

Abstract
Evolutionary conservation of substructure architecture between yeast iso-1-cytochrome c and the well-characterized horse cytochrome c is studied with limited proteolysis, the alkaline conformational transition and global unfolding with guanidine-HCl. Mass spectral analysis of limited proteolysis cleavage products for iso-1-cytochrome c show that its least stable substructure is the same as horse cytochrome c. The limited proteolysis data yield a free energy of 3.8 ± 0.4 kcal mol−1 to unfold the least stable substructure compared with 5.05 ± 0.30 kcal mol−1 for global unfolding of iso-1-cytochrome c. Thus, substructure stabilities of iso-1-cytochrome c span only ∼1.2 kcal mol−1 compared with ∼8 kcal mol−1 for horse cytochrome c. Consistent with the less cooperative folding thus expected for the horse protein, the guanidine-HCl m-values are ∼3 kcal mol−1M−1 versus ∼4.5 kcal mol−1M−1 for horse versus yeast cytochrome c. The tight free energy spacing of the yeast cytochrome c substructures suggests that its folding has more branch points than for horse cytochrome c. Studies on a variant of iso-1-cytochrome c with an H26N mutation indicate that the least and most stable substructures unfold sequentially and the two least stable substructures unfold independently as for horse cytochrome c. Thus, important aspects of the substructure architecture of horse cytochrome c, albeit compressed energetically, are preserved evolutionally in yeast iso-1-cytochrome c.
Keywords: protein folding, limited proteolysis, protein substructures, evolutionary conservation, folding cooperativity, alkaline transition
Introduction
Many small single-domain proteins behave as if they are rigorously two-state based on kinetic and thermodynamic analysis.1 The cooperativity inherent in the two-state transition from the native to the denatured state is of considerable interest for both fundamental and practical reasons. Some proteins display distinct cooperative substructures indicating that protein folding is not rigorously two-state for all small single domain proteins.2–5 Because some misfolding diseases have been attributed to the enhanced accessibility of partially unfolded forms (PUFs) of proteins,6 in other words a loss of cooperativity, understanding the factors that modulate cooperativity is important.
Horse heart cytochrome c (hh cytc) is a paradigm for a protein with substructures.3–5,7–11 Native-state hydrogen exchange (NSHX) shows that hh cytc has five substructures (see Fig. 1). From lowest to highest in energy these are an Ω-loop that runs from residues 40 to 57 (gray in Fig. 1), an Ω-loop that runs from residues 71–85 (red in Fig. 1), a short segment of β-sheet (residues 37–39, 58–61) (yellow in Fig. 1), an Ω-loop that runs from the end of the N-terminal helix to residue 36 plus the 60's helix (green in Fig. 1) and finally, the most stable substructure, the N- and C-terminal helices (blue in Fig. 1). For brevity, we will refer to these substructures from least stable to most stable as infrared (shown in gray in Fig. 1), red, yellow, green, and blue.9 Equally complete substructure characterization is unavailable for other mitochondrial cytochromes c. Thus, the degree of evolutionary conservation of the substructure architecture of cytc is unclear.
Figure 1.
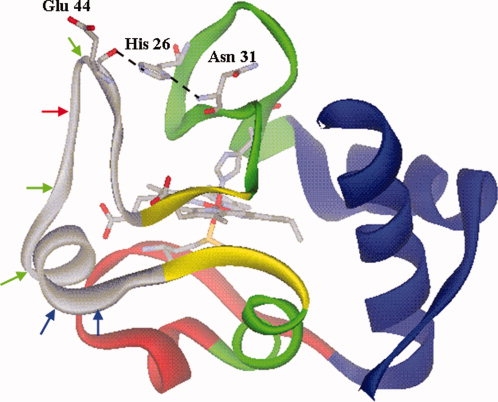
Structure of yeast iso-1-cytc in the oxidized state (2YCC)12 showing the substructures observed for hh cytc4,5 using color coding described in the text. The heme and its ligands, Met 80 and His 18, are shown as stick models color coded by atom type. His 26, Glu 44, and Asn 31 are shown as stick models color coded by atom type with the hydrogen bonds between the side chain of His 26 and the main chain carbonyl of Glu 44 and the amide NH of Asn 31 shown with dashed lines. The red arrow shows the initial cleavage site of proteinase K for WT iso-1cytc. The additional proteinase K cleavage sites observed for the TM variant are shown in green. Blue arrows show the initial trypsin cleavage sites observed for WT iso-1-cytc and the TM variant.
Comparison of RNase H from a mesophile and a thermophile does show evolutionary conservation of substructure architecture.13 The free energy spacing between PUFs is smaller in the less stable mesophilic protein. If this phenomenon is general, it suggests that less stable homologs of a protein should exhibit more cooperative folding.
Iso-1-cytochrome c (iso-1-cytc) from Saccharomyces cerevisiae is significantly less stable14,15 than hh cytc.16,17 The yeast protein (108 a.a.) has a five amino acid extension at the N-terminus and the horse protein (104 a.a.) has one extra amino acid at the C-terminus. The sequence identity is 59.6% for the 109 a.a. alignment.18 We have demonstrated that the behavior of the alkaline conformational transition of iso-1-cytc (involves replacement of the Met 80 heme ligand with lysines from the red substructure) has properties consistent with unfolding of the red substructure,19–21 an observation that had been confirmed with hh cytc.10 We have also found that stability effects of mutations are the same for the alkaline conformational transition and global unfolding (blue substructure) of iso-1-cytc, consistent with sequential unfolding of substructures as for hh cytc.22
Although our previous data suggest that iso-1-cytc has similar substructures to hh cytc, the available data for iso-1-cytc are limited to the behavior of the red and blue substructures. Thus, additional characterization of iso-1-cytc is necessary to assess the degree of evolutionary conservation between iso-1-cytc and hh cytc and the extent to which the stability range of the PUFs is compressed in this less stable cytc. NMR-detected NSHX,2,3 the standard method for characterizing substructures, is difficult to apply to less stable proteins.23,24 Thus, we use limited proteolysis, a method that detects the least stable part of a protein25 and can quantitatively evaluate its stability.26 We use global unfolding with gdnHCl to monitor the stability of the blue substructure and the alkaline conformational transition to monitor the red substructure. Finally, we probe the effect of a H26N mutation, which is expected to destabilize the least stable infrared substructure by removing an intersubstructural His 26/Glu 44 hydrogen bond (see Fig. 1), to assess whether the order of unfolding of the substructures is preserved between iso-1-cytc and hh cytc.
Results
Iso-1-cytochromes c expressed in E. coli
The WT iso-1-cytc from S. cerevisiae used in this investigation was expressed in Escherichia coli.27 Cys 102 has been replaced with serine (C102S) in our WT iso-1-cytc to avoid intermolecular disulfide dimerization during physical studies. When iso-1-cytc is expressed in S. cerevisiae, Lys 72 is trimethylated at the ɛ-amino group. This posttranslational modification is not made in E. coli permitting Lys 72 to act as a heme ligand in the alkaline conformer of iso-1-cytc, in addition to lysines 73 and 79.27
The TM variant15 of iso-1-cytc was also expressed in E. coli. In addition to the C102S mutation, this variant contains the mutations H26N, H33N, and H39Q, all of which are evolutionally allowed substitutions.18 The H33N and H39Q mutations do not affect the stability of iso-1-cytc.15,28 Mutation of His 26 is known to destabilize various cytochromes c.15,28–30 Thus, the H26N mutation is expected to dominate changes in the stability of the TM variant.
Limited proteolysis
Much like NSHX,2–5 limited proteolysis probes the stability of portions of a protein that require less energy to unfold than for global unfolding.25,26 There are two key distinctions between the methods. First, for a protein with substructures, limited proteolysis only provides information about the least stable substructure.25 Second, limited proteolysis is less sensitive to small conformational changes because a significant portion of the protein must unfold to permit proteolytic cleavage (see Fig. 2).25,31 Otherwise, the kinetic scheme is identical to that for NSHX and leads to an observed rate constant with the same form [Eq. (1), rates constants defined in Fig. 2]. The same limits apply to Eq. (1) as for NSHX. When kcl << kint, kobs is given by Eq. (2) (EX1 case)
| (1) |
and the rate of cleavage is limited by the rate of unfolding (kop) of the protease cleavage site. In
| (2) |
the limit where kcl >> kint (EX2 case) kobs reduces to Eq. (3) and kobs is controlled by the product of K′op and kint. K′op is the equilibrium constant for opening the protease-resistant folded state into
| (3) |
the cleavable state. The rate constant for intrinsic cleavage, kint, can be obtained using peptide models for the cleavage site.26 Thus, under EX2 conditions, K′op and the free energy for opening, 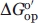 ( = −RT ln K′op), can be obtained for the least stable substructure.
( = −RT ln K′op), can be obtained for the least stable substructure.
Figure 2.
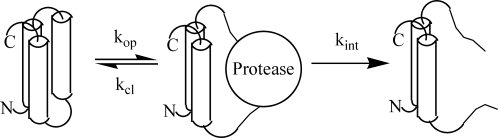
Kinetic scheme for the cleavage of a protein under conditions of limited proteolysis.
Characterization of initial protease cleavage sites
We have used two different proteases, proteinase K and trypsin, to define the least stable part of WT iso-1-cytc and the TM variant. Proteinase K is a broad specificity protease, and trypsin, cleaves on the C-terminal side of lysine and arginine residues.
Figure 3 compares cleavage patterns observed by MALDI-TOF mass spectrometry for WT iso-1-cytc and the TM variant subjected to proteinase K digestion for 1 min at 0°C. Two differences are immediately evident: the extent of cleavage is much greater for the TM variant and many additional fragments are observed for this variant. The +2 ion of the intact protein serves as a convenient internal standard for both WT iso-1-cytc (expected, 6326.1 m/z, observed, 6324.7 m/z in Fig. 3) and the TM variant (expected, 6298.6 m/z, observed, 6296 m/z in Fig. 3). For the WT protein, two additional peaks are observed (see Fig. 3) corresponding to a single cleavage site between Tyr 46 and Ser 47 (N-46 expected, 6055.7 m/z, observed, 6052.2 m/z; and 47-C, expected, 6615.6 m/z, observed, 6010 m/z). For the TM variant, multiple pairs of peaks are observed (see Fig. 3), however, the dominant peak also corresponds to cleavage between Tyr 46 and Ser 47 (N-46 expected, 6000.6 m/z, observed, 5999 m/z; and 47-C, expected, 6615.6 m/z, observed, 6614 m/z). The data also show that cleavage, albeit less favorable, occurs between Glu 44 and Gly 45, between Tyr 48 and Thr 49, and between Asp 50 and Ala 51 for this variant.
Figure 3.
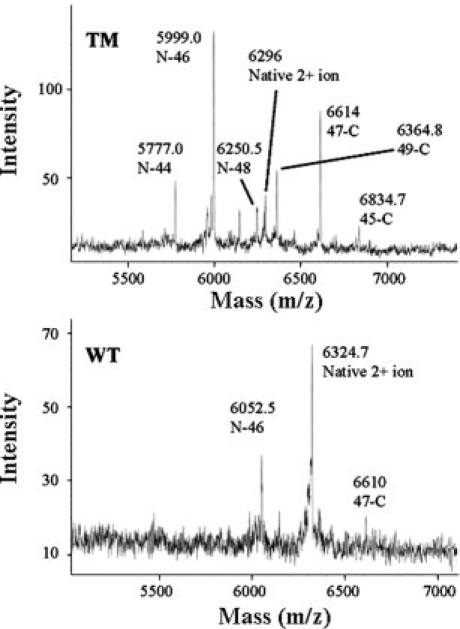
Initial proteinase K cleavage sites for WT iso-1-cytc and the TM variant. MALDI-TOF mass spectra are performed after digestion with proteinase K at a 1:500 protease:protein ratio for 1 min at 0°C. The major peaks are labeled N-X or Y-C, where N-X corresponds to a fragment from the N-terminus to sequence position X and Y-C corresponds to a fragment from sequence position Y to the C-terminus. For the major cleavage site between Tyr 46 and Ser 47, the expected masses are given in the text. For other fragments observed for the TM variant, the expected masses are: N-44, 5780.4; N-48, 6250.9; 49-C, 6365.3, 45-C, 6835.8 m/z. The unmarked peak just below N-48 is attributable to 51-C (expected, 6149.1 m/z) and the small unmarked peak at <6500 m/z is attributable to N-50 (expected, 6467.1 m/z).
The multiple cleavage sites observed for the TM variant could be due to other initial cleavage sites or they could be secondary cleavage sites after initial cleavage between Tyr 46 and Ser 47. Two arguments favor assignment as initial cleavage sites. First, the relative intensities of the N- and C-terminal fragments of each pair are similar. Second, though secondary cleavage could produce an N-44 fragment after initial cleavage between Tyr 46 and Ser 47, it could not generate the 45-C fragment which complements the N-44 fragment.
If the WT protein is digested with proteinase K at room temperature for 2 min, the major fragment pair is still due to cleavage between Tyr 46 and Ser 47. An additional peak is observed that is consistent with an N-44 fragment. No corresponding 45-C fragment is observed, so secondary cleavage cannot be ruled out in this case (Supporting Information Figure 1).
We also analyzed limited proteolysis by trypsin at room temperature for WT iso-1-cytc and the TM variant with mass spectrometry (Supporting Information Figure 2). Iso-1-cytc has 19 possible cleavage sites for trypsin. Only cleavage on the C-terminal side of Lys 54 and Lys 55 is observed.
It is apparent from the limited proteolysis data that both WT iso-1-cytc and the TM variant show initial cleavage in the same region of the protein (see Fig. 1). Removing the His 26/Glu 44 hydrogen bond in the TM variant facilitates cleavage in this region (see Fig. 3). The results with the TM variant also demonstrate that more facile cleavage in the infrared substructure leads to less specificity in the initial cleavage site (see Fig. 3).
Kinetics of limited proteolysis of WT and TM iso-1-cytc by proteinase K
We pursued quantitative kinetic analysis using proteinase K because it has a dominant cleavage site. We first evaluate kint (see Fig. 2) using a peptide, based on the sequence of the preferred Tyr 46/Ser 47 cleavage site and then use SDS-PAGE to monitor cleavage of WT iso-1-cytc and the TM variant as a function of time. All data were acquired at room temperature (22 ± 1°C).
The synthetic peptide used to measure kint has the sequence, Glu-Tyr-Ser-Tyr-Lys-NH2, and is labeled with a FRET pair at the N-terminus and at Lys. Low peptide concentration was used so that the kinetics of cleavage would be pseudo-first-order with the rate constant, kobs = (kcat/KM) [E]t, where [E]t is total enzyme concentration. As the peptide is cleaved, an increase in fluorescence is observed that fits well to a single exponential equation (Supporting Information Figure 3), consistent with pseudo-first-order conditions. We obtain kcat/KM = 6.0 ± 2.5 × 105M−1 s−1 for this cleavage site, which is similar to values of ∼1 × 106M−1 s−1 observed for other peptide substrates of proteinase K near room temperature.32 The kcat/KM allows evaluation of kint at any enzyme concentration as long as substrate concentration is well below KM.
Proteolysis of the WT and TM proteins was followed by SDS-PAGE (Supporting Information Figure 4). The native protein band is gradually converted to a well-resolved band approximately half the mass of the native protein, consistent with cleavage in the infrared substructure, as seen in the mass spectral data. An iso-1-cytc concentration of 0.5 mM was used which is comparable with the KM of ∼0.25 mM reported near room temperature for peptide substrates.32 Because only a small fraction of the protein is expected to be cleavable under EX2 conditions, the assumption that pseudo-first-order conditions apply (KM >> [S]) is valid as long as K′op is small (≤0.01). Typical data are shown in Figure 4(A). As expected for pseudo-first-order conditions, the data fit well to a single exponential function.
Figure 4.
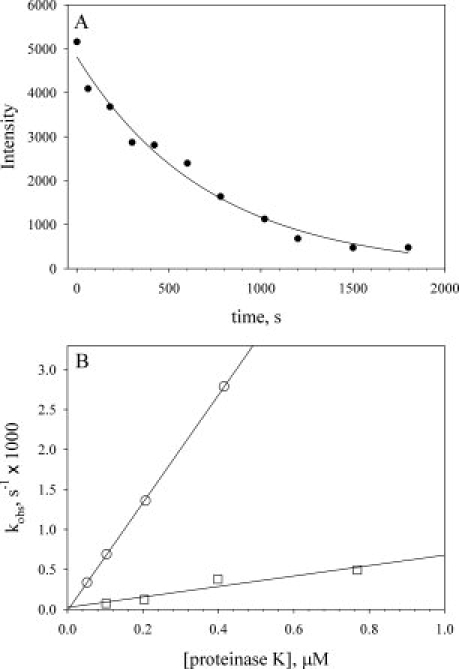
Kinetics of limited proteolysis by proteinase K from SDS-PAGE data for WT iso-1-cytc and the TM variant. A: Determination of kobs from SDS-PAGE data for the intensity of the native uncut band as a function of time (Supporting Information Figure 4). Data were collected at room temperature with a TM variant concentration of 0.5 mM and a proteinase K concentration of 200 nM. The solid curve is a fit of the data to a single exponential decay function. B: kobs versus proteinase K concentration for the TM (○) and WT (□) iso-1-cytochromes c at 22 ± 1°C. The solid curves are linear fits to the data yielding slope = K′op × (kcat/KM).
When substrate concentration is low, kint = (kcat/KM) × [E]t. Substituting this expression in Eq. (3), we obtain Eq. (4). Thus, under EX2 conditions, the rate of proteolysis should be linearly
| (4) |
dependent on the concentration of protease, allowing K′op × (kcat/KM) to be obtained from a plot of kobs versus proteinase K concentration.
Both WT iso-1-cytc and the TM variant give linear plots of kobs versus proteinase K concentration, confirming EX2 conditions [Fig. 4(B)]. The slope of the kobs versus proteinase K concentration plot is much shallower for WT iso-1-cytc compared with the TM variant, indicating that it is more resistant to proteolysis. The kobs versus proteinase K concentration plots do not level out at high-proteinase K concentration; therefore, EX1 conditions are not approached at high-proteinase K concentrations. Thus, the EX2 limit applies for both TM and WT iso-1-cytochromes c allowing both K′op and to be obtained from the slopes, (kcat/KM)K′op in Figure 4(B) (Table I). We note that the K′op values are both ≤0.01, validating the assumption that KM >> [S] made in data analysis.
Table I.
Kinetic and Thermodynamic Parameters Obtained from Limited Proteolysis of WT and TM Iso-1-Cytochromes c at 22 ± 1°C and pH 7.0
| Variant |
||
|---|---|---|
| WTa | TMb | |
| (kcat/KM) × Kop′ (M−1 s−1)c | 1000 ± 500 | 6600 ± 500 |
| Kop′d | 0.0017 ± 0.0011 | 0.011 ± 0.004 |
|
(kcal mol−1)e
|
3.8 ± 0.4 | 2.6 ± 0.3 |
Data from three independent trials.
Data from two independent trials.
Error in (kcat/KM) × Kop′ is taken as the larger of the standard deviation of the two (TM) or three (WT) independent trials or the average of the standard errors reported by SigmaPlot for the slopes of plots of kobs versus proteinase K concentration in the individual trials.
kcat/KM = 6 × 105M−1 s−1 was used to calculate Kop′. Reported error is from standard propagation of the error in (kcat/KM) × Kop′ and kcat/KM.
Reported error is from standard propagation of the error in Kop′.
Probing the red and blue substructures
GdnHCl denaturation shows that the TM variant is somewhat less stable than WT iso-1-cytc (Supporting Information Figure 5, Table II). Previously reported data for these same variants expressed in yeast (yWT and yTM) are also given in Table II. It is evident that lack of trimethylation of Lys 72 has only a small effect on the stability of WT iso-1-cytc and no effect on the stability of the TM variant. Comparison of the 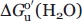 (unfolding of the blue substructure) in Table II with in Table I shows that is smaller for both WT iso-1-cytc and the TM variant. Thus, limited proteolysis is clearly detecting a portion of the protein that unfolds at lower free energy than necessary for global unfolding.
(unfolding of the blue substructure) in Table II with in Table I shows that is smaller for both WT iso-1-cytc and the TM variant. Thus, limited proteolysis is clearly detecting a portion of the protein that unfolds at lower free energy than necessary for global unfolding.
Table II.
Thermodynamic Parameters from gdnHCl Denaturation of WT and TM Iso-1-Cytochromes c at 22 ± 1°C and pH 7.5a
| Variant |
(H2O) (kcal mol−1) |
m (kcal mol−1M−1) | Cm (M) |
|---|---|---|---|
| WT | 5.05 ± 0.30 | 4.24 ± 0.13 | 1.19 ± 0.04 |
| yWTb | 5.77 ± 0.40 | 5.11 ± 0.36 | 1.13 ± 0.02 |
| TM | 4.00 ± 0.02 | 3.83 ± 0.10 | 1.04 ± 0.02 |
| yTMb | 3.95 ± 0.22 | 4.02 ± 0.19 | 0.98 ± 0.01 |
Reported errors are the standard deviation of three independent trials.
yWT and yTM indicate that these variants were expressed in yeast (Lys 72 is trimethylated). Data are from Ref. 15.
The alkaline conformational transition allows us to probe the effect of the H26N mutation on the red substructure of iso-1-cytc.11,22 The alkaline transition for WT iso-1-cytc and the TM variant (Supporting Information Figure 6) yield the apparent pKa, pKapp, and number of protons released in the conformational change, n (Table III). Comparison with previously reported data for WT (C102T) iso-1-cytc expressed in E. coli.27 shows that Ser versus Thr at position 102 has a negligible effect on the alkaline conformational transition (Table III). The H26N mutation causes the pKapp to decrease by 0.44 units, consistent with destabilization of the red substructure.
Table III.
Thermodynamic Parameters from the Alkaline Conformational Transition of WT and TM Iso-1-Cytochromes c at 22 ± 1°C and in 0.1M NaCl
All variants are expressed in E. coli, thus Lys 72 is not trimethylated.
n is the number of protons released in the alkaline conformational transition.
Data are from Ref.27. Data were fit assuming n = 1.
Discussion
The least stable substructure of iso-1-cytc
Both proteinase K and trypsin cut WT and TM iso-1-cytc in the Ω-loop corresponding to the infrared substructure of hh cytc (arrows in Fig. 1). Limited proteolysis of hh cytc,33 yielded proteinase K cleavage sites in this loop, as well. The exact cleavage sites are different from iso-1-cytc due to sequence differences in the horse protein (hh cytc has Phe 46 and Thr 47). The infrared loop also shows the least HX protection in yeast iso-1-cytc in aqueous solution34 and the lowest order parameters (S2) based on 15N NMR relaxation data.35 Thus, our data are consistent with data from other methods indicating that this segment of yeast iso-1-cytc is the most dynamic and least stable part of the protein.
It is noteworthy that cleavage sites closer to the N-terminal boundary of the substructure (Ser 40) are not observed (see Fig. 1), consistent with the larger unfolding necessary for cleavage by a protease than for NSHX.31 Similarly, Arg 38 is not observed as an initial trypsin cleavage site, indicating that this residue is not part of the infrared substructure in either WT iso-1-cytc or the less stable TM variant. Thus, the range of cleavage sites, residues 44–55, show that the extent of the infrared substructure observed for hh cytc (residues 40–57) is well preserved in the evolutionally distant yeast protein.
Spacing of substructures in yeast iso-1-cytc versus hh cytc
Limited proteolysis data yield = 3.8 ± 0.4 kcal mol−1 for the infrared substructure of WT iso-1-cytc (Table I). Because limited proteolysis requires larger conformational changes than NSHX,31 this likely corresponds to unfolding of the full infrared substructure and, at minimum, is a lower limit for this free energy. Comparison with HX data in aqueous solution for WT iso-1-cytc is not straightforward. The data were collected in D2O at pH* = 4.6.34 HX can be observed for 4 of 18 amides in the infrared substructure. All yield ∼ 2.7 kcal mol−1. This is lower than the we observe at pH 7, however, the infrared substructure of hh cytc is destabilized at pH 4.5 relative to pH 7.8
GdnHCl denaturation data gives a global stability (blue substructure) of 5.05 ± 0.30 kcal mol−1. Thus, the spacing between the lowest and highest energy substructure of iso-1-cytc is at most ∼1.2 kcal mol−1. For hh cytc, this spacing is ∼7.8 kcal mol−1 (12.8–5.0 kcal mol−1 at pD = 7).4,5,7,8 Thus, the stability range of the PUFs of iso-1-cytc is approximately the same as the 1 kcal mol−1 spacing between the infrared and red substructures of hh cytc at pH 7.5,8,9 The close spacing of the substructure stabilities should yield more cooperative unfolding of iso-1-cytc compared to hh cytc. Typically, m-values of 3.0 ± 0.1 kcal mol−1M−1 are observed for hh cytc at 25°C and neutral pH.16,17,36 The denaturant m-value observed for the blue substructure of hh cytc by NSHX is 4.7 kcal mol−1M−1.8 Because m-values are depressed by the presence of intermediates,37 the optically monitored gdnHCl unfolding of hh cytc is clearly not fully cooperative. The m-value observed here for global unfolding of WT iso-1-cytc expressed in E. coli is 4.24 ± 0.13 kcal mol−1M−1. For yWT iso-1-cytc, the m-value ranges from 4.6 to 5.1 kcal mol−1M−1,15,36 very close to the m-value observed for the blue substructure of hh cytc. Thus, the tight free energy spacing of the substructures of iso-1-cytc versus hh cytc leads to more cooperative equilibrium unfolding.
Is the distribution of stabilizing energy conserved across evolution?
Studies on RNase H show that the substructure energies are compressed in the protein from E. coli compared with the protein from Thermus thermophilus.13 The substructures of E. coli RNase H were destabilized in proportion to the decrease in global stability relative to T. thermophilus RNase H, suggesting that the distribution of stabilizing energy in a protein is conserved across evolution. If conservation of the distribution of stabilizing energy is rigorously true, then the m-values and of the hh cytc substructures can be used to predict the stabilities of the substructures of c-type cytochromes with different global stabilities (for the infrared substructure of yeast, 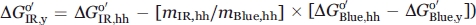 . Thus, for iso-1-cytc one would predict
. Thus, for iso-1-cytc one would predict 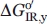 to be ∼2.5 kcal mol−1. Pseudomonas aeruginosa (PA) cytochrome c551, a more distant homolog of hh cytc than iso-1-cytc with intermediate stability (∼8 kcal mol−1 at pH 6) and some differences in the details of its substructures,38 would be predicted to have a least stable substructure stability of ∼3.5 kcal mol−1. The observed stability of the least stable substructure of PA cytc551 is ∼5.3 kcal mol−1.38
to be ∼2.5 kcal mol−1. Pseudomonas aeruginosa (PA) cytochrome c551, a more distant homolog of hh cytc than iso-1-cytc with intermediate stability (∼8 kcal mol−1 at pH 6) and some differences in the details of its substructures,38 would be predicted to have a least stable substructure stability of ∼3.5 kcal mol−1. The observed stability of the least stable substructure of PA cytc551 is ∼5.3 kcal mol−1.38
Interestingly, the least stable E-helix of the T. thermophilus RNase H is actually more stable in the E. coli protein. Thus, it appears that conserving the distribution of stabilizing energy across evolution is balanced against other factors such as a minimum threshold for the stability39 for the least stable substructure which would be important to protect against susceptibility of proteins to aggregation and proteolysis.
Implications of substructure dynamic range for the folding mechanism of a protein
For hh cytc, the PUFs are well separated and thus can be discretely populated. For iso-1-cytc, the lowest and highest free energy PUFs are separated by, at most, 1.2 kcal mol−1. Therefore, the probabilities of populating each PUF of iso-1-cytc differ over a range of only a factor of 10, meaning that discrete occupancy of each PUF cannot occur. Yet, our previous22 and current data (see next session of Discussion) support sequential unfolding of iso-1-cytc substructures. However, our methods do not probe the green and the yellow substructures. The NMR-detected hydrogen exchange data available for iso-1-cytc in buffer indicate that the stabilities of the blue and green substructures are similar.34 Thus, it is reasonable to question whether the folding pathway is as well defined for iso-1-cytc as for hh cytc. Pulsed hydrogen-deuterium exchange kinetic experiments are consistent with most hh cytc molecules forming an early intermediate that has structured N- and C-terminal helices,40 as expected for formation of the blue substructure. In contrast, recent work on the folding kinetics of yeast iso-1-cytc indicates that an intermediate corresponding to the blue substructure is no more that 25% populated early in folding.41 Similarly, for PA cytc551, which is intermediate in stability between hh cytc and iso-1-cytc, kinetic data indicate the presence of parallel pathways, that do not all involve formation of contacts between the N- and C-terminal helices.42–44 Thus, it is likely that the folding pathway of iso-1-cytc with its very narrow band of PUF stabilities, as well as those of other less stable c-type cytochromes, have more optional branching points than for hh cytc.10
Effect of the H26N mutation on substructure stability
Comparison of of the infrared substructure for WT iso-1-cytc and the TM variant shows that removal of the His 26/Glu 44 intersubstructural hydrogen bond decreases the stability of the infrared substructure by ∼1.2 kcal mol−1 (Table I). The effect of this mutation on overall stability is ∼1.1 kcal mol−1 (Table II). Thus, the loss of stability to the infrared and blue substructures is similar, consistent with sequential unfolding of these substructures, as observed for hh cytc.
On the other hand, the H26N mutation decreases the pKapp of the alkaline transition by 0.44 units which corresponds to a decrease in stability of ∼0.6 kcal mol−1 in the red substructure. Thus, destabilization of the infrared substructure by the H26N mutation does not impact the red substructure fully, consistent with the observation that the red and infrared substructures unfold independently for hh cytc.8,9 This differential effect on stability is even more pronounced for a H26Y variant of iso-1-cytc (C102T background).45 The 0.47 unit drop in the pKapp for the alkaline transition (∼0.65 kcal mol−1) is much less than the ∼1.8 kcal mol−1 decrease in the global stability caused by this mutation. Data on a H26V variant of rat cytc lead to a similar conclusion.29 This mutation decreases the global stability of the rat protein by nearly 4 kcal mol−1, but only decreases pKapp for the alkaline transition by 0.9 units (∼1.2 kcal mol−1). Thus, data for different substitutions for His 26 in both rat and yeast cytochromes c all support independent unfolding of the infrared and red substructures.
Materials and Methods
Preparation of proteins
Expression of WT iso-1-cytc and the TM variant was done from E. coli BL21(DE3) host cells. WT iso-1-cytc was expressed from the pBTR1 vector,46 which coexpresses the heme lyase from S. cerevisiae allowing attachment of heme in the cytoplasm of E. coli.27 The TM variant (H26N, H33N, H39Q, C102S) was expressed from the pRbs_BTR1 vector.47 This vector replaces the ribosomal binding site from the T7 phage gene 10 leader sequence in the pBTR1 vector with an optimized ribosomal binding site to enhance cytc expression.48 Methods for growth and isolation of iso-1-cytc from bacteria have been described previously.49
HPLC-purified protein was oxidized with potassium ferricyanide (5 mg mg−1 of protein) for 1 h at 4°C. Ferricyanide was removed by gel filtration (sephadex G-25) using the experimental buffer (40 mM NaCl, 20 mM Tris, pH 7.0) and concentrated to 0.5 mM.
Protein stability measurements
GdnHCl denaturation of WT iso-1-cytc and the TM variant monitored by CD was carried out at pH 7.5 (20 mM Tris, 40 mM NaCl) and 25°C, as previously described.25 The data were fit assuming a linear free energy relationship to extract the free energy of unfolding in the absence of denaturant, , and the m-value, as previously described.28
Alkaline conformational transition
Iso-1-cytc was oxidized as described earlier except the G-25 column was equilibrated and run with 0.2M NaCl. The protein concentration was adjusted to 400 μM in 0.2M NaCl (2× protein solution) and the starting sample was made by mixing 500 μL of 2× protein solution and 500 μL of deionized water with a stir bar in a semimicro quartz cuvette suitable for use with a magnetic stirrer (Hellma Cat. #: 109.004F). The pH was measured with an Accumet calomel combination microelectrode (Fisher Cat. #: 13-620-95). The spectrum was measured from 750 to 550 nm with a Beckman DU 800 UV–Vis spectrometer. The pH was adjusted by adding 1 μL of 2× protein and 1 μL of an acid or base solution of appropriate concentration. Titrations were carried out at room temperature (22 ± 1°C). Data plotted at 695 nm were fit to a modified form of the Henderson–Hasselbalch equation to obtain pKapp and the number of protons, n, released.20
Proteolysis conditions
Proteinase K and trypsin concentrations were adjusted to 3 mg mL−1 (E280 1% = 14.2,50 and 14.4, respectively) in a 40 mM NaCl, 20 mM Tris, pH 7.0 buffer. Digestions were performed at room temperature (22 ± 1°C) and on ice. The enzyme was added to the protein reaction vessel and mixed with a Pipetman for 5 s. Aliquots of 10 μL of the reaction mixture were removed at various times, ranging from 15 s to 8 h, the enzymes deactivated with 5 μL of 10 mM PMSF and the aliquots stored at −20°C.
Mass spectrometry
MALDI-TOF mass spectrometry (Bruker Reflex IV MALDI-TOF/MS) was used to analyze proteolytic cleavage products using a sinapic acid matrix. The mass accuracy of the Bruker Reflex IV is about 10 Da for a protein with a mass of 10,000 Da. Expected masses of potential cleavage products were calculated at http://ca.expasy.org/tools/peptide-mass.html.
SDS-PAGE analysis
Three micrograms of the protein sample were added to a tricine loading buffer (Bio-Rad, 200 mM Tris-HCl, pH 6.8, 2% SDS, 40% glycerol, 0.04% Coomassie blue G-250), denatured at 95°C for 5 min and centrifuged to remove particulates. Samples were electrophoresed on a 16.5% Tris/Tricine preformed gel (Bio-Rad) at 200 V for 35–40 min using 100 mM Tris, 100 mM tricine, 0.1% SDS, pH 8.3 running buffer (Bio-Rad). After fixing, gels were stained with Coomassie blue and then destained.
Gels were digitized with an HP scanner (Scanjet 3500C). The intensities of the protein bands were quantified using ImageJ software (http://rsbweb.nih. gov/ij/). Gel samples with masses ranging from 0.1 to 3 μg of undigested iso-1-cytochrome c were found to be linear across the entire mass range. For digestion with proteinase K, the intensity change of the native band versus time was fit to a single exponential decay equation.
Intrinsic cleavage rate
A custom peptide, 5-FAM-Glu-Tyr-Ser-Tyr-Lys(QXL 520)-CONH2 (AnaSpec, San Jose, CA, 5-FAM is 5-carboxyfluorescein, QXL™-520 is a quencher with maximal absorbance near 520 nm) was used to determine the intrinsic cleavage properties of proteinase K. The peptide concentration was measured at pH 9.0 using the extinction coefficient of 79,000 M−1 cm−1 at 520 nm for the 5-FAM/QXL-520 pair (as per AnaSpec).
Enzyme reactions were carried out at 200 nM peptide and ∼10 nM proteinase K at 22 ± 1°C in 20 mM Tris, pH 7.0, 40 mM NaCl. Fluorescence as a function of time was measured with a SPEX Fluorolog II spectrofluorometer (ex: 490 nm, em: 520 nm). The pseudo-first-order rate constant, kobs, was determined by fitting the fluorescence change to a single exponential rise to maximum equation.
Conclusions
Limited proteolysis of iso-1-cytc by proteinase K and trypsin demonstrates that the infrared substructure is preserved evolutionally as the least stable substructure between yeast iso-1-cytc and hh cytc. Additionally, the dimension of the infrared substructure in iso-1-cytc appears to be robust to mutation. The for the infrared substructure demonstrates that the PUFs of iso-1-cytc occur in a much tighter band than for hh cytc, consistent with the equilibrium folding of the yeast protein being more cooperative. This conclusion is validated by the denaturant m-values for global unfolding of the horse and yeast proteins. Our results add to the available data indicating that the stability range of substructures in less stable proteins is compressed and suggests that folding pathways in less stable proteins are likely to have more optional branch points. The H26N mutation destabilizes the infrared and blue substructures by the same amount consistent with sequential unfolding of these two substructures of iso-1-cytc. However, the red substructure is destabilized less by this mutation indicating that the infrared and red substructures unfold independently. Thus, the substructure architecture of cytc, while compressed energetically for iso-1-cytc, is largely conserved evolutionally between horse and yeast cytochromes c.
Acknowledgments
The authors thank Melisa Cherney and Swati Bandi for their helpful discussions in the preparation of this manuscript.
Glossary
Abbreviations:
free energy of unfolding in the absence of denaturant
- CD
circular dichroism
- cytc
cytochrome c
- gdnHCl
guanidine-HCl
- HX
hydrogen deuterium exchange
-
K′op,
equilibrium constant and free energy, respectively, of opening to a protease cleavable state
- m-value
rate of change of free energy of unfolding as a function of denaturant concentration
- NSHX
native-state hydrogen deuterium exchange
- pH*
uncorrected pH meter reading in D2O
- PUF
partially unfolded form
- WT
wild type.
References
- 1.Jackson SE. How do small single-domain proteins fold? Fold Des. 1992;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 2.Wildes D, Marqusee S. Hydrogen exchange strategies applied to energetics of intermediate processes in protein folding. Methods Enzymol. 2004;380:328–349. doi: 10.1016/S0076-6879(04)80015-6. [DOI] [PubMed] [Google Scholar]
- 3.Englander SW, Mayne L, Rumbley JN. Submolecular cooperativity produces multi-state protein unfolding and refolding. Biophys Chem. 2002;101:57–65. doi: 10.1016/s0301-4622(02)00190-4. [DOI] [PubMed] [Google Scholar]
- 4.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishna MMG, Lin J, Rumbley JN, Englander SW. Cooperative omega loops in cytochrome c: role in folding and function. J Mol Biol. 2003;331:29–36. doi: 10.1016/s0022-2836(03)00697-1. [DOI] [PubMed] [Google Scholar]
- 6.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 7.Maity H, Maiti M, Englander SW. How cytochrome c folds and why: submolecular folding units and their stepwise sequential stabilization. J Mol Biol. 2004;343:223–233. doi: 10.1016/j.jmb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Krishna MMG, Maity H, Rumbley JN, Lin Y, Englander SW. Order of steps in the cytochrome c folding pathway: evidence for a sequential stabilization mechanism. J Mol Biol. 2006;359:1410–1419. doi: 10.1016/j.jmb.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 9.Krishna MMG, Maity H, Rumbley JN, Englander SW. Branching in the sequential folding pathway of cytochrome c. Protein Sci. 2007;16:1946–1956. doi: 10.1110/ps.072922307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoang L, Maity H, Krishna MMG, Lin Y, Englander SW. Folding units govern the cytochrome c alkaline transition. J Mol Biol. 2003;331:37–43. doi: 10.1016/s0022-2836(03)00698-3. [DOI] [PubMed] [Google Scholar]
- 11.Maity H, Rumbley JN, Englander SW. Functional role of a foldon—an Ω-loop folding controls the alkaline transition of ferricytochrome c. Proteins. 2006;63:349–355. doi: 10.1002/prot.20757. [DOI] [PubMed] [Google Scholar]
- 12.Berghuis AM, Brayer GD. Oxidation state-dependent conformational changes in cytochrome c. J Mol Biol. 1992;223:959–976. doi: 10.1016/0022-2836(92)90255-i. [DOI] [PubMed] [Google Scholar]
- 13.Hollien J, Marqusee S. Structural distribution of stability in a thermophilic enzyme. Proc Natl Acad Sci USA. 1999;96:13674–13678. doi: 10.1073/pnas.96.24.13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Godbole S, Bowler BE. Effect of pH on formation of a nativelike intermediate on the unfolding pathway of a Lys 73→His variant of yeast iso-1-cytochrome c. Biochemistry. 1999;38:487–495. doi: 10.1021/bi981698k. [DOI] [PubMed] [Google Scholar]
- 15.Godbole S, Hammack B, Bowler BE. Measuring denatured state energetics: deviations from random coil behavior and implications for the folding of iso-1-cytochrome c. J Mol Biol. 2000;296:217–228. doi: 10.1006/jmbi.1999.3454. [DOI] [PubMed] [Google Scholar]
- 16.Knapp JA, Pace CN. Guanidine hydrochloride and acid denaturation of horse, cow, and Candida krusei cytochrome c. Biochemistry. 1974;13:1289–1294. doi: 10.1021/bi00703a036. [DOI] [PubMed] [Google Scholar]
- 17.McLendon G, Smith M. Equilibrium and kinetic studies of protein folding. J Biol Chem. 1978;253:4004–4008. [PubMed] [Google Scholar]
- 18.Dickerson RE, Timkovich R. Cytochromes c. In: Boyer PD, editor. 3rd ed. Vol. 11. New York: Academic Press; 1975. pp. 397–547. The enzymes. [Google Scholar]
- 19.Godbole S, Dong A, Garbin K, Bowler BE. A lysine 73→histidine variant of yeast iso-1-cytochrome c: evidence for a native-like intermediate on the unfolding pathway and implications for m-value effects. Biochemistry. 1997;36:119–126. doi: 10.1021/bi961915m. [DOI] [PubMed] [Google Scholar]
- 20.Nelson CJ, Bowler BE. pH dependence of formation of a partially unfolded state of a Lys 73 → His variant of iso-1-cytochrome c: implications for the alkaline conformational transition of cytochrome c. Biochemistry. 2000;39:13584–13594. doi: 10.1021/bi0017778. [DOI] [PubMed] [Google Scholar]
- 21.Nelson CJ, LaConte MJ, Bowler BE. Direct detection of heat and cold denaturation for partial unfolding of a protein. J Am Chem Soc. 2001;123:7453–7454. doi: 10.1021/ja016144a. [DOI] [PubMed] [Google Scholar]
- 22.Kristinsson R, Bowler BE. Communication of stabilizing energy between substructures of a protein. Biochemistry. 2005;44:2349–2359. doi: 10.1021/bi048141r. [DOI] [PubMed] [Google Scholar]
- 23.Bai Y, Englander SW. Future directions in folding: the multi-state nature of protein structure. Proteins. 1996;24:145–151. doi: 10.1002/(SICI)1097-0134(199602)24:2<145::AID-PROT1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 24.Fuentes EJ, Wand AJ. Local dynamics and stability of apocytochrome b562 examined by hydrogen exchange. Biochemistry. 1998;37:3687–3698. doi: 10.1021/bi972579s. [DOI] [PubMed] [Google Scholar]
- 25.Fontana A, Polverino de Laureto P, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 26.Park C, Marqusee S. Probing the high energy states in proteins by proteolysis. J Mol Biol. 2004;343:1467–1476. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 27.Pollock WBR, Rosell FI, Twitchett MB, Dumont ME, Mauk AG. Bacterial expression of a mitochondrial cytochrome c. Trimethylation of lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition. Biochemistry. 1998;37:6124–6131. doi: 10.1021/bi972188d. [DOI] [PubMed] [Google Scholar]
- 28.Hammack BN, Smith CR, Bowler BE. Denatured state thermodynamics: residual structure, chain stiffness and scaling factors. J Mol Biol. 2001;311:1091–1104. doi: 10.1006/jmbi.2001.4909. [DOI] [PubMed] [Google Scholar]
- 29.Qin W, Sanishvili R, Plotkin B, Schejter A, Margoliash E. The role of histidines 26 and 33 in the structural stabilization of cytochrome c. Biochim Biophys Acta. 1995;1252:87–94. doi: 10.1016/0167-4838(95)00124-d. [DOI] [PubMed] [Google Scholar]
- 30.Fetrow JS, Dreher U, Wiland DJ, Schaak DL, Boose TL. Mutagenesis of histidine 26 demonstrates the importance of loop-loop and loop-protein interactions for the function of iso-1-cytochrome c. Protein Sci. 1998;7:994–1005. doi: 10.1002/pro.5560070417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubbard SJ, Eisenmenger F, Thornton JM. Modeling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci. 1994;3:757–768. doi: 10.1002/pro.5560030505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kristjánsson MM, Magnusson ÓT, Gudmunsson HM, Alfredsson GÁ, Matsuzawa H. Properties of a subtilisin-like proteinase from a psychrotrophic Vibrio species. Comparison with proteinase K and aqualysin I. Eur J Biochem. 1999;260:752–760. doi: 10.1046/j.1432-1327.1999.00205.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Chen RX, Kallenbach NR. Proteolysis as a probe of thermal unfolding of cytochrome c. Proteins. 1998;30:435–44. [PubMed] [Google Scholar]
- 34.Baxter S, Fetrow J. Hydrogen exchange behavior of [U-15N]-labeled oxidized and reduced iso-1-cytochrome c. Biochemistry. 1999;38:4493–4503. doi: 10.1021/bi982742z. [DOI] [PubMed] [Google Scholar]
- 35.Baxter S, Fetrow J. Assignment of 15N chemical shifts and 15N relaxation measurements for oxidized and reduced iso-1-cytochrome c. Biochemistry. 1999;38:4480–4492. doi: 10.1021/bi9827417. [DOI] [PubMed] [Google Scholar]
- 36.Betz SF, Pielak GJ. Introduction of a disulfide bond in cytochrome c stabilizes a compact denatured state. Biochemistry. 1992;31:12337–12344. doi: 10.1021/bi00164a007. [DOI] [PubMed] [Google Scholar]
- 37.Pace CN. The stability of globular proteins. CRC Crit Rev Biochem. 1975;3:1–43. doi: 10.3109/10409237509102551. [DOI] [PubMed] [Google Scholar]
- 38.Michel LV, Bren KL. Submolecular unfolding units of Pseudomonas aeruginosa cytochrome c-551. J Biol Inorg Chem. 2008;13:837–845. doi: 10.1007/s00775-008-0370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pielak GJ, Auld DS, Beasley JR, Betz SF, Cohen DS, Doyle DF, Finger SA, Fredericks ZL, Hilgen-Willis S, Saunders AJ, Trojak SK. Protein thermal denaturation, side-chain models, and evolution: amino acid substitutions at a conserved helix-helix interface. Biochemistry. 1995;34:3268–3276. doi: 10.1021/bi00010a017. [DOI] [PubMed] [Google Scholar]
- 40.Roder H, Elöve GA, Englander SW. Structural characterization of folding intermediates in cytochrome c by H-exchange labeling and protein NMR. Nature. 1988;335:700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pletneva EV, Gray HB, Winkler JR. Snapshots of cytochrome c folding. Proc Natl Acad Sci USA. 2005;102:18397–18402. doi: 10.1073/pnas.0509076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gianni S, Travaglini-Allocatelli C, Cutruzzolà F, Brunori M, Shastry MCR, Roder H. Parallel pathways in cytochrome c551 folding. J Mol Biol. 2003;330:1145–1152. doi: 10.1016/s0022-2836(03)00689-2. [DOI] [PubMed] [Google Scholar]
- 43.Travaglini-Allocatelli C, Gianni S, Brunori M. A common folding mechanism in the cytochrome c family. Trends Biochem Sci. 2004;29:535–541. doi: 10.1016/j.tibs.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Borgia A, Bonivento D, Travaglini-Allocatelli C, Di Matteo A, Brunori M. Unveiling a hidden folding intermediate in c-type cytochomes by protein engineering. J Biol Chem. 2006;281:9331–9336. doi: 10.1074/jbc.M512127200. [DOI] [PubMed] [Google Scholar]
- 45.Sinibaldi F, Piro MC, Howes BD, Smulevich G, Ascoli F, Santucci R. Rupture of the hydrogen bond linking two Ω-loops induces the molten globule state at neutral pH in cytochrome c. Biochemistry. 2003;42:7604–7610. doi: 10.1021/bi034132r. [DOI] [PubMed] [Google Scholar]
- 46.Rosell FI, Mauk AG. Spectroscopic properties of a mitochondrial cytochrome c with a single thioether bond to the heme prosthetic group. Biochemistry. 2002;41:7811–7818. doi: 10.1021/bi016060e. [DOI] [PubMed] [Google Scholar]
- 47.Kurchan EW. 2005. Kinetics and thermodynamics in the denatured state of iso-1-cytochrome c, Ph.D. Thesis, University of Denver, Denver, CO.
- 48.Rumbley JN, Hoang L, Englander SW. Recombinant equine cytochrome c in Escherichia coli: high-level expression, characterization, and folding and assembly mutants. Biochemistry. 2002;41:13894–13901. doi: 10.1021/bi026543y. [DOI] [PubMed] [Google Scholar]
- 49.Kurchan E, Roder H, Bowler BE. Kinetics of loop formation and breakage in the denatured state of iso-1-cytochrome c. J Mol Biol. 2005;353:730–743. doi: 10.1016/j.jmb.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 50.Ebeling W, Hennrich N, Klockow M, Metz H, Orth HD, Lang H. Proteinase K from Tritirachium album limber. Eur J Biochem. 1974;47:91–97. doi: 10.1111/j.1432-1033.1974.tb03671.x. [DOI] [PubMed] [Google Scholar]