

Abstract
Objective
Mitogen-activated protein kinase p38 (p38 MAPK) is part of an intracellular signaling pathway activated by environmental stress and inflammatory factors. Since in vitro studies show that inhibiting p38 activity leads to a reduction of degenerative metalloproteinase release from chondrocytes, it was speculated that inactivation of p38 in vivo may be chondroprotective. We test this hypothesis by examining the morphology of adult mice that express a dominant-negative (DN) p38 MAPK transgene in a cartilage-specific manner.
Methods
The in vivo effects of the genetic inhibition of p38 MAPK activity in cartilage was investigated in one-year old p38 DN heterozygote (+/−) transgenic (TG) mice (n=10) using morphologic measurements, micro-computed tomography (micro-CT) scanning, biomechanical testing, and histology. Results were compared to wild-type (WT) littermates (n=9).
Results
Adult DN p38 (+/−) TG mice exhibited 50% p38 MAPK activity in articular chondrocytes when compared to WT mice. They were significantly shorter in overall body length and in the length of the femur and tibia. There were no differences in bone material or mechanical properties between the TG and WT mice. Surprisingly, the TG mice had higher osteoarthritis grades at the knee joint.
Conclusion
Genetic inhibition of p38 MAPK activity in cartilage results in shortened limb length and defects in the articular cartilage of knee joints in adult mice. This study demonstrates that chronic lifelong reduction of p38 MAPK activity may be harmful to joint health, and suggests that the timing of p38 inhibition for chondroprotection in vivo is an important variable that warrants further investigation.
Keywords: p38 MAP kinase, bone, osteoarthritis, cartilage
INTRODUCTION
Mitogen-activated protein kinase p38 (p38 MAPK) is part of an intracellular signaling pathway activated by proinflammatory cytokines and environmental stressors including IL-1, TNF-α, mechanical loading, and osmotic shock,1,2,3,4 all of which have been linked to osteoarthritis pathogenesis. Upon activation, p38 MAPK phosphorylates transcriptional factors and transduces signals into the nucleus that alter gene expression.5 It has been shown that p38 MAPK is intimately involved in activating matrix metalloproteinases responsible for collagen cleavage and osteoarthritis pathogenesis.6,7,8,9,10 In vitro studies have suggested that p38 inhibition may serve as a potential therapeutic target for osteoarthritis.11–26 Furthermore, increased p38 activity is accompanied by type X collagen staining in osteochondrocytes and marginal synovial cells in a mouse model of osteoarthritis,27 which was created by transecting the anterior cruciate ligament and medial meniscus in adult mice. However, in vivo effects of p38 activity reduction in cartilage on osteoarthritis pathogenesis are not known.
There are at least three families of MAP kinases which differ in the sequence and size of the activation loop by phosphorylation: the extracellular regulated kinases (ERKs) have a TEY motif, the c-Jun NH2 terminal kinases or stress activated protein kinases (JNKs or SAP kinases) which have a TPY motif, and the p38 family which have a TGY motif.10 As indicated by previous research, increased levels of p38 MAPK activities lead to heightened expression of the hypertrophic cartilage phenotype, while decreased levels of p38 MAPK activities lead to diminished chondrocyte proliferation and hypertrophy.27,28,29 Thus, a clear correlation between the level of p38 activity and the process of cartilage hypertrophic differentiation during skeletal development has been indicated. However, it is not clear whether genetic inhibition of p38 MAPK activity in adult articular cartilage affects skeletal properties or its homeostasis. It is believed that the MAPK cascades consist of a core MAPK module, which has no less than three enzymes activated in series by phosphorylation cascades: 1) a MAPK, 2) an immediate upstream kinase (Known as Mitogen-Activated Protein Kinase Kinase or MAPKK), and 3) an additional kinase upstream of the MAPKK (Known as Mitogen-Protein Kinase Kinase Kinase or MAPKKK); these regulatory cascades not only convey information to the target effectors, but also coordinate incoming information from parallel signaling pathways.30 The cascade is known to be activated by receptor tyrosine kinase activity induced by growth factors including IL-1,31 EGF,32 FGF,33 PTH,24 TGF-β,34 and growth differentiation factor-5 (GDF-5)35 in chondrocytes.1 Upon activation by upstream MAPKK including MKK3 and MKK6, p38 MAPK in turn activates down stream target proteins by phosphorylating them,5 which affect cell proliferation, differentiation, and aging. 36,37,38 A mechanism of specific activation of p38 MAP kinase is the selective formation of complexes between MAPKK (MKK3 or MKK6) and different p38 MAPKs. Dual phosphorylation on theronine and tyrosine by MAPKK is required for p38 MAPK activation. It has been shown that a p38 MAPK mutant that harbors point mutations in its phosphorylation site fails to activate downstream target proteins, thereby exerting a dominant negative effect on the activation of p38 MAPK pathway to block its response to environmental stimuli and/or stress signals.39
Taking advantage of this activation property of p38 MAPK, in this study, we inhibited p38 MAPK activity in articular cartilage in vivo by expressing dominant negative p38 MAPK in transgenic mice under the control of Col II promoter. This study was performed to determine whether genetic reduction of p38 MAPK activity in cartilage leads to phenotypic changes in bone and/or cartilage in adult mice.
