |
| ALPSbase | Published mutations in Fas | http://research.nhgri.nih.gov/alps/ |
| STAT3base | Published mutations in STAT3 | http://www3.niaid.nih.gov/labs/aboutlabs/lcid/stat3base/default.htm |
| IDbases | Databases for immunodeficiency-causing mutations | http://bioinf.uta.fi/base_root/ |
| RESCUE-ESE | Exonic splicing enhancer predictor | http://genes.mit.edu/burgelab/rescue-ese/ |
| ESE-Finder | Exonic splicing enhancer predictor | http://rulai.cshl.edu/tools/ESE2/index.html |
dbSNP, single nucleotide polymorphism database; ESE, exonic splicing enhancer; IL2RG, IL-2 receptor, gamma; OMIM, Online Mendelian Inheritance in Man; SNP, single nucleotide polymorphism; STAT3, signal transducer and activator of transcription 3.
Case study 2
This case is that of an 8-year-old female patient with a history of recurrent pyogenic bacterial infections and normal immunoglobulin levels. An additional laboratory analysis demonstrates that the patient’s peripheral blood mononuclear cells (PBMCs) fail to respond to lipopolysaccharide (LPS) or IL-1β stimulation, thereby establishing that the toll-like receptor (TLR) pathway is defective. These data suggest that the most likely genetic cause for this clinical picture is a defect in IL-1 receptor-associated kinase 4 (IRAK4) (OMIM 606883) or myeloid differentiation primary response gene 88 (MYD88) (OMIM 602170), both of which encode for adaptor proteins in the TLR signaling pathways.
On the basis of these data, genomic DNA from the patient was sent to a commercial laboratory for sequencing of IRAK4. A heterozygous nonsense change in exon 8 (c.877C-T, p.Q293X) was identified in the patient’s sample and also confirmed in a sample from the father. Although IRAK4 deficiency is an autosomal recessive disorder [8], no other nucleotide change was detected in this patient or in her mother. The strength of the clinical story and laboratory data resulted in follow-up testing to determine whether a second molecular defect had been missed via standard genomic DNA sequencing due to a failure to amplify both alleles.
The 877C-T change detected in the initial sequencing is predicted to cause premature termination of the message and should result in nonsense-mediated decay of the IRAK4 mRNA encoded by that allele [9]. To confirm this, and look for potential splice mutations affecting the patient’s second allele that could have been missed by exon-specific directed genomic sequencing, we performed reverse transcription of the RNA to produce cDNA. This was followed by amplification of full-length cDNA for IRAK4. Figure 2 compares the results of the genomic and cDNA sequence analyses. Whereas genomic sequencing data (Fig. 2, middle panel) identified a heterozygous single base change when compared with the wild-type control sample (Fig. 2, top panel), the cDNA sequencing data (Fig. 2, lower panel) showed only the paternal allele harboring the 877C-T mutation and no evidence of the maternal allele at the mRNA level.
Figure 2. Alignment of wild-type genomic DNA (top), patient genomic DNA (middle), and patient cDNA (bottom) sequencing data for IL-1 receptor-associated kinase 4.
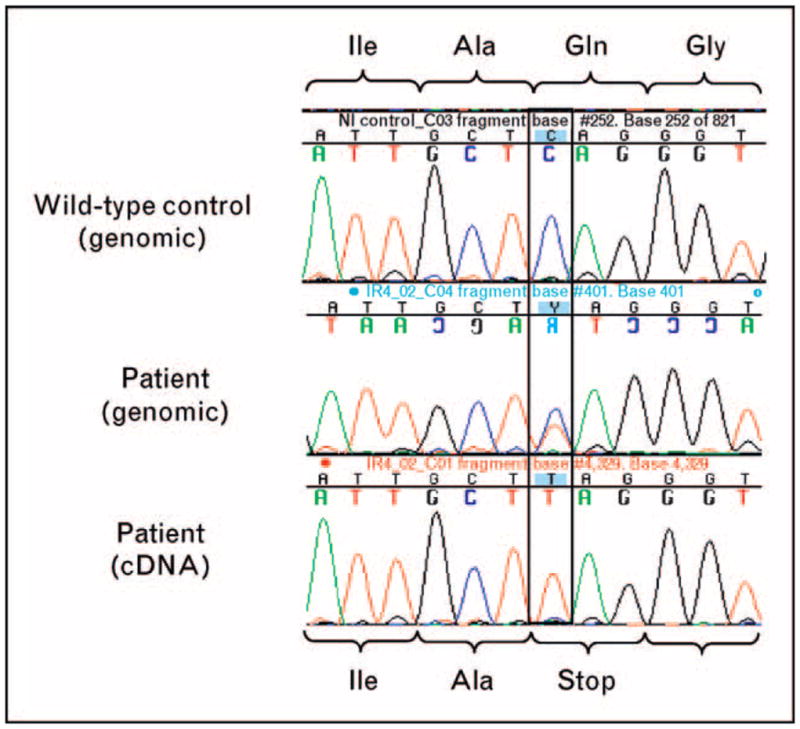
Patient genomic DNA demonstrates both wild-type and mutant alleles, whereas patient cDNA only shows the mutant allele.
