

Abstract
Background
The effect of multiple integrated stimuli on vascular wall expression of matrix metalloproteinases (MMPs) remains unknown. Accordingly, this study has examined the influence of the vasoactive peptide angiotensin II (AngII) on wall tension-induced promoter activation of MMP-2, MMP-9, and membrane type-1 MMP (MT1-MMP).
Methods and Results
Thoracic aortic rings harvested from transgenic reporter mice containing the MMP-2, MMP-9, or MT1-MMP promoter sequence fused to a reporter gene were subjected to three hours of wall tension at 70, 85, or 100 mmHg with or without 100nM AngII. Total RNA was harvested from the aortic rings, and reporter gene transcripts were quantified by QPCR to measure MMP promoter activity. MT1-MMP promoter activity was increased at both 85 and 100 mmHg compared to baseline tension of 70 mmHg, while treatment with AngII stimulated MT1-MMP promoter activity to the same degree at all tension levels (p<0.05). Elevated tension and AngII displayed a potential synergistic enhancement of MMP-2 promoter activation at 85 and 100mmHg, while the same stimuli caused a decrease in MMP-9 promoter activity (p<0.05) at 100 mmHg.
Conclusions
This study has demonstrated that exposure to a relevant biological stimulus (AngII) in the presence of elevated tension modulated MMP promoter activation. Furthermore, these data suggest that a mechanical-molecular set point exists for the induction of MMP promoter activation, and that this set point can be adjusted up or down by a secondary biological stimulus. Together, these results may have significant clinical implications toward the regulation of hypertensive vascular remodeling.
Keywords: Metalloproteinases, Aorta, Wall stress, Angiotensin
Introduction
While the development and progression of vascular remodeling, such as that which can occur with hypertension and with aneurysmal disease, is a multifactorial process, it is now becoming evident that the activation of key proteolytic pathways contributes to the structural changes that occur within the extracellular matrix. One mechanism by which extracellular remodeling within the vascular wall is regulated is through the induction and activation of the matrix metalloproteinases (MMPs).(1) The MMPs constitute a large family of enzymes that are proteolytically diverse and cause-effect relationships exist between the induction of certain MMP types and pathological vascular remodeling with regard to atherosclerosis, hypertension and aortic aneurysm formation.(2-4) For example, a class of MMPs generically termed the gelatinases and consisting of primarily MMP-2 and MMP-9, have been implicated in vascular remodeling in several disease states.(2, 3) Since MMPs cause dynamic and significant changes within the extracellular space, it follows that these enzymes are tightly controlled at the transcriptional, translational, and post-translational levels.(5) However, it remains unclear to what degree mechanical and/or biological stimuli modulate the induction of MMP promoter activity.
In a recent report from this laboratory, aortic rings harvested from transgenic mice expressing MMP promoter-reporter gene constructs were utilized in an ex vivo ring apparatus to examine the role of isolated tension on MMP-2 and MMP-9 promoter activation.(6) The study demonstrated that a physiologically relevant mechanical stimulus was sufficient to induce MMP-2 promoter activity while the MMP-9 promoter was found to be unresponsive. Furthermore, these initial data suggested that a mechanical-molecular set point may exist for induction of MMP-2, which may have a significant impact on the prognostic and therapeutic management of hypertensive patients.
In order to further examine the role of a mechanical stimulus on the regulation of MMP promoter activation, the present study explored two objectives. First, it tested the hypothesis that promoter activity of a known activator of MMP-2, the membrane type 1-MMP (MT1-MMP) gene,(7) should be co-regulated with MMP-2 in response to mechanical stimuli. The MT-MMPs, of which MT1-MMP is considered prototypical, proteolytically process a wide range of extracellular proteins, cytokines and growth factors - all relevant to the vascular remodeling process.(8) In addition, increased levels of MT1-MMP have been identified in vascular remodeling in both animal and clinical studies.(9, 10) Thus, the induction of MT1-MMP would not only cause increased proteolytic potential, but could also amplify extracellular remodeling through post-translational modification of other MMPs such as MMP-2.
In addition to hemodynamic forces, MMP expression may be induced by neurohormonal stimuli and one of the most extensively studied vasoactive peptides is angiotensin II (AngII). This hormone has been shown to contribute to numerous cardiovascular disease states, including atherosclerosis, hypertension, and left ventricular dysfunction,(11-13) by eliciting endothelial dysfunction(14) and/or a potent mitogenic and hypertrophic response in vascular smooth muscle cells.(15, 16) Evidence also suggests that AngII can directly contribute to matrix remodeling by inducing MMP expression.(13, 17) Understanding how these discrete forces may be integrated to initiate and propagate vascular remodeling may identify novel therapeutic strategies, however, very little is known about the collective influence of mechanical and hormonal stimuli. Accordingly, the second objective of this study was to evaluate the regulation of MMP promoter activation under the influence of both increased wall tension and AngII.