MATERIALS AND METHODS
Transgenic mice
This study was approved by our institutional review board (CMTT #:0304-02). All procedures were IACUC approved. All mice in this study were from the B6D2F/1/C57BL/6 background. All mice were housed in Rhode Island Hospital animal facility. Food and water were provided ad libitum, and all care given to the mice was in compliance with the NIH guidelines for the care and use of laboratory animals. DN p38 MAPK transgenic mice were generated using a collagen type IIa-DNp38 expression vector as previously described.40 Briefly, DN p38 cDNA was generated by replacing phosphorylation residues Thr180 and Tyr182 by Ala and Phe in wildtype p38 MAPK cDNA, respectively. It has been shown that transfection of this DNp38 resulted in defective endogenous p38 MAPK activation, which is not responsive to upstream stimuli including UV or MKK6.39 DN p38, hGh, and the cartilage-specific enhancer in the first intron of COL II gene were amplified respectively by PCR and then ligated with 5’-flanking COL2A promoter.41 To create transgenic mice, linearized and purified DNA was injected directly into the pronuclei of fertilized mouse eggs. Skin and tail samples were collected from each animal after birth for genotyping. Integration of the transgene into the genome was demonstrated by Southern Blot and PCR. Three strains of transgenic mice were generated and all of them exhibited similar phenotypes. Mice were of two groups: a) DN p38 MAPK (+/-) transgenic mice (n=10, 5 males/5 females) sacrificed at an average age of 53 ± 9.28 weeks, and b) wild-type controls (−/−) (n=9, 4 males/5 females) sacrificed at 52 weeks of age. No mice died prematurely. The DNp38 MAPK (+/+) homozygote transgenic mice die shortly after birth due to diminished endochondral bone formation.40 However, DN p38 MAPK (+/−) heterozygote mice (TG) lived a normal lifespan as the wildtype littermates (WT). As mice cease long bone growth at 8 weeks, all animals in this study were considered fully developed and skeletally mature.42
Morphological characterization
After sacrifice of mice, specimens were weighed, their overall lengths were measured (rostral snout to base of tail), and they were x-rayed in both posterior-anterior and medial-lateral views. Spine lengths and cranial diameters were measured from posterior-anterior radiographs. Both femurs and tibias from each mouse were harvested under microscopy. Special care was taken to maintain the integrity of the articular cartilage at the knee joint. Bones were cleaned of extraneous soft tissues and photographed, and a digital caliper was used to measure length (superior femoral head to inferior femoral condyles, superior tibial articular surface to inferior tibial articular surface) and the anterior-posterior and medial-lateral diameters at the midshaft.
Micro-CT Technique
The properties of hind-limb long bones were compared in wild-type (WT, −/−) and DN p38 (+/−) mice. Cross-sectional areas and moments of inertia were calculated from micro-computed tomography (CT) scans taken at the mid-diaphysis of the left femur from each animal (CT 20, Scanco Medical, Bassensdorf, Switzerland). During scanning the bones were oriented in the scanner such that their sagittal, frontal and transverse planes were aligned with the scanner's built-in coordinate system. Ten serial transverse slice images were acquired using machine settngs of 70 kV and 160 μA. The resolution of the scanned images was 9.0 μm (isometric voxel size). The original scanned images were filtered to reduce noise and then segmented using a fixed threshold, yielding binary (white) images of the bone cross-sections on a black background. Bone area and moment of inertia (about the mediolateral axis) were calculated for each of the ten segmented cross-sections, after which they were averaged to yield single, mean values for each bone.
Mechanical testing
After measurement and imaging, the femurs were tested to failure in three-point bending using a servohydraulic materials testing system (Instron Corporation, Canton, MA) fitted with custom-made fixtures. The three-point bending test has long been used in the evaluation of bone strength and studies have shown that breaking force (peak load) and stiffness, as well as intrinsic parameters, such as stress are good indicators of mechanical strength of cortical bone.43,44 The bones were positioned with their posterior surfaces on the lower supports (7mm apart) and the load applied to the anterior surface. Testing was performed to failure at a rate of 0.1 mm/sec; during testing the load and displacement data was acquired at 100 Hz with a digital data acquisition system. The raw load-displacement data was reduced to yield values for stiffness, peak load, and energy to failure. Peak stress was calculated from the experimentally derived values using following equation
where F is applied load, L is the span of the support points (7.0mm), c is the half-diameter of the midshaft in the load direction derived from the micro-CT analysis, I is the cross-sectional moment of inertia defined by the micro-CT analysis.44 Morphologic and biomechanical properties were compared with standard two-tailed t-tests assuming unequal variances. P < 0.05 was considered to be statistically significant.
Western blot analysis
Using the femoral head cartilage of the tested bones, the extent of p38 MAPK activity reduction was evaluated with a western blot for p38 to confirm mice genotypes and to strengthen correlations with phenotype. To address the specificity of the p38 transgene, we performed western blot of two other MAP kinases to show that the activities of ERK and JNK were not affected by DNp38 transgene. Analysis was performed with cartilage extracts from all mice using a polyclonal antibody sc-7975R against p-p38 activity (Santa Cruz, CA), p-JNK sc-12882R (Santa Cruz, CA), and a monoclonal antibody sc-7383 against p-ERK (Santa Cruz, CA).
While it was expected that phosphorylated-p38 would be decreased in dominant-negative mice, the protein expression levels of p38 and other MAP kinases were not expected to change from control. This was confirmed as follows. After mechanical testing, the femoral head articular cartilage was harvested under a dissecting microscope and washed twice in PBS. Cartilage was homogenized in 8 M urea, 50 mM sodium acetate at pH 5.8. The protein content in the supernatant was quantified with BCA Protein Assay Reagent Kit (Pierce, Rockford, IL). For each sample, 15 μg of total protein was electrophoresed in 10% SDS PAGE under reducing conditions before it was blotted and probed with monoclonal antibody against p-p38 activity. At the same time, the protein levels were determined by western blot analysis with polyclonal Ab sc-535 against p38 MAP kinase, polyclonal Ab sc-571 against JNK, and monoclonal Ab sc-153 against ERK (Santa Cruz, CA). All of the antibodies were diluted (1:1000) in PBS-T containing 1% bovine serum albumin. Horseradish peroxidase-conjugated goat anti-rabbit IgG (H+L) (Bio-Rad Laboratories, Richmond, CA) were diluted (1:3000) in PBS-T, and used as secondary antibodies. Visualization of immunoreactive proteins was achieved by using ECL Western blotting detection reagents (Amersham, Arlington Heights, IL) and by subsequent exposure of the membrane to Kodak X-Omat AR film. MAPK activities were determined by quantifying the intensity of phospho-MAPK bands on western blot using image acquisition and analysis software (UVP bioimaging systems, Upland, CA).
Immunohistochemistry
Sections were digested with bovine testicular hyaluronidase (4,000 U/ml in PBS; Sigma, St. Louis, MA, USA) for 30 min at 37°C. After washing in PBS, the sections were incubated with 5% normal goat serum for 30 min at room temperature before being incubated for 1 hr at room temperature with either antibody against p38 MAP kinase or antibody against p-p38 MAP kinase as described above (Santa Cruz, CA). Affinity-purified fluorescein-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) were then applied at 1:100 for 1 hr at room temperature.
