

Abstract
Irreversibility of thermally denatured proteins due to aggregation limits thermodynamic characterization of proteins and also confounds the identification of thermostable mutants in protein populations. Identification of mutations that prevent the aggregation of unfolded proteins provides insights into folding pathways. In a lipase from Bacillus subtilis, evolved by directed evolution procedures, the irreversibility due to temperature-mediated aggregation was completely prevented by a single mutation, M137P. Though the parent and the mutants unfold completely on heating, mutants having substitutions M137P, along with M134E and S163P, completely or partially prevent the formation of aggregation-prone intermediate(s) at 75°C. The three mutants show only a marginal increase in free energy of unfolding (ΔGH2O), however, the profiles of the residual activity with temperature shows remarkable shift to higher temperature compared to parent. The intermediate(s) were characterized by enhanced binding of bis-ANS, a probe to titrate surface hydrophobicity, aggregation profiles and by estimation of soluble protein. Inclusion of salt in the refolding conditions prevents the reversibility of mutant having charge substitution, while the reversibility of mutant with the introduction of proline was unaffected, indicating the role of charge mediated interaction in M134E in preventing aggregation. Partial prevention of thermal aggregation in wild-type lipase with single substitution, M137P, incorporated by site-directed mutagenesis, suggests that the affect of M137P is independent of the intrinsic thermostability of lipase. Various effects of the mutations suggest their role is in prevention of the formation of aggregation prone intermediate(s). These mutations, describe yet another strategy to enhance the thermotolerance of proteins, where their influence is observed only on the denatured ensemble.
Keywords: lipase, site-saturation mutagenesis, thermal denaturation, aggregation, intermediate, bis-ANS binding, denatured ensemble
Introduction
Thermally denatured proteins are poor solutes in water. Exposure of occluded hydrophobic amino acids in proteins, upon denaturation, places them in incompatible solvent, water, leading to aggregation and precipitation.1–6 It is well documented that denatured state of proteins are not random coil without side group interactions but is an ensemble of conformers.7,8 Recent measurements using NMR have revealed persistent structures in thermally denatured proteins.9,10 Various measurements indicated that thermally unfolded proteins are 20% more compact than the chemically denatured proteins.11,12 These structures are partly driven by hydrophobic clustering and to some extent by hydrogen bonds. Aggregation of thermally denatured proteins due to existence of aggregation prone residual structures prevents measurements of thermodynamic parameters on these proteins.9 Denaturation of proteins, either by temperature or chaotropes, tremendously expands the conformational space on withdrawal of denaturing forces, leads to online refolding to native structure or off line refolding to aggregates.13 Increasingly, it is realized that for detailed structural and energetic characterization, denatured states of proteins has become essential to completely describe the free energy changes during protein folding.1 Stability of a protein, defined by free energy difference between unfolded and folded states, ΔG, could be enhanced by either lowering the free energy of the folded state or by increasing the free energy of the unfolded state. A number of mutations have been reported, including replacement of amino acids with proline or charge amino acids, which affect the ΔG by altering the denatured state ensemble.12,14–16
When estimated by activity measurements, thermostability of enzymes is operationally defined as the activity remaining at ambient temperature (residual activity) after exposure to high temperatures. Such a description does not accurately reflect protein thermostability because it is biased toward efficient refolders. Many proteins or protein variants would be identified as unstable by this criterion, even though they may show increased ΔG compared to its reference protein. Studies on thermally denatured proteins remains technically challenging because of the tendency of thermally unfolded proteins to aggregate.9 Hence, data on thermodynamic and kinetic parameters of proteins and role of mutations are conveniently studied using chemical denaturants viz. urea or guanidinium chloride. Strategies to identify mutations that influence thermal denaturation pathways are important.1 Also, biotechnological processes based on enzymes involving temperature cycling would be largely benefited if the enzyme recovery is complete after heat treatments.
Methods of directed (or in vitro) evolution were proved to be very successful in altering those properties of proteins that are determined by numerous weak interactions, viz. thermostability, substrate binding, allostery etc.17–21 Using directed evolution methods, thermostability of proteins has been improved cumulatively, over several generations, by addition of hydrogen or ionic bonds or by replacing surface non-polar amino acids.22–24 Reversibility of thermal denaturation as a property has not been targeted in any mutagenesis programs that attempt to alter the protein properties. As large number of conformers are present in denatured ensemble, native structure based approaches are not applicable to identify amino acid positions that may have influence on the denatured ensemble or refolding pathway. Methods of directed evolution and screening procedures offer a scope to identify such rare mutations by judiciously screening large populations. From our directed evolution studies on a lipase from Bacillus subtilis, we have identified few amino acid positions that did not show significant increase in free energy of unfolding, but show excellent recovery in activity compared to its parent after thermal denaturation.25,26 By performing saturation mutagenesis at three key locations, we have identified a mutation that completely blocks the aggregation upon thermal denaturation thus reverses the otherwise irreversible unfolding to a significant extent.