Although the cDNA sequence did not reveal a specific mutation, this approach confirmed that the patient’s maternal allele is not expressed at the RNA level in the patient’s cells (but is present in the genomic DNA sample, at least at the site of the 877C-T mutation). This could be caused by a partial gene deletion, as reported in Lafora disease [10] or by other mutations preventing the allele from being expressed [11]. Nonetheless, the additional cDNA analysis established the presence of a compound, heterozygous mutation in IRAK4 and established IRAK4 deficiency as the cause of this patient’s recurrent infections.
Several approaches are available to further pursue the specific change inherited from the patient’s mother that could explain the failure of mRNA expression for IRAK4. The first is full sequencing of IRAK4 using the mother’s genomic DNA to perform single nucleotide polymorphism (SNP) haplotype analysis. If heterozygous SNPs are not detectable in a given region, this could indicate the presence of a large deletion or insertion that prevented exon-specific amplification of that allele. Microarray-based comparative genomic hybridization (array CGH) may also provide supporting evidence of a submicroscopic, genomic deletion (1 kb–10 Mb). Detection of heterozygous SNPs in other regions would indicate the presence of two alleles and may provide clues about the boundaries of the insertion or deletion, and allow long-range PCR either upstream or downstream from the SNP to be performed followed by direct sequencing. The PCR would likely yield a smaller or larger than expected amplicon in the respective case of either a deletion or insertion, provided the defect was entirely contained within the PCR amplicon. Fluorescent in-situ hybridization (FISH) analysis could also be used to determine whether there is a large insertion. This case demonstrates the importance of evaluating all of the available data. To summarize, the first step is to evaluate the clinical presentation of the patient, focusing on the infection history so as to develop a differential diagnosis including potential genetic causes. The next step is to obtain specific laboratory studies, including immunologic function evaluation, and based on these findings, to proceed with DNA sequencing of one or several selected candidate genes. At this point, it is critical to remember that genomic DNA sequencing has the aforementioned limitations, and that alternative follow-up approaches may be necessary to establish the molecular basis of the primary immunodeficiency in a given patient.
Case study 3
This case is that of a 3-month-old boy with failure to thrive, chronic diarrhea, and lymphopenia. Flow cytometric evaluation demonstrates profoundly decreased levels of T cells and natural killer (NK) cells with normal levels of B cells. Given this male patient’s T−B+NK— presentation consistent with severe combined immuno-deficiency (SCID), the most frequent genetic cause would be a mutation in IL-2 receptor, gamma (IL2RG) that encodes the cytokine receptor common gamma chain [12]. Genomic DNA was isolated, and the X-linked SCID gene, IL2RG (OMIM 308380), was sequenced to look for a hemizygous mutation. The results of this study demonstrated that the sequence of all eight exons and flanking splice sites of this gene were identical to wild type. Furthermore, sequence of genomic DNA from the mother was also identical to the reference wild-type sequence.
However, flow cytometric evaluation of Epstein–Barr virus (EBV)-transformed B cells from the patient indicated that the cell surface expression of IL-2 receptor, gamma chain (CD132) was markedly decreased. In our experience, B-cell lines normally express CD132, and this finding suggested that a defect in this protein was likely the cause of SCID in this patient. In light of the CD132 expression data, additional studies were undertaken looking at mRNA by northern blot, which demonstrated reduced levels of multiple abnormally sized mRNA species in the patient sample. Taken together, these two pieces of data suggested that the underlying cause of SCID in this patient is due to a mutation in IL2RG that was not identified using standard genomic DNA sequencing.