Histology
To determine the influence of p38 on tissue phenotype, the right tibia from each mouse was used for histology. Each tibia was cut with a scalpel approximately 3 mm inferior to the superior articular surface and fixed in 4% formalin for 24 hrs. Following fixation, samples were decalcified in 0.2M EDTA for 3 weeks, dehydrated with 70%, 95%, and 99.9% ethanol, cleared with methylbenzoate and xylene, and embedded in parafin for sectioning. Eight sagittal sections were cut from each bone at 6 μm intervals. The sections were mounted on glass slides and deparaffinized in xylene (3 changes, 5 minutes each). Slides were rehydrated with tap water, then placed through 3 changes (1 minute each) of absolute ethanol, and 1 change (1 minute) of 95% ethanol. Slides were again washed with tap water. Slides were washed with Gill III hematoxylin solution for 5 minutes, washed with tap water for 5 minutes and then with 1% aqueous lithium carbonate for 1 minute. Slides were again washed in running tap water for 2 minutes and then rinsed briefly in 95% ethanol. Slides were then rinsed with 1% eosin Y, 0.2% phloxine, and 80% ethanol for 1 minute and then dehydrated in 1 change of 95% ethanol and 3 changes of absolute ethanol (30 seconds each). Slides were then cleared in xylene with 3 changes (1 minute each). Finally, a coverslip was mounted onto slides with Micromount mounting medium (Surgipath Medical Industries, Richmond, Ill.) and they were analyzed using a light microscope.
For Safranin-O/Fast Green staining, 5μm paraffin-embedded sections of tibia from mice were counter-stained with Hematoxylin before being stained with 0.02% aqueous Fast Green for 4 min (followed by 3 dips in 1% acetic acid) and then 0.1% Safranin-O for 6 min. The slides were then dehydrated and mounted with crystal mount medium.
The articular surfaces of all 8 slices from each sample were analyzed by two blinded observers. Each of the slices from each sample was graded on a 0 to 4 scale.45 A grossly intact articular cartilage surface was rated a grade 0. Fissuring and fibrillation at the joint surface were rated a grade 1. Well-demarcated defects not extending below the tidemark were rated a grade 2. Defects reaching into the calcified cartilage were rated a grade 3. Finally, extensive defects reaching into the bony layer, with signs of eburnation, and prominent osteophytes were rated a grade 4. The osteoarthritis (OA) grade for a given tibia was defined to be the highest grade for any of the eight sections. This process was repeated for each animal, and the average OA grade of the DN p38 group was compared to the WT controls using a standard two-tailed t-test assuming unequal variances.
RESULTS
Activity of p38 in Dominant-Negative (DN) Mice
Western blot analysis showed the amount of activated, or phosphorylated, p38 in the cartilage of 53 week-old DN p38 (+/−) mice was 0.476+/−0.075 unit as compared to WT (−/−) controls (1 unit) (Figure 1A). Immunohistochemical analysis showed a reduction of intensity of p-p38 staining in the growth plate cartilage in new-born DN p38 (+/−) mice in comparison to WT (−/−) controls (Figure 1B). The activities of ERK and JNK were not affected by DNp38 transgene (Figure 2A). The skeletal structure of DN p38 (+/−) mice appeared to be shorter than that of the WT (−/−) control mice, but otherwise normal in X-ray radiographs (Figure 2B).
Figure 1. MAP kinase activities in DNp38 transgenic mice.
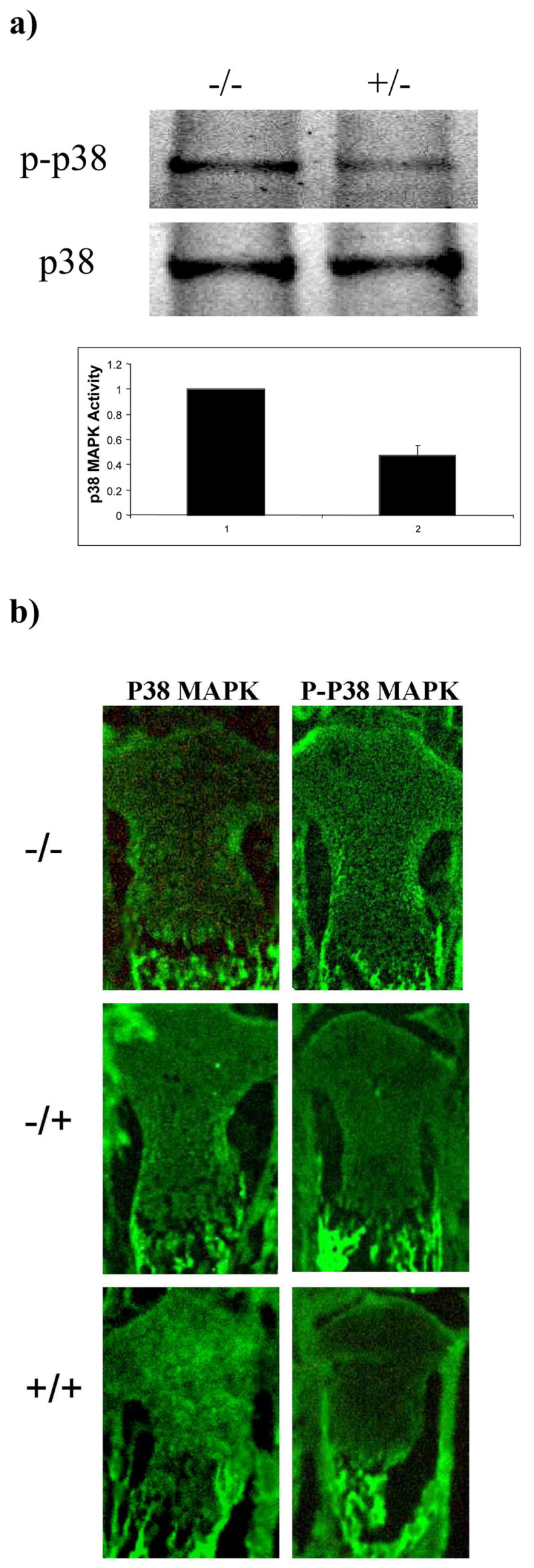
a) Reduction of phosphorylated p38 MAPK activity in cartilage of DNp38 transgenic mice (+/−) in comparison to that of WT mice (−/−) at 53 weeks of age. Western blot analysis of mouse cartilage extract was performed with an antibody against phosphorylated p38 MAPK (p-p38) and an antibody against p38 protein (p38), respectively. Equal amount of protein was loaded in each lane. Densitometry quantification of the protein bands was shown (error bars indicated standard deviation).
b) Reduction of phosphorylated p38 MAPK activity in growth plate cartilage of DNp38 transgenic mice (+/−) in comparison to that of WT mice (−/−) at birth. Immunohistochemical analysis of mouse growth plate cartilage was performed using an antibody against phosphorylated p38 MAPK (p-p38) and an antibody against p38 protein (p38), respectively.