Material and Methods
MMP reporter strains
Male and female reporter mice carrying a transgene construct consisting of an MMP promoter fragment linked to a reporter gene were utilized. A 5-kb fragment of the rat MMP-2 gene extending from −1686 to the second exon was linked to the β-galactosidase gene lacZ to create the transgenic CD-1[MMP-2:β-gal] reporter mouse line.(18) The rabbit MMP-9 gene segment from base pairs −522 to +19 was fused to lacZ to generate the CD-1[MMP-9:β-gal] reporter mice.(19) Alternatively, the MT1-MMP reporter strain linked the firefly luciferase gene Luc to the human MT1-MMP promoter genomic fragment spanning from −3364 base pairs to the first intron. This construct was subsequently inserted on an FVB background. In the MMP-2 and MMP-9 reporter strains, transgene expression was confirmed by reaction of tail tissue with the β-galactosidase substrate X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside). Additionally, PCR amplification of tail-clip DNA with primers specific for lacZ or Luc was performed in all three reporter strains to verify transgene expression. A concordant response to elevated tension between the native MMP promoter and the MMP promoter-reporter gene construct in these mouse strains has been previously established.(6)
Experimental Design
Wall tension was applied to segments of murine descending thoracic aorta utilizing a vascular ring ex vivo method as previously described.(12, 20) Wall tension values consistent with mildly hypertensive mean arterial pressures were applied to excised aortic rings which were concomitantly treated with AngII. Aortic tissue was homogenized and lacZ or Luc gene expression was measured by quantitative real-time polymerase chain reaction (QPCR) to directly evaluate MMP promoter activity. All mice were maintained according to the National Institutes of Health Guide to the Care and Use of Laboratory Animals (National Research Council, Washington, DC, 1996) and this animal protocol was approved by the MUSC Institutional Animal Care and Use Committee.
Preparation of aortic rings
Following induction of general anesthesia with 2% isoflurane, the mice underwent a left posterolateral thoracotomy. The descending thoracic aorta was harvested and immediately placed in cold Krebs-Hanseleit buffer (118mM NaCl, 4.6mM KCl, 1.2mM KH2PO4, 25mM NaHCO3, 2.5mM CaCl2, 0.5mM Na2-EDTA, 11mM Glucose, 1.2mM MgSO4, pH 7.4). Extraneous connective tissue was removed and aortas were denuded of endothelium to minimize the potential for altered shear stress to influence gene expression. The vessel was then divided into 3 mm long segments and mounted on parallel wires in a water jacketed tissue bath system (Radnoti, Monrovia, CA; 25ml) maintained at 37°C and connected to an isometric force transducer (Radnoti, Monrovia, CA). The vessels were equilibrated on the ring apparatus for 30 minutes at zero tension and the aerated buffer (95% O2/5% CO2) was exchanged every 30 minutes. Micrometer measurements at zero tension and calculations set forth by He et al., and Lalli et al.,(21, 22) were used to derive the mean internal circumference and cross-sectional area of the vessels. For each aortic ring, tension was digitally measured and graphically recorded (BioBench; National Instruments, Austin, TX).
Defining optimal tension and experimental pressure gradients
Before applying tension to the aortic rings to examine promoter activity, the native tension parameters of the tissue were defined. The passive, active, and optimal tension values for the MMP-2 and MMP-9 reporter strains have been previously defined.(6) The same experimental procedure was performed to define the optimal tension for the MT1-MMP reporter strain. Briefly, to quantify the passive tension, vessel segments (n=8) were sequentially stretched in 0.1 g increments and allowed to equilibrate, during which time the ECM microfibrils would adapt and relax, demonstrating a decay in the transduced vessel tension. The percent decline in passive tension at each level of applied tension was compared to a standard of 10% using a one-sample t-test. The passive tension of the vessel was identified when the transduced vessel tension decayed less than 10%. Similarly, active tension was then defined in fresh aortic rings stabilized at a given tension increment by stimulating contraction with 100 mM KCl (n=8). Peak tension generation was recorded, and comparisons among active tension values were conducted by one-way ANOVA with a post-hoc Tukey’s multiple comparison test.
The point at which all passive tension was overcome and the smooth muscle cells were able to maximally contract, was identified as the optimal tension. This value varies among mouse strains.(23, 24) Therefore, optimal tension was converted to a pressure measurement to ensure equivalent application of tension across aortic rings from each mouse strain.(21, 22) The optimal tension for the MMP-2 and MMP-9 reporter mouse strains has been previously defined at 0.7g, and normalization for cross-sectional area led to a pressure value of 70 mmHg.(6) The MT1-MMP reporter mice had a cross-sectional area of 0.28±0.01mm2 and ring surface area of 7.33±0.05mm2. Using these measurements, optimal tension for the MT1-MMP reporter mouse line was also found to be equivalent to 70 mmHg.
