

Introduction
All naturally-occurring proteins are chiral, that is, they have a distinct “handedness”. This is important biologically, but also has implications for their study by X-ray crystallography. Molecules in general can crystallize in 230 different ways (i.e., with 230 different symmetries or “space groups”). Chiral molecules can only crystallize in 65 of these packing arrangements. The remaining 165 are “centrosymmetric” and contain “centers of inversion” or “centers of symmetry” which require that the molecule and its mirror image be present in equal numbers in the crystal, or that the molecule itself be centrosymmetric. Neither of these is possible for naturally-occurring proteins.
As is now universally appreciated, the determination of any crystal structure requires not only the measurement of the amplitudes of scattering, Fo(hkl), from each set of Bragg planes (hkl) but also the determination of the phase angle α(hkl) for each X-ray reflection. For centrosymmetric crystals each phase angle is restricted to one of two possible values, 0° or 180°. In non-centrosymmetric crystals α(hkl) can have any value from 0° to 360°. For the same reason the determination of structures in centrosymmetric crystals is easier than those in non-centrosymmetric ones. In the classical isomorphous replacement method, for example, one heavy-atom replacement is sufficient to determine the structure of a centrosymmetric crystal whereas two heavy-atom replacements are required in the non-centrosymmetric case.
A New Racemic Protein Crystal Structure
In this issue of Protein Science, Kent and coworkers1 describe the structure of a crystal formed from a racemic mixture of d- and l-plectasin. l-plectasin is a naturally-occurring 40-amino acid protein with potent antimicrobial activity. d-plectasin is the mirror image of l-plectasin. It is unknown in nature and devoid of any biological activity. In the experiment of Mandal et al.,1 both l- and d-plectasin were obtained by chemical synthesis. The mixture of the two racemic forms crystallized in space group P1. In this space group the unit cell has an oblique shape but the molecular packing is extremely simple. Typically there are just two molecules in the unit cell and they are related to each other by a center of inversion. This is exactly the situation with the racemic crystal of plectasin. The unit cell contains one l-plectasin plus one d-plectasin molecule, with the center of symmetry located midway between them.
The feasibility of obtaining racemic crystals of a protein was pioneered by Zawadzke and Berg2 using a rubredoxin that contained 45 amino acids. They pointed out that if sufficiently high resolution data could be measured, the restriction of the phase angles to 0° or 180° would facilitate structure determination by direct methods (i.e., using only the intensity measurements from the native crystals). This was, in fact, the method used by Mandal et al.1 to determine the structure of plectasin.
Limitations on the Use of Anomalous Scattering
It might be noted, however, that racemic crystals complicate the use of anomalous scattering data in determining the structure of the protein. If a protein crystal contains heavy atoms such as mercury, iodine or selenium the Friedel-related reflections Fo(hkl) and 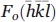 will differ slightly in intensity and these differences can be used to obtain information on the phase angles. In a centrosymmetric crystal, however, Fo(hkl) and have equal amplitudes and no phase information can be obtained. Recently, Pentelute et al.3 described a way around this difficulty by making a quasi-racemic mixture of snow flea antifreeze protein in which an additional -SeCH2- group was incorporated into the l-enantiomer. The presence of this symmetry-breaking group still allowed the quasi-racemic mixture to crystallize with quasi-centrosymmetric symmetry. Because the crystals were no longer centrosymmetric, conventional multiwavelength anomalous dispersion (MAD) phasing could be used to solve the structure. In contrast, since the structure was not truly centrosymmetric, the phases were no longer 0° or 180° and one of the main advantages of the racemic approach was lost. Pentelute et al.3 were driven to this approach not as an easy route to solve the structure but because it was the only way they could obtain crystals (see below).