Figure 2. Specificity of the effect of DNp38 transgene.
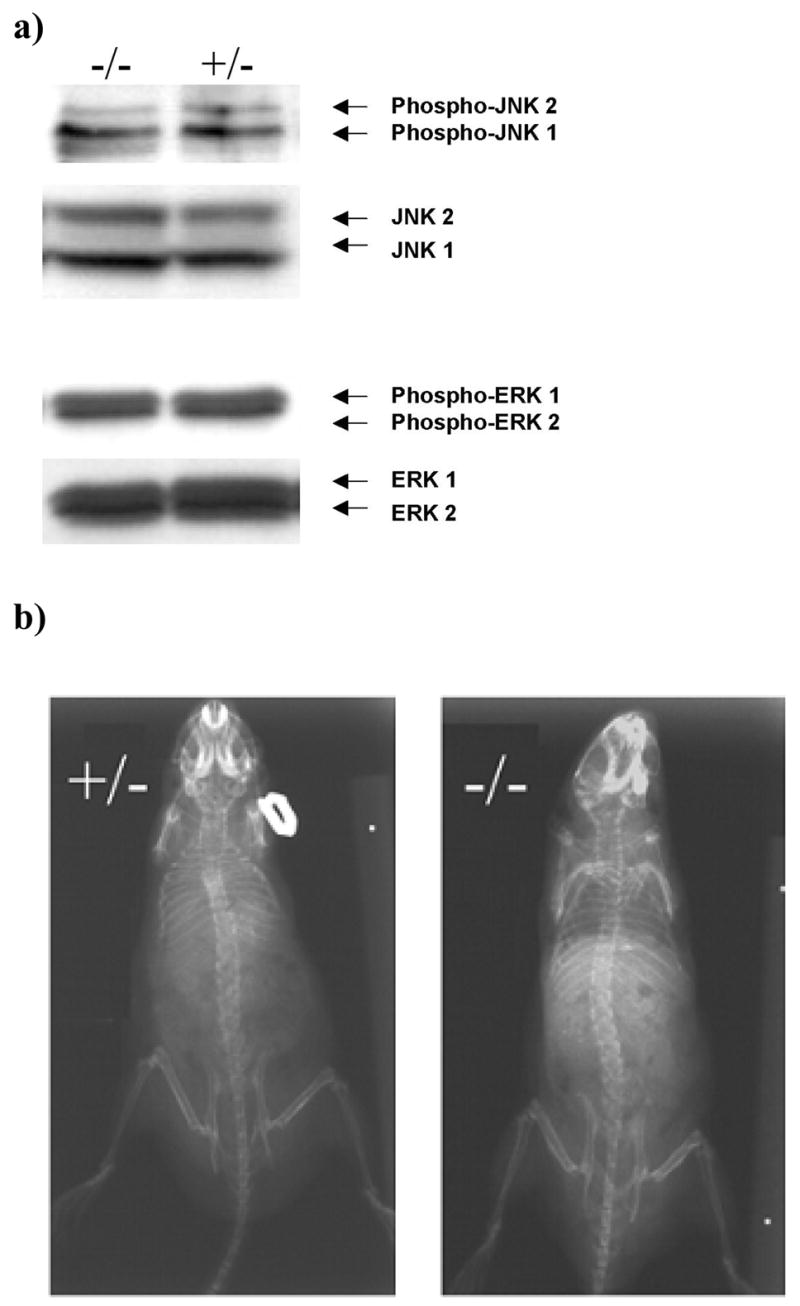
a) Activities of ERK and JNK were not affected by DNp38 transgene. Western blot analysis of mouse cartilage extract was performed using an antibody against phosphorylated JNK (phospho-JNK) and an antibody against JNK protein (JNK), respectively; or was performed using an antibody against phosphorylated ERK (phospho-ERK) and an antibody against ERK protein (ERK), respectively. Equal amount of protein was loaded in each lane. Densitometry quantification of the protein bands showed no significant difference of phospho-JNK or phospho-ERK between DNp38 mice (−/−) and that of WT mice (+/+) respectively.
b) No apparent difference in skeletal mineralization between DN p38 (+/−) and WT (−/−) mice. Mice were x-rayed after sacrifice in posterior-anterior view.
Overall Morphology
Average overall height was 8.8 cm in DN p38 (+/−) mice, compared to 9.5 in WT (−/−) controls (p=0.03) (Figure 3A). Detail measurement revealed that DN p38 (+/−) mice were significantly shorter in overall femur length (14.62mm vs 15.86mm, p<0.001), (Figure 3B and 3C) and tibia length (17.59mm vs 18.84mm, p<0.001) (Figure 3C) and spine length (4.8cm vs 5.8cm, P<0.001) when compared to WT (−/−) controls. However, differences between DN p38 (+/−) mice and WT (−/−) controls in overall animal weights (34.4gm vs 34.1gm, p=0.92), tibia thickness (1.38mm vs 1.35mm, p=0.74), femur thickness (1.47mm vs 1.47mm, p=0.96), and cranial diameter (2.5cm vs 2.4cm, p=.09) were not statistically significant.
Figure 3. Reduced long bone length in DNp38 transgenic mice.
