

Abstract
Ric-8 is a highly conserved cytosolic protein (MW 63 KDa) initially identified in C. elegans as an essential factor in neurotransmitter release and asymmetric cell division. Two different isoforms have been described in mammals, Ric-8A and Ric-8B; each possess guanine nucleotide exchange activity (GEF) on heterotrimeric G-proteins, but with different Gα subunits specificities. To gain insight on the mechanisms involved in Ric-8 cellular functions it is essential to obtain some information about its structure. Therefore, the aim of this work was to create a structural model for Ric-8. In this case, it was not possible to construct a model based on comparison with a template structure because Ric-8 does not present sequence similarity with any other protein. Consequently, different bioinformatics approaches that include protein folding and structure prediction were used. The Ric-8 structural model is composed of 10 armadillo folding motifs, organized in a right-twisted α-alpha super helix. In order to validate the structural model, a His-tag fusion construct of Ric-8 was expressed in E. coli, purified by affinity and anion exchange chromatography and subjected to circular dichroism analysis (CD) and thermostability studies. Ric-8 is approximately 80% alpha helix, with a Tm of 43.1°C, consistent with an armadillo-type structure such as α-importin, a protein composed of 10 armadillo repeats. The proposed structural model for Ric-8 is intriguing because armadillo proteins are known to interact with multiple partners and participate in diverse cellular functions. These results open the possibility of finding new protein partners for Ric-8 with new cellular functions.
Keywords: Ric −8, GEF, threading, armadillo, bioinformatics
Introduction
Ric-8 proteins have been implicated in two different main cellular functions: signaling regulation through G-proteins and asymmetric cell division during embryonic development.1,2 Genetic analyses performed in C.elegans, have shown that Ric-8 is essential for synaptic transmission3 and that its encoded protein is a conserved novel protein required for Gαq and Gαo signaling.1 Also it was observed that Ric-8 is involved in the regulation of centrosome movements during early embryogenesis.2 The mechanism of action of Ric-8 was unknown until 2003, when Tall et al.,4 used two-hybrid screening to isolate two mammalian isoforms of Ric-8, Ric-8A, and Ric-8B, that possessed the ability to interact with G protein α subunits. In vitro biochemical characterization of Ric-8A showed that it is able to stimulate the GDP/GTP exchange of Gαi and Gαq, but not of Gαs, acting therefore as a receptor-independent GEF for heterotrimeric G proteins.4 At the same time, our group reported the interaction of human Ric-8B with Gαs and Gαq and demonstrated its translocation to the plasma membrane in response to the activation of the corresponding G protein.5 Interestingly, in contrast to the mechanism used by GPCRs that act on the heterotrimeric form of G proteins, Ric-8 interacts and stimulates the GDP/GTP exchange on the dissociated Gα subunit in the absence of Gβγ.4
In C. elegans embryos, Ric-8 participates controlling the spindle position by modulating Gα activity6 and the molecular machinery involved in this process is constituted by a complex formed between Ric-8, the nuclear mitotic apparatus protein NμMA, Gαi-GDP and PINS.7 In this work it was found that Ric-8A induces the dissociation of the complex formed by Gαi-GDP/LGN/NμMA in vitro, releasing activated Gαi-GTP and NμMA from LGN. Based on these observations, the authors proposed that Ric-8A could be participating in regulating microtubule pulling forces on centrosomes during cell division by catalyzing the dissociation of the Gαi-GDP/PIN/NμMA complex. This hypothesis was confirmed, through studies conducted in D. melanogaster and C.elegans which showed that interaction between Ric-8 and Gαi1 was essential for cortical localization of Gαi1 and asymmetric cell division.6,8–11 In addition, studies performed in dividing mammalian cells demonstrated that over expression of LGN and Gαi3 affect the cortical position of the mitotic spindle in symmetrically dividing cells.12
Demonstration of the role of Ric-8A and Ric-8B in G protein signaling regulation came from in vivo studies performed by different groups, which showed that Ric-8 proteins are able to potentiate Gαq, Gαolf and Gαs-mediated signal transduction by acting downstream of their corresponding GPCRs.13–16
These observations are consistent with Ric-8 acting as a multifunctional protein with the ability to interact with many other proteins besides Gα subunits. Therefore, in order to better understand the cellular functions described for Ric-8, it is essential to know its structure, which at present is unknown. For this reason, the aim of this study was to construct a structural model for Ric-8, through the application of different bioinformatics methods that include folding recognition (threading). The results obtained indicate the presence of an α-alpha super helix folding, which represents the armadillo structure according to SCOP classification.17 Based on this initial folding prediction, we constructed a refined model using proteins with known armadillo structures as templates.18,19 To corroborate our structural model, we expressed Xenopus laevis Ric-8 in E.coli and the purified protein was subjected to circular dichroism (CD) spectroscopy analysis and thermo stability studies. The spectrum obtained clearly indicated a high level of alpha helix secondary structure and was similar to other proteins composed of armadillo domains.20,21
Results and Discussion
Ric-8 is an armadillo type protein
To construct a structural model for Ric-8, we first performed a BLAST analysis comparing the Xenopus laevis Ric-8 (xRic-8) amino acid sequence (AAZ23806) with protein data bases (Swissprot and PDB). This analysis revealed that Ric-8 belongs to a unique protein family with no homology to other proteins, making difficult to find a template structure to construct a protein model. The second analysis carried out was a secondary structure prediction study, which showed an exceptionally high level of α-helix content for xRic-8 [Fig. 1(B)]. Based on the fact that proteins with different amino acid sequences can share conserved structures, we hypothesized that xRic-8 might be structured by a type of folding previously reported for other proteins. To test our hypothesis, we followed the folding recognition approach and analyzed xRic-8 primary sequence through five different Web servers. Table I shows the five methods used to perform the folding recognition analysis and the top five proteins identified by each method. Interestingly, almost all of them, especially the ones with rank 1 or 2, propose an armadillo-type folding for xRic-8, which is in accordance with our secondary structure prediction analysis [Fig. 1(B)]. The armadillo fold, is composed by a right-handed super helical structure formed by three alpha helices folded in a triangle shaped structure, repeated several times and stacked to form a compact domain. Numerous proteins are known to display armadillo folding, with β-catenin the best known and representative example of this protein family. One characteristic of armadillo proteins is their capacity to interact with multiple protein partners and consequently to be involved in a broad range of biological processes such as transcription regulation, cell adhesion, tumor suppressor activity and nucleo-cytoplasmic transport.19 Interestingly, this feature, of forming protein complexes with several other proteins, has also been observed for xRic-8 by our group (unpublished data). Using xRic-8 as bait, we have identified through a yeast two-hybrid screening two very attractive proteins called DasRa and INCENP, which are constituents of the chromosome passenger complex (CPC), a protein complex that controls chromosome segregation and cytokinesis.22
Figure 1.
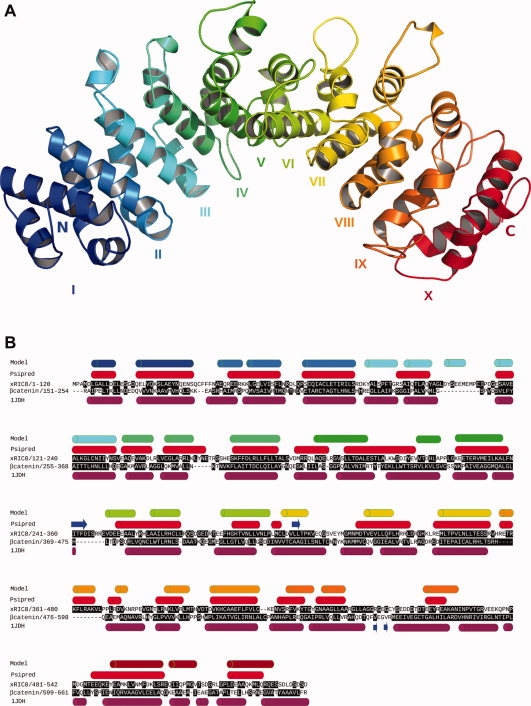
3D model of xRic-8 (A) Lateral view of xRic-8 predicted structure showing the 10 armadillo domains repeated in tandem along the protein. The amino and carboxylic ends of the protein are indicated by the capital letters N and C, respectively. Each armadillo repeat is designated by roman numbers and illustrated with different colors. (B) Comparative primary and secondary structure of xRic-8 and β-catenin. Predicted secondary structure of xRic-8 obtained with Psipred is shown in red cylinders (Psipred) and of the 3D model is shown in different color cylinders (Model), in which each color corresponds to a different armadillo repeat as shown in panel A. β-catenin secondary structure (based on its known tertiary structure, 1JDH is shown in purple cylinders. Buried aminoacids are highlighted in black boxes.
Table I.
Methods for Folding Recognition
| Methods | |||||
|---|---|---|---|---|---|
| Ranking | HHPRED | I-TASSER | 3D-JURY | PSIPRED | SP4 |
| 1 | β-catenin (2z6h) | α-importin (1ejl) | β-catenin (1jdh) | α-importin (2jdq) | plakofilin 1 (1xm9) |
| 2 | α-importin (1y2a) | β-catenin (1jdh) | β-catenin (1i7w) | plakofilin 1 (1xm9) | α-importin (1ee4) |
| 3 | HSPBP1 (1xqr) | α-importin (1ee4) | α-importin (1q1s) | RSR (2nvo) | β-catenin (1jdh) |
| 4 | β-catenin (1jdh) | plakofilin 1 (1xm9) | α-importin (1ial) | PR65a (1b3u) | 6-PGDase (1wdk) |
| 5 | α-importin (1jdq) | β-adaptin (1gw5) | α-importin (2jdq) | α-importin (2c1m) | β-adaptin (1gw5) |
The table show the top 5 folding obtain from each method. Protein PDB codes are shown in parentheses. Bold indicates proteins with armadillo folding.
Ric-8 3D model generation
After an armadillo type of folding was discovered for xRic-8, and in the absence of a template to build a homology model, two different approaches were followed to face this problem. First, a refined model was constructed using as template the human β-catenin tertiary structure. The xRic-8 structural model was build using the MODELLER Web server,23 by aligning the secondary structures of both proteins and conserving the main chain trace. The model was then refined with the SCWRL program,24 which corrected the positions of the side chains and placed them in the most favorable location, according to the local backbone conformation. This analysis allowed us to eliminate side chain steric clashes, by changing the energetically incorrect conformation of several amino acids and improving the hydrogen bond network. Finally, energy minimization was performed using GROMACS with Gromos 9643b1 force field,25 which allowed us to relax the structure, improving the interactions in the core of the protein model and minimizing any error.
The second strategy, was to build a structural model using the web server I-Tasser,26 which is a hierarchical protein structure modeling approach based on the secondary-structure enhanced Protein-Profile threading Alignment (PPA). The I-Tasser algorithm consists of three consecutive steps of threading, fragment assembly and iteration. In this method, the target sequence is first threaded through the PDB structure library to identify appropriate local fragments, which will be adopted for further structural reassembly. The unaligned regions (mainly loops) are built by ab initio modeling and finally all fragments are reassembled and the representative structure of each cluster is optimized and ordered by lowest energy.
Through both methodologies, we obtained similar results: a structural model for xRic-8 with 10 armadillo domains [Fig. 1(A)]. Analysis of these models with Ramachandran plot, showed that more than 90% of the residues are in a favorable conformation, and energetic analysis performed with Prosa27 showed that the I-Tasser model was energetically more favorable (data not shown).
Another analysis that we made in order to validate our model, was to do an alignment comparison between the secondary structure of our xRic-8 structural model and β-catenin. As can be seen in Figure 1(B), very similar arrangements along the sequences of the a-helixes that form the different domains are exhibited. The buried aminoacids, highlighted in black boxes, are also well conserved in both structures. For β-catenin, the percentage of buried aminoacids that are hydrophobic is 64% (as deduced from its known tertiary structure) and for xRic-8 is 59% (as deduced from our model), and hydrophilic exposed aminoacids are also very similar in both proteins, being 67% for β-catenin and 76% for xRic-8. In summary, all these features together strongly support and validate our structural model for xRic-8 and allow us to propose it as a member of the armadillo family.
Circular dichroism analysis confirms bioinformatics predictions
To test the proposed model we expressed recombinant His-tagged xRic8 in E. coli and subjected the purified protein (see Fig. 2) to biophysical analysis. The high alpha helical content, previously found by secondary structure prediction and fold recognition analysis, was confirmed by circular dichroism. As illustrated in Figure 3(A), the CD spectrum obtained for xRic-8 corresponds to the typical curve displayed by α-helical proteins, showing a minimum at 208 and 222 nm. Quantification of xRic-8 alpha helical content, by deconvolution of the circular dichroism spectra, gave a value of 82%. These results support the threading 3D model obtained by us for xRic-8 and are in good agreement with reported biophysical characteristics of polypeptides containing different number of armadillo domains, β-catenin and α-importin, which displayed an alpha helical content between 40% and 85%, in concordance with the amount of armadillo motifs present in the protein.28 Interestingly, this kind of folding has also been reported for another GEF protein.29
Figure 2.
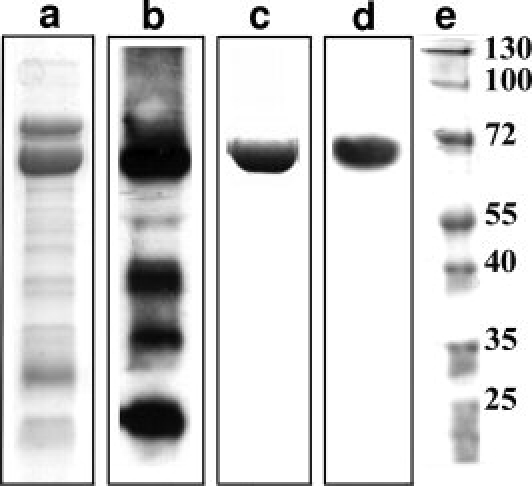
SDS-PAGE and Western Blot analysis of purified xRic-8 Lanes a and c show SDS-PAGE analysis for xRic-8 pooled fractions collected from Ni2+-agarose affinity chromatography and HiLoad-Q anion exchange chromatography, respectively. Lanes b and d show Western Blot analysis for the proteins observed in lanes a and c respectively. Lane e contains protein molecular weight standards. The protein show in c was used for CD spectroscopic analysis.
Figure 3.
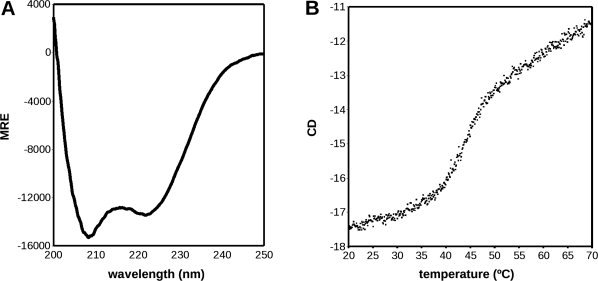
CD Spectrum of xRic-8 and Thermostability assay A) The CD spectrum shown is typical of a protein with high level of α-helix content, with well-defined minimums at 208 and 222 nm. The deconvolution of the spectrum with SOMCD software indicated 80% of helix content. B) The Thermostability assay show that xRic-8 is a protein with two structural states (folded and unfolded) with a Tm of 43.1 ± 0.1°C
To better characterize xRic-8, we also studied its thermo stability by measuring its ellipticity at the minimum of 222 nm in a range of temperature between 20 and 70°C [Fig. 3(B)]. The denaturation curve that was obtained is typical of a protein with two structural states with a Tm of 43.1 ± 0.1°C and a ΔHVH of 394.6 Kcal/mol. The reversibility of the folding was 75% (data not shown). Interestingly, the Tm obtained for xRic-8 is almost identical to that obtained for α-importin by Parmeggiani et al., which was 43°C.28 This result strongly supports our structural model because α-importin possesses 10 armadillo domains, the same number we are proposing for xRic-8. Parmeggiani et al.28 in their study, also suggest a direct relationship between the number of armadillo domains and the Tm of the protein. Consistent with this proposal, β-catenin, that contains 12 armadillo repeats, shows a Tm of 58°C.
In summary, the CD spectroscopic analysis and the thermo-stability assay strongly support the structural model we have obtained for xRic-8; consequently we can propose it as a new member of the armadillo family of proteins. The fact that Ric-8 is a member of this family is exciting, because armadillo proteins have been shown to act as “scaffold proteins,” interacting with a diverse set of partners and participating in many signalling pathways.
Methodology
Expression and purification of xRic-8
In order to express xRic-8 in bacteria, the corresponding cDNA was subcloned into the pQE81-L vector (Quiagen, Valencia, CA U.S.A.) fused to a hexa-histidine tag at its amino terminus. E.coli BL-21(DE3), harboring the plasmid pQE81-L/xRic-8, were grown in 4 L of LB medium with 100 μg/mL ampicillin at 25°C to an OD600 of 0.4. Protein expression was then induced by the addition of 50 μM IPTG and incubated for 2 h at the same temperature. After centrifugation, the cell pellet was resuspended in 30 mL of buffer A (50 mM phosphate, pH 8.0, 500 mM NaCl, 5 mM β-mercaptoethanol, 20 mM imidazole, and a cocktail of protease inhibitors (Complete, Roche)) and disrupted by sonication. The crude cell lysate was then cleared by centrifugation at 100,000g for 60 min. and the supernatant loaded onto a 3 mL Ni2+-nitrilotriacetic acid-agarose resin column (NI-NTA, Novagen), equilibrated in buffer A. After washing the column with 5 volumes of buffer A, the bound proteins were eluted with buffer B (50 mM phosphate, pH 8.0, 500 mM NaCl, 5 mM β-mercaptoethanol, 100 mM imidazole and cocktail of protease inhibitors). Ten 1 mL fractions were collected and 5 μL of each analyzed by SDS-PAGE and stained with Coomassie blue. To confirm the expression of recombinant xRic-8, fractions were also subjected to Western blot analysis, using an anti-His antibody (Clontech). xRic-8 containing fractions were pooled and concentrated by centrifugation in a Centricon tube (Millipore) and imidazole removed by performing several dilutions and centrifugations in buffer 20 mM HEPES, pH 8.0, 250 mM NaCl, 5 mM 2-mercaptoethanol and 1 mM PMSF. Partially purified xRic-8 was then loaded onto a HiLoad Q sepharose (GE) ion exchange chromatography column, equilibrated in a buffer containing 20 mM HEPES, pH 8.0, 250 mM NaCl, 5 mM 2-mercaptoethanol and 1 mM PMSF. After washing the column with two volumes of the same buffer, the protein was eluted with a linear gradient between 250 and 500 mM NaCl with a flux of 1 mL/min during 100 min. Fractions were analyzed by SDS-PAGE, stained with Coomassie blue and also by western blotting using an anti-His antibody. Fresh purified protein was used in all spectral analysis.
CD spectroscopy
Circular dichroism (CD) spectra was performed over a range of 200–250 nm with 0.1 nm increments at a scan speed of 50 nm/min in a Jasco J-715 CD spectropolarimeter. The final concentration of xRic-8 in the analysis was 2.33 mg/mL in buffer 50 mM phosphate pH 8.0 and 200 mM NaCl. Three different samples were scanned in a 1 mm quartz cuvette and the data transformed to MRE (Mean Residue Ellipticity). The contribution of the buffer was subtracted from each value. To estimate the percentage of secondary structure, the CD spectrum was analyzed with the SOMCD software.30
To study the thermo stability of xRic-8, a heat denaturation curve was performed by measuring the CD signal at 222 nm in the same buffer used for the CD spectra, at a temperature between 20 and 70°C. The temperature was raised at 0.1°C intervals at a ramp rate of 2°/min, using a Peltier-effect temperature controller and a in-cell temperature monitor. The unfolding melting temperature (Tm) and van't Hoff enthalpy (ΔHVH) were calculated with EXAM.31
Folding recognition
In order to gain some insights on xRic-8 structure, its amino acid sequence was analyzed with five different Web servers for folding-recognition: HHPRED,32 I-TASSER,26 3D-Jury,33 PSIPRED34 and SP4.35 All programs were used with the default parameters.
3D model generation
Two strategies were used to generate a 3D model of xRic-8:
Using the MODELLER program on MODELLER servers,33 utilizing the human β-catenin protein structure as template (PDB code: 1JDH). The model was first refined with the SCWRL program24 to construct the side chains and subsequently energy-minimized with GROMACS.25 The PROCHECK program was used for stereochemical analysis of the model.36
Using the web server I-Tasser, which is a method were the target sequence is first threaded through the PDB structure library to identify appropriate local fragments. After, the unaligned regions (mainly loops) are built by ab initio modeling and finally all fragments are reassembled and the representative structure of each cluster is optimized and ordered by energy minimization.
Both models were evaluated by Ramachandran plot,36 Prosa server27 and by calculating the characteristics of buried and exposed amino acids in the structure. An aminoacid was defined as exposed, when the accessible surface area was higher than 25%.
References
- 1.Miller KG, Emerson MD, McManus JR, Rand JB. Ric-8 (synembryn): a novel conserved protein that is required for G(q) alpha signaling in the C.elegans nervous system. Neuron. 2000;27:289–299. doi: 10.1016/s0896-6273(00)00037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller KG, Rand JB. A role for RIC-8 (synembryn) and GOA-1 (G(o)α) in regulating a subset of centrosome movements during early embryogenesis in Caenorhabditis elegans. Genetics. 2000;156:1649–1660. doi: 10.1093/genetics/156.4.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller KG, Alfonso A, Nguyen M, Crowell JA, Johson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci USA. 1996;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tall GG, Krumins AM, Gilman AG. Mammalian Ric-8A (Synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J Biol Chem. 2003;278:8356–8362. doi: 10.1074/jbc.M211862200. [DOI] [PubMed] [Google Scholar]
- 5.Klattenhoff C, Montecino M, Soto X, Guzmán L, Romo X, García MD, Mellstrom B, Naranjo JR, Hinrichs MV, Olate J. Human synembryn interacts with Gsα and Gqα and is translocated to the plasma membrane in response to isoproterenol and carbachol. J Cell Physiol. 2003;195:151–157. doi: 10.1002/jcp.10300. [DOI] [PubMed] [Google Scholar]
- 6.Couwenbergs C, Spilker AC, Gotta M. Control of embryonic spindle positioning and Gα activity by C.elegans RIC-8. Curr Biol. 2004;14:1871–1876. doi: 10.1016/j.cub.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 7.Tall GG, Gilman AG. Resistance to inhibitors of cholinesterase 8A catalyzes release of Gαi-GTP and nuclear mitotic apparatus protein (NuMA) from NuMA/LGN/Gαi-GDP complexes. Proc Natl Acad Sci USA. 2005;102:16584–16589. doi: 10.1073/pnas.0508306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Afshar K, Willard FS, Colombo K, Johnston CA, McCudden CR, Siderovski DP, Gonczy P. RIC-8 is required for GPR-1/2-dependent Galpha function during asymmetric division of C.elegans embryos. Cell. 2004;119:219–230. doi: 10.1016/j.cell.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 9.David NB, Martin CA, Segalen M, Rosenfeld F, Schweisguth F, Bellaiche Y. Drosophila Ric-8 regulates Galphai cortical localization to promote Galphai-dependent planar orientation of the mitotic spindle during asymmetric cell division. Nat Cell Biol. 2005;7:1083–1090. doi: 10.1038/ncb1319. [DOI] [PubMed] [Google Scholar]
- 10.Hampoelz B, Hoeller O, Bowman SK, Dunican D, Knoblich JA. Drosophila Ric-8 is essential for plasma-membrane localization of heterotrimeric G proteins. Nat Cell Biol. 2005;7:1099–1105. doi: 10.1038/ncb1318. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Ng KH, Qian H, Siderovski DP, Chia W, Yu F. Ric-8 controls Drosophila neural progenitor asymmetric division by regulating heterotrimeric G proteins. Nat Cell Biol. 2005;7:1091–1098. doi: 10.1038/ncb1317. [DOI] [PubMed] [Google Scholar]
- 12.Blumer JB, Kuriyama R, Gettys TW, Lanier SM. The G-protein regulatory (GPR) motif-containing Leu-Gly-Asn-enriched protein (LGN) and Giα3 influence cortical positioning of the mitotic spindle poles at metaphase in symmetrically dividing mammalian cells. Eur J Cell Biol. 2006;85:1233–1240. doi: 10.1016/j.ejcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Malik S, Ghosh M, Bonacci TM, Tall GG, Smrcka AV. Ric-8 enhances G protein βγ-dependent signaling in response to βγ-binding peptides in intact cells. Mol Pharmacol. 2005;68:129–136. doi: 10.1124/mol.104.010116. [DOI] [PubMed] [Google Scholar]
- 14.Von Dannecker LE, Mercadante AF, Malnic B. Ric-8B, an olfactory putative GTP exchange factor, amplifies signal transduction through the olfactory-specific G-protein Gαolf. J Neurosci. 2005;25:3793–3800. doi: 10.1523/JNEUROSCI.4595-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishimura A, Okamoto M, Sugawara Y, Mizuno N, Yamauchi J, Itoh H. Ric-8A potentiates Gq-mediated signal transduction by acting downstream of G protein-coupled receptor in intact cells. Genes Cells. 2006;11:487–498. doi: 10.1111/j.1365-2443.2006.00959.x. [DOI] [PubMed] [Google Scholar]
- 16.Romo X, Pastén P, Martinez S, Soto X, Lara P, Ramirez A, Torrejón M, Montecino M, Hinrichs M, Olate J. xRic-8 is a GEF for Gsα and participates in maintaining meiotic arrest in Xenopus laevis oocytes. J Cell Physiol. 2008;214:673–680. doi: 10.1002/jcp.21257. [DOI] [PubMed] [Google Scholar]
- 17.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucl Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peifer M, Ber S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell. 1994;76:789–791. doi: 10.1016/0092-8674(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 19.Coates JC. Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol. 2003;13:463–471. doi: 10.1016/s0962-8924(03)00167-3. [DOI] [PubMed] [Google Scholar]
- 20.Hirschl D, Bayer P, Müller O. Secondary structure of an armadillo single repeat from the APC protein. FEBS Lett. 1996;383:31–36. doi: 10.1016/0014-5793(96)00215-3. [DOI] [PubMed] [Google Scholar]
- 21.Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 22.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers:conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 23.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 24.Canutescu AA, Shelenkov AA, Dunbrack RL. A graph theory algorithm for protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. http://dunbrack.fccc.edu/SCWRL3.php. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J ChemTheory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. http://www.gromacs.org. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;23:9–40. doi: 10.1186/1471-2105-9-40. http://zhang.bioinformatics.ku.edu/I-TASSER. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. 35(Web Server issue) 2007:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parmeggiani F, Pellarin R, Larsen AP, Varadamsetty G, Stumpp MT, Zerbe O, Caflisch A, Plückthun A. Designed armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J Mol Biol. 2008;376:1282–1304. doi: 10.1016/j.jmb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 29.Vikis HG, Stewart S, Guan KL. SmgGDS displays differential binding and exchange activity towards different Ras isoforms. Oncogene. 2002;21:2425–2432. doi: 10.1038/sj.onc.1205306. [DOI] [PubMed] [Google Scholar]
- 30.Unneberg P, Merelo JJ, Chacón P, Morán F. SOMCD: method for evaluating protein secondary structure from UV circular dichroism spectra. Proteins Struct Funct Genet. 2001;42:460–470. doi: 10.1002/1097-0134(20010301)42:4<460::aid-prot50>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 31.Kirchoff W. EXAM: a two-state thermodynamic analysis program. Maryland: Gaithersburg; 1993. [Google Scholar]
- 32.Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucl Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. http://toolkit.tuebingen.mpg.de/hhpred. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. http://meta.bioinfo.pl/submit_wizard.pl. [DOI] [PubMed] [Google Scholar]
- 34.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. http://bioinf.cs.ucl.ac.uk/psipred/psiform.html. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Zhang C, Liang S, Zhou Y. Fold recognition by concurrent use of solvent accessibility and residue depth. Proteins. 2007;68:636–645. doi: 10.1002/prot.21459. http://sparks.informatics.iupui.edu/SP4. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereo chemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]