Inducing MMP promoter activity
Aortic rings were stabilized at zero tension and equilibrated at optimal tension for 30 minutes each. Tension was then applied for 3 hours at 70, 85, or 100 mmHg with buffer exchange every 30 minutes (n=6 for each). Rings were treated with 100nM AngII at initiation of the zero tension stabilization phase and 100nM AngII treatment was repeated with each buffer exchange over the 3 hour duration of the experiment. This concentration of AngII was chosen based upon previous studies which reported that 100nM would cause a minimal contractile response of the murine thoracic aorta, but would still be a functional ligand toward activating the type-1 AngII receptor (AT1R) and initiating molecular signaling in an organ bath ex vivo system.(25, 26) Each aortic ring was subsequently stored for 24-48 hours in RNAlater (Ambion, Austin, TX) then snap-frozen and stored at −80°C. Approximately 5% of aortic rings were discarded from the study due to evidence of tissue destruction observed while mounting vessel segments or during the three-hour treatment period.
QPCR amplification of lacZ and Luc
Aortic rings underwent rotor-stator homogenization in Buffer RLT (QIAGEN, Valencia, CA), total RNA was extracted using the RNeasy Fibrous Tissue MiniKit (QIAGEN, Valencia, CA), and reverse transcription to cDNA was conducted using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA). QPCR utilizing TaqMan chemistry with specific primers/probes was performed with either lacZ (TaqMan Gene Expression Assays on Demand catalog # 185757894; Applied Biosystems, Foster City, CA) or Luc primers (Custom designed: forward-5′-AAGATTCAAAGTGCGCTGCTGGTG-3′; reverse–5′-TTGCCTGATACCTGGC AGATGGAA-3′; probe–5′-/6-FAM/TACACGAAATTGCTTCTGGTGGCGCT/ BHQ_1/−3′; Integrated DNA Technologies, Coralville, IA). Results were compared to 18s rRNA used as an internal standard (TaqMan Gene Expression Assays on Demand catalog # 4333760 F; Applied Biosystems, Foster City, CA). The BioRad MyiQ Single Color Real Time PCR Detection System was used (BioRad, Hercules, CA), with an initial denaturing cycle consisting of 10 minutes at 95°C, followed by 45 cycles of 30 seconds at 95°C and 1 minute at 60°C. The fold gene expression of lacZ or Luc was calculated by the ΔΔCt method with promoter activity at optimal tension (70 mmHg) considered to be baseline.
Statistical Analysis
Fold expression of lacZ or Luc was compared to baseline promoter activity by a two-sided one-sample t-test versus the fixed value of one. Comparisons among treatment groups were conducted by one-way and two-way analysis of variance (ANOVA). All values are reported as a mean +/− SEM. Stata 8 statistical software (Intercooled, College Station, TX) was utilized for all calculations and values were significant at p<0.05.
Results
Optimal tension for the MT1-MMP reporter mouse strain was 0.6 g, which was equivalent to 70 mmHg. When aortic rings extracted from these mice were subjected to elevated pressure, promoter activity was significantly increased at both 85 and 100 mmHg (Figure 1). Upon adding 100nM AngII, MT1-MMP promoter activity at the baseline optimal tension was increased approximately 2-fold and remained at this level despite the addition of elevated tension (Figure 1). A co-regulatory interaction was identified between tension and AngII for the MT1-MMP promoter (p=0.01).
Figure 1.

MT1-MMP promoter activity was determined by QPCR for Luc mRNA in murine thoracic aortic rings subjected to 70, 85, or 100 mmHg in the presence or absence of 100nM AngII. At both 85 and 100 mmHg, MT1-MMP promoter activity was increased compared to that at 70 mmHg, the baseline optimal tension (*p<0.05). Aortic rings treated with tension and AngII demonstrated increased MT1-MMP promoter activity compared to baseline, but displayed significantly less promoter activity than 85 mmHg alone (§p<0.05).
It was previously demonstrated that an elevated pressure of 100mmHg caused a 1.5-fold increase in MMP-2 promoter activity.(6) With the addition of 100nM AngII to aortic rings, promoter activity was significantly augmented at both 85 and 100 mmHg as compared to baseline optimal tension. MMP-2 promoter activation at 85 mmHg in the presence of AngII displayed a 1.5-fold increase, whereas treatment with AngII at 100 mmHg affected approximately a 7-fold increase (Figure 2).
Figure 2.

MMP-2 promoter activity was determined by QPCR for lacZ mRNA, in thoracic aortic rings subjected to 70, 85, or 100 mmHg in the presence or absence of 100nM AngII. Aortic rings receiving combined therapy displayed significantly increased MMP-2 promoter activity at 85 and 100 mmHg (*p<0.05). Additionally, promoter activity in response to combined therapy at 100 mmHg was elevated as compared to 100 mmHg alone (†p<0.05).
Similarly, it was reported in a previous study that aortic rings from the MMP-9 promoter-reporter mouse strain were unaffected by elevated pressure alone.(6) However, at 100 mmHg the addition of 100nM AngII resulted in a reduction of MMP-9 promoter activity compared to both baseline optimal tension and 100 mmHg alone (Figure 3).
Figure 3.

MMP-9 promoter activity was determined by QPCR for lacZ mRNA in thoracic aortic rings subjected to 70, 85, or 100 mmHg in the presence or absence of 100nM AngII. MMP-9 promoter activity was decreased from baseline (*p<0.05) with the combined treatment of 100 mmHg and 100nM AngII when compared to 100 mmHg alone (†p<0.05).