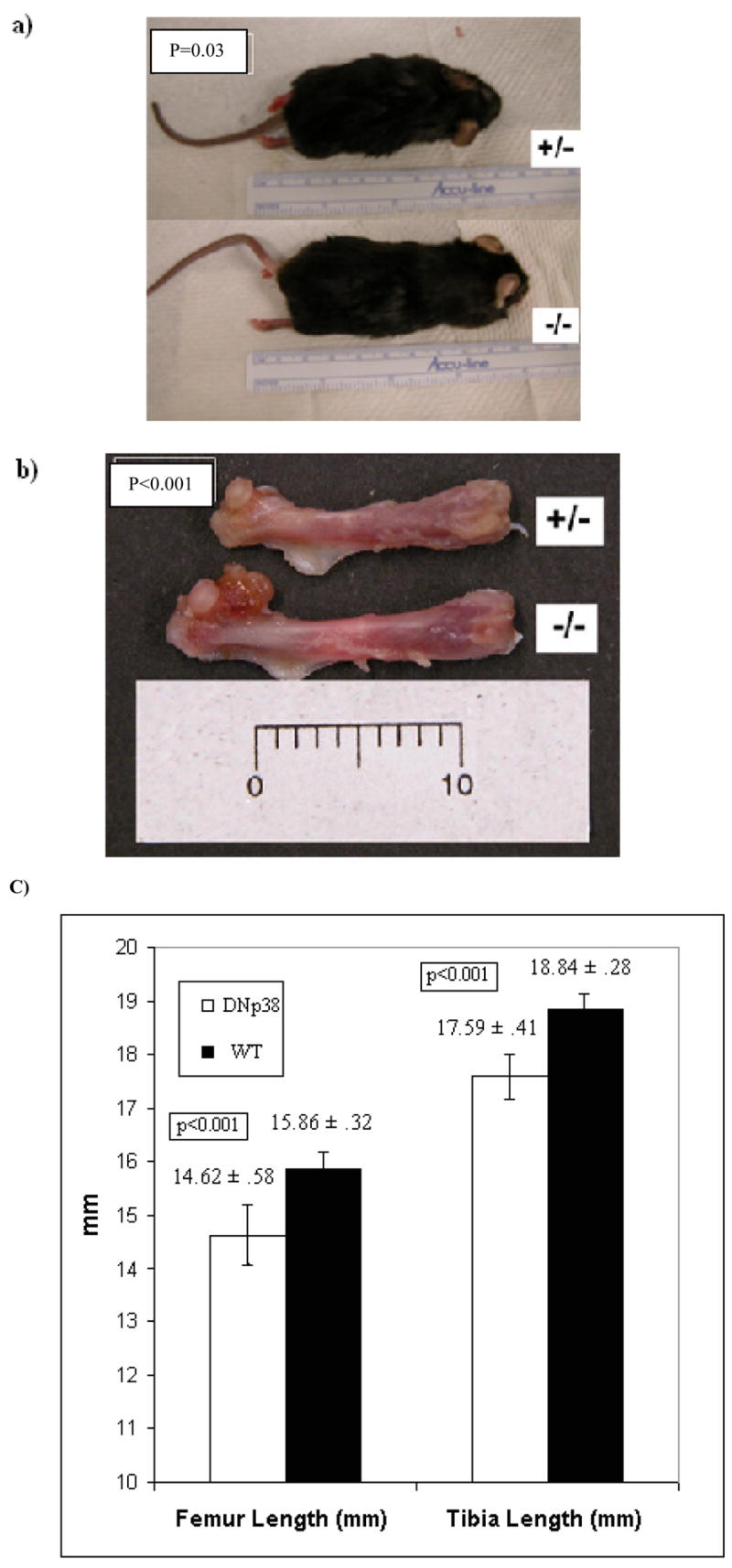
a) Reduced overall length in DN p38 (+/−) mice.
b)Shorter femur length in the DN p38 (+/−) mice. Each unit = 1mm.
c) Reduced femur and tibia lengths in DN p38 (+/−) mice (error bars indicate standard deviation).
Bone
Although femur and tibia lengths were notably shorter in DN p38 (+/−) mice, the material properties of the femurs remained the same with WT (−/−) controls. Biomechanical testing revealed no significant differences in peak load, stiffness, energy to peak, or peak stress between DN p38 (+/−) and WT (−/−) controls (Table 1). Micro-CT scanning of the femurs indicated no statistically significant difference in moments of inertia or overall cortical areas in the midshaft region between DNp38 mice and WT controls (Table 2).
Table 1.
No significant differences in whole bone and tissue properties
| DNp38 (N=10) | WT (N=9) | P-value | |
|---|---|---|---|
| Peak Load (N) | 20.89 ± 1.89 | 20.69 ± 3.89 | 0.74 |
| Stiffness (N/mm) | 155.9 ± 23.97 | 151.82 ± 29.6 | 0.20 |
| Energy to Peak (Nmm) | 3.26 ± 0.38 | 3.16 ± 1.18 | 0.18 |
| Stress (N/mm^2) | 181.0 ± 33.5 | 160.9 ± 45.6 | 0.39 |
Table 2.
No significant differences in MOI and Cross-section areas
| DNp38 (N=10) | WT (N=9) | P-value | |
|---|---|---|---|
| Moment of Inertia (mm^4) | 0.55 ± 0.10 | 0.52 ± 0.06 | 0.63 |
| Area (mm^2) | 0.97 ± 0.16 | 0.92 ± .07 | 0.35 |
Articular Cartilage
The mean osteoarthritis grade of the knee joints in DN p38 (+/−) mice (n=10) was 2.50 +/− 1.06, which was significantly higher than the mean grade of the WT (−/−) controls (n=9), which was 1.13 +/− 0.74 (p = 0.004). In particular, by both Hematoxylin/Eosin staining (Figure 4a) and Safranin-O/Fast Green staining (Figure 4b), we observed increased fissuring and fibrillation, and degeneration of the superficial and middle zones in the knee joint cartilage of DNp38 (+/−) mice. Comparison of the blinded grading revealed only one small discrepancy between the two blinded graders; that sample was not included in the data analysis.
Figure 4. Defects in the articular cartilage of knee joints in adult mice.

a) Micrographs of H&E histology showing damage at the articular surface in knee joints of DN p38 (+/−) mice (black arrow) and a corresponding region in WT (−/−) mice (red arrow). Bar = 1mm.
b) Micrographs of Safranin-O staining showing greater damage to the articular cartilage in knee joints of DN p38 (+/−) mice (black arrow) compared to WT (−/−) mice (red arrow). Bar = 1mm.
DISCUSSION
In this study we evaluated the in vivo effect of cartilage-specific reduction of p38 MAPK activity on the bones and joints of one year-old adult mice. Previously, deletion of the p38α MAPK gene from mouse genome resulted in embryonic lethality.46 To study the effect of genetic inhibition of p38 MAPK activity on articular cartilage, we created transgenic mice harboring a dominant negative p38 transgene under the regulation of type II collagen gene promoter. Although DNp38 MAPK/Col II homozygote mice died shortly after birth, the heterozygote mice lived a normal lifespan with reduced p38 MAPK basal activity in joint cartilage. We show that, while reduced p38 MAPK activity leads to diminished limb length, the biomechanical properties of long bones are unchanged. The limb length is determined by endochondral ossification of the cartilage anlagen that expresses collagen type II. Since the DNp38 MAPK transgene is under the regulation of Col II promoter, the reduction of p38 MAPK activity in chondrocytes may result in reduced bone growth from the cartilage anlagen. We speculate that the “normal” level of p38 MAPK activity in cartilage is necessary for normal longitudinal growth of the bone. The data suggests that mice with partially reduced (~50%) p38 activity in cartilage lack the ability to optimize longitudinal growth. A limitation of the study is that part of the transgenic mice phenotype may be induced through the expression of the DNp38 MAPK transgene where it is normally not expressed due to its expression under the control of the Col II promoter. However, the lack of difference in bone material and biomechanical properties between DN p38 (+/−) and WT (−/−) mice indicates that partial reduction of p38 MAPK activity in cartilage is not critical to postnatal bone mineralization. It also reflects the fact that the p38 MAPK activity is not pertubed in bone because the DNp38 transgene is expressed specifically in cartilage but not in the bone. Important to note, the lack of significant difference in overall weight as well as some other morphologic characteristics may be a result of a lack of adequate statistical power.