It has been described previously that mutations causing alternative splicing may be missed by standard genomic DNA sequencing in a number of genes, including fibrillin 1 (FBN1) [13] and fibrinogen beta chain (FGB) [14]. The finding of abnormally sized mRNA species in this patient made this a strong possibility. Therefore, we performed reverse transcriptase-PCR (RT-PCR) amplification of the coding portion of the cDNA generated from the patient’s mRNA to look for possible intronic mutations including cryptic splice sites that could allow the inclusion of an additional exon. However, no intronic mutations were detected in the patient’s or his mother’s cDNA.
As mutations in regulatory regions have been identified in several other genes including polyA site mutations in β-globin [15] and forkhead box P3 (FOXP3) [16] and minimal promoter regions of cytochrome b-245, beta polypeptide (CYBB) [17], we attempted to rule out an RNA processing mutation by amplifying and sequencing the regulatory regions of IL2RG including the promoter and polyA signal sequence. The minimal promoter region did not differ from the consensus sequence; however, the polyA signal sequence demonstrated a single base change from the consensus of AATAAA to AATAAG. Cloning and sequencing of full length IL2RG transcripts from the patient, utilizing an oligo dT primer to include the full 3′ untranslated region (UTR), demonstrated not only normal coding sequence for the gene but also multiple aberrant transcripts that continued past the mutated polyA site and utilized a polyA consensus sequence several kilobases downstream (Fig. 3). The additional sequence included in the transcripts was spliced at various locations throughout the extended 3′ UTR. Splicing occurring after the normal stop codon results in nonsense-mediated decay [9] of much but not all of the transcript, accounting for the multiple sized RNA species identified by northern blot and the trace amount of CD132 found on the surface of the patient’s B-cell line.
Figure 3. A schematic figure demonstrating the processing of extended IL2RG transcript due to polyadenylation site mutation.
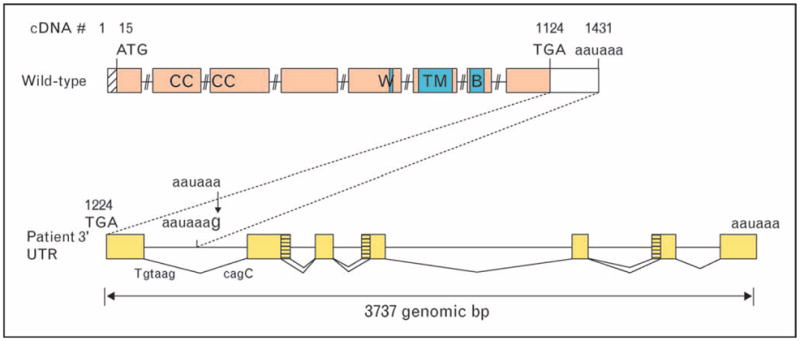
bp, base pairs; UTR, untranslated region.
This case emphasizes the importance of a combination of assays when genomic DNA sequencing does not reveal a defect in the setting of markedly diminished expression of the protein suspected as associated with the disease. In this case, use of the genomic sequencing data or the cDNA sequencing data either alone or together would still have led to a false-negative result.
Case study 4
This case is that of a 15-month-old male referred with a history of recurrent bacterial pneumonias and a clinical presentation that suggested anhidrotic ectodermal dysplasia with immune deficiency (EDA-ID; OMIM 300291). This disorder is associated with mutations in the X-linked gene encoding the nuclear factor (NF)-κB essential modulator (NEMO) [18•]. As a part of the evaluation, genomic DNA samples from the patient and his mother were PCR-amplified and sequenced with NEMO exon-specific primers. A hemizygous missense NEMO mutation (c.1075C-T, p.R359W) was detected in the patient, and the same mutation was detected in a heterozygous state in the mother (Fig. 4). As the NEMO R359W mutation has not been described in the literature, we analyzed this mutation using bioinformatics tools, including ConSeq [19], Polymorphism Phenotyping (PolyPhen) [20], and Sorting Intolerant From Tolerant (SIFT) [21]. Given a set of 20 proteins homologous to NEMO in the form of a multiple sequence alignment (MSA), ConSeq calculated the evolutionary rate at each amino acid site, thus predicting that R359 is highly conserved, exposed, and functional. Consistently, both PolyPhen and SIFT predicted that substitution of R359 by W would have a deleterious effect on protein function. Due to the presence of a NEMO pseudogene, the mutation noted above was confirmed by sequencing cDNA.