Discussion
Vascular remodeling results from the complicated interplay of numerous mechanical and neurohormonal stimuli, many of which have been shown to stimulate MMP production, thereby driving extracellular matrix degradation and reorganization. Careful experimental derivation of the effect of individual stimuli has created a vast network of data concerning vascular physiology and pathophysiology. It is important to note, however, that physiologic vascular biology must integrate the influence of multiple pro-remodeling stimuli in the regulation of gene expression within the vessel wall, but very little evidence exists concerning the collaborative effects of such stimuli. Therefore, we first examined how elevated tension effected promoter activity for individual members of the MMP family, and then evaluated how those promoter sequences would respond to concomitant treatment with a neurohormonal stimulus, the vasoactive peptide AngII. The MT1-MMP promoter demonstrated increased activity in response to elevated tension alone, and more importantly this effect was triggered at a smaller pressure amplitude than previously seen in the MMP-2 promoter,(6) further defining the differential effect of wall tension on MMP promoter activation. The combination of tension and AngII stimulated MT1-MMP promoter activity and resulted in a consistent level of expression across all tension levels. AngII amplified the tension-induced increase in MMP-2 promoter activity and contributed to decreased MMP-9 promoter activity at elevated tension levels. These results have provided evidence not only that a particular mechanical-molecular set point exists to trigger MMP promoter activity and subsequent matrix remodeling, but also that this set point varies among members of the MMP family and can be modulated in the presence of a relevant biological stimulus. Taken together these data suggest that particular MMP profiles may be induced in early hypertensive remodeling, and identify potential sites of therapeutic intervention (Figure 4).
Figure 4.
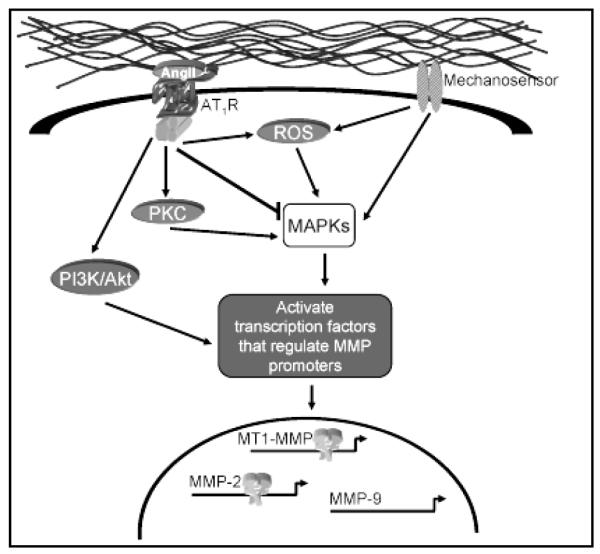
Potential signal transduction pathways for mechanical stress and AngII. Mechanosensors can transmit signals through reactive oxygen species (ROS) to activate the mitogen-activated protein kinases (MAPKs) which subsequently activate transcription factors that regulate MMP promoter activity. Likewise, AngII signaling through the AT1R may stimulate MAPK-dependent MMP promoter activation with or without the generation of ROS. AngII can also activate nonreceptor tyrosine kinases and/or negative regulators of MAPK signaling, that may stimulate alternative signaling cascades that regulate MMP transcription.
Signal transduction of mechanical stress
The continuous influence of shear stress and wall tension not only modulates normal vascular function but is suspected to contribute to vascular dysfunction by regulating differential gene expression. Pertinent mechanosensors include the integrins, receptor tyrosine kinases, and G protein-coupled receptors (Figure 4).(27) By creating a physical connection between the ECM and the intracellular space, integrins transduce mechanical stress to initiate signaling cascades resulting in activation of the mitogen-activated protein kinases (MAPKs).(28) Although the receptor tyrosine kinases and G protein-coupled receptors possess ligand specificity, members of both families have demonstrated an ability to be activated by biomechanical stress alone with subsequent MAPK activation.(29) Additionally, evidence suggests that mechanical stress may directly stimulate the generation of reactive oxygen species (ROS), also capable of activating MAPKs.(30, 31) As the MAPKs have been directly implicated in the activation of various transcription factors, it therefore follows that MAPK activation in response to divergent signaling pathways may differentially activate unique subsets of transcription factors, thereby differentially regulating MMP promoter activation.
Signal transduction for Angiotensin II
With regard to the long term biological effects of AngII in the vascular system, such as hypertrophy and ECM deposition, the AT1R, a G protein-coupled receptor, is the major mediator.(32) Through the immediate activation of protein kinase C (PKC), several nonreceptor tyrosine kinases become phosphorylated and trigger cascades resulting in MAPK activation (Figure 4).(33-36) Interestingly, the AT1R can also stimulate production of ROS with subsequent activation of MAPKs.(16, 37, 38) The nuclear translocation and regulation of inducible transcription factors resulting from MAPK activation plays a major role in AngII stimulated gene expression and cellular growth.(39) Remarkably, in addition to fostering protein synthesis through the MAPK signaling cascades, AngII also induces key negative feedback mechanisms including activation of the primary negative regulator of MAPKs, MAPK phosphatase-1 (MKP-1).(40) AT1R can also directly stimulate phosphorylation of numerous tyrosine kinases, such as phosphoinositol-3 kinase (PI3K), that activate signaling cascades with implications in gene expression.(32, 41) Therefore, similar to the signaling cascades evoked by mechanical stress, AngII may trigger signaling cascades that result in the activation of a unique set of key transcription elements capable of regulating MMP gene expression.