We demonstrated, for the first time, the effect of genetic inhibition of p38 MAPK activity in cartilage on the development of osteoarthritis in mice. Surprisingly, contrary to our hypothesis, we found that reduced p38 MAPK activity in cartilage was associated with increased severity of osteoarthritis in one year-old DN p38 (+/−) mice. Previously, in vitro studies have found that inhibition of p38 MAPK activity in chondrocytes resulted in an inhibition of chondrocyte hypertrophic differentiation and cell death, while increased p38 MAPK activity resulted in the elevated levels of osteoarthritis markers. 6–26 Murata et al, in their investigation of signaling pathways in human articular chondrocytes, showed that hypoxia-induced VEGF production in osteoarthritic chondrocytes was abolished by p38 MAPK inhibitors.47 Wei and colleagues found that inhibition of p38 MAPK activity abolishes anti-CD95 induced chrondrocyte death by inhibiting the activities of activating transcription factor-2 (ATF-2) and caspase-3. 12
Our findings do not necessarily contradict with the conclusions from these in vitro studies, but instead serve to highlight the importance of the timing and the period of inhibition of p38 MAPK activity in cartilage for chondroprotection. There are several potential explanations for our findings.
First, as the dominant negative p38 transgene is under the regulation of type II collagen promoter, it is expressed in cartilage during skeletal development. Thus, the adult transgenic mice may show phenotypic effects of reduced p38 activity on cartilage during the early developmental period. In vitro studies suggest the p38 MAPK pathway is integral in the differentiation of mesenchymal stem cells during chondrogenesis. Lim et al show that p38 MAPK signaling is necessary for chondrogenesis induced by actin cytoskeleton disruption of mesenchymal cells derived from chick embryonic limb buds.48 In addition, they show that inhibition of p38 MAPK with SB203580 blocked chondrogenesis. It can be speculated that p38 MAPK activity is necessary for normal cartilage anlagen formation during development. As the articular cartilage is a remnant of these anlagen, defective cartilage differentiation during development may lead to osteoarthritic degenerative changes later in life.
Second, the p38 MAPK activity in chondrocytes may be necessary for homeostasis of extracellular matrix in cartilage. For example, it has been shown that p38 MAPK activity in chondrocytes is involved in the regulation of aggrecan gene expression in response to IL-1.49 Thus, diminished p38 MAPK activity may lead to inadequate chondrocyte homeostasis thereby contributing to degeneration of cartilage matrix during osteoarthritis. Our mouse model, however, did exclude the possibility that the increased cartilage matrix degeneration in DNp38 MAPK transgenic mice was due to the alteration of the mechanical properties of subchondral bone. Our analysis indicated that the material and mechanical properties of the bones in transgenic mice were not altered.
Finally, p38 activity may be required for a chondrocyte to respond to environmental stress. Several reports have indicated activation of p38 in response to inflammatory stimuli and have found this response to be related to increased NF-kappa B recruitment.50 It is possible that without this capability, chrondocytes are unable to react favorably to local environmental stressors. Although it remains to be tested which, if any, of these mechanisms is responsible for the more severe cartilage degeneration in DNp38 transgenic mice, these mice provide excellent models for the effect of genetic inhibition of p38 MAPK activity on cartilage.
Our study suggests that while p38 MAPK may still hold promise as a therapeutic target for osteoarthritis, its inhibition throughout the lifespan leads to worsened pathology at the articular surface in adult mice. As a result, the timing of p38 inhibition in preventing or delaying osteoarthritis may be critical. Our data indicate that the p38 MAPK pathway’s role in osteoarthritis development in vivo is much more complex than initially thought. Further investigation, using an inducible promoter or a controllable delivery device to achieve a temporal inhibition of p38 MAPK activity in cartilage, is necessary to determine whether a short period of p38 inhibition is effective in reducing osteoarthritis pathogenesis in vivo.
Acknowledgments
Supported by grants from the NIH (AG-14399, AG-17021, RR024484, and AR052479)
We thank Kathy Cheah for providing Col II promoter and enhancer cDNA constructs, and Roger Davis for providing dominant negative p38 cDNA construct.
References
- 1.Yosimichi G, Nakanishi T, Nishida T, Hattori T, Takano-Yamamoto T, Takigawa M. CTGF/Hcs24 induces chondrocyte differentiation through a p38 mitogen-activated protein kinase (p38MAPK), and proliferation through a p44/42 MAPK/extracellular-signal regulated kinase (ERK) Eur J Biochem. 2001 Dec;268(23):6058–65. doi: 10.1046/j.0014-2956.2001.02553.x. [DOI] [PubMed] [Google Scholar]
- 2.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem. 2001 Dec 14;276(50):46707–13. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 3.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994 Sep 23;78(6):1027–37. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 4.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, et al. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–6. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 5.Wei L, Chen Q. 0329- Regulation of chondrocyte death during endochondral ossification by p38 mitogen-activated protein kinase: mechanism of coupling hypertrophy and apoptosis. Transactions: Orthopaedic Research Society Annual Meeting; San Francisco, CA. 2001. [Google Scholar]
- 6.Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TF, Taylor DJ, DeWitt DL, Saklatvala J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J Immunol. 1997;158:3165–73. [PubMed] [Google Scholar]
- 7.Ravanti L, Heino J, Lopez-Otin C, Kahari VM. Induction of collagenase-3 (MMP-13) expression in human skin fibroblasts by three-dimensional collagen is mediated by p38 mitogen-activated protein kinase. J Biol Chem. 1999;274:2446–55. doi: 10.1074/jbc.274.4.2446. [DOI] [PubMed] [Google Scholar]
- 8.Ravanti L, Hakkinen L, Larjava H, Saarialho-Kere U, Foschi M, Han J, et al. Transforming growth factor-beta induces collagenase-3 expression by human gingival fibroblasts via p38 mitogen-activated protein kinase. J Biol Chem. 1999;274:37292–300. doi: 10.1074/jbc.274.52.37292. [DOI] [PubMed] [Google Scholar]
- 9.Simon C, Goepfert H, Boyd D. Inhibition of the p38 mitogen-activated protein kinase by SB 203580 blocks PMA-induced Mr 92,000 type IV collagenase secretion and in vitro invasion. Cancer Res. 1998;58:1135–9. [PubMed] [Google Scholar]
- 10.Adams JL, Badger AM, Kumar S, Lee JC. P38 MAP kinase: molecular target for the inhibition of pro-inflammatory cytokines. Prog Med Chem. 2001;38:1–60. doi: 10.1016/s0079-6468(08)70091-2. [DOI] [PubMed] [Google Scholar]
- 11.Dai SM, Shan ZZ, Nakamura H, Masuko-Hongo K, Kato T, Nishioka K, et al. Catabolic stress induces features of chondrocyte senescence through overexpression of Caveolin 1. Arthritis Rheum. 2006;54(3):818–31. doi: 10.1002/art.21639. [DOI] [PubMed] [Google Scholar]
- 12.Wei L, Sun X, Wang Z, Chen Q. CD95-induced osteoarthritic chondrocyte apoptosis and necrosis: dependency on p38 mitogen-activated protein kinase. Arthritis Res Ther. 2006;8(2):R37. doi: 10.1186/ar1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whiteman M, Spencer JPE, Zhu YZ, Armstrong JS, Schantz JT. Peroxynitrite-modified collagen-II induces p38/ERK and NF-kB-dependent synthesis of prostaglandin E2 and nitric oxide in chondrogenically differentiated mesenchymal progenitor cells. Osteoarthritis Cartilage. 2006;14:460–70. doi: 10.1016/j.joca.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Wada Y, Shimada K, Sugimoto K, Kimura T, Ushiyama S. Novel p38 mitogen-activated protein kinase inhibitor R-130823 protects cartilage by down-regulating matrix metalloproteinase-1,-13 and prostaglandin E2 production in human chondrocytes. Int Immunopharmacol. 2006 Feb;6(2):144–55. doi: 10.1016/j.intimp.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Ea HK, Uzan B, Rey C, Liote F. Octacalcium phosphate crystals directly stimulate expression of inducible nitric oxide synthase through p38 and JNK mitogen-activated protein kinases in articular chondrocytes. Arthritis Res Ther. 2005;7(5):R915–26. doi: 10.1186/ar1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boileau C, Pelletier JP, Tardif G, Fahmi H, Laufer S, Lavigne M, et al. The regulation of human MMP-13 by licofelone, an inhibitor of cyclo-oxygenases and 5-lipoxygenase, in human osteoarthritic chondrocytes is mediated by the inhibition of the p38 MAP kinase signalling pathway. Ann Rheum Dis. 2005 Jun;64(6):891–8. doi: 10.1136/ard.2004.026906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malemud CJ. Protein kinases in chondrocyte signaling and osteoarthritis. Clin Orthop Relat Res. 2004 Oct;427(Suppl):S145–51. doi: 10.1097/01.blo.0000143802.41885.50. [DOI] [PubMed] [Google Scholar]
- 18.Masuko-Hongo K, Berenbaum F, Humbert L, Salvat C, Goldring MB, Thirion S. Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004 Sep;50(9):2829–38. doi: 10.1002/art.20437. [DOI] [PubMed] [Google Scholar]
- 19.Martel-Pelletier J, Mineau F, Jovanovic D, Di Battista JA, Pelletier JP. Mitogen-activated protein kinase and nuclear factor kappaB together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (MAPKAPK) Arthritis Rheum. 1999 Nov;42(11):2399–409. doi: 10.1002/1529-0131(199911)42:11<2399::AID-ANR19>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 20.Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–44. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 21.Cecil DL, Johnson K, Rediske J, Lotz M, Schmidt AM, Terkeltaub R. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J Immunol. 2005 Dec 15;175(12):8296–302. doi: 10.4049/jimmunol.175.12.8296. [DOI] [PubMed] [Google Scholar]
- 22.Yudoh K, Nakamura H, Masuko-Hongo K, Kato T, Nishioka K. Catabolic stress induces expression of hypoxia-inducible factor (HIF)-1 alpha in articular chondrocytes: involvement of HIF-1 alpha in the pathogenesis of osteoarthritis. Arthritis Res Ther. 2005;7(4):R904–14. doi: 10.1186/ar1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HA, Lee KB, Bae SC. The mechanism of low-concentration sodium nitroprusside-mediated protection of chondrocyte death. Arthritis Res Ther. 2005;7(3):R526–35. doi: 10.1186/ar1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhen X, Wei L, Wu Q, Zhang Y, Chen Q. Mitogen-activated protein kinase p38 mediates regulation of chondrocyte differentiation by parathyroid hormone. J Biol Chem. 2001 Feb 16;276(7):4879–85. doi: 10.1074/jbc.M004990200. [DOI] [PubMed] [Google Scholar]
- 25.Berenbaum F. Signaling transduction: target in osteoarthritis. Curr Opin Rheumatol. 2004 Sep;16(5):616–22. doi: 10.1097/01.bor.0000133663.37352.4a. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen J, Gogusev J, Knapnougel P, Bauvois B. Protein tyrosine kinase and p38 MAPK pathways involved in stimulation of matrix metalloproteinase-9 by TNF-α in human monocytes. Immunol Lett. 2006 Jul 15;106(1):34–41. doi: 10.1016/j.imlet.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Seto H, Kamekura S, Miura T, Yamamoto A, Chikuda H, Ogata T, et al. Distinct Roles of SMAD pathways and p38 pathways in cartilage-specific gene expression in synovial fibroblasts. J Clin Invest. 2004 Mar;113(5):718–26. doi: 10.1172/JCI19899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berenbaum F, Humbert L, Bereziat G, Thirion S. Concomitant recruitment of ERK1/2 and p38 MAPK signaling pathway is required for activation of cytoplasmic phospholipase A2 via ATP in articular chondrocytes. J Biol Chem. 2003 Apr 18;278(16):13680–7. doi: 10.1074/jbc.M211570200. [DOI] [PubMed] [Google Scholar]
- 29.Weston AD, Sampaio AV, Ridgeway AG, Underhill TM. Inhibition of p38 MAPK signaling promotes late stages of myogenesis. J Cell Sci. 2003;116(14):2885–93. doi: 10.1242/jcs.00525. [DOI] [PubMed] [Google Scholar]
- 30.Maroni PD, Koul S, Meacham RB, Koul HK. Mitogen activated protein kinase signal transduction pathways in the prostate. Cell Commun Signal. 2004 Jun 25;2(1):5. doi: 10.1186/1478-811X-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robbins JR, Thomas B, Tan L, Choy B, Arbiser JL, Berenbaum F, et al. Immortalized human adult articular chondrocytes maintain cartilage-specific phenotype and responses to interleukin-1beta. Arthritis Rheum. 2000 Oct;43(10):2189–201. doi: 10.1002/1529-0131(200010)43:10<2189::AID-ANR6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 32.Alessi DR, Ceunda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 33.Murakami S, Kan M, McKeehan WL, de Crombrugghe B. Up-regulation of the chondrogenic sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Prox Natl Acad Sci USA. 2000;91:1113–8. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yonekura A, Osaki M, Hirota Y, Tsukazaki T, Miyazaki Y, Matsumoto T, et al. Transforming growth factor-beta stimulates articular chondrocyte cell growth through p44/42 MAP kinase (ERK) activation. Endocr J. 46:545–53. doi: 10.1507/endocrj.46.545. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura K, Shirai T, Morishita S, Uchida S, Saeki-Miura K, Makishima F. p38 mitogen-activated protein kinase functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells. Exp Cell Res. 1999;250:351–63. doi: 10.1006/excr.1999.4535. [DOI] [PubMed] [Google Scholar]
- 36.Thomas B, Thirion S, Humbert L, Tan L, Goldring MB, Bereziat G, et al. Differentiation regulates interleukin-1beta-induced cyclo-oxygenase-2 in human articular chondrocytes: role of p38 mitogen-activated protein kinase. Biochem J. 2002 Mar 1;362(Pt 2):367–73. doi: 10.1042/0264-6021:3620367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang S, Jung M, Kim CW, Shin DY. Inactivation of p38 kinase delays the onset of senescence in rabbit articular chondrocytes. Mech Ageing Dev. 2005 May;126(5):591–7. doi: 10.1016/j.mad.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Jin EJ, Lee SY, Choi YA, Jung JC, Bang OS, Kang SS. BMP-2-enhanced chondrogenesis involves p38 MAPK-mediated down-regulation of Wnt-7a pathway. Mol Cells. 2006 Dec 31;22(3):353–9. [PubMed] [Google Scholar]
- 39.Enslen H, Brancho DM, Davis RJ. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 2000 Mar 15;19(6):1301–11. doi: 10.1093/emboj/19.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun X, Wei L, Cheah K, Bronson S, Chen Q. Abolition of endochondral bone formation by a dominant negative p38 MAP kinase transgene in a transgenic mouse model; Transactions: Orthopaedic Research Society Annual Meeting; New Orleans, LA: 2003. [Google Scholar]
- 41.Cheah KS, Levy A, Trainor PA, Wai AW, Kuffner T, So CL, Leung KK, Lovell-Badge RH, Tam PP. Human COL2A1-directed SV40 T antigen expression in transgenic and chimeric mice results in abnormal skeletal development. J Cell Biol. 1995;128( 1–2):223–37. doi: 10.1083/jcb.128.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiltunen A, Vuorio E, Aro HT. A standardized experimental fracture in the mouse tibia. J Orthop Res. 1993 Mar;11(2):305–12. doi: 10.1002/jor.1100110219. [DOI] [PubMed] [Google Scholar]
- 43.Jamsa T, Jalovaara P, Peng Z, Vaananen HK, Tuukkanen J. Comparison of three-point bending test and peripheral quantitative computed tomography analysis in the evaluation of the strength of mouse femur and tibia. Bone. 1998 Aug;23(2):155–61. doi: 10.1016/s8756-3282(98)00076-3. [DOI] [PubMed] [Google Scholar]
- 44.Turner CH, Burr DB. Basic biomechanical measurements of bone: a tutorial. Bone. 1993 Jul-Aug;14(4):595–608. doi: 10.1016/8756-3282(93)90081-k. [DOI] [PubMed] [Google Scholar]
- 45.Maier I, Wilhelmi G. Osteoarthrosis-like disease in mice: effects of anti-arthrotic and anti-rheumatic agents. In: Lott DJ, et al., editors. Studies in osteoarthrosis: pathogenesis, intervention and assessment. John Wiley and Sons Chichester; New York, Bisbane, Toronto, Singapore: 1987. pp. 75–83. [Google Scholar]
- 46.Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA. Deficiency of the stress kinase p38alpha results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med. 2000 Mar 6;191(5):859–70. doi: 10.1084/jem.191.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murata M, Yudoh K, Nakamura H, Kato T, Inoue K, Chiba J, et al. Distinct signaling pathways are involved in hypoxia- and IL-1-induced VEGF expression in human articular chondrocytes. J Orthop Res. 2006 Jul;24(7):1544–54. doi: 10.1002/jor.20168. [DOI] [PubMed] [Google Scholar]
- 48.Lim YB, Kang SS, An WG, Lee YS, Chun JS, Sonn JK. Chondrogenesis induced by actin cytoskeleton disruption is regulated via protein kinase C-dependent p38 mitogen-activated protein kinase signaling. J Cell Biochem. 2003 Mar 1;88(4):713–8. doi: 10.1002/jcb.10389. [DOI] [PubMed] [Google Scholar]
- 49.Radons J, Bosserhoff AK, Grassel S, Falk W, Schubert TE. p38MAPK mediates IL-1-induced down-regulation of aggrecan gene expression in human chondrocytes. Int J Mol Med. 2006 Apr;17(4):661–8. [PubMed] [Google Scholar]
- 50.Ulivi V, Tutolo G, Mallein-Gerin F, Daga A, Cancedda R, Cancedda FD. A common pathway in differentiation and inflammation: p38 mediates expression of the acute phase SIP24 iron binding lipocalin in chondrocytes. J Cell Physiol. 2006 Mar;206(3):728–37. doi: 10.1002/jcp.20511. [DOI] [PubMed] [Google Scholar]