will differ slightly in intensity and these differences can be used to obtain information on the phase angles. In a centrosymmetric crystal, however, Fo(hkl) and have equal amplitudes and no phase information can be obtained. Recently, Pentelute et al.3 described a way around this difficulty by making a quasi-racemic mixture of snow flea antifreeze protein in which an additional -SeCH2- group was incorporated into the l-enantiomer. The presence of this symmetry-breaking group still allowed the quasi-racemic mixture to crystallize with quasi-centrosymmetric symmetry. Because the crystals were no longer centrosymmetric, conventional multiwavelength anomalous dispersion (MAD) phasing could be used to solve the structure. In contrast, since the structure was not truly centrosymmetric, the phases were no longer 0° or 180° and one of the main advantages of the racemic approach was lost. Pentelute et al.3 were driven to this approach not as an easy route to solve the structure but because it was the only way they could obtain crystals (see below).
Assuming that a true racemic crystal of a protein contains heavy atoms, there is a way in which anomalous scattering X-ray measurements can be used to determine the structure. (The heavy atoms could be incorporated, for example, via selenomethionine substitution or standard heavy-atom soaks.) As noted above, for such a crystal the amplitudes of the reflections Fo(hkl) and are equal and provide no phase information. If, however, measurements are made at two or more different wavelengths there will be a change in the anomalous scattering of the heavy atoms which in turn will alter the amplitude of Fo(hkl), The method has not been tested for a racemic crystal, but the amplitude changes should allow location of the heavy atoms and determination of the phases for the protein as a whole. The wavelength-dependent differences would in general be small, but could be optimized by appropriate choice of the wavelengths used. The small changes in the amplitude of Fo(hkl) would be offset by the central advantage of racemic crystallography, namely that the phase determination for a given reflection simply requires discriminating between the two alternatives, 0° or 180°.
Ease of Crystallization
It was noted above that one of the advantages of racemic crystallography may be ease of crystallization. In a remarkable prescient article Wukovitz and Yeates4 addressed the question “Why do protein crystals favor some space groups over others?”. They proposed that the favored space groups are those which allow the molecules more rigid-body degrees of freedom and therefore allow crystal contacts to be formed in a greater number of ways. On this basis they predicted that “P1 will be the most frequently observed space group for racemic protein mixtures”. The degree to which subsequent studies have verified this prediction has been remarkable. Six of the eight reported structures of racemic mixtures of proteins or peptides have space group P1,1–3,5–7 the seventh, dl-monellin,8 is pseudo P1, and the eighth, a scorpin toxin,9 is in a space group that is centrosymmetric but not P1.
Wukovitz and Yeates4 also predicted that because space group P1 is significantly more probable than other symmetries, racemic mixtures of proteins would be easier to crystallize than the single biological enantiomer. This has also been supported by experimental observation.2,3,8,9
Refinement of centrosymmetric crystal structures is easier than for structures in chiral space groups. Because the phases are restricted to 0° or 180° an initial model of limited quality can still provide a very powerful starting point. In the case of rubredoxin, for example, a poor starting model (R = 57%), still correctly predicted 80% of the phase angles of the medium and strong reflections.2 Use of such data is expected to give a very good electron density map.
Because the structure of dl-plectasin is both centrosymmetric and to high resolution, it provides an opportunity to address a nagging concern in X-ray crystallography, namely, why are the R-factors of many refined crystal structures so high? The R-factor measures the average discrepancy between the scattering amplitudes Fc(hkl) calculated from the refined model and those measured experimentally (Fo(hkl)). The large majority of structures in the Protein Data Bank (PDB) have R-values in the range 15–25%. Such values are perhaps three times larger than the experimental error in the measurement of Fo(hkl). This discrepancy clearly indicates that there must be deficiencies in the refined protein models. Such deficiencies are not surprising for structures refined at low to moderate resolution (e.g., up to about 1.5 Å resolution) because the number of experimental observations is insufficient to define all of the parameters needed to define a detailed model. As the resolution gets higher (e.g., 1.0 Å or better) the ratio of observations to parameters becomes more favorable, but structures refined at this resolution still often have relatively high R-factors. The 1.0 Å resolution structure of dl-plectasin, for example, has an R-factor of 20.2%. (An R-factor of 20.2% for a centrosymmetric structure corresponds to about 13% for an acentric one.10)
A standard way to check for possible errors in the refined model of a protein is to calculate a difference Fourier map with amplitudes (Fo(hkl)-Fc(hkl)) and phases from the refined model. If the model perfectly represents the structure in the crystal the map will be uniformly zero. If, however, there are deficiencies in the model such as atoms misplaced or solvent molecules missed, these will be indicated by characteristic positive or negative features in the map. For normal protein structures in chiral space groups the difference maps are imperfect because the calculation does not allow for differences in phases between the model and the crystal structure. For a centrosymmetric structure, however, the phases of the model and the crystal will usually be correct, and should provide a more accurate difference map.