Potential sites of signaling pathway interactions
Evidence outlining the intracellular signal transduction pathways for mechanical stimuli and AngII has implicated activation of particular profiles of transcription factors, many of which have binding sequences within the MMP promoters evaluated in this study, potentially inducing or repressing MMP promoter activity (Figure 4).
In the case of MT1-MMP, tension-induced activation of MAPK can lead to activation of Ets and NF-κB, transcription factors which both have binding elements within the MT1-MMP promoter region.(42) Accordingly, the activation of MAPK may therefore contribute to the increased promoter activity seen at 85 and 100 mmHg. On the other hand, AngII can stimulate production of early growth response genes including Egr-1, an element whose binding site in the MT1-MMP promoter has previously been shown to regulate transcription,(43) and may explain the increased promoter activity seen at baseline optimal tension in aortic rings treated with AngII. Alternatively, AngII has been shown to activate MKP-1,(40) allowing for the activation of AngII signaling to inhibit tension-induced transcription altogether, thus resulting in the consistent upregulation of MT1-MMP promoter activity through an AngII-dependent transcriptional response. This is consistent with our results showing a stable amount of MT1-MMP promoter activity across all tension levels in the presence of AngII. With regard to early hypertensive remodeling, increased MT1-MMP transcriptional activity suggests that this enzyme contributes to initiation of proteolytic remodeling of the vessel wall.
The MMP-2 promoter responded to tension alone at 100 mmHg.(6) The significant increase in MMP-2 promoter activity quantified at both 85 and 100 mmHg when aortic rings were treated with AngII suggests a synergistic effect. This response may be mediated through the activation of MAPK and transcription factors compatible with the MMP-2 promoter region such as AP-1, Ets and c-myc.(44) The amplified effect demonstrated by addition of AngII may be attributed to increased input into a single signaling pathway, potentially bypassing the negative feedback initiated by AngII. Alternatively, the AT1R can phosphorylate PI3K, initiating a pathway that activates Akt, a kinase subsequently shown to activate AP-2, a pertinent element in MMP-2 transcriptional activity.(44) Therefore, in addition to amplifying the MAPK pathway, AngII may activate the PI3K/Akt pathway to enhance AP-2 activity and produce a large increase in MMP-2 promoter activity over that of tension alone. A third potential pathway for heightened MMP-2 promoter activation may be derived from evidence that increasing amplitudes of cardiac myocyte stretch elicited increasing amounts of ROS generation.(45) Likewise, greater tension application to the thoracic aorta may amplify ROS production, ultimately stimulating MMP-2 transcriptional activity. The marked elevation in MMP-2 promoter activity documented with elevated tension in the presence of AngII, a potent hypertensive stimulus, strongly suggests that this protease may play a major role in initiating hypertensive remodeling.
While not effected by tension alone,(6) the combination of elevated tension and AngII reduced MMP-9 promoter activity. Previous studies have demonstrated an increase in MMP-9 transcription in response to mechanical stress or AngII.(2, 46, 47) Each stimulus has the potential to activate several transcription factors capable of binding the MMP-9 promoter region, including NF-κB, AP-1, and Ets.(29) The lack of response by the MMP-9 promoter in this model implies that either additional regulatory elements are required for promoter activation, or that transcriptional repressors are also induced by these stimuli to inhibit MMP-9 transcription. In early hypertensive remodeling, therefore, the role for MMP-9 appears to be limited.
Limitations
The goal of this project was to evaluate a single aspect of MMP regulation, gene promoter activation, as it is influenced by integrated mechanical and neurohormonal stimuli. The net MMP proteolytic activity ultimately driving vascular remodeling is influenced at both the transcriptional and translational levels, in addition to propeptide activation, and endogenous inhibition. The present study demonstrates that mechanical and neurohormonal stimuli induce transcriptional activity using a promoter-reporter system. Previous work from this laboratory and others, has shown that the MMP promoter-reporter gene constructs utilized in this study do respond to these stimuli in similar fashion to the native genes.(6, 18, 19) Additionally, it has been well documented that MMP mRNA, protein levels, and zymographic activity increase concomitantly in various cardiovascular tissue systems.(10, 48-50) While this may imply that MMPs are primarily regulated at the transcriptional level, it must be noted that this study has only assessed changes in promoter activation. Unfortunately, the small size of the murine thoracic aortic rings, coupled with the short duration of treatment, prohibited protein quantification. Therefore, the contribution of post-transcriptional regulation to overall MMP activity in this ex vivo could not be assessed. Nevertheless, employing stimuli known to induce vascular remodeling, revealed two MMPs that undergo early transcriptional activation, and identified these proteases as potential targets to attenuate ECM restructuring. To ascertain whether the protein levels of these targets follow the observed changes in transcriptional activation will require further investigation.
Summary and Clinical Implications
This project has demonstrated that two discrete stimuli, elevated wall tension and AngII, can interact to regulate MMP promoter activity, and ultimately induce transcription of a subset of enzymes, including MMP-2 and MT1-MMP, which are suspected to play a major role in initiating hypertensive vascular remodeling. Understanding the integrated influence of numerous vascular remodeling stimuli has significant therapeutic implications. Interference in the renin-angiotensin system with angiotensin-converting enzyme inhibitors and AngII receptor blockers has already improved morbidity among patients suffering from diabetes mellitus, numerous forms of heart disease, and peripheral vascular disease.(11) Clarification of the interaction between AngII and a mild hypertensive state to initiate vascular remodeling may identify new indications for AngII inhibitory therapy or new targets for pharmacologic intervention.
Acknowledgments
Funding Source This study was supported by NIH/NHLBI grant R01 HL075488-05 (JSI) and the Ralph H. Johnson Veterans Affairs Medical Center, Charleston, SC, through a Merit Award (FGS) and CDA-2 Career Award (JAJ).
References
- 1.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 2.Lehoux S, Lemarie CA, Esposito B, Lijnen HR, Tedgui A. Pressure-induced matrix metalloproteinase-9 contributes to early hypertensive remodeling. Circulation. 2004;109:1041–1047. doi: 10.1161/01.CIR.0000115521.95662.7A. [DOI] [PubMed] [Google Scholar]
- 3.Eagleton MJ, Ballard N, Lynch E, Srivastava SD, Upchurch GR, Jr., Stanley JC. Early increased MT1-MMP expression and late MMP-2 and MMP-9 activity during Angiotensin II induced aneurysm formation. J Surg Res. 2006;135:345–351. doi: 10.1016/j.jss.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 4.Abbruzzese TA, Guzman RJ, Martin RL, Yee C, Zarins CK, Dalman RL. Matrix metalloproteinase inhibition limits arterial enlargements in a rodent arteriovenous fistula model. Surgery. 1998;124:328–334. discussion 334-325. [PubMed] [Google Scholar]
- 5.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- 6.Ruddy J, Jones J, Stroud R, Mukherjee R, Spinale F, Ikonomidis J. Differential Effect of Wall Tension on Matrix Metalloproteinase Promoter Activation in the Thoracic Aorta. J Surg Res. 2009 doi: 10.1016/j.jss.2008.12.033. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 8.Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206:1–8. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 9.Nollendorfs A, Greiner TC, Nagase H, Baxter BT. The expression and localization of membrane type-1 matrix metalloproteinase in human abdominal aortic aneurysms. J Vasc Surg. 2001;34:316–322. doi: 10.1067/mva.2001.115962. [DOI] [PubMed] [Google Scholar]
- 10.Sinha I, Hannawa KK, Eliason JL, Ailawadi G, Deogracias MP, Bethi S, Ford JW, Roelofs KJ, Grigoryants V, Henke PK, Stanley JC, Upchurch GR., Jr. Early MT-1 MMP expression following elastase exposure is associated with increased cleaved MMP-2 activity in experimental rodent aortic aneurysms. Surgery. 2004;136:176–182. doi: 10.1016/j.surg.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 11.Dzau VJ. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047–1052. doi: 10.1161/01.hyp.37.4.1047. [DOI] [PubMed] [Google Scholar]
- 12.Coker ML, Jolly JR, Joffs C, Etoh T, Holder JR, Bond BR, Spinale FG. Matrix metalloproteinase expression and activity in isolated myocytes after neurohormonal stimulation. Am J Physiol Heart Circ Physiol. 2001;281:H543–551. doi: 10.1152/ajpheart.2001.281.2.H543. [DOI] [PubMed] [Google Scholar]
- 13.Browatzki M, Larsen D, Pfeiffer CA, Gehrke SG, Schmidt J, Kranzhofer A, Katus HA, Kranzhofer R. Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-kappaB and activator protein 1 in a redox-sensitive manner. J Vasc Res. 2005;42:415–423. doi: 10.1159/000087451. [DOI] [PubMed] [Google Scholar]
- 14.Wang M, Takagi G, Asai K, Resuello RG, Natividad FF, Vatner DE, Vatner SF, Lakatta EG. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41:1308–1316. doi: 10.1161/01.HYP.0000073843.56046.45. [DOI] [PubMed] [Google Scholar]
- 15.Rosen B, Barg J, Zimlichman R. The effects of angiotensin II, endothelin-1, and protein kinase C inhibitor on DNA synthesis and intracellular calcium mobilization in vascular smooth muscle cells from young normotensive and spontaneously hypertensive rats. Am J Hypertens. 1999;12:1243–1251. doi: 10.1016/s0895-7061(99)00158-2. [DOI] [PubMed] [Google Scholar]
- 16.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem. 1996;271:23317–23321. doi: 10.1074/jbc.271.38.23317. [DOI] [PubMed] [Google Scholar]
- 17.Luchtefeld M, Grote K, Grothusen C, Bley S, Bandlow N, Selle T, Struber M, Haverich A, Bavendiek U, Drexler H, Schieffer B. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochem Biophys Res Commun. 2005;328:183–188. doi: 10.1016/j.bbrc.2004.12.152. [DOI] [PubMed] [Google Scholar]
- 18.Mukherjee R, Mingoia JT, Bruce JA, Austin JS, Stroud RE, Escobar GP, McClister DM, Jr., Allen CM, Alfonso-Jaume MA, Fini ME, Lovett DH, Spinale FG. Selective spatiotemporal induction of matrix metalloproteinase-2 and matrix metalloproteinase-9 transcription after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H2216–2228. doi: 10.1152/ajpheart.01343.2005. [DOI] [PubMed] [Google Scholar]
- 19.Mohan R, Rinehart WB, Bargagna-Mohan P, Fini ME. Gelatinase B/lacZ transgenic mice, a model for mapping gelatinase B expression during developmental and injury-related tissue remodeling. J Biol Chem. 1998;273:25903–25914. doi: 10.1074/jbc.273.40.25903. [DOI] [PubMed] [Google Scholar]
- 20.Swafford AN, Jr., Harrison-Bernard LM, Dick GM. Knockout mice reveal that the angiotensin II type 1B receptor links to smooth muscle contraction. Am J Hypertens. 2007;20:335–337. doi: 10.1016/j.amjhyper.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 21.He GW, Angus JA, Rosenfeldt FL. Reactivity of the canine isolated internal mammary artery, saphenous vein, and coronary artery to constrictor and dilator substances: relevance to coronary bypass graft surgery. J Cardiovasc Pharmacol. 1988;12:12–22. doi: 10.1097/00005344-198807000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Lalli J, Harrer JM, Luo W, Kranias EG, Paul RJ. Targeted ablation of the phospholamban gene is associated with a marked decrease in sensitivity in aortic smooth muscle. Circ Res. 1997;80:506–513. doi: 10.1161/01.res.80.4.506. [DOI] [PubMed] [Google Scholar]
- 23.Brandes RP, Kim D, Schmitz-Winnenthal FH, Amidi M, Godecke A, Mulsch A, Busse R. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000;35:231–236. doi: 10.1161/01.hyp.35.1.231. [DOI] [PubMed] [Google Scholar]
- 24.Freay AD, Curtis SW, Korach KS, Rubanyi GM. Mechanism of vascular smooth muscle relaxation by estrogen in depolarized rat and mouse aorta. Role of nuclear estrogen receptor and Ca2+ uptake. Circ Res. 1997;81:242–248. doi: 10.1161/01.res.81.2.242. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Y, Dirksen WP, Babu GJ, Periasamy M. Differential vasoconstrictions induced by angiotensin II: role of AT1 and AT2 receptors in isolated C57BL/6J mouse blood vessels. Am J Physiol Heart Circ Physiol. 2003;285:H2797–2803. doi: 10.1152/ajpheart.00466.2003. [DOI] [PubMed] [Google Scholar]
- 26.Stacy LB, Yu Q, Horak K, Larson DF. Effect of angiotensin II on primary cardiac fibroblast matrix metalloproteinase activities. Perfusion. 2007;22:51–55. doi: 10.1177/0267659106074793. [DOI] [PubMed] [Google Scholar]
- 27.Shaw A, Xu Q. Biomechanical stress-induced signaling in smooth muscle cells: an update. Curr Vasc Pharmacol. 2003;1:41–58. doi: 10.2174/1570161033386745. [DOI] [PubMed] [Google Scholar]
- 28.Oktay M, Wary KK, Dans M, Birge RB, Giancotti FG. Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J Cell Biol. 1999;145:1461–1469. doi: 10.1083/jcb.145.7.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C, Xu Q. Mechanical stress-initiated signal transductions in vascular smooth muscle cells. Cell Signal. 2000;12:435–445. doi: 10.1016/s0898-6568(00)00096-6. [DOI] [PubMed] [Google Scholar]
- 30.Hishikawa K, Oemar BS, Yang Z, Luscher TF. Pulsatile stretch stimulates superoxide production and activates nuclear factor-kappa B in human coronary smooth muscle. Circ Res. 1997;81:797–803. doi: 10.1161/01.res.81.5.797. [DOI] [PubMed] [Google Scholar]
- 31.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation. 2003;108:1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- 32.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 33.Ishida M, Marrero MB, Schieffer B, Ishida T, Bernstein KE, Berk BC. Angiotensin II activates pp60c-src in vascular smooth muscle cells. Circ Res. 1995;77:1053–1059. doi: 10.1161/01.res.77.6.1053. [DOI] [PubMed] [Google Scholar]