Results and Discussion
A variant of Bacillus subtilis lipase, 4D3, evolved by in vitro evolution methods, having nine mutations as compared to wild-type protein, was used to investigate the influence of mutations on the reversibility of thermal unfolding of the protein.26 During the course of the in vitro evolution over several generations, three positions were identified viz. M134, M137, and S163, which may have a positive contribution to protein stability. The identified mutations at these positions, all on the surface, contributed very little to the thermostability of the protein but have occurred frequently in population to warrant their importance.26 Because error-prone PCR method used in earlier studies did not sample all the possible amino acid substitutions, site-saturation mutagenesis was performed on three locations to investigate the influence of all substitutions on thermostability. The mutant population was generated by overlap-extension PCR using mutagenic primers encoding target position by degenerate codon NNK. The mutagenic library so obtained was used for periplasmic expression of protein in E. coli. Screening was performed using high-throughput assay format in 96-well plates, upon thermal challenge at elevated temperature. Approximately 1500 clones, 500 clones for each position, were screened for thermostability after exposure to 70°C for 20 min, a temperature at which parent 4D3 show less than 20% residual activity. After screening, clones which were displaying significantly higher residual activities compared with parent (>50%) were further tested in tube assays. Thermal inactivation profiles of mutants were made by monitoring residual activities in crude culture supernatant upon incubation over a range of temperature. The temperature at which 50% of activity is lost (T50) was used as thermostability index. At position 134, substitution of methionine (ATG) with glutamate (GAG) increases T50 by 5.8 degrees, while at position 137, replacement of methionine (ATG) with proline (CCT) increases T50 by 6.8 degrees. At position 163, substitution of serine (AGC) with proline (CCG) increases T50 by 1.8 degrees. Thus, the best substitutions identified at these three positions are M134E, M137P, and S163P which are present in mutants 5-A, 5-B, and 5-D, respectively.
All the three lipase mutants along with parent 4D3 were cloned in expression vector and protein was purified using standard procedures. The far- and near-UV CD profiles, intrinsic tryptophan fluorescence, and binding of the fluorescent probe bis-ANS were all identical in the parent and the mutants indicating unaltered secondary and tertiary structure as well as surface hydrophobicity (Supporting Information).
Thermostability of lipase variants was tested by monitoring temperature induced unfolding using CD [Fig. 1(A)]. All the four proteins show single melting transition with increase in apparent melting temperature (Tmapp) by 1–3°C, compared with parent (Table I). The maximum increase in Tmapp was observed with mutant 5-B, having M137P mutation, which is 2.9 degrees higher than parent 4D3. Conformational stability of lipase mutants was investigated by monitoring equilibrium unfolding in the presence of GdmCl using fluorescence and CD spectroscopy. The shift in tryptophan fluorescence emission (λmax) and ellipticity at 222 nm (θ222) were monitored with increasing concentration of denaturant [Fig. 1(B)]. The denaturation curves were sigmoidal indicating absence of any populating unfolding intermediates. Free energy of unfolding in the absence of denaturant, ΔGH2O, was calculated by the procedure as described by Pace et al.27A marginal decrease in ΔGH2Ocompared to the parent was observed in case of 5-A, while 5-B and 5-D show an increase in ΔGH2Ocompared to parent (Table I). Identical results were obtained when observations were made using both far-UV CD and intrinsic tryptophan fluorescence of the protein, indicating that the denaturation transition behaves in a two-state manner (data not shown). Linear extrapolation method was used to calculate the m-values from the denaturation profiles, which does not show any significant change between the parent and the mutants (Table I). m-values, which are indicative of the accessible surface area of the protein, remains largely unaltered in the mutants. The invariant near- and far- UV CD spectra of the mutants and parent also indicate (vide supra) that the three mutants have unaltered secondary and tertiary conformation.
Figure 1.