An analysis of the Fo(hkl)-Fc(hkl) difference map for the refined model of plectasin (Mandal et al.1; PDB code 3E7R) is shown in Figure 1. The histogram shows the average and root-mean-square difference density throughout the unit cell calculated a function of the distance to the closest atom of the model (see the figure caption for details). The idea behind this calculation was to search, in a systematic way, for specific deficiencies in the model. The plot shows that the rms value of the difference density increases in the region that is about 2.8 Å or more from any atom in the model (i.e., in the bulk solvent region). Also the average value of the difference density in this region is substantially positive. This indicates that there is additional electron density in the bulk solvent region that is not accounted for in the model. Because this positive density begins at the hydrogen-bonding distance of 2.8 Å it suggests that there are water molecules in the bulk solvent that are hydrogen-bonded to atoms in the model. These solvent molecules could be at sites which are of low occupancy, or high mobility and not normally considered as viable candidates for inclusion in a crystallographic model. The figure also shows, however, that the number of electron density grid points in the bulk solvent region is small, relative to the whole unit cell. This is consistent with the unusually low solvent content of these crystals (13–15%).1 Therefore, any deficiencies of the model cannot be attributed exclusively to the bulk solvent region. The other region where the rms difference density increases is close to the centers of atoms. This could, perhaps, suggest a systematic error in the crystallographic B-factors. Again, however, there are relatively few density points at such sites. Overall the plot suggests that there is room for improvement within the protein itself as well as the solvent region.
Figure 1.
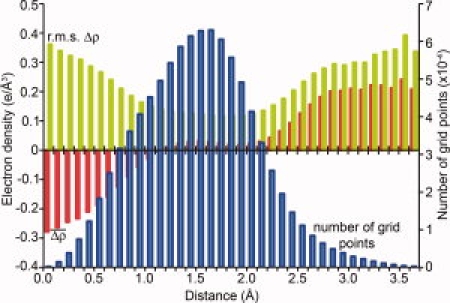
Average and root-mean-square values of the difference electron density for plectasin plotted as a function of the distance from the nearest atom in the model. The difference map (Δρ) has coefficients (Fo−Fc), phases from the refined model of plectasin (PDB code 3E7R), and was calculated on a 0.1 Å grid. For each grid point, a search was made for the nearest atom in the model, including ordered solvent. The density values were binned according to the distance to the nearest atom and the average (red) and rms (green) values plotted in the figure. The histogram also shows the number of grid points in each bin (blue). The plot shown is for the difference density map calculated at 1.5 Å resolution. This is not the outer limit of the data but was used because the X-ray observations are very complete and most of the phases should be correct. Maps at higher resolution give fairly similar histograms as do SigmaA-weighted Fo−Fc maps (not shown).
Recently, there have been some protein refinements at very high resolution where the R-factors have been reduced substantially below the 15–25% range. According to Nishibori et al.11 there are more than 600 structures in the PDB with data to a resolution of 1.2 Å or better and of these 50 or so have R-values below 10%. In order to achieve such values the model needs to allow for the following three features: (1) Anisotropic motion of individual atoms, (2) inclusion of hydrogen atoms, and (3) individual side-chains having more than one distinct conformation. Modeling of these features is not justified in many cases, but they are clearly bona fide aspects of protein structures in crystals, and should be taken into account in situations where very high resolution data are available.
Summary
As protein synthetic techniques become more precise and more routine, racemic crystallography will become a powerful and more attractive alternative to conventional protein structure determination. Theory suggests and experimental evidence is beginning to accumulate that crystallization of a racemic mixture is easier than crystallization of the single enantiomer. In centrosymmetric space groups, structure determination is simpler and more powerful than in chiral ones. The conventional MAD method cannot be used but structure determination is feasible by several different approaches including the use of a single heavy-atom derivative, by collecting data at different wavelengths, or by direct methods.
Acknowledgments
The author thank Dr. Stephen Kent and his group for making their X-ray data available before publication. Also the help of Dr. Lijun Liu in performing the electron density analysis and preparing the figure was indispensable.
References
- 1.Mandal K, Pentelute B, Tereshko V, Thammavongsa V, Schneewind O, Kossiakoff A, Kent S. Racemic crystallography of synthetic protein enantiomers used to determine the X-ray structure of plectasin by direct methods. Protein Sci. 2009;18:1146–1154. doi: 10.1002/pro.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zawadzke LE, Berg JM. The structure of a centrosymmetric protein crystal. Proteins. 1993;16:301–305. doi: 10.1002/prot.340160308. [DOI] [PubMed] [Google Scholar]
- 3.Pentelute BL, Gates ZP, Tereshko V, Dashnau JL, Vanderkooi JM, Kossiakoff AA, Kent SBH. X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J Am Chem Soc. 2008;130:9695–9701. doi: 10.1021/ja8013538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wukovitz SW, Yeates TO. Why protein crystals favour some space-groups over others. Nat Struct Biol. 1995;2:1062–1067. doi: 10.1038/nsb1295-1062. [DOI] [PubMed] [Google Scholar]
- 5.Doi M, Ishibe A, Shinozaki H, Urata H, Inoue M, Ishida T. Conserved and novel structural characteristics of enantiomorphic Leu-enkephalin. Int J Peptide Proteins Res. 1994;41:325–331. doi: 10.1111/j.1399-3011.1994.tb00526.x. [DOI] [PubMed] [Google Scholar]
- 6.Toniolo C, Peggion C, Crisma M, Formaggio F, Shui X, Eggleston DS. Structure determination of racemic trichogin A IV using centrosymmetric crystals. Structural Biol. 1994;1:908–914. doi: 10.1038/nsb1294-908. [DOI] [PubMed] [Google Scholar]
- 7.Patterson WR, Anderson DH, DeGrado WF, Cascio D, Eisenberg D. Centrosymmetric bilayers in the 0.75 Å resolution structure of a designed alpha-helical peptide. D,L-Alpha-1 Protein Sci. 1999;8:1410–1422. doi: 10.1110/ps.8.7.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung L-W, Kohmura M, Ariyoshi Y, Kim S-H. Structural differences in D and L-monellin in the crystals of racemic mixture. J Mol Biol. 1999;285:311–321. doi: 10.1006/jmbi.1998.2308. [DOI] [PubMed] [Google Scholar]
- 9.Mandal K, Pentelute BL, Tereshko V, Kossiakoff AA, Kent SBH. X-ray structure of a native scorpion toxin BmBKTx1 by racemic protein crystallography using direct methods. J Am Chem Soc. 2009;131:1362–1363. doi: 10.1021/ja8077973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luzzati PV. Traitement statistique des erreurs dans la determination des structures cristallines. Acta Cryst. 1952;5:802–810. [Google Scholar]
- 11.Nishibori E, Nakamura T, Arimoto M, Aoyagi S, Ago H, Miyano M, Ebisuzaki T, Sakata M. Application of maximum-entropy maps in the accurate refinement of a putative acylphosphatase using 1.3 Å X-ray diffraction data. Acta Cryst D. 2008;64:237–247. doi: 10.1107/S0907444907065663. [DOI] [PubMed] [Google Scholar]