- 34.Sabri A, Govindarajan G, Griffin TM, Byron KL, Samarel AM, Lucchesi PA. Calcium- and protein kinase C-dependent activation of the tyrosine kinase PYK2 by angiotensin II in vascular smooth muscle. Circ Res. 1998;83:841–851. doi: 10.1161/01.res.83.8.841. [DOI] [PubMed] [Google Scholar]
- 35.Duff JL, Berk BC, Corson MA. Angiotensin II stimulates the pp44 and pp42 mitogen-activated protein kinases in cultured rat aortic smooth muscle cells. Biochem Biophys Res Commun. 1992;188:257–264. doi: 10.1016/0006-291x(92)92378-b. [DOI] [PubMed] [Google Scholar]
- 36.Govindarajan G, Eble DM, Lucchesi PA, Samarel AM. Focal adhesion kinase is involved in angiotensin II-mediated protein synthesis in cultured vascular smooth muscle cells. Circ Res. 2000;87:710–716. doi: 10.1161/01.res.87.8.710. [DOI] [PubMed] [Google Scholar]
- 37.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 38.Hanna IR, Taniyama Y, Szocs K, Rocic P, Griendling KK. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002;4:899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- 39.Blume A, Herdegen T, Unger T. Angiotensin peptides and inducible transcription factors. J Mol Med. 1999;77:339–357. doi: 10.1007/s001090050360. [DOI] [PubMed] [Google Scholar]
- 40.Hiroi Y, Hiroi J, Kudoh S, Yazaki Y, Nagai R, Komuro I. Two distinct mechanisms of angiotensin II-induced negative regulation of the mitogen-activated protein kinases in cultured cardiac myocytes. Hypertens Res. 2001;24:385–394. doi: 10.1291/hypres.24.385. [DOI] [PubMed] [Google Scholar]
- 41.Saward L, Zahradka P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ Res. 1997;81:249–257. doi: 10.1161/01.res.81.2.249. [DOI] [PubMed] [Google Scholar]
- 42.Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 43.Haas TL, Stitelman D, Davis SJ, Apte SS, Madri JA. Egr-1 mediates extracellular matrix-driven transcription of membrane type 1 matrix metalloproteinase in endothelium. J Biol Chem. 1999;274:22679–22685. doi: 10.1074/jbc.274.32.22679. [DOI] [PubMed] [Google Scholar]
- 44.Park MJ, Kwak HJ, Lee HC, Yoo DH, Park IC, Kim MS, Lee SH, Rhee CH, Hong SI. Nerve growth factor induces endothelial cell invasion and cord formation by promoting matrix metalloproteinase-2 expression through the phosphatidylinositol 3-kinase/Akt signaling pathway and AP-2 transcription factor. J Biol Chem. 2007;282:30485–30496. doi: 10.1074/jbc.M701081200. [DOI] [PubMed] [Google Scholar]
- 45.Pimentel DR, Amin JK, Xiao L, Miller T, Viereck J, Oliver-Krasinski J, Baliga R, Wang J, Siwik DA, Singh K, Pagano P, Colucci WS, Sawyer DB. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res. 2001;89:453–460. doi: 10.1161/hh1701.096615. [DOI] [PubMed] [Google Scholar]
- 46.Pan CH, Wen CH, Lin CS. Interplay of angiotensin II and angiotensin(1-7) in the regulation of matrix metalloproteinases of human cardiocytes. Exp Physiol. 2008;93:599–612. doi: 10.1113/expphysiol.2007.041830. [DOI] [PubMed] [Google Scholar]
- 47.Raffetto JD, Qiao X, Koledova VV, Khalil RA. Prolonged increases in vein wall tension increase matrix metalloproteinases and decrease constriction in rat vena cava: Potential implications in varicose veins. J Vasc Surg. 2008;48:447–456. doi: 10.1016/j.jvs.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spruill LS, Lowry AS, Stroud RE, Squires CE, Mains IM, Flack EC, Beck C, Ikonomidis JS, Crumbley AJ, McDermott PJ, Spinale FG. Membrane-type-1 matrix metalloproteinase transcription and translation in myocardial fibroblasts from patients with normal left ventricular function and from patients with cardiomyopathy. Am J Physiol Cell Physiol. 2007;293:C1362–1373. doi: 10.1152/ajpcell.00545.2006. [DOI] [PubMed] [Google Scholar]
- 49.Ailawadi G, Knipp BS, Lu G, Roelofs KJ, Ford JW, Hannawa KK, Bishop K, Thanaporn P, Henke PK, Stanley JC, Upchurch GR., Jr. A nonintrinsic regional basis for increased infrarenal aortic MMP-9 expression and activity. J Vasc Surg. 2003;37:1059–1066. doi: 10.1067/mva.2003.163. [DOI] [PubMed] [Google Scholar]
- 50.Schmoker JD, McPartland KJ, Fellinger EK, Boyum J, Trombley L, Ittleman FP, Terrien C, 3rd, Stanley A, Howard A. Matrix metalloproteinase and tissue inhibitor expression in atherosclerotic and nonatherosclerotic thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2007;133:155–161. doi: 10.1016/j.jtcvs.2006.07.036. [DOI] [PubMed] [Google Scholar]