Figure 4. Alignment of nuclear factor-κB essential modulator genomic DNA sequencing data: wild-type control (top), male patient (middle), and female carrier (bottom).
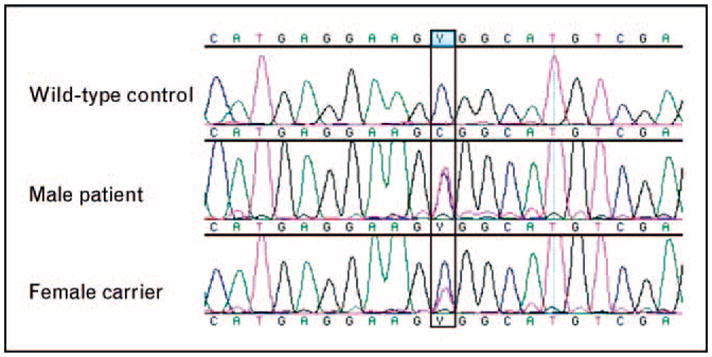
Male patient demonstrates one mutant NEMO allele and one pseudo-gene allele, whereas the female carrier demonstrates both wild-type and mutant NEMO alleles as well as two copies of the pseudogene. NEMO, nuclear factor-κB essential modulator.
When sequencing NEMO, it is important to be aware of the presence of a highly homologous NEMO pseudogene (ΔNEMO). ΔNEMO maps to the X-chromosome within a 35.5-kb duplicated fragment opposite to NEMO in orientation [22]. It spans NEMO exons 3 through 10 including intronic regions; therefore, genomic DNA primers for NEMO exons 3–10 will also amplify ΔNEMO. Two cases of molecular misdiagnosis have been reported that demonstrate the need to discriminate between the NEMO and ΔNEMO mutations [23]. Thus, to avoid pseudogene-derived false-positive results, it is critical to sequence cDNA to confirm any mutation in the presence of a pseudogene. A pseudogene also makes it difficult to distinguish hemizygosity from true heterozygosity, as the chromatogram for genomic DNA sequencing will also contain the sequence of the pseudogene. This is illustrated by the data in Fig. 4. Note how the male patient’s mutation appears heterozygous, as he has one copy of NEMO and one copy of ΔNEMO, and the mother’s mutant peak appears weaker, as she has two copies of NEMO and two copies of ΔNEMO. As a mutant allele would only constitute about 25% of the signal intensity in the sequencing data for autosomal genes and in cases of female carrier testing for X-linked disorders, a pseudogene may also lead to false-negative results due to the current detection limit of automated sequencers. Thus, in the presence of a pseudogene, it is preferable to sequence cDNA to avoid false positives and negatives leading to misdiagnosis. If only genomic DNA is available, the PCR product should be sequenced in both directions.
Case study 5
This case is that of a 12-year-old patient with a history of nonmalignant lymphadenopathy and splenomegaly starting in early childhood and idiopathic thrombocytopenic purpura (ITP) that has recurred and has been difficult to manage with standard therapy. Laboratory findings in this patient include an increase in the level of T cells that do not express CD4 or CD8 cell but do express the α-β T-cell antigen receptor [double-negative α/β T cells (DNTs)]. This cell is characteristically elevated in patients with the autoimmune lymphoproliferative syndrome (ALPS; OMIM 601859).
To confirm the diagnosis of ALPS, genomic DNA was extracted by standard methods from PBMCs. The DNA was PCR-amplified and sequenced using tumor necrosis factor superfamily, member 6 (TNFRSF6) (Fas) exon-specific primers, but no Fas mutation was detected. This left the possibility that this patient has an ALPS associated with either an alternative mutation not involving Fas (type Ib or type II), a noncharacterized form of ALPS (type III), or a Fas somatic mutation causing ALPS (type 1a–s) [24,25]. In view of the increased number of DNTs together with high levels of vitamin B12 and IL-10 found in this patient, the last alternative was viewed as very likely (I. Caminha et al., in preparation).
To examine this possibility, follow-up sequencing was performed using DNA extracted from isolated DNTs. This revealed a heterozygous splice site mutation at the invariant ag splice acceptor of Fas intron 7 (Fig. 5). Generally, the detection limit of current automated sequencers requires that the mutant signal must reach approximately 20% of total signal intensity (J.E. Niemela and K.C. Dowdell, unpublished observations). A mutant peak in the chromatogram obtained with the genomic DNA from the patient PBMCs was not distinguishable from baseline noise even upon re-review. This is most likely due to the fact that the DNTs represented approximately 8% of the total lymphocytes that is below the limit of mutation detection in our experience. Therefore, this somatic mutation was only detectable by sequencing DNA from isolated DNTs, the cells primarily affected by this process.
Figure 5. Alignment of genomic DNA sequencing data at the Fas exon 8 acceptor splice site: wild-type control (top), patient peripheral blood mononuclear cells (middle), and patient purified double-negative α/β T-cell receptor-positive T cells (bottom).
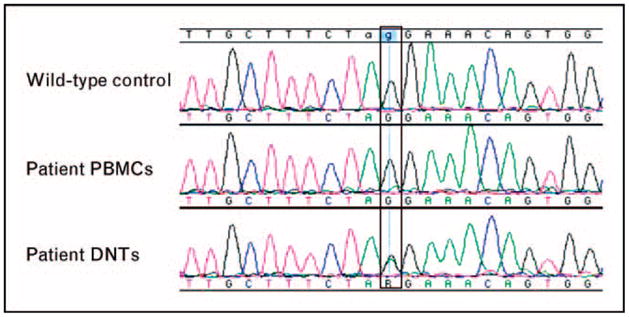
The mutant peak in the patient’s PBMCs is not distinguishable from baseline noise, and the mutation is only detectable in the chromatogram for the isolated DNTs. DNTs, double-negative α/β T cells; PBMCs, peripheral blood mononuclear cells.
This somatic splice site mutation is similar, but not identical, to a previously described somatic splice site mutation that resulted in exon 8 skipping [25]. Although this mutation would also be predicted to cause exon skipping, confirmation would be beneficial and could be accomplished by sequencing of the RT-PCR product and demonstrating both a wild-type sequence and an abnormal product missing exon 8. This case study highlights the role of somatic mutations in the pathogenesis of nonmalignant conditions that is also well described in a variety of clonal malignancies. Moreover, it demonstrates the need to consider the origin of the DNA sample when interpreting DNA sequencing results.
Discussion
Mutation analysis in the characterization of primary immunodeficiencies is now an important component of the complete evaluation of a patient particularly as affected genes associated with this group of diseases continue to increase. One important caveat to molecular mutation detection is that, even if an alteration is identified at the genomic level, it must be demonstrated to be deleterious, unless the specific mutation has been previously reported in association with the relevant disease. For many genes, there are mutation databases such as IL2RGbase for X-linked SCID, ALPSbase for mutations in Fas, and STAT3base for mutations in signal transducer and activator of transcription 3 (STAT3)-deficient hyper-IgE syndrome (see Table 1). Although these databases are useful tools, they may not be current or comprehensive, which remains a challenge as new disease-causing mutations continue to be identified for many of the primary immunodeficiencies.
If the identified change has not been previously reported in a mutation registry, it is critical to verify that the change is not a normal variant, as SNPs erroneously thought to be disease-causing changes would lead to false-positive results. To accomplish this, one can refer to the SNP database (dbSNP) (see Table 1), the National Center for Biotechnology Information (NCBI) curated site for identified population-based polymorphisms. If the nucleotide change detected in a patient has also been detected in apparently healthy controls, it represents a SNP and is unlikely to be the cause of disease. However, if the change is not reported in dbSNP, it may be necessary to perform population screening for the variation detected in the patient sample by direct sequencing of 100 alleles to rule out that the finding is actually a normal variation. Although most SNPs are ubiquitous allelic variants that exist in all or nearly all populations, private polymorphisms are allelic variants found only within a restricted population. Therefore, when performing a mutation analysis for an individual from a restricted population, one may also have to consider the possibility of private polymorphism.
If the identified nucleotide change is not a known SNP, is not detected among 100 wild-type alleles, and has not been previously identified in association with the relevant disease, then one must evaluate the effect of the nucleotide change either at the amino acid level or at the mRNA level in terms of expression and splicing fidelity. Bioinformatics tools such as ConSeq [19], PolyPhen [20], and SIFT [21] may be useful in predicting the effects of amino acid substitutions, providing precise information about sequence conservation, protein stability, structural disorder, contacts, and electrostatic potential. For example, ConSeq is a tool for the identification of structurally and functionally important residues in protein sequences. It calculates the evolutionary rate at each amino acid site in an MSA of homologous proteins. The assumption is that slowly evolving sites are often biologically important. The MSA is also used to predict the relative solvent accessibility state of each site (i.e. buried vs. exposed) to determine which of these sites are important for maintaining protein structure and which are functionally important. With the occasional exception [e.g. hypervariable peptide-binding sites in major histo-compatibility complex (MHC) molecules], functionally important amino acids, such as those that participate in ligand or DNA binding and protein–protein interactions, are often evolutionarily conserved and are most likely to be solvent accessible, whereas highly conserved amino acids within the protein core are likely to have a role in maintaining the protein’s conformation. It should be noted, however, that this generalization is problematic in cases in which a given amino acid has both a functional and a structural role. PolyPhen is a tool, which predicts the possible impact of an amino acid substitution on the structure and function of a human protein using physical and comparative considerations that estimate the impact of an amino acid replacement on the three-dimensional structure and function of the protein. SIFT predicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids. Given the correlation between structure and function, these tools may provide overlapping results, adding value to the predictions.
When bioinformatics analyses do not reveal useful information about a novel mutation, further experiments must be carried out to determine the effect. At this point, evaluation of the consequence of a mutation on the splicing of RNA should be considered. In fact, it has been suggested that point mutations should be routinely analyzed at the mRNA level before drawing conclusions about the importance of the affected amino acid, as there is increasing evidence that exonic point mutations may affect pre-mRNA splicing [26]. In the absence of mRNA analysis, a synonymous or nonsynonymous point mutation of an exonic splicing enhancer (ESE) or exonic splicing silencer (ESS) element may be misclassified as an SNP or missense mutation, when it actually disrupts critical ESE or ESS motifs, affecting normal splicing. For example, Pfarr et al. [27] identified a homozygous A to G transition in exon 10 of C5 (OMIM 120900) that predicted a lysine to arginine substitution in a patient with C5 deficiency (OMIM 609536). This amino acid substitution was not expected to result in a complete C5 deficiency; however, the transition changed an ESE and resulted in complete skipping of exon 10, leading to a frameshift and unstable mRNA in the affected individual. This case linking C5 deficiency to an ESE mutation and other studies of known ESE mutations that affect breast cancer 2, early onset (BRCA2) [28], growth hormone 1 (GH1) [29–31], and cystic fibrosis transmembrane conductance regulator (CFTR) [32] underscore the fact that diseases may result from mutations that influence any one of the diverse aspects of mRNA production and processing. Although beyond the scope of our review, appreciation of the varied mechanisms involved [33] should promote further investigation and thereby facilitate accurate mutation analysis. Such an investigation would begin by direct cDNA sequencing to look for splicing defects followed by the determination whether a sequence change alters a putative ESE motif by using, for example, RESCUE-ESE [34] or ESEfinder [35]. Moreover, an apparently silent third base change in the first or last base of an exon may result in a splice defect based on changes in the consensus splice site recognition sequence. Additionally, the creation of cryptic splice sites that alter normal mRNA processing can occur and should be considered [36].
It is also important to note that synonymous point mutations should not be summarily dismissed, as they can not only impact splicing, but a recent report has described a new mechanism in which a synonymous nucleotide change to a rare codon results in altered protein structure and function, possibly by affecting the timing of cotranslational folding [37]. Although this mechanism of altered translational efficiency results in similar mRNA and protein levels, it highlights the importance of including synonymous nucleotide changes in the patient report.
If the sequencing data do not reveal a mutation in a candidate gene, the result should be reported as ‘No mutation detected in gene X’, as opposed to ‘Gene X was normal’ or ‘Gene X mutation analysis was negative’. This reporting approach recognizes the possibility that the patient may have a mutation that was not detectable by standard genomic DNA sequencing (see cases 2–5 above). It must be kept in mind that when a mutation analysis result is inconclusive or inconsistent with the patient’s clinical phenotype, additional follow-up should be undertaken using alternative molecular methods and/or specific functional testing to clarify the underlying defect responsible for the immunodeficiency. In addition, novel sequence changes not previously associated with a disease require additional functional testing and/or bioinformatics analyses to help clarify the potential role of the finding on the pathogenesis of the disease.
Conclusion
The application of mutation analysis is becoming an integral part of the complete evaluation of patients with primary immunodeficiencies, and as such, clinicians caring for these patients must develop a better understanding of the utility and challenges of this important laboratory technology. A complete report from the clinical laboratory providing mutation testing should include the analysis of the mutation(s) identified as well as the presence of SNPs and synonymous point mutation findings. It is critical to ascertain that any sequence change identified has a deleterious impact prior to reporting this as the genetic basis of the patient’s immunodeficiency. The case studies presented identify situations in which the initial evaluation could lead to a false-negative or positive conclusion. We have outlined approaches to resolve these situations and avoid misdiagnosis. Finally, as a part of a genetic evaluation, a genetic counselor is extremely important to help ensure that the patients understand to the best of their ability the underlying diagnosis and the genetic information they request including the impact on themselves and their family.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 575).
- 1.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith LM, Sanders JZ, Kaiser RJ, et al. Fluorescence detection in automated DNA sequence analysis. Nature. 1986;321:674–679. doi: 10.1038/321674a0. [DOI] [PubMed] [Google Scholar]
- 3.Ronaghi M, Uhlén M, Nyrén P. A sequencing method based on real-time pyrophosphate. Science. 1998;281:363–365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 4.Drmanac S, Chui G, Diaz R, et al. Sequencing by hybridization (SBH): advantages, achievements, and opportunities. Adv Biochem Eng Biotechnol. 2002;77:75–101. doi: 10.1007/3-540-45713-5_5. [DOI] [PubMed] [Google Scholar]
- 5.Bajorath J, Seyama K, Nonoyama S, et al. Classification of mutations in the human CD40 ligand, gp39, that are associated with X-linked hyper IgM syndrome. Protein Sci. 1996;5:531–534. doi: 10.1002/pro.5560050316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thusberg J, Vihinen M. The structural basis of hyper IgM deficiency: CD40L mutations. Protein Eng Des Sel. 2007;20:133–141. doi: 10.1093/protein/gzm004. [DOI] [PubMed] [Google Scholar]
- 7.Karpusas M, Hsu YM, Wang J, et al. 2 å crystal structure of an extracellular fragment of human CD40 ligand. Structure. 1995;3:1031–1039. doi: 10.1016/s0969-2126(01)00239-8. [DOI] [PubMed] [Google Scholar]
- 8.Picard C, Puel A, Bonnet M, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 9.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 10.Lohi H, Turnbull J, Zhao XC, et al. Genetic diagnosis in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology. 2007;68:996–1001. doi: 10.1212/01.wnl.0000258561.02248.2f. [DOI] [PubMed] [Google Scholar]
- 11.Bekri S, May A, Cotter PD, et al. A promoter mutation in the erythroid-specific 5-aminolevulinate synthase (ASAS2) gene causes X-linked sideroblastic anemia. Blood. 2003;102:698–704. doi: 10.1182/blood-2002-06-1623. [DOI] [PubMed] [Google Scholar]
- 12.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–655. doi: 10.1146/annurev.immunol.22.012703.104614. [DOI] [PubMed] [Google Scholar]
- 13.Guo DC, Gupta P, Tran-Fadulu V, et al. An FBN1 pseudoexon mutation in a patient with Marfan syndrome: confirmation of cryptic mutations leading to disease. J Hum Gene. 2008;53:1007–1011. doi: 10.1007/s10038-008-0334-7. [DOI] [PubMed] [Google Scholar]
- 14.Davis RL, Homer VM, George PM, Brennan SO. A deep intronic mutation in FGB creates a consensus exonic splicing enhancer motif that results in afibrinogenemia caused by aberrant mRNA splicing, which can be corrected in vitro with antisense oligonucleotide treatment. Hum Mutat. 2009;30:221–227. doi: 10.1002/humu.20839. [DOI] [PubMed] [Google Scholar]
- 15.Jacquette A, Le Roux G, Lacombe C, et al. Compound heterozygosity for two new mutations in the beta-globin gene [codon 9 (+TA) and polyadenylation site (AATAAA–>AAAAAA)] leads to thalassemia intermedia in a Tunisian patient. Hemoglobin. 2004;28:243–248. doi: 10.1081/hem-120040304. [DOI] [PubMed] [Google Scholar]
- 16.Bennett CL, Brunkow ME, Ramsdell F, et al. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics. 2001;53:435–439. doi: 10.1007/s002510100358. [DOI] [PubMed] [Google Scholar]
- 17.Defendi F, Decleva E, Martel C, et al. A novel point mutation in the CYBB gene promoter leading to a rare X minus chronic granulomatous disease variant: impact on the microbicidal activity of neutrophils. Biochim Biophys Acta. 2009;1792:201–210. doi: 10.1016/j.bbadis.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 18•.Hanson EP, Monaco-Shawver L, Solt LA, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–1177. doi: 10.1016/j.jaci.2008.08.018. This study links genotype and phenotype in NEMO deficiency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berezin C, Glaser F, Rosenberg J, et al. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20:1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- 20.Ramensky V, Bork P, Sunyaev S. Human nonsynonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng PC, Henikoff S. Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm. Genome Res. 2001;11:863–874. [Google Scholar]
- 22.Aradhya S, Bardaro T, Galgóczy P, et al. Multiple pathogenic and benign genomic rearrangements occur at a 35 kb duplication involving the NEMO and LAGE2 genes. Hum Mol Genet. 2001;10:2557–2567. doi: 10.1093/hmg/10.22.2557. [DOI] [PubMed] [Google Scholar]
- 23.Bardaro T, Falco G, Sparago A, et al. Two cases of misinterpreation of molecular results in contintinentia peigmenti, and a PCR-based method to discriminate NEMO/IKKgamma gene deletion. Hum Mutat. 2003;21:8–11. doi: 10.1002/humu.10150. [DOI] [PubMed] [Google Scholar]
- 24.Fleisher TA. The autoimmune lymphoproliferative syndrome: an experiment of nature involving lymphocyte apoptosis. Immunol Res. 2008;40:87–92. doi: 10.1007/s12026-007-8001-1. [DOI] [PubMed] [Google Scholar]
- 25.Helzelova E, Vonarbourg C, Stolzenberg MC, et al. Autoimmune lymphopro-liferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–1418. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- 26.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 27.Pfarr N, Prawitt D, Kirschfink M, et al. Linking C5 deficiency to an exonic splicing enhancer mutation. J Immunol. 2005;174:4172–4177. doi: 10.4049/jimmunol.174.7.4172. [DOI] [PubMed] [Google Scholar]
- 28.Fackenthal JD, Cartegni L, Krainer AR, Olopade OI. BRCA2 T2722R is a deleterious allele that causes exon skipping. Am J Hum Genet. 2002;71:625–631. doi: 10.1086/342192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cogan JD, Ramel B, Lehto M, et al. A recurring dominant negative mutation causes autosomal dominant growth hormone deficiency: a clinical research center study. J Clin Endocrinol Metab. 1995;80:3591–3595. doi: 10.1210/jcem.80.12.8530604. [DOI] [PubMed] [Google Scholar]
- 30.Cogan JD, Prince MA, Lekhakula S, et al. A novel mechanism of aberrant premRNA splicing in humans. Hum Mol Genet. 1997;6:909–912. doi: 10.1093/hmg/6.6.909. [DOI] [PubMed] [Google Scholar]
- 31.Moseley CT, Mullis PE, Prince MA, Phillips JA., III An exon splice enhancer mutation causes autosomal dominant GH deficiency. J Clin Endocr Metab. 2002;87:847–852. doi: 10.1210/jcem.87.2.8236. [DOI] [PubMed] [Google Scholar]
- 32.Aznarez I, Chan EM, Zielenski J, et al. Characterization of disease-associated mutations affecting an exonic splicing enhancer and two cryptic splice sites in exon 13 of the cystic fibrosis transmembrane conductance regulator gene. Hum Mol Genet. 2003;12:2031–2040. doi: 10.1093/hmg/ddg215. [DOI] [PubMed] [Google Scholar]
- 33.Mendell JT, Dietz HC. When the message goes awry: disease producing mutations that influence mRNA content and performance. Cell. 2001;107:411–414. doi: 10.1016/s0092-8674(01)00583-9. [DOI] [PubMed] [Google Scholar]
- 34.Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 35.Cartegni L, Wang J, Zhu Z, et al. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acid Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartmann L, Theiss S, Niederacher D, Schaal H. Diagnostics of pathogenic splicing mutations: does bioinformatics cover all bases? Front Biosci. 2008;13:3252–3272. doi: 10.2741/2924. [DOI] [PubMed] [Google Scholar]
- 37.Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]