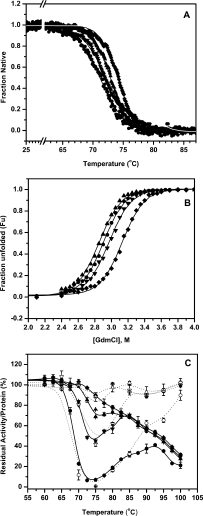
(A) Thermal unfolding profiles of lipase mutants 4D3 (•), 5-A (▴), 5-B (♦), and 5-D (▾) as monitored by circular dichroism. (B) Equilibrium unfolding of lipase mutants 4D3 (•), 5-A (▴), 5-B (♦), and 5-D (▾) in the presence of GdmCl monitored using CD. Fraction unfolded was calculated based on change in ellipticity at 222 nm (θ222) with GdmCl concentration. (Identical transitions were obtained when fraction unfolded was calculated using shift in tryptophan fluorescence emission, λmax, not shown here for the sake of clarity). (C) Thermal inactivation profiles of lipase mutants along with the fraction of protein recovered upon thermal challenge. Residual activity and residual protein in solution are shown in solid and open symbols for mutants 4D3 (•, ○), 5-A (▴, ▵), 5-B (♦, ⋄), and 5-D (▾, ▿), respectively.
Table I.
Thermostability and Equilibrium Unfolding Parameters of Lipase Mutants
|
t(1/2) (min)b |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutant | Mutationa | 55 °C | 75 °C | 85 °C | T(50)c (°C) | Tm(app)d (°C) | ΔG(H2O)e (kcal/mol) | mf (kcal/mol) | ΔG/m (M) |
| wild-type | — | 2.8 | — | — | 53.3 | 56.0 | 11.39 ± 0.72 | 2.61±0.16 | 2.05 |
| 4D3 | — | — | 4.4 | 15.2 | 68.0 | 71.2 | 13.75 ± 0.40 | 4.74 ± 0.14 | 2.90 |
| 5-A | M134E | — | 38.8 | 46.9 | 93.0 | 72.9 | 14.41 ± 0.35 | 5.04 ± 0.12 | 2.86 |
| 5-B | M137P | — | 101.2 | 54.3 | 93.0 | 74.1 | 14.73 ± 0.37 | 4.71 ± 0.37 | 3.13 |
| 5-D | S163P | — | 22.2 | 49.9 | 72.0 | 72.2 | 13.50 ± 0.43 | 4.55 ± 0.15 | 2.96 |
Given mutations are in background of 4D3, which has nine mutations compared to wild-type protein.24
Half-life of thermal inactivation.
Temperature at which enzyme looses 50% activity upon incubation for 20 min.
Mid-point of thermal unfolding transition as calculated by circular dichroism spectroscopy.
ΔG relates to the difference beyween the free energies of folded and unfolded states of protein.
m indicates the extent of change in free energy as a function of denaturant (GdmCl) concentration.
A very interesting behaviour of mutants against resistance to irreversible thermal inactivation was observed [Fig. 1(C)]. In contrast to the thermal melting [Fig. 1(A)], where the protein was heated at a rate of 1°C/min, resistance to irreversible thermal inactivation was monitored by measuring residual activity upon incubation at various temperatures for 20 min. After cooling back to room temperature, activity measurements were performed. Parent lipase 4D3 show a very sharp decline with complete loss in residual activity by 75°C but, surprisingly, show 40% of the residual activity when incubated directly at 90°C for 20 min. The loss in residual activity at 75°C was abrogated in mutants to various extents. In 5-B mutant, having M137P mutation, the loss of activity at 75°C was completely abolished and the activity was lost monotonically from 75 to 100°C, whereas in the mutants 5-A and 5-D, (having mutation M134E and S167P, respectively), the loss of activities at 75°C were 30% and 50%, respectively [Fig. 1(C); Table II]. It is apparent that the process of denaturation of the parent was different at 75 and 95°C. Upon denaturation of the parent at 75°C, possibly the protein unfolds into an intermediate that was prone to aggregation. At 95°C accumulation of the residual structure was limited. The mutations may have an influence on the formation of the residual structure at 75°C. Exclusive occurrence of the intermediate at 75°C in the parent was further demonstrated by the residual protein in the supernatant after heat treatment [Fig. 1(C)]. The soluble protein after heat treatment traces the activity profile at each temperature indicating that the loss in activity was primarily due to aggregation of the intermediate occurring at 75°C.
Table II.
Residual Activity and Soluble Protein Recovered After Incubation of Lipase Mutants at Elevated Temperatures
| 75°Cb |
||||||||
|---|---|---|---|---|---|---|---|---|
| Thermal transitionsa |
1M NaCl |
85°Cb |
||||||
| Mutant | RA (%) | RP (%) | RA (%) | RP (%) | RA (%) | RP (%) | RA (%) | RP (%) |
| 4D3 | 1 | 5 | 8 | 5 | 2 | 4 | 30 | 32 |
| 5-A | 42 | 88 | 72 | 80 | 19 | 20 | 68 | 98 |
| 5-B | 51 | 84 | 92 | 90 | 88 | 90 | 68 | 95 |
| 5-D | 30 | 43 | 48 | 45 | 2 | 4 | 67 | 98 |
Residual Activity (RA) and Residual soluble protein (RP) left in solution after thermal unfolding and refolding scans from 25°C to 95°C and back at a rate of 1°C/min.
After incubating proteins at indicated temperatures for 20 min.
The atypical trend of irreversible thermal denaturation was further confirmed by monitoring denaturation kinetics upon exposure to higher temperatures, which is one way to know the kinetic barriers to unfolding. At 75°C all mutants show decreased kinetics of inactivation compared to parent 4D3 (Table I). While parent 4D3 has a half-life (t1/2) of 4.4 min, the same was 101 min for mutant 5-B. However, when compared at 85°C the denaturation rates of all the mutants were similar but were significantly reduced as compared to the parent. From 15 min for the parent, t1/2 has increased to more than 45 min in each of the mutants. It is interesting to observe that the half-life of parent 4D3 has increased more than threefold when temperature of incubation was increased from 75 to 85°C. The changes in the Tmapp, ΔGH2O, and the inactivation kinetics in all the three mutants compared to the parent indicate that the thermal stability and the kinetic stability of the mutants have increased.
In addition to improved thermostability, all mutants show enhanced catalytic efficiencies, as shown by the enzyme kinetic parameters (Table III). The catalytic efficiency as determined by kcat/Km has increased 2–6 fold for the mutants compared to wild-type enzyme. The three positions are close to the active site and the local changes in the conformation may have induced improved binding of substrate and facilitated the dissociation of the product. The most significant change was observed in case of 5-A, having mutation M134E, with decrease in Km from 0.98 to 0.28 mM and increase in kcat from 220 to 380 min−1 compared to wild-type, thus increasing the overall efficiency by more than sixfold. This observation is in contrast to the observations from several thermophilic enzymes and is probably due to the conditions used during screening in which the clones displaying improved thermostability along with improved catalytic efficiency were selected.
Table III.
Kinetic Parameters of Lipase Mutants
| Mutant | Kma (mM) | kcat (mM) | kcat/Km (mM/min) |
|---|---|---|---|
| wild-type | 0.98 | 220 | 224 |
| 4D3 | 0.79 | 290 | 370 |
| 5-A | 0.28 | 380 | 1383 |
| 5-B | 0.67 | 565 | 845 |
| 5-D | 0.70 | 637 | 914 |
Kinetic parameters were estimated from assays conducted at 25°C using PNPA as substrate.
The unusual trend of thermal inactivation of parent 4D3 and differential effect of the three mutations on thermal inactivation [Fig. 1(C)] was further probed by monitoring thermal unfolding and refolding of all the four proteins using CD (see Fig. 2). Unfolding was monitored by measuring ellipticity at 222 nm while heating the protein at a rate of 1°C/min from 25–95°C. Refolding was monitored by reversing the scan under identical conditions. As shown in Figure 2, the parent protein 4D3 undergoes thermal unfolding beyond 70°C while the CD signal almost reaches to baseline upon completion of unfolding transition. Moreover, the CD signal does not reverse upon cooling, which remains near to baseline suggesting the loss of signal is primarily due to the loss of soluble protein from the solution [Fig. 2(A)]. In the case of mutant 5-A and 5-B having mutation M134E and M137P, respectively, unfolding starts beyond 70°C and was complete by 80°C. Moreover, upon cooling to room temperature more than 85% of the signal was recovered suggesting efficient refolding of these mutants [Fig. 2(B,C)]. On the other hand, mutant 5-D, having mutation S163P, displays partial recovery of the CD signal suggesting partial refolding of protein [Fig. 2(D); Table II]. Thermal unfolding was further monitored by collecting far-UV CD spectrum, ellipticity at 222 nm and HT voltage as a function of temperature (Supporting Information). A clear isosbestic point with no apparent increase in HT voltage was observed in case of mutant 5-A and 5-B, suggestive of reversible and apparent two-state unfolding transition.28 On the other hand, no clear isosbestic point was observed for 5-D and parent 4D3, in which unfolding transition was accompanied by an increase in HT voltage indicating increase in turbidity of the solution upon heating. These observations show the irreversible nature of thermal unfolding by parent protein 4D3 which reverses to different extent by the incorporation of three mutations.
Figure 2.
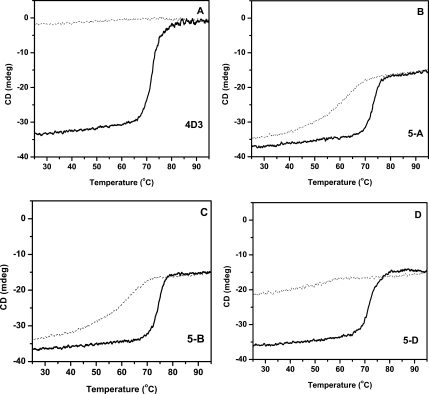
Thermal unfolding and refolding transitions of lipase mutants monitored by CD. Unfolding and refolding transitions of 4D3, 5-A, 5-B, and 5-D are shown in A–D. Thick solid line represent unfolding, while thin dot line show refolding transitions.
Refolding of thermally unfolded proteins was further probed by far-UV CD, residual activity measurements and by quantitation of soluble protein left upon incubation at elevated temperatures [Fig. 3; Table II]. All the three mutants along with parent 4D3 were incubated at 75 and 85°C for 20 min followed by cooling to room temperature and equilibrated for 15 min. Spectra of native proteins were recorded before heating and after cooling upon incubation at high temperatures. At 75°C, parent 4D3 does not show any recovery upon refolding while mutant 5-A and 5-B show almost complete recovery of far-UV CD spectra. Mutant 5-D, on the other hand displays only partial recovery [Fig. 3(A)]. In contrast to the data obtained at 75°C, all the three mutants show complete recovery of far-UV CD spectra upon incubation at 85°C, a temperature far above the melting temperature of all the mutants [Fig. 3(B)]. It is interesting to observe that parent 4D3, which hardly show any recovery upon incubation at 75°C also display partial recovery upon incubation at 85°C. This suggest that all mutants except 5-D refolds back to regain secondary structure completely upon incubation at 75°C, while 5-D and 4D3 failed to do so, however, all mutants completely refolds back upon cooling, if the temperature of incubation was increased to 85°C. This data indicates poor refolding of parent 4D3 upon thermal denaturation and the efficiency of which was largely determined by the type of mutation occurred.
Figure 3.
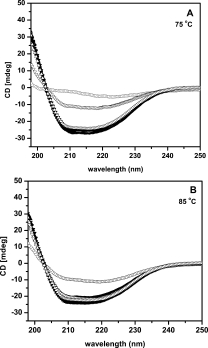
Reversibility of thermal unfolding of lipase mutants as monitored by far UV-CD spectra upon incubation at 75°C (A) and 85°C (B). Far-UV CD spectra of lipase mutants at 25°C are shown in solid symbols, while spectra of refolded proteins after incubation at desired temperature for 20 min are shown in open symbols; 4D3 (•, ○), 5-A (▴, ▵), 5-B (♦, ⋄), and 5-D (▾, ▿).
Aggregation of a protein is primarily driven due to poor solubility of the denatured protein in water. Loss of structure due to denaturation decreases the entropy of system by exposing occluded hydrophobic amino acids to water.2 The system attempts to increase the entropy by initiating inter- or intra-protein interactions leading to occlusion of hydrophobic amino acids, which leads to formation of aggregates. Occlusion of exposed hydrophobicity was more systematically used in oligomerization of monomers and is one of the strategies to increase the thermostability in many proteins.29 Hydrophobicity thus is the driving force for the behaviour of denatured proteins. Hydrophobic dye 1-anilinonaphthalene-8-sulfonate and its derivatives have been widely used to titrate surface hydrophobicity changes in proteins.30 Higher bis-ANS binding is indicative of enhanced surface hydrophobicity of a protein. In order to probe the exposed hydrophobic surface upon unfolding, bis-ANS binding of the mutants was monitored upon incubation of protein in the range of 25–95°C (see Fig. 4). Intense fluorescence due to bound bis-ANS to the parent was observed at 75°C while mutant 5-D shows significantly less binding. The other two mutants 5-A and 5-B show significantly poor binding to hydrophobic dye at the same temperature. The occurrence of a peak due to strong binding of bis-ANS to the protein was maximum for the parent. Only mutant 5-D show significant binding at 75°C consistent with its inability to completely refold after thermal denaturation. Interestingly outside the temperature range of 65 to 85°C, bis-ANS binding was remarkably low. The surface hydrophobicity, as titrated by bis-ANS remarkably reflects the residual activity profile shown in Figure 1(C). During unfolding upon heating, protein generates a mixture of partially unfolded species at various stages of unfolding, which have increased surface hydrophobicity and are prone to aggregation. These aggregates do not precipitate out of solution. Bis-ANS binds to the population of both denatured and partially aggregated protein. To account for the effects of aggregation on bis-ANS binding same experiment was performed in the presence of 20% glycerol, in which all the mutants including parent 4D3 undergoes unfolding, identical in extent as in the absence of glycerol, however does not aggregate. The occurrence of strong bis-ANS binding species in the parent, which on prolonged incubation (>20 min) and centrifugation leads to precipitation, as observed in Figure 1(C), indicates the accumulation of intermediate species that strongly binds to bis-ANS and has strong tendency to aggregate.
Figure 4.
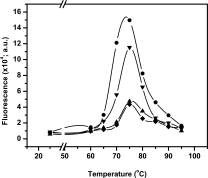
Binding of fluorophore bis-ANS to lipase mutants 4D3 (•), 5-A (▴), 5-B (♦), and 5-D (▾) as a function of temperature.
To further confirm that aggregation is the cause of irreversibility of thermal unfolding shown by parent and mutant 5-D upon incubation at 75°C, aggregation of all the mutants including parent 4D3 was monitored by static light scattering at 75 and 85°C (see Fig. 5). Aggregation was rapid and was completed in less than a minute in parent 4D3 at both the temperatures. At 75°C, all the mutants show imperceptible scatter due to aggregation of protein except 5-D which show significant light scattering compared to parent 4D3 but with delayed kinetics. However, at 85°C all the three mutants including 5-D show imperceptible aggregation. This observation is in agreement with previous observations that mutant 5-D and parent 4D3 fails to completely refold back upon incubation at 75°C due to the accumulation of partially unfolded species which aggregates, thus making the process of unfolding irreversible.
Figure 5.
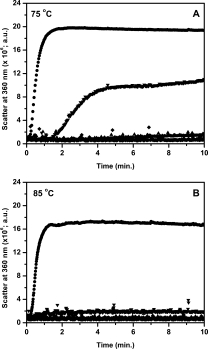
Aggregation of lipase mutants 4D3 (•), 5-A (▴), 5-B (♦), and 5-D (▾), as monitored by static light scattering, upon heating at 75°C (A) and 85°C (B).
Thermal denaturation of the parent 4D3 can be modeled as follows. Parent lipase thermally unfolds, passing through intermediate state(s) in a temperature specific manner, leading to population of denatured ensemble, prone to aggregation as shown in Scheme 1. The nature and the number of intermediates are not clear and the data does not preclude the occurrence of multiple intermediates in the folding or unfolding pathway.
Scheme 1.
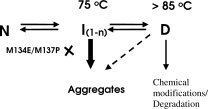
Schematic showing the probable path of wild type lipase and its mutants on thermal denaturation.
The intermediate(s) were characterized by its occurrence at 75°C, by enhanced bis-ANS binding and loss of activity and soluble protein due to aggregation. Upon further increase in temperature, (>85°C), the intermediate state(s) unfolds further to denatured state D, with little residual structure and highly reduced propensity for aggregation. It is possible that the three mutations impose constraints on conformational flexibility either by energy barriers or by electrostatic effects preventing the formation of denatured conformations that are aggregation prone. Based on CD data, the three mutants completely unfold but also refold because they do not aggregate. Several observations acknowledge the fact that in the absence of off-pathways, such as aggregation, most of the monomeric proteins show reversible unfolding.31,32 Strategies on protein engineering for altered properties rely exclusively on the motifs and interactions observed in the native structure. Non-native interactions occurring during the folding and in the denatured state are crucial for the formation of the native structure. The knowledge base on non-native interactions is emerging for few proteins,33,34 but designing a knowledge based strategy based on the denatured ensemble is a challenge. Random mutagenesis approaches panning large mutant populations, as used in this study, may identify mutants that have influences on denatured state.
Mutation M137P, located in a loop connecting β7 strand and αE helix brings stability mainly due to the constraints imposed by the proline in the backbone conformation (see Fig. 6). Several examples are reported wherein substitution with proline has increased the stability of proteins.35–38 Proline contributes to stability by reducing the conformational space available for the polypeptide to fold. Secondly, proline profoundly influences both unfolding and refolding of the protein due to energy barrier between the two isomers of proline. Because isomerization of proline is slow, refolding of unfolded peptides require facilitation by isomerases.39 The effect of mutation M137P in mutant 5-B appears to be primarily in retarding the formation of aggregation prone intermediate observed in parent 4D3. Though mutation M137P increases ΔG compared to the parent, large shift in the midpoint of residual activity vs. temperature curves was primarily due to its ability to keep the protein soluble by preventing the aggregate formation allowing the soluble protein to refold normally. The near complete refolding observed with CD spectrum, lack of binding with bis-ANS and recovery of nearly 90% activity after heating to 95°C suggest that proline primarily prevents the formation of aggregation prone intermediate during thermal unfolding responsible for the loss of protein due to aggregation.
Figure 6.
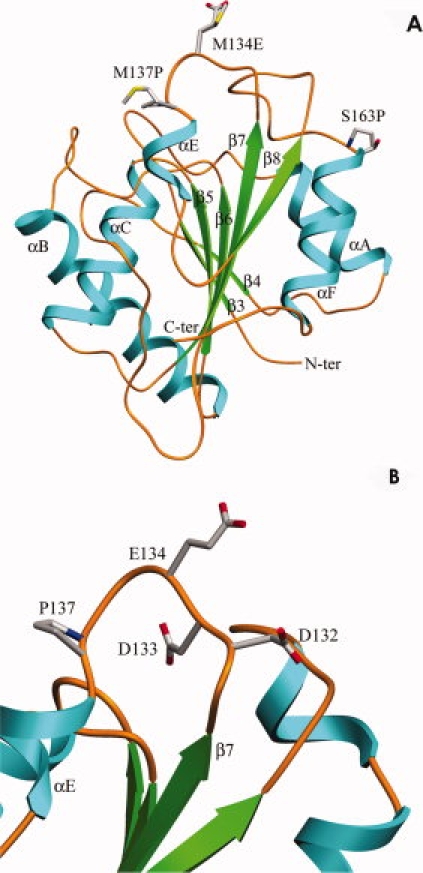
(A) Ribbon diagram of lipase mutant 4D3, indicating the relative position of three mutations. (B) Location of E134, which is adjacent to two negatively charged residues, D132 and D133. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Strategies to stabilize proteins received wide attention due to the importance of protein stability in biological and industrial context. Excellent reports based on comparative genomics, proteomics and site-directed approaches revealed a number of mechanisms to stabilize proteins.23,40 The choice of a stabilization strategy appears to be circumstantial (specific to the protein fold), plural and no ‘generalities' were arrived at. The particular strategy demonstrated by mutation M137P, i.e., prevention of the formation of denatured species prone to aggregation, has not been demonstrated as a method to stabilize the proteins. This strategy, in true sense, is not stabilization of native structure but a novel method to prevent the loss of protein due to the properties of the denatured state. Methodologically, thermostabilization of enzymes involves exposure of proteins to screening temperatures and cooling to room temperature for measurement of activity. This treatment would potentially miss many “positive” mutants due to their inability to refold because the mutation did not influence the denatured ensemble. Description of denatured state of proteins, in recent past, has shifted from a “highly solvated nonstructured polymer” with no interactions between the side chains.11 The inter conversion among the conformers in the denatured ensemble could be profoundly influenced by mutations which may not influence the native state.16 Proline is a strong example given its isomerization energy barrier. Expectedly, the location of proline plays a crucial role because the second mutation S163P, located at the N-terminal of the helix αF [Fig. 6(A)], could only partially influence the aggregation of the denatured ensemble. Stabilization effects brought about by the introduction of proline residues may also be brought about (or even be enhanced) by the introduction of proline derivatives or other non-natural conformationally constrained cyclic amino acid residues.41–43 However, the extent of stabilization obtained will critically depend on the proline analogue's preferred conformation mimicking that of the native state and the conformation adopted in the denatured state.
Mutation M134E is located in a loop connecting β7 strand and αE helix [Fig. 6(B)]. Stability of this mutant is lower than the parent, probably due to the presence of two negative charges at Asp133 and Asp132, thus leading to a repulsive strain in the region. Mutation M134E has significant influence on the formation of the aggregation prone intermediate during thermal denaturation. Its influence is equal to M137P in all the tested parameters, however, the influence of this mutation is probably based on the charge repulsion among the denatured conformers preventing the formation of the hydrophobic intermediate that aggregates.44,45 In order to confirm the electrostatic influence of mutation M134E in imparting reversibility by inhibiting aggregation, influence of ionic strength on the aggregation of the four proteins was determined. The data presented in Figure 7 show the aggregation profiles of all proteins in the presence of 1M NaCl and activity and recovery of soluble protein upon heating at 75°C for 20 min in the presence and absence of 1M NaCl. Influence of NaCl on the aggregation [Fig. 7(A)] and recovery of activity and soluble protein upon thermal denaturation [Fig. 7(B)] was drastic in case of mutant 5-A having mutation M134E, indicating that electrostatic interactions are critical for the prevention of the aggregation probe intermediate. Importantly, introduction of negative charge at position 134 (M134D) was found to contribute to protein thermotolerance in an independent study.46,47 Equally significant observation is the absence of the effect of ionic strength on aggregation and recovery of soluble protein and activity in case of mutant 5-B, having mutation M134P, clearly demonstrating that the role of the mutation is on the formation of aggregation prone intermediate by imposing conformational constraints.
Figure 7.
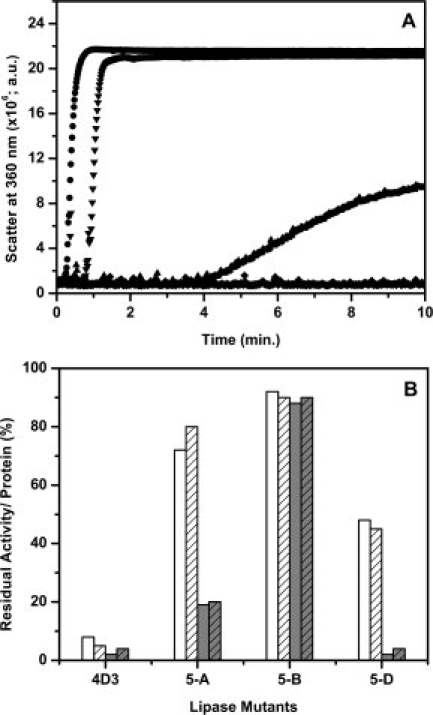
Effect of salt on the aggregation and residual activity of lipase mutants upon incubation at elevated temperature. (A) Aggregation profile of lipase mutants 4D3 (•), 5-A (▴), 5-B (♦), and 5-D (▾) upon incubation at 75°C in the presence of 1M NaCl. (B) Residual activity of lipase mutants in the absence (open bar) and presence (gray bar) of 1M NaCl upon incubation at 75°C for 20 min. Fraction of soluble protein recovered in solution in the absence (open bars with strips) and presence (gray bars with strips) of 1M NaCl under identical conditions.
To affirm the role of mutation M137P, this replacement was introduced in the wild-type lipase by site-directed mutagenesis. The wild-type lipase is different from the parent 4D3 used in these studies, which was evolved by directed evolution, has 9 mutations and melting temperature 15 degrees higher than wild-type protein.26 It is possible that the influence of mutation M137P is “context’ dependent i.e. only in the background of 4D3. The mutant having only M137P mutation in wild-type (wt-M137P) has Tmapp 6 degrees higher than wild-type (Tmapp of wild-type and wt-M137P was 56 and 62°C, respectively) with significant reversibility (>50%) as measured by far-UV CD upon incubation at 70°C for 10 min (Supporting Information). This suggests that mutation M137P in wild-type background also could bring about the recovery of the structure, albeit partially. Thus the influence of M137P is, to that extent, independent of the intrinsic thermostability of the protein.
Stability of proteins could be enhanced either by decrease in GN or by an increase in GD, where ΔG = GD-GN.2 We have no direct proof on the effect of these mutants on the thermally denatured state of the four lipase proteins. The reported data focuses on the unfolding and refolding of the thermally denatured protein. The mutations, either introduction of proline or charge, remarkably prevent the formation of the off-pathway intermediate during unfolding of thermally unfolded protein. The significant effect of proline in preventing the formation of intermediate, and its marginal contribution to ΔG, implicates proline isomerization. Due to partial double bond character of the peptide bond the isomerization between the two isomers has high energy barrier (20 kcal/mol), which may kinetically prevent the accumulation of some conformers.39 The role of proline in folding in model and biological systems is well documented (same as above references). Similarly, presence of a stretch of charges in mutant 5-A (with mutation M134E) also may have preventive influence on the similar off-pathway intermediates. Changes in pH could charge a protein and these charges may also prevent aggregation. We have recently reported that wild-type Bacillus subtilis lipase show a very strong pH dependent temperature-mediated aggregation.48 At pH <6, though lipase loses the structure completely, does not aggregate, whereas at pH >6 the lipase rapidly aggregates upon heating. This property is abrogated in the presence of salt suggesting the involvement of ionic interactions for aggregation. Detailed pH titrations indicated that protonation of histidine residues is biased in the denatured ensemble towards nonaggregation states. In another study it was shown that a K12M mutation in the N-terminal domain of the ribosomal protein N9 has increased the stability over the parent by disrupting the interactions in denatured state.16 Thus, at present irrefutable evidence is available to demonstrate that the mutations do alter the structure and the energetics of the denatured state of a protein. Mutations reported in this study are able to avoid the intermediate by way of preventing or delaying the formation of structures leading to the intermediate(s) formation. Because the mutations are at a position, the overall pathway of folding may be largely unaltered. Many reports have demonstrated that denatