

Abstract
When cancer cells develop resistance to chemotherapeutics, it is frequently conferred by the ATP-dependent efflux pump P-glycoprotein (MDR1, P-gp, ABCB1). P-gp can efflux a wide range of cancer drugs; thus its expression confers cross-resistance, termed multidrug resistance (MDR), to a wide range of drugs. Strategies to overcome this resistance have been actively sought for over 30 years, yet no clinical solutions exist. A less understood aspect of MDR is the hypersensitivity of resistant cancer cells to other drugs, a phenomenon generally known as collateral sensitivity (CS). This review highlights the extent of this effect for the first time, discusses hypotheses such as ROS generation to account for the underlying generality of this phenomenon, and proposes the exploitation of CS as a strategy to improve response to chemotherapy.
Introduction
Considerable effort has been devoted to improving cancer treatment by identifying targets in cancer cells deriving directly or indirectly from molecular alterations that promote unregulated cell growth, invasion and metastasis [1]. Whatever strategy is employed for cancer treatment, the development of drug resistance is a likely outcome. Clinical multidrug resistance (MDR) is mediated via a range of cellular alterations including reduced drug accumulation, changes in the level of protein targets, mutations that diminish the binding of drug to target, increased trapping of drug in acidic vesicles, altered metabolism of drugs, increased tolerance of cellular damage and diminished apoptotic signaling [2]. Reduction in cellular accumulation of drugs occurs through reduced expression of cell surface passive importers, and increased expression of ATP-binding cassette (ABC) efflux transporters that extrude a broad range of amphiphilic compounds against the concentration gradient in an energy-dependent fashion. The archetypal ABC transporter is P-glycoprotein (P-gp, MDR1, encoded by the ABCB1 gene, Box 1, Figure 1). Its expression in a number of malignancies has been shown to correlate with poor chemotherapeutic response and prognosis [3].
Box 1. P-glycoprotein.
P-glycoprotein is a cell surface glycoprotein composed of 12 transmembrane helices arranged into two interleaved domains (Figure 1). Appended to each domain on the cytosolic side are two ATP-binding sub-units. A wealth of mechanistic work has explored the range of efflux substrates recognized by P-gp, and the steps involved in efflux [86]. Drug substrate extrusion occurs via a drug-binding site accessible from the lipid bilayer, and competition assays with substrates have demonstrated that multiple distinct drug binding sites exist, probably spatially separate but overlapping areas of a large contiguous drug recognition site. A recent crystal structure of mouse P-glycoprotein revealed two ‘portals’ up to 9 Å wide, opening up into an internal drug binding cavity of ~6000 Å3 and able to accommodate more than one molecule [87]. The internal cavity has an array of inward facing residues capable of accommodating a range of spatial intermolecular bonding modes, allowing cross-recognition of small molecules irrespective of the spatial distribution of non-covalent bonding partners. By building drug extrusion into the lipid bilayer, P-gp acts as a membrane vacuum cleaner, intercepting drugs before they reach high affinity targets within the cell, thereby precluding their build-up within the cell [88].
While a number of detailed mechanisms have been proposed for mechanistic steps of drug efflux, a plausible hypothesis is that when ATP binds to an ATP-binding domain, the two domains are brought together, initiating ATP hydrolysis and release of ADP and Pi [86]. The hydrolysis of one ATP molecule enables substrate efflux, and the transporter resets to once again sequester substrate. A highly lipophilic molecule may return to the lipid bilayer readily to re-bind to P-gp (as has been reported for verapamil), resulting in a cycle of constant efflux, termed ‘futile cycling’, which may consume a relatively large amount of ATP (Figure 1). Some inhibitors of P-gp also bind tightly to the drug binding site, and in so doing ‘block’ the pump, preventing efflux of substrates. This can be measured as an inhibition of ATPase activity of the transporter.
Figure 1.

The efflux cycle of P-gp described in Box 1.
An extensive literature on the development of multidrug resistance (MDR) in vitro and in vivo exists. MDR is typified by the broad cross-resistance that P-gp confers to structurally dissimilar cytotoxic agents – in other words, the development of resistance by a cell to one drug produces similar resistance to other drugs (Figure 2a) [3,4]. Other ABC transporters have also been found to increase in expression in response to drug selection in cell culture, primarily ABCG2 (also termed mitoxantrone resistance protein, MXRP, or breast cancer resistance protein, BCRP) and the drug-conjugate transporter ABCC1 (multidrug resistance protein 1, MRP1). While there is substantial overlap in substrate recognition among these transporters, and their endogenous expression has been shown, ABCG2 and ABCC1 have not yet been definitively demonstrated to contribute to MDR in patients. A range of strategies have been explored to ameliorate P-gp-mediated MDR, both directly and indirectly [3]. Chief among these is the development of several generations of P-gp inhibitors, though clinical trials have been disappointing (see Box 2).
Figure 2.

Collateral sensitivity defined schematically. (a) Representative dose-response curve of a parental cell line (solid line, center). Development of resistance to a drug, and concomitant cross-resistance to a variety of cytotoxic agents conferred by ABC transporters such as P-gp, results in a loss of sensitivity of possibly several orders of magnitude (dotted line, right). Inhibitors of P-gp (so-called reversal agents) inhibit the efflux function of P-gp, restoring cellular accumulation and re-sensitizing cross-resistant cells to levels approaching that of the original parental cells. A small number of agents have been demonstrated to sensitize multidrug-resistant cells to a greater degree than the original parental cells (dashed line, left). This property is termed collateral sensitivity (CS). (b) The determination of collateral sensitivity (≤0.5) and multidrug resistance (≥2) as defined in this review. It is important to emphasize that a lack of cross-resistance to an agent—the same drug response for a parental and resistant line—is not collateral sensitivity, but merely a lack of resistance. This occurs, for example, with the P-gp substrate vinblastine against cisplatin-resistant 7407-CP human liver carcinoma cells which do not express P-gp as part of their resistance phenotype [112].
Box 2. Resolving MDR.
Recognizing that the calcium channel blocker verapamil and the immunosuppressant cyclosporin A (CsA) overcome the drug accumulation defect in multidrug-resistant cells [18], clinical trials assessed whether they could reverse the MDR phenotype in patients (reviewed in Ref. [89]). A rich literature on verapamil’s P-gp inhibitory properties exists, though clinical trials failed and the drug gave way to new-generation inhibitors with greater specificity and efficacy [89].
Second-generation semi-synthetic analogs (such as the CsA analog valspodar, PSC833) were developed that retained their P-gp inhibitory properties while abrogating their primary pharmacological activities. A series of third-generation inhibitors were subsequently designed de novo for their highly specific P-gp inhibitory properties, typified by tariquidar (XR9576). The in vitro effectiveness of these inhibitors has not translated to the clinic despite a number of clinical trials employing inhibitors in combination with chemotherapeutics [89]. There are a number of reasons for these trial failures; early inhibitors elicited side-effects related to their pharmacological properties (e.g., verapamil caused dose-limiting cardiotoxicity), the non-specific inhibition of physiological P-gp and other ABC transporter function alters drug pharmacokinetics and increases drug AUC resulting in unpredictable toxicities, and the lack of careful demonstration of P-gp expression in tumors and little or no determination of the effectiveness of P-gp inhibition at the tumor site(s).
A number of other strategies have been explored to engage, evade and exploit P-gp-mediated multidrug resistance [3]. Dose-escalation to overcome P-gp efflux narrows the therapeutic window and adds further selection pressure on already MDR tumor cells, and detergent co-administered to enhance cellular accumulation has been largely unsuccessful. Direct disruption of functional expression through approaches such as siRNA [90,91], gene therapy, or antibodies [92] are still being translated to the clinic. The promising strategy of identifying non-substrates is now a standard approach in the design of new chemotherapeutics. For example, while doxorubicin, epirubicin and daunorubicin are ineffective in P-gp-expressing HB8065/R human hepatoma cells, the structural analog aclarubicin retains activity similar to the parental line [93]. In this review, we highlight ways to exploit expression of P-gp to specifically kill MDR cells.
A phenomenon in drug-resistant cells (prokaryotic and eukaryotic) identified during in vitro studies is that the development of resistance to one agent can confer greater sensitivity to an alternate agent than seen in the original (parental) line (Figure 2). The term for this, ‘collateral sensitivity’ (CS), was first used in a study by Szybalski and Bryson in 1952 to describe the hypersensitivity of drug-resistant Escherichia coli to other unrelated agents [5]. The authors described CS as ‘the result of a selective process’ and suggested that this phenomenon could lead to drugs that are highly effective against drug-resistant bacteria [1]. CS can be considered a kind of ‘synthetic lethality’ in which the genetic alteration that confers resistance to a drug sensitizes it to other drugs. CS can be assessed most easily in vitro by determining the cytotoxicity (IC50) of a compound against a parental line and its MDR sub-line (Figure 2). A compound displaying cross-resistance will show lower efficacy against the MDR line than the parental line, and therefore yield a resistance ratio >1 (RR, determined by dividing the IC50 against a resistant line by the IC50 against a parental line). Conversely, a CS agent will show greater toxicity against the MDR line than the parental line, and therefore the RR will be <1. In the case of both CS and cross-resistance, at least a two-fold effect is probably required to be considered of significance.
This review is intended to summarize observations on the CS demonstrated by MDR cells expressing the drug efflux transporter P-gp. A range of small molecules have been shown to demonstrate CS activity, and these are discussed in the context of possible mechanisms conferring this activity. Given the ongoing challenge of resolving cancer MDR in the clinic, it is hoped this review will bring attention to the long-standing but immature field of CS.
Collateral Sensitivity
The first full report of CS in MDR cancer cells was made by Bech-Hansen and coworkers in 1976 [6]. Using a series of MDR sub-lines derived from the Chinese hamster ovary (CHO) line AuxB1 selected in increasing amounts of colchicine, agents were examined in an effort to understand the extent of cross-resistance in these lines (now shown to express hamster P-gp). The lines were also found to demonstrate CS to a series of local anesthetics, steroid hormones and lipophilic nonionic Triton X detergents. The highly-resistant CHRC5 cells, a sub-line of CHO, showed the greatest degree of CS, suggesting that increased CS is tied directly to increased P-gp expression. However, the magnitude of CS was significantly lower (10-fold CS at most) than the 184-fold resistance to colchicine demonstrated by the cells.
Several reports followed showing that cells selected with other cytotoxic agents also exhibited CS. A CHO line selected in colcemid (a colchicine analog) that presumably did not express P-gp was sensitive to paclitaxel [7], and vinblastine- and paclitaxel-resistant CHO cells were sensitive to bleomycin, cisplatin and cytarabine [8]. It has also been shown that CS is not solely the domain of P-gp-expressing MDR cell lines; cisplatin-resistant cells (with a range of cellular alterations to confer resistance to platinum drugs including decreased drug influx transporters and increased thiol levels, and not expressing P-gp [9]) are hypersensitized to the camptothecin analog SN-38. Even human ovarian carcinoma cells selected for resistance to radiation demonstrate collateral sensitivity to cisplatin, methotrexate and 5-FU [10].
Many compounds known to cause CS were initially identified through ad hoc observations, collectively demonstrating that an Achilles heel may exist for cell lines resistant to virtually any clinical therapeutic agent. This concept is not limited to older ‘shotgun’ cytotoxics (which, as their name suggests, induce non-specific cytotoxicity rather than acting against a specific cellular target), but also cell lines resistant to newer therapeutics such as histone deacetylase inhibitors (HDAC’s) or the kinase inhibitor gefitinib [11,12]. CS observations with cancer chemotherapeutic drugs are summarized in Table 1. While many of the drugs are P-gp substrates, others (e.g., cisplatin and nucleoside analogs) are not. Along with the cisplatin-resistant lines (which do not develop resistance via P-gp [9]), several other cellular alterations that confer resistance to an original selection agent—such as other ABC transporters, microtubule alterations or even plasma membrane changes [13]—can sensitize cells to a second agent.
Table 1.
Summary of collateral sensitivity reported with clinical cancer drugs in the literaturea
| Selection agent ⇒ | Microtubule assembly | Microtubule disassembly | Microtubule disruption | Topo I Inhibitor | Topo II Inhibitor | Topo II Inhibitor and Intercalator | DNA Alkylator | Antimetabolite | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vincristine | Vinblastine | Paclitaxel (Paclitaxel) | Colcemid | Colchicine | Camptothecin | EtopoSide (VP-16) | Teniposide (VM-26) | Daunorubicin (Daunomycin) | Doxorubicin (Adriamycin) | Cisplatin | Carmustine (BCNU) | Cytarabine (Ara-C) | 5-Fluorouracil | |
| Treatment agent⇓ | ||||||||||||||
| Vincristine | [98, [113] | [14] | [114] | |||||||||||
| Vinblastine | [98, [113] | |||||||||||||
| Paclitaxel | [115] | [7] | [16, [100, [113] | [14] | ||||||||||
| Colcemid | ||||||||||||||
| Colchicine | [7] | |||||||||||||
| Camptothecin | [116] | |||||||||||||
| Irinotecan | [99] | |||||||||||||
| SN-38 | [99] | [116] | ||||||||||||
| Etoposide | [14] | [14] | [116] | |||||||||||
| Teniposide | [14] | [14] | [14] | |||||||||||
| Daunorubicin | [116] | |||||||||||||
| Doxorubicin | [14] | [14] | ||||||||||||
| Bleomycin | [8] | [8] | [117] | [116] | ||||||||||
| Cisplatin | [116] | [8] | [8,16] | [14, [118] | [14] | [119] | [120] | |||||||
| Carmustine | ||||||||||||||
| Melphalan | [121, [122] | |||||||||||||
| Lomustine | [123] | |||||||||||||
| Cytarabine | [8] | [8] | [14, [118] | [96] | [14] | [122] | [14] | [14] | ||||||
| Gemcitabine | [97] | [96] | [14,96, [124] | [97] | [14] | |||||||||
| 5-Fluorouracil | [8] | [125, [126] | [98] | |||||||||||
| Hydroxyurea | [123] | [14] | [14] | |||||||||||
| Methotrexate | [123] | [127] | ||||||||||||
Cell lines selected for resistance to a range of drugs (listed across top of table) and the drugs reported to induce collateral sensitivity against them (highlighted with references). Drugs are clustered by their generally accepted mechanism of action. Collateral sensitivity would not be expected to be observed in a cell line selected with a drug that shares the same mechanism of action (grey areas).
Consequently, along with the P-gp induced CS that is the subject of this review, CS can also be caused by other readily explainable gene expression alterations (Box 3). As such, selection with a given drug may lead to unpredictable cross-resistance/collateral sensitivity profiles depending on the gene expression alterations induced during adaptation. This may account for the fact that CS is not always consistently observed for a given selection (i.e., CS agent combination in various cell lines). Irrespective of this, Table 1 serves to demonstrate the extent of CS observed with clinical cancer drugs, and the potential for its exploitation.
Box 3. Alternative gene targets for CS in MDR cells.
Alongside the P-gp-mediated CS observed in MDR cells, and described here, a range of other gene expression alterations can hypersensitize MDR cells to drugs that target a particular alteration.
CS to antimetabolites such as cytarabine and gemcitabine in cell lines selected for resistance to topoisomerase inhibitors is not directly related to P-gp or lowered topoisomerase expression [94,95], but to increased expression of deoxycytidine kinase which activates the antimetabolites [96,97]. Cisplatin-resistant cells expressing increased Topo I levels [98] show CS to the topo I inhibitor SN-38 [99], and cisplatin-resistant IGROV-1 cells showed CS to paclitaxel conferred by p53 mutation [100]. Similarly, cells adapt to inhibitors of topoisomerase II (Top II) by lowering its expression. To compensate for the loss of function, cells up-regulate Top I expression, thus cells resistant to Top II are hypersensitized to Top I inhibitors [14].
2-deoxy-D-glucose (2-DG) is a glucose analog originally employed as a mechanistic probe for glycolysis [101]. This antimetabolite enters cells via the glucose importer GLUT-1, inhibiting the glycolysis pathway that generates ATP anaerobically by competing with glucose for uptake and utliization [102]. Glycolysis inhibition was considered to be a viable strategy in tumors, as they were known to use aerobic glycolysis rather than oxidative phosphorylation to generate their energy—a phenomenon known as the ‘Warburg effect’ [102], though several clinical trials were unsuccessful. This effect has been utilized to develop the 18F-labelled 2-DG PET ligand 2-fluoro-deoxy-D-glucose (FDG) that hyperaccumulates in metabolically demanding tumor cells to enable tumor imaging [103].
Following on observations that doxorubicin-resistant MCF7 ADR cells display a 3-fold increase in the rate of glycolysis [104], 2-DG was tested and displayed CS towards MCF7 ADR cells, and 31P NMR of cells revealed phosphorylated 2-DG was produced more rapidly in resistant cells along with complete ATP depletion [105]. The sensitivity of cells to 2-DG was inverse to [3H]-2-DG accumulation; yet 2-DG is not a P-gp substrate, but the glucose transporter GLUT-1 was shown to decrease with increasing P-gp accounting for the diminished accumulation. CS is probably conferred through the increased nutrient demand of P-gp-expressing cells and exacerbated by the diminished capacity to import glucose, meaning lower 2-DG levels are required to compete with glucose for uptake [106]. 2-DG also inhibits N-linked glycosylation, and the N-linked glycosylation inhibitor tunicamycin also shows CS [107]. Warr has proposed that the reduced level of GLUT-1 normally seen in MDR cells is due to ‘reduced glycosylation of GLUT-1 as a consequence of the high glycosylation demands imposed on the cell by P-gp’ [107]. Which of the two pathways is responsible for CS remains to be determined, though FDG is known to be a poor N-linked glycosylation inhibitor and would help to delineate the mechanism.
A small number of systematic studies have been published in which drug-resistant cell lines were screened against a panel of drugs; one screened vinblastine- (VinR) and paclitaxel (TaxR-2)-resistant lines derived from CHO cells against 37 anti-cancer compounds [8], and another screened seven drug-resistant small-cell cancer cell lines to search for drugs that induced CS as suitable clinical drug partners to chemotherapeutics [14]. These studies give some indication of the extent of cross-resistance and CS of MDR cell lines, revealing a relatively low number of CS agents observed in each case (average 11%), compared to the more extensive and well characterized cross-resistance (average 43% of compounds showed cross-resistance). Correlation analysis revealed that cisplatin was the most efficacious drug against VP-16 (etoposide)-resistant cells, and doxorubicin was most effective against cisplatin-resistant cells (VP-16 and cisplatin were examined as they were the drugs of choice against small-cell lung cancer (SCLC) at the time). The study also sought to identify drug pairs least likely to develop cross-resistance to one another. The drug pairs found least likely to develop cross-resistance to one another were carmustine (BCNU)-paclitaxel and paclitaxel-cisplatin. This latter relationship has been the subject of recent attention given the use of this combination against ovarian cancers [15]. A systematic analysis of 137 platinum- or taxane-resistant cell lines revealed that 68% of cisplatin-resistant lines were sensitive to paclitaxel and 67% of paclitaxel-resistant lines were sensitive to cisplatin. While CS was not explicitly calculated in the analysis, CS was observed in a significant number of these lines [16].
Verapamil—a case study
Of the known CS compounds, verapamil has received the most attention. Verapamil is a phenylalkylamine L-type calcium channel antagonist, and has a number of pharmacological applications based on this activity [17]. It was first reported as one of several calcium channel blockers able to increase accumulation of vincristine and doxorubicin in P-gp-expressing P388 tumor cells [18], being an avid P-gp substrate at low concentrations, and inhibiting it at high concentrations (Box 2). Experiments showed vincristine-resistant CHO cells were hypersensitive to verapamil, and that the dose-response curves were biphasic—after an initial response that reduced plating efficiency to about 3%, the remaining cells survived much higher concentrations of verapamil [19]. The calcium channel blockers diltiazem [20], nicardipine [20], bepridil [21] and nifedipine [21] were subsequently shown to also be more toxic to CHO MDR cell lines compared to parental CHO cells, along with quinidine sulfate [20] and trifluoperazine [21], which do not interfere with calcium channel function, but all of which are avid substrates for P-gp. MDR cells that show increased sensitivity to verapamil accumulate less verapamil than parental cells [22]. A number of observations have ruled out calcium channel-blocking as being responsible for CS, including accumulation studies showing that 45Ca2+ levels are equivalent in CHO parental and MDR cells [22], and that these levels are unaffected by verapamil [19].
Verapamil induces CS in cells expressing mouse [23] and human MDR1 [24], whereas drug-resistant lines not expressing ABC transporters are not hypersensitive [25]. Furthermore, ‘revertant’ cells that had lost their MDR phenotype after a prolonged period of growth without drug selection lost their hypersensitivity to verapamil. A series of increasingly resistant MDR cell lines showed a corresponding increase in CS to verapamil and nicardipine [26]. Importantly, while cells expressing a 43-fold increase in MDR1 mRNA showed high sensitivity, cells with a 7-fold increase showed little to no response to verapamil, suggesting that low-grade resistance may not sensitize cells to verapamil.
In concert with its selective toxicity, verapamil has been shown to down-regulate P-gp expression; in K562/ADR cells, P-gp protein loss was maximal (35% of control) after 72 h incubation, and was maintained over a period of 9 days, but that expression could be restored within 24 h of verapamil removal [24]. MDR1 mRNA levels were lowered after only 16 h, suggesting that a complex regulatory mechanism is involved. The effect on P-gp expression is elicited at sub-toxic concentrations, which along with the relative rapidity of the effects on message and protein levels indicates that the effect is one of suppression rather than selection. A detailed follow-up study with K562/ADR cells found a maximal 6-fold lowering of MDR1 mRNA by verapamil, and post-transcriptional MDR1 mRNA degradation was not increased in verapamil-treated cells [27]. Nuclear transcription run-on assays in the same study demonstrated that verapamil elicited a 4-fold decrease in MDR1 transcription, suggesting a transcriptional mechanism for MDR1 down-regulation, an observation confirmed in cells transiently transfected with a plasmid containing the MDR1 proximal promoter upstream of chloramphenicol acetyltransferase (CAT) that showed reduced activity after exposure to verapamil.
The ROS hypothesis for CS
Given that verapamil is an avid substrate of P-gp at lower concentrations, cellular ATP consumption in the presence of azide (to stop further ATP generation) was compared in parental (A2780) and drug-resistant (A2780AD) human ovarian carcinoma cells—ATP was depleted at twice the rate in P-gp-expressing A2780AD cells exposed to verapamil, bepridil and nifedipine [28]. The ATP depletion was ascribed to the rapid cyclical efflux, re-entry and efflux of verapamil in the cells (known as ‘futile cycling’) [28]. This ATP depletion was accompanied by a 12% increase in glycolysis (measured as lactate production) in A2780AD cells; however, no alteration in oxygen consumption (i.e., respiration, that would indicate ATP generation through oxidative phosphorylation in the mitochondrion) was observed [29].
One group noted that verapamil causes cell death by apoptosis [30], and that CS can be abrogated when P-gp function is inhibited (which halts ATP consumption, Box 1) [30]. When parental AUXB1 and MDR CHRC5 cells were exposed to verapamil, only CHRC5 were found to produce elevated levels of the reactive oxygen species superoxide (O2-), and reduced levels of glutathione. This effect was abrogated when verapamil was co-incubated with the P-gp inhibitor PSC833 [30]. The hypothesis that followed is that as the cell constantly replenishes the ATP consumed by the futile efflux of verapamil, repletion of ATP from ADP (at least in part) by oxidative phosphorylation generates reactive oxygen species (ROS) [30]. This cycling leads to inefficiencies in oxidative phosphorylation, leading to reduction of dioxygen to superoxide (O2-), and reaction with H2O2 to produce hydroxyl radicals (·OH), known collectively as ROS. Adding to this argument are the observations that verapamil toxicity increases with increasing P-gp expression [31] (so more P-gp efflux produces greater ROS stress on the cell), and other ROS generating agents have also been shown to down-regulate P-gp expression, as observed for verapamil [32]. ROS-mediated CS may also account for the fact that cell lines expressing low levels of P-gp do not suffer from CS [31,33]; there is in effect a cut-off in expression before CS is observed, which might represent the base-line level of ROS a cell can accommodate.
An apparent weakness in the hypothesis is that only increased glycolysis was observed in A2780AD cells exposed to verapamil, whereas oxidative phosphorylation is the main source of ROS (though ROS production is increased in mitochondria when electron transport is reduced as cells shift to glycolysis [34]). Unfortunately other examples of respiration experiments are limited; however, isolated rainbow trout hepatocytes showed an 18.5% and 25.7% increase in oxygen consumption when exposed to 5 and 10 μM, respectively, of the P-gp substrate Rhodamine 123 [35], and L1210 mouse leukemia cells selected for resistance to vincristine and expressing P-gp showed an increase in O2 consumption, but not lactate production, when effluxing vinblastine [36]. Thus, individual MDR cell lines may vary in their ATP production depending on variation in adapatation, cellular stress and tissue of origin.
Verapamil causes CS against MRP1
CS also occurs with verapamil in MRP1-expressing NCI-H69 small cell lung cancer cells selected for resistance to doxorubicin [37]. The action of verapamil (and an analog) against baby hamster kidney BHK-21 cells transfected with both wild-type and non-functional MRP1-expressing plasmids was explored. Only wt-MRP1 cells were hypersensitized, indicating functional MRP1 is required for CS [38]. While verapamil is not a strong substrate of MRP1, it stimulates glutathione (GSH) efflux [38] with a concomitant increase in ATP consumption [39]. Furthermore, verapamil reduced GSH levels dramatically in MRP1-transfected cells, causing ROS generation and caspase-dependent apoptosis, which could be prevented by the addition of exogenous GSH [40]. Apigenin, which also stimulates MRP1-mediated GSH efflux, was found to have a similar effect [40]. In the case of MRP1, CS appears to be caused by a combination of transport-mediated cellular stress combined with the removal of GSH that normally helps to overcome this oxidative stress [41]. Consistent with this idea, cellular GSH depletion with the γ-glutamylsynthase inhibitor buthionine sulfoximine (BSO) also induces CS.
Given the CS observed for verapamil and other substrates, a general mechanism for substrate-induced CS has been proposed, where the futile cycling of a non-toxic substrate induces toxicity as a result of the drain on cellular energy and the damaging ROS generated as the cell continuously repletes energy levels. As such, it might be proposed that any non-toxic compound that is an avid substrate for an ABC transporter may induce CS in its own right, and serve a useful purpose in sensitizing MDR cells in concert with other drugs, though there are few examples in the literature. Whether this approach could prove clinically useful depends on whether levels of ABC transporters in vivo in MDR cancer cells achieve the high protein levels needed to sensitize them to non-toxic substrates, and whether healthy tissues that express P-gp will be similarly sensitized.
Indirect P-gp-mediated CS
A number of compounds have been reported that produce CS, but are not themselves substrates of P-gp. By correlating the gene expression of MDR1 in the National Cancer Institute NCI-60 cell line panel with the cytotoxicity profiles of 1,429 compounds, predicted substrates of P-gp were reported [42]. Along with these, a number of CS agents were identified as drugs to which P-gp-expressing cells were more sensitive, and the isatin-β-thiosemicarbazone NSC73306 was validated against MDR cell lines. NSC73306 CS increased with functional P-gp expression, and inhibition of P-gp abrogated this selectivity [43]. Down-regulation of P-gp with siRNA did likewise, and long-term selection of P-gp-expressing HCT-15 colon carcinoma cells resulted in loss of P-gp and sensitization to drugs that are P-gp substrates. The increased toxicity of NSC73306 is specific to P-gp-expressing cells, with no increased toxicity towards ABCG2- and MRP1-expressing cells and their parental lines. In fact, despite being a substrate for ABCG2, and demonstrating reduced accumulation as a result, NSC73306 shows no altered activity against ABCG2-expressing cells [44].
While thiosemicarbazones tested in the clinic as iron-chelating ribonucleotide reductase inhibitors are known to be P-gp substrates [45], NSC73306 is neither a substrate nor an inhibitor of P-gp in contrast to the CS agents described above, despite the fact that functional P-gp is required for activity. SAR analysis of a series of analogs revealed that the functional groups capable of binding metal ions are essential for the CS activity of NSC73306 [46]. A number of other thiosemicarbazones have subsequently been reported to demonstrate CS [46]. A study of pyridoxal isonicotinyl hydrazone analogs identified the cytotoxic thiosemicarbazone Dp44mT, which was selectively active against KB-V1 cells (Figure 3) [47]. Dp44mT showed good activity against mouse xenografts, but while DNA strand breaks, topoisomerase IIA inhibition and apoptosis induction have been subsequently demonstrated [48], the CS activity requires elaboration. Analogs of Dp44mT have been shown to bind iron avidly [49], and the electrochemistry and ascorbate oxidation activity of their iron complexes indicates the potential for redox cycling between oxidation states.
Figure 3.
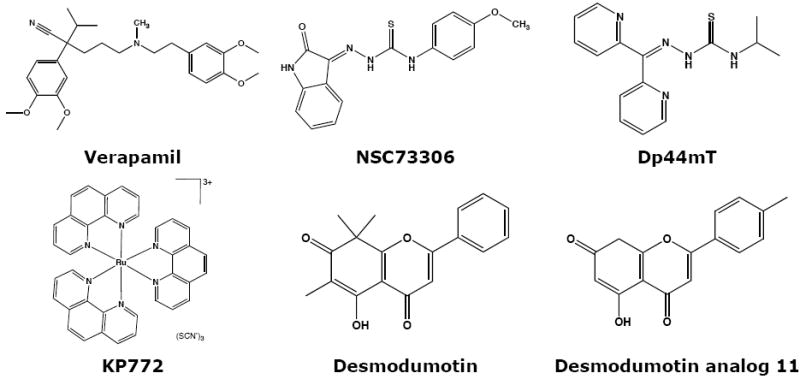
Structures of compounds described in the text.
Along with thiosemicarbazones, a number of other compounds capable of interacting with iron in cells have been reported. The lanthanum tris-phenanthroline complex KP772 preferentially kills P-gp-expressing cells with 2-fold selectivity compared to parental cells [50, [51]. Similar to NSC73306, KP772 does not inhibit P-gp function, nor is it a substrate and long-term exposure resulted in the loss of P-gp expression [50]. 1,10-phenanthroline is known to confer its cytotoxicity through interaction with copper and iron, resulting in ROS generation and cell death [52-54], and the ligand alone has been shown to confer CS on MDR KB-V1 cells, suggesting that the phenanthroline complexes identified are carriers that can release the active free ligand [55]. The copper complex of N-(2-hydroxy acetophenone) (CuNG) [56] generates ROS in MDR cells, depletes glutathione and down-regulates P-gp and MDR1 mRNA in a similar way to hydrogen peroxide treatment [57,58].
How can a set of metal-interacting compounds that do not appear to interact with P-gp directly elicit CS? These compounds (ligands) are able to complex with endogenous intracellular metals such as the labile iron pool [59, [60]. Metal ions with two accessible redox states under physiological conditions, such as iron (II/III) and copper (I/II), can cycle between these redox states through oxidation and reduction [61]. While the compounds are not shown to interact directly with P-gp, it may be that redox cycling occurs that is additive to other cellular stress in MDR cells. Thus, it is possible that increased ROS generation is a common feature for CS agents that are P-gp substrates and some that are not.
Other CS agents and the CS discovery pathway
A number of other drug classes have been identified that display CS. The steroids 5b-pregnan-3,20-dione, deoxycorticosterone and 1-dehydrotestosterone showed CS against CHRC5 Chinese hamster ovary cells [6]—prednisolone [62] and dexamethasone [63] followed. Progesterone and analog megestrol acetate were reported to be selectively-active towards a series of P-gp-expressing cells, and inhibited P-gp efflux at high concentrations [64]. While suggestive of substrate-mediated CS, megestrol acetate was shown to enhance the binding of [3H]-azidopine to P-gp indicative of an allosteric effect on P-gp, and progesterone is not a substrate of P-gp [65,66]. More work is required to understand the mechanistic basis of this CS.
A screen of 1,266 compounds against the RPMI 8226 myeloma cell line and doxorubicin-resistant 8226/Dox40 cells identified 33 compounds that displayed CS [67]. The most pronounced cluster were the glucocorticoids, which bind to the glucocorticoid receptor and transactivate gene transcription. Microarray analysis revealed glucocorticoid receptor NR3C1 expressed at greater levels in the 8226/Dox40 cells, and CS of the drug-resistant cells was abolished when the glucocorticoids were administered in the presence of the glucocorticoid receptor antagonist RU486 [67]. This highlights a complication of CS; the glucocorticoids are substrates for P-gp [68], but also bind the glucocorticoid receptor and alter gene expression at a global level. For this reason, the glucocorticoids lost CS activity in other MDR cell line pairs that did not display differential glucocorticoid receptor expression. In a similar fashion, estrogen down-regulates P-gp expression in the estrogen-receptor α (ER-α) positive MDR breast cancer cell lines MCF-7/MDR and T-47D/MDR, but not ER-α negative cells MDA-MB-231/MDR and NCI/ADR-RES (which has been shown not to be a breast cancer cell line [69,70]). Various drug-resistant cell lines will have other gene expression alterations along with P-gp, and several cell line pairs, or preferably a cell line transfected with the MDR1 gene, should be examined to confirm that P-gp mediated CS exists.
CS has also been reported for the local anesthetics procaine, tetracaine, and xylocaine (lidocaine) [6]. While some inhibit P-gp substrate efflux [71], data indicates that lidocaine is not transported by P-gp [72,73]. CS was shown for the analgesics pentazocine, naloxone, pethidine (meperidine) and morphine [74]. Morphine is a known substrate [75], while meperidine is not [76]. It has been proposed that the CS induced by local anesthetics and narcotics may be caused by lipid bilayer changes that may be required to accommodate the presence of large quantities of P-gp [77]. Decreased surface hydrophobicity and reduced lipid mobility have been reported [13], though similar alterations are induced in non-P-gp drug-resistant cell lines that do not display CS [78]. It is suggested that membrane perturbing properties may sensitize MDR cells preferentially through increased entry, though work is needed to confirm this.
A limitation of many of the CS agents described is that they possess an alternative pharmacological modality that could result in multiple bystander side effects if used in vivo. To this end, a number of reports on the development of novel CS agents exist. Natural product pyranocoumarins [79], diallyl sulfide [80] (DAS) and indole-3-carbinol [81] have been discovered to display CS and down-regulate P-gp expression. The need to assess the specificity of new CS agents is highlighted by the fact that DAS increased mouse glutathione-S-transferase and MRP2 expression in renal brush-border membranes, potentially altering the chemotherapeutic response for the worse [82].
Conclusions
The fact that drug-resistant cells may indeed be hypersensitized to alternative cytotoxic agents in the process of developing resistance is clearly an attractive prospect, particularly given that it could lead to systematic re-sensitization of MDR tumors. The common phenotype for P-gp-mediated CS agents seems to be that P-gp is down-regulated at sub-toxic concentrations prior to selective cell killing of MDR cells at higher concentrations. The precise mechanism of this effect and its relationship to the increased cytotoxicity of CS compounds remains to be determined. In this review, we noted the current hypothesis that production of ROS may be a feature of CS, either through hyperstimulation of high levels of P-gp by substrates or by independent mechanisms. One difficulty in demonstrating this mechanism is that the time required for a chronic ROS effect far outstrips the short-term assays currently in use. However, alternative mechanisms must also exist, including altered intracellular drug trafficking and enhanced uptake of CS agents, and it is possible that a number of pathways associated with the over-expression of P-gp may be associated with CS. There is clearly a great deal of work required to resolve the issue, and irrespective of mechanism, there is no concrete animal or clinical example describing CS induced by P-gp (Box 4) and it remains to be seen whether P-gp levels in tumors are adequate for CS.
Box 4. The prospects for collateral sensitivity in the clinic.
There are few reports utilizing CS in pre-clinical or clinical studies, though some with xenograft mouse models are known [108,109]. One can conceive of a see-saw administration strategy for parrying each new resistance mechanism that arises from a given therapeutic [110]. Clinical CS may be further complicated by the use of combination chemotherapy in the clinic, leading to multi-factorial drug selection phenotypes and confounding rational CS agent selection. Clinical analysis can provide clues to optimal CS agents. Bosanquet and Bell tested 38 drugs against lymphocytes isolated from patients both untreated (n=216) and previously treated (n=188) with known chemotherapy combinations [111]. Patient samples demonstrated the expected cross-resistance for previously treated patients. However, CS was observed for most treatment regimens. For example, lymphocytes from patients treated with chlorambucil showed a 10-fold CS to prednisolone. Patients with exposure to multiple prior chemotherapeutics showed broad cross-resistance to the agents tested, but steroids and 6-mercaptopurine both induced CS. Much work remains to be accomplished in this area.
One challenge in developing CS agents is improving their selectivity. Flavonoid analogs of Desmosdumotin B have been reported with >100-fold selectivity towards KB-V1 cells (Figure 3) [83,84], though flavonoids are known to have wide-ranging biological activity and selectivity needs to be demonstrated [85]. There is great potential for the screening and discovery of new CS agents against MDR cells, including those expressing other transporters. As highlighted here, a compound that is an avid substrate for more than one transporter can elicit CS against cells expressing any or all of them. Although much work remains to characterize the mechanism of action of agents that cause CS, exploiting mechanisms of drug resistance to specifically kill MDR cells is an exciting challenge for clinical development.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute. The authors would like to acknowledge Dr. Gergely Szakacs, who began the work on CS in our laboratory, and Dr. Elias Georges, who has worked extensively on verapamil CS and first reported the ROS hypothesis for CS. We would like to thank members of the Laboratory of Cell Biology for helpful discussions and George Leiman and Kyle R. Brimacombe for editorial assistance. The authors also acknowledge the contributions from the referees, whose critical comments enhanced this review.
References
- 1.Luo J, et al. Principles of Cancer Therapy: Oncogene and Non-Oncogene Addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottesman MM. Mechanisms of Cancer Drug Resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 3.Szakacs G, et al. Targeting Multidrug Resistance in Cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 4.Sheps JA, Ling V. Preface: The Concept and Consequences of Multidrug Resistance. Pflugers Arch - Eur J Physiol. 2007;453:545–553. doi: 10.1007/s00424-006-0115-0. [DOI] [PubMed] [Google Scholar]
- 5.Szybalski W, Bryson V. Genetic Studies on Microbial Cross Resistance to Toxic Agents. I Cross Resistance of Escherichia Coli to Fifteen Antibiotics. J Bacteriol. 1952;64:489–499. doi: 10.1128/jb.64.4.489-499.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bech-Hansen NT, et al. Pleiotropic Phenotype of Colchicine-Resistant Cho Cells: Cross-Resistance and Collateral Sensitivity. J Cell Physiol. 1976;88:23–31. doi: 10.1002/jcp.1040880104. [DOI] [PubMed] [Google Scholar]
- 7.Warr JR, et al. Mutants of Chinese Hamster Ovary Cells with Altered Sensitivity to Taxol and Benzimidazole Carbamates. Cell Biol Int Rep. 1982;6:455–460. doi: 10.1016/0309-1651(82)90117-5. [DOI] [PubMed] [Google Scholar]
- 8.Gupta RS. Cross-Resistance of Vinblastine- and Taxol-Resistant Mutants of Chinese Hamster Ovary Cells to Other Anticancer Drugs. Cancer Treat Rep. 1985;69:515–521. [PubMed] [Google Scholar]
- 9.Hall MD, et al. The Role of Cellular Accumulation in Determining Sensitivity to Platinum-Based Chemotherapy. Annu Rev Pharmacol Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 10.Dempke WC, et al. Expression of Collateral Sensitivity to Cisplatin, Methotrexate, and Fluorouracil in a Human Ovarian Carcinoma Cell Line Following Exposure to Fractionated X-Irradiation in Vitro. Semin Oncol. 1992;19:66–72. [PubMed] [Google Scholar]
- 11.Ando K, et al. Enhancement of Sensitivity to Tumor Necrosis Factor Alpha in Non-Small Cell Lung Cancer Cells with Acquired Resistance to Gefitinib. Clin Cancer Res. 2005;11:8872–8879. doi: 10.1158/1078-0432.CCR-05-0811. [DOI] [PubMed] [Google Scholar]
- 12.Fiskus W, et al. Molecular and Biologic Characterization and Drug Sensitivity of Pan-Histone Deacetylase Inhibitor-Resistant Acute Myeloid Leukemia Cells. Blood. 2008;112:2896–2905. doi: 10.1182/blood-2007-10-116319. [DOI] [PubMed] [Google Scholar]
- 13.Callaghan R, et al. A Comparison of Membrane Properties and Composition between Cell Lines Selected and Transfected for Multi-Drug Resistance. Br J Cancer. 1992;66:781–786. doi: 10.1038/bjc.1992.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen PB, et al. In Vitro Cross-Resistance and Collateral Sensitivity in Seven Resistant Small-Cell Lung Cancer Cell Lines: Preclinical Identification of Suitable Drug Partners to Taxotere, Taxol, Topotecan and Gemcitabin. Br J Cancer. 1997;75:869–877. doi: 10.1038/bjc.1997.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markman M. Antineoplastic Agents in the Management of Ovarian Cancer: Current Status and Emerging Therapeutic Strategies. Trends Pharmacol Sci. 2008;29:515–519. doi: 10.1016/j.tips.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Stordal B, et al. A Systematic Review of Platinum and Taxane Resistance from Bench to Clinic: An Inverse Relationship. Cancer Treat Rev. 2007;33:688–703. doi: 10.1016/j.ctrv.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Prisant LM. Verapamil Revisited: A Transition in Novel Drug Delivery Systems and Outcomes. Heart Dis. 2001;3:55–62. doi: 10.1097/00132580-200101000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Tsuruo T, et al. Increased Accumulation of Vincristine and Adriamycin in Drug-Resistant P388 Tumor Cells Following Incubation with Calcium Antagonists and Calmodulin Inhibitors. Cancer Res. 1982;42:4730–4733. [PubMed] [Google Scholar]
- 19.Warr JR, et al. Verapamil Hypersensitivity of Vincristine Resistant Chinese Hamster Ovary Cell Lines. Cell Biol Int Rep. 1986;10:389–399. doi: 10.1016/0309-1651(86)90011-1. [DOI] [PubMed] [Google Scholar]
- 20.Warr JR, et al. Properties of Verapamil-Hypersensitive Multidrug-Resistant Chinese Hamster Ovary Cells. Cancer Res. 1988;48:4477–4483. [PubMed] [Google Scholar]
- 21.Schuurhuis GJ, et al. Differential Sensitivity of Multi-Drug-Resistant and -Sensitive Cells to Resistance-Modifying Agents and the Relation with Reversal of Anthracycline Resistance. Int J Cancer. 1990;46:330–336. doi: 10.1002/ijc.2910460232. [DOI] [PubMed] [Google Scholar]
- 22.Cano-Gauci DF, Riordan JR. Action of Calcium Antagonists on Multidrug Resistant Cells. Specific Cytotoxicity Independent of Increased Cancer Drug Accumulation. Biochem Pharmacol. 1987;36:2115–2123. doi: 10.1016/0006-2952(87)90139-0. [DOI] [PubMed] [Google Scholar]
- 23.Croop JM, et al. Genetics of Multidrug Resistance: Relationship of a Cloned Gene to the Complete Multidrug Resistant Phenotype. Cancer Res. 1987;47:5982–5988. [PubMed] [Google Scholar]
- 24.Muller C, et al. Verapamil Decreases P-Glycoprotein Expression in Multidrug-Resistant Human Leukemic Cell Lines. Int J Cancer. 1994;56:749–754. doi: 10.1002/ijc.2910560523. [DOI] [PubMed] [Google Scholar]
- 25.Vickers SE, et al. Relationship between Multidrug Resistance, Hypersensitivity to Resistance Modifiers and Cell Volume in Chinese Hamster Ovary Cells. Cell Biol Int Rep. 1993;17:477–485. doi: 10.1006/cbir.1993.1088. [DOI] [PubMed] [Google Scholar]
- 26.Stow MW, Warr JR. Amplification and Expression of Mdr Genes and Flanking Sequences in Verapamil Hypersensitive Hamster Cell Lines. Biochim Biophys Acta. 1991;1092:7–14. doi: 10.1016/0167-4889(91)90171-s. [DOI] [PubMed] [Google Scholar]
- 27.Muller C, et al. Evidence for Transcriptional Control of Human Mdr1 Gene Expression by Verapamil in Multidrug-Resistant Leukemic Cells. Mol Pharmacol. 1995;47:51–56. [PubMed] [Google Scholar]
- 28.Broxterman HJ, et al. Induction by Verapamil of a Rapid Increase in Atp Consumption in Multidrug-Resistant Tumor Cells. FASEB J. 1988;2:2278–2282. doi: 10.1096/fasebj.2.7.3350243. [DOI] [PubMed] [Google Scholar]
- 29.Broxterman HJ, et al. Glycolysis in P-Glycoprotein-Overexpressing Human Tumor Cell Lines. Effects of Resistance-Modifying Agents. FEBS Lett. 1989;247:405–410. doi: 10.1016/0014-5793(89)81380-8. [DOI] [PubMed] [Google Scholar]
- 30.Karwatsky J, et al. A Mechanism for P-Glycoprotein-Mediated Apoptosis as Revealed by Verapamil Hypersensitivity. Biochemistry. 2003;42:12163–12173. doi: 10.1021/bi034149+. [DOI] [PubMed] [Google Scholar]
- 31.Biedler JL, Spengler BA. Collateral Sensitivity in Multidrug Resistance. In: Kellen JA, editor. Reversal of Multidrug Resistance in Cancer. CRC Press; 1993. pp. 21–46. [Google Scholar]
- 32.Cai Y, et al. Reactive Oxygen Species Contribute to Cell Killing and P-Glycoprotein Downregulation by Salvicine in Multidrug Resistant K562/A02 Cells. Cancer Biol Ther. 2007;6:1794–1799. doi: 10.4161/cbt.6.11.4860. [DOI] [PubMed] [Google Scholar]
- 33.Warr JR, et al. Gain and Loss of Hypersensitivity to Resistance Modifiers in Multidrug Resistant Chinese Hamster Ovary Cells. Cancer Lett. 1995;98:115–120. [PubMed] [Google Scholar]
- 34.Wallace DC. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bains OS, Kennedy CJ. Alterations in Respiration Rate of Isolated Rainbow Trout Hepatocytes Exposed to the P-Glycoprotein Substrate Rhodamine 123. Toxicology. 2005;214:87–98. doi: 10.1016/j.tox.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Polekova L, et al. Adaptation of Mouse Leukemia Cells L1210 to Vincristine. Evidence for Expression of P-Glycoprotein. Neoplasma. 1992;39:73–77. [PubMed] [Google Scholar]
- 37.Twentyman PR, et al. Drug Resistance in Human Lung Cancer Cell Lines: Cross-Resistance Studies and Effects of the Calcium Transport Blocker, Verapamil. Int J Radiat Oncol Biol Phys. 1986;12:1355–1358. doi: 10.1016/0360-3016(86)90170-7. [DOI] [PubMed] [Google Scholar]
- 38.Trompier D, et al. Verapamil and Its Derivative Trigger Apoptosis through Glutathione Extrusion by Multidrug Resistance Protein Mrp1. Cancer Res. 2004;64:4950–4956. doi: 10.1158/0008-5472.CAN-04-0143. [DOI] [PubMed] [Google Scholar]
- 39.Rothnie A, et al. Mechanistic Differences between GSH Transport by Multidrug Resistance Protein 1 (Mrp1/Abcc1) and GSH Modulation of Mrp1-Mediated Transport. Mol Pharmacol. 2008;74:1630–1640. doi: 10.1124/mol.108.049080. [DOI] [PubMed] [Google Scholar]
- 40.Laberge RM, et al. Modulation of Gsh Levels in Abcc1 Expressing Tumor Cells Triggers Apoptosis through Oxidative Stress. Biochem Pharmacol. 2007;73:1727–1737. doi: 10.1016/j.bcp.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Lopez-Mirabal HR, Winther JR. Redox Characteristics of the Eukaryotic Cytosol. Biochim Biophys Acta. 2008;1783:629–640. doi: 10.1016/j.bbamcr.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 42.Szakacs G, et al. Predicting Drug Sensitivity and Resistance: Profiling Abc Transporter Genes in Cancer Cells. Cancer Cell. 2004;6:129–137. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 43.Ludwig JA, et al. Selective Toxicity of Nsc73306 in Mdr1-Positive Cells as a New Strategy to Circumvent Multidrug Resistance in Cancer. Cancer Res. 2006;66:4808–4815. doi: 10.1158/0008-5472.CAN-05-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu C-P, et al. Evidence for Dual Mode of Action of a Thiosemicarbazone, Nsc73306: A Potent Substrate of the Multidrug Resistance–Linked Abcg2 Transporter. Mol Cancer Ther. 2007;6:3287–3296. doi: 10.1158/1535-7163.MCT-07-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rappa G, et al. Overexpression of the Multidrug Resistance Genes Mdr1, Mdr3, and Mrp in L1210 Leukemia Cells Resistant to Inhibitors of Ribonucleotide Reductase. Biochem Pharmacol. 1997;54:649–655. doi: 10.1016/s0006-2952(97)00210-4. [DOI] [PubMed] [Google Scholar]
- 46.Hall MD, et al. Synthesis, Activity and Pharmacophore Development for Isatin-Β-Thiosemicarbazones with Mdr1-Selective Activity. J Med Chem. 2009;52:3191–3204. doi: 10.1021/jm800861c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitnall M, et al. A Class of Iron Chelators with a Wide Spectrum of Potent Antitumor Activity That Overcomes Resistance to Chemotherapeutics. Proc Natl Acad Sci U S A. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao VA, et al. The Iron Chelator Dp44mt Causes DNA Damage and Selective Inhibition of Topoisomerase Iialpha in Breast Cancer Cells. Cancer Res. 2009;69:948–957. doi: 10.1158/0008-5472.CAN-08-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richardson DR, et al. 2-Acetylpyridine Thiosemicarbazones Are Potent Iron Chelators and Antiproliferative Agents: Redox Activity, Iron Complexation and Characterization of Their Antitumor Activity. J Med Chem. 2009;52:1459–1470. doi: 10.1021/jm801585u. [DOI] [PubMed] [Google Scholar]
- 50.Heffeter P, et al. Multidrug-Resistant Cancer Cells Are Preferential Targets of the New Antineoplastic Lanthanum Compound Kp772 (Ffc24) Biochem Pharmacol. 2007;73:1873–1886. doi: 10.1016/j.bcp.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heffeter P, et al. Anticancer Activity of the Lanthanum Compound [Tris(1,10-Phenanthroline)Lanthanum(Iii)]Trithiocyanate (Kp772; Ffc24) Biochem Pharmacol. 2006;71:426–440. doi: 10.1016/j.bcp.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 52.Burkitt MJ, et al. 1,10-Phenanthroline Stimulates Internucleosomal DNA Fragmentation in Isolated Rat-Liver Nuclei by Promoting the Redox Activity of Endogenous Copper Ions. Biochem J. 1996;313(Pt 1):163–169. doi: 10.1042/bj3130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai X, et al. Copper-1,10-Phenanthroline-Induced Apoptosis in Liver Carcinoma Bel-7402 Cells Associates with Copper Overload, Reactive Oxygen Species Production, Glutathione Depletion and Oxidative DNA Damage. Biometals. 2007;20:1–11. doi: 10.1007/s10534-006-9008-0. [DOI] [PubMed] [Google Scholar]
- 54.de Avellar IG, et al. Reevaluating the Role of 1,10-Phenanthroline in Oxidative Reactions Involving Ferrous Ions and DNA Damage. Biochim Biophys Acta. 2004;1675:46–53. doi: 10.1016/j.bbagen.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 55.Turk D, et al. Identification of Mdr1-Inverse Agents That Selectively Kill Multidrug Resistant Cancer Cells. Cancer Res. 2009 in press. [Google Scholar]
- 56.Majumder S, et al. Synthesis, Characterization and Biological Properties of a Novel Copper Complex. Eur J Med Chem. 2003;38:893–898. doi: 10.1016/j.ejmech.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Majumder S, et al. Reversal of Drug Resistance in P-Glycoprotein-Expressing T-Cell Acute Lymphoblastic Cem Leukemia Cells by Copper N-(2-Hydroxy Acetophenone) Glycinate and Oxalyl Bis (N-Phenyl) Hydroxamic Acid. Cancer Lett. 2006;244:16–23. doi: 10.1016/j.canlet.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 58.Mookerjee A, et al. A Novel Copper Complex Induces Ros Generation in Doxorubicin Resistant Ehrlich Ascitis Carcinoma Cells and Increases Activity of Antioxidant Enzymes in Vital Organs in Vivo. BMC Cancer. 2006;6:267. doi: 10.1186/1471-2407-6-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ankel E, Petering DH. Iron-Chelating Agents and the Reductive Removal of Iron from Transferrin. Biochem Pharmacol. 1980;29:1833–1837. doi: 10.1016/0006-2952(80)90146-x. [DOI] [PubMed] [Google Scholar]
- 60.Antholine W, et al. Studies of the Reaction of 2-Formylpyridine Thiosemicarbazone and Its Iron and Copper Complexes with Biological Systems. Mol Pharmacol. 1977;13:89–98. [PubMed] [Google Scholar]
- 61.Richardson DR. Iron Chelators as Therapeutic Agents for the Treatment of Cancer. Crit Rev Oncol Hematol. 2002;42:267–281. doi: 10.1016/s1040-8428(01)00218-9. [DOI] [PubMed] [Google Scholar]
- 62.Diddens H, et al. Characterization of Actinomycin-D-Resistant Cho Cell Lines Exhibiting a Multidrug-Resistance Phenotype and Amplified DNA Sequences. Int J Cancer. 1987;40:635–642. doi: 10.1002/ijc.2910400511. [DOI] [PubMed] [Google Scholar]
- 63.Barancik M, et al. Overcoming of Vincristine Resistance in L1210/Vcr Cells by Several Corticosteroids. Collateral Sensitivity of Resistant Cells Neoplasma. 1993;40:21–25. [PubMed] [Google Scholar]
- 64.Fleming GF, et al. Megestrol Acetate Reverses Multidrug Resistance and Interacts with P-Glycoprotein. Cancer Chemother Pharmacol. 1992;29:445–449. doi: 10.1007/BF00684845. [DOI] [PubMed] [Google Scholar]
- 65.Barnes KM, et al. Steroid Transport, Accumulation, and Antagonism of P-Glycoprotein in Multidrug-Resistant Cells. Biochemistry. 1996;35:4820–4827. doi: 10.1021/bi952380k. [DOI] [PubMed] [Google Scholar]
- 66.Naito M, et al. Steroid Hormones Inhibit Binding of Vinca Alkaloid to Multidrug Resistance Related P-Glycoprotein. Biochem Biophys Res Commun. 1989;158:1066–1071. doi: 10.1016/0006-291x(89)92830-1. [DOI] [PubMed] [Google Scholar]
- 67.Rickardson L, et al. Screening of an Annotated Compound Library for Drug Activity in a Resistant Myeloma Cell Line. Cancer Chemother Pharmacol. 2006;58:749–758. doi: 10.1007/s00280-006-0216-7. [DOI] [PubMed] [Google Scholar]
- 68.Gruol DJ, Bourgeois S. Chemosensitizing Steroids: Glucocorticoid Receptor Agonists Capable of Inhibiting P-Glycoprotein Function. Cancer Res. 1997;57:720–727. [PubMed] [Google Scholar]
- 69.Scudiero DA, et al. Cell Line Designation Change: Multidrug-Resistant Cell Line in the Nci Anticancer Screen. J Natl Cancer Inst. 1998;90:862. doi: 10.1093/jnci/90.11.862. [DOI] [PubMed] [Google Scholar]
- 70.Mutoh K, et al. Estrogen-Mediated Post Transcriptional Down-Regulation of P-Glycoprotein in Mdr1-Transduced Human Breast Cancer Cells. Cancer Sci. 2006;97:1198–1204. doi: 10.1111/j.1349-7006.2006.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carlsen SA, et al. Modulation of Membrane Drug Permeability in Chinese Hamster Ovary Cells. Biochim Biophys Acta. 1976;455:900–912. doi: 10.1016/0005-2736(76)90059-6. [DOI] [PubMed] [Google Scholar]
- 72.Aanismaa P, Seelig A. P-Glycoprotein Kinetics Measured in Plasma Membrane Vesicles and Living Cells. Biochemistry. 2007;46:3394–3404. doi: 10.1021/bi0619526. [DOI] [PubMed] [Google Scholar]
- 73.Kakumoto M, et al. Mdr1-Mediated Interaction of Digoxin with Antiarrhythmic or Antianginal Drugs. Biol Pharm Bull. 2002;25:1604–1607. doi: 10.1248/bpb.25.1604. [DOI] [PubMed] [Google Scholar]
- 74.Callaghan R, Riordan JR. Collateral Sensitivity of Multidrug Resistant Cells to Narcotic Analgesics Is Due to Effects on the Plasma Membrane. Biochim Biophys Acta. 1995;1236:155–162. doi: 10.1016/0005-2736(95)00042-2. [DOI] [PubMed] [Google Scholar]
- 75.Yousif S, et al. Effect of Chronic Exposure to Morphine on the Rat Blood-Brain Barrier: Focus on the P-Glycoprotein. J Neurochem. 2008;107:647–657. doi: 10.1111/j.1471-4159.2008.05647.x. [DOI] [PubMed] [Google Scholar]
- 76.Dagenais C, et al. Variable Modulation of Opioid Brain Uptake by P-Glycoprotein in Mice. Biochem Pharmacol. 2004;67:269–276. doi: 10.1016/j.bcp.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 77.Cano-Gauci DF, Riordan JR. Collateral Sensitivity of Multidrug-Resistant Cells. In: Roninson IB, editor. Molecular and Cellular Biology of Multidrug Resistance in Tumor Cells. Plenum Press; 1991. pp. 337–347. [Google Scholar]
- 78.Ferte J. Analysis of the Tangled Relationships between P-Glycoprotein-Mediated Multidrug Resistance and the Lipid Phase of the Cell Membrane. Eur J Biochem. 2000;267:277–294. doi: 10.1046/j.1432-1327.2000.01046.x. [DOI] [PubMed] [Google Scholar]
- 79.Wu JY, et al. Reversal of Multidrug Resistance in Cancer Cells by Pyranocoumarins Isolated from Radix Peucedani. Eur J Pharmacol. 2003;473:9–17. doi: 10.1016/s0014-2999(03)01946-0. [DOI] [PubMed] [Google Scholar]
- 80.Arora A, et al. Reversal of P-Glycoprotein-Mediated Multidrug Resistance by Diallyl Sulfide in K562 Leukemic Cells and in Mouse Liver. Carcinogenesis. 2004;25:941–949. doi: 10.1093/carcin/bgh060. [DOI] [PubMed] [Google Scholar]
- 81.Arora A, et al. Modulation of P-Glycoprotein-Mediated Multidrug Resistance in K562 Leukemic Cells by Indole-3-Carbinol. Toxicol Appl Pharmacol. 2005;202:237–243. doi: 10.1016/j.taap.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 82.Demeule M, et al. Diallyl Disulfide, a Chemopreventive Agent in Garlic, Induces Multidrug Resistance-Associated Protein 2 Expression. Biochem Biophys Res Commun. 2004;324:937–945. doi: 10.1016/j.bbrc.2004.09.141. [DOI] [PubMed] [Google Scholar]
- 83.Nakagawa-Goto K, et al. Antitumor Agents 260. New Desmosdumotin B Analogues with Improved in Vitro Anticancer Activity. J Med Chem. 2008;51:3297–3303. doi: 10.1021/jm701208v. [DOI] [PubMed] [Google Scholar]
- 84.Nakagawa-Goto K, et al. Total Synthesis and Bioactivity of Unique Flavone Desmosdumotin B and Its Analogs. Bioorg Med Chem Lett. 2005;15:3016–3019. doi: 10.1016/j.bmcl.2005.04.070. [DOI] [PubMed] [Google Scholar]
- 85.Cnubben NH, et al. Metabolism of Atp-Binding Cassette Drug Transporter Inhibitors: Complicating Factor for Multidrug Resistance. Expert Opin Drug Metab Toxicol. 2005;1:219–232. doi: 10.1517/17425255.1.2.219. [DOI] [PubMed] [Google Scholar]
- 86.Ambudkar SV, et al. Biochemical, Cellular, and Pharmacological Aspects of the Multidrug Transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 87.Aller SG, et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gottesman MM, et al. Structure of a Multidrug Transporter. Nat Biotechnol. 2009;27:546–547. doi: 10.1038/nbt0609-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McHugh K, Callaghan R. Clinical Trials on Mdr Reversal Agents. In: Colabufo NA, editor. Multidrug Resistance: Biological and Pharmaceutical Advance in Antitumour Treatment. Research Signpost; 2008. pp. 321–353. [Google Scholar]
- 90.Li L, et al. Reversal of Mdr1 Gene-Dependent Multidrug Resistance Using Low Concentration of Endonuclease-Prepared Small Interference Rna. Eur J Pharmacol. 2006;536:93–97. doi: 10.1016/j.ejphar.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 91.Stierle V, et al. Modulation of Mdr1 Gene Expression in Multidrug Resistant Mcf7 Cells by Low Concentrations of Small Interfering Rnas. Biochem Pharmacol. 2005;70:1424–1430. doi: 10.1016/j.bcp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 92.Watanabe T, et al. Regression of Established Tumors Expressing P-Glycoprotein by Combinations of Adriamycin, Cyclosporin Derivatives, and Mrk-16 Antibodies. J Natl Cancer Inst. 1997;89:512–518. doi: 10.1093/jnci/89.7.512. [DOI] [PubMed] [Google Scholar]
- 93.Lehne G, et al. Human Hepatoma Cells Rich in P-Glycoprotein Are Sensitive to Aclarubicin and Resistant to Three Other Anthracyclines. Br J Cancer. 1996;74:1719–1729. doi: 10.1038/bjc.1996.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hait WN, et al. Sensitivity of K562 Human Chronic Myelogenous Leukemia Blast Cells Transfected with a Human Multidrug Resistance Cdna to Cytotoxic Drugs and Differentiating Agents. J Clin Invest. 1993;91:2207–2215. doi: 10.1172/JCI116447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Triest B, et al. Cross-Resistance to Thymidylate Synthase Inhibitors in P-Glycoprotein and Non-P-Glycoprotein Cell Lines. Adv Exp Med Biol. 1994;370:189–193. doi: 10.1007/978-1-4615-2584-4_41. [DOI] [PubMed] [Google Scholar]
- 96.Bergman AM, et al. Collateral Sensitivity to Gemcitabine (2’,2’-Difluorodeoxycytidine) and Cytosine Arabinoside of Daunorubicin- and Vm-26-Resistant Variants of Human Small Cell Lung Cancer Cell Lines. Biochem Pharmacol. 2001;61:1401–1408. doi: 10.1016/s0006-2952(01)00627-x. [DOI] [PubMed] [Google Scholar]
- 97.Bergman AM, et al. Increased Sensitivity to Gemcitabine of P-Glycoprotein and Multidrug Resistance-Associated Protein-Overexpressing Human Cancer Cell Lines. Br J Cancer. 2003;88:1963–1970. doi: 10.1038/sj.bjc.6601011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moritaka T, et al. Cisplatin-Resistant Human Small Cell Lung Cancer Cell Line Shows Collateral Sensitivity to Vinca Alkaloids. Anticancer Res. 1998;18:927–933. [PubMed] [Google Scholar]
- 99.Kotoh S, et al. Increased Expression of DNA Topoisomerase I Gene and Collateral Sensitivity to Camptothecin in Human Cisplatin-Resistant Bladder Cancer Cells. Cancer Res. 1994;54:3248–3252. [PubMed] [Google Scholar]
- 100.Perego P, et al. Ovarian Cancer Cisplatin-Resistant Cell Lines: Multiple Changes Including Collateral Sensitivity to Taxol. Ann Oncol. 1998;9:423–430. doi: 10.1023/a:1008265012435. [DOI] [PubMed] [Google Scholar]
- 101.Wick AN, et al. Localization of the Primary Metabolic Block Produced by 2-Deoxyglucose. J Biol Chem. 1957;224:963–969. [PubMed] [Google Scholar]
- 102.Kurtoglu M, et al. Differential Toxic Mechanisms of 2-Deoxy-D-Glucose Versus 2-Fluorodeoxy-D-Glucose in Hypoxic and Normoxic Tumor Cells. Antioxid Redox Signal. 2007;9:1383–1390. doi: 10.1089/ars.2007.1714. [DOI] [PubMed] [Google Scholar]
- 103.Weinstein JN, et al. Neural Computing in Cancer Drug Development: Predicting Mechanism of Action. Science. 1992;258:447–451. doi: 10.1126/science.1411538. [DOI] [PubMed] [Google Scholar]
- 104.Lyon RC, et al. Glucose Metabolism in Drug-Sensitive and Drug-Resistant Human Breast Cancer Cells Monitored by Magnetic Resonance Spectroscopy. Cancer Res. 1988;48:870–877. [PubMed] [Google Scholar]
- 105.Kaplan O, et al. Effects of 2-Deoxyglucose on Drug-Sensitive and Drug-Resistant Human Breast Cancer Cells: Toxicity and Magnetic Resonance Spectroscopy Studies of Metabolism. Cancer Res. 1990;50:544–551. [PubMed] [Google Scholar]
- 106.Bentley J, et al. 2-Deoxy-D-Glucose Toxicity and Transport in Human Multidrug-Resistant Kb Carcinoma Cell Lines. Oncol Res. 1996;8:77–84. [PubMed] [Google Scholar]
- 107.Bentley J, et al. The Human Kb Multidrug-Resistant Cell Line Kb-C1 Is Hypersensitive to Inhibitors of Glycosylation. Cancer Lett. 1997;115:221–227. doi: 10.1016/s0304-3835(97)04739-3. [DOI] [PubMed] [Google Scholar]
- 108.Demidova NS, et al. Decreased Sensitivity of Multidrug-Resistant Tumor Cells to Cisplatin Is Correlated with Sorcin Gene Co-Amplification. Neoplasma. 1995;42:195–201. [PubMed] [Google Scholar]
- 109.Fichtner I, et al. Characterization of Four Drug-Resistant P388 Sublines: Resistance/Sensitivity in Vivo, Resistance-and Proliferation-Markers, Immunogenicity. Anticancer Res. 1994;14:1995–2003. [PubMed] [Google Scholar]
- 110.Aisner J, et al. Sequencing Topotecan and Etoposide Plus Cisplatin to Overcome Topoisomerase I and Ii Resistance: A Pharmacodynamically Based Phase I Trial. Clin Cancer Res. 2003;9:2504–2509. [PubMed] [Google Scholar]
- 111.Bosanquet AG, Bell PB. Novel Ex Vivo Analysis of Nonclassical. Pleiotropic Drug Resistance and Collateral Sensitivity Induced by Therapy Provides a Rationale for Treatment Strategies in Chronic Lymphocytic Leukemia. Blood. 1996;87:1962–1971. [PubMed] [Google Scholar]
- 112.Shen DW, et al. Characterisation of High-Level Cisplatin-Resistant Cell Lines Established from a Human Hepatoma Cell Line and Human Kb Adenocarcinoma Cells: Cross-Resistance and Protein Changes. Br J Cancer. 1995;71:676–683. doi: 10.1038/bjc.1995.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Parekh H, Simpkins H. Cross-Resistance and Collateral Sensitivity to Natural Product Drugs in Cisplatin-Sensitive and -Resistant Rat Lymphoma and Human Ovarian Carcinoma Cells. Cancer Chemother Pharmacol. 1996;37:457–462. doi: 10.1007/s002800050412. [DOI] [PubMed] [Google Scholar]
- 114.Martin-Aragon S, et al. Cytosine Arabinoside (Ara-C) Resistance Confers Cross-Resistance or Collateral Sensitivity to Other Classes of Anti-Leukemic Drugs. Anticancer Res. 2000;20:139–150. [PubMed] [Google Scholar]
- 115.Brewer F, Warr JR. Verapamil Reversal of Vincristine Resistance and Cross-Resistance Patterns of Vincristine-Resistant Chinese Hamster Ovary Cells. Cancer Treat Rep. 1987;71:353–359. [PubMed] [Google Scholar]
- 116.Ishii M, et al. Growth Inhibitory Effect of a New Camptothecin Analog, Dx-8951f, on Various Drug-Resistant Sublines Including Bcrp-Mediated Camptothecin Derivative-Resistant Variants Derived from the Human Lung Cancer Cell Line Pc-6. Anticancer Drugs. 2000;11:353–362. doi: 10.1097/00001813-200006000-00005. [DOI] [PubMed] [Google Scholar]
- 117.Tanaka T, et al. Increased Glutathione Level Is Not Involved in Enhanced Bleomycin Sensitivity in Cisplatin-Resistant 2780cp Cells. Anticancer Res. 2008;28:2663–2668. [PubMed] [Google Scholar]
- 118.Taki T, et al. In Vivo Etoposide-Resistant C6 Glioma Cell Line: Significance of Altered DNA Topoisomerase Ii Activity in Multi-Drug Resistance. J Neurooncol. 1998;36:41–53. doi: 10.1023/a:1005718912236. [DOI] [PubMed] [Google Scholar]
- 119.Dhar S, et al. Relationship between Cytotoxic Drug Response Patterns and Activity of Drug Efflux Transporters Mediating Multidrug Resistance. Eur J Pharmacol. 1998;346:315–322. doi: 10.1016/s0014-2999(98)00058-2. [DOI] [PubMed] [Google Scholar]
- 120.Liang Y, et al. Enhanced in Vitro Invasiveness and Drug Resistance with Altered Gene Expression Patterns in a Human Lung Carcinoma Cell Line after Pulse Selection with Anticancer Drugs. Int J Cancer. 2004;111:484–493. doi: 10.1002/ijc.20230. [DOI] [PubMed] [Google Scholar]
- 121.Dhar S, et al. Anti-Cancer Drug Characterisation Using a Human Cell Line Panel Representing Defined Types of Drug Resistance. Br J Cancer. 1996;74:888–896. doi: 10.1038/bjc.1996.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jonsson-Videsater K, et al. Doxorubicin-Resistant, Mrp1-Expressing U-1285 Cells Are Sensitive to Idarubicin. Ther Drug Monit. 2003;25:331–339. doi: 10.1097/00007691-200306000-00014. [DOI] [PubMed] [Google Scholar]
- 123.Breier A, et al. Cytotoxic Activity of Several Unrelated Drugs on L1210 Mouse Leukemic Cell Sublines with P-Glycoprotein (Pgp) Mediated Multidrug Resistance (Mdr) Phenotype. A Qsar Study Neoplasma. 2000;47:100–106. [PubMed] [Google Scholar]
- 124.Bergman AM, et al. Increased Sensitivity to Gemcitabine of P-Gp and Mrp Overexpressing Human Non-Small Cell Lung Cancer Cell Lines. Adv Exp Med Biol. 1998;431:591–594. doi: 10.1007/978-1-4615-5381-6_114. [DOI] [PubMed] [Google Scholar]
- 125.van Triest B, et al. Cross-Resistance to Antifolates in Multidrug Resistant Cell Lines with P-Glycoprotein or Multidrug Resistance Protein Expression. Biochem Pharmacol. 1997;53:1855–1866. doi: 10.1016/s0006-2952(97)82448-3. [DOI] [PubMed] [Google Scholar]
- 126.Warr JR, et al. The Preferential Induction of Apoptosis in Multidrug-Resistant Kb Cells by 5-Fluorouracil. Cancer Lett. 2002;175:39–44. doi: 10.1016/s0304-3835(01)00721-2. [DOI] [PubMed] [Google Scholar]
- 127.Matsuo K, et al. Growth Inhibitory Effects of Antifolates against an Adriamycin-Resistant Human Small Cell Lung Cancer Cell Line. Acta Med Okayama. 1997;51:121–127. doi: 10.18926/AMO/30795. [DOI] [PubMed] [Google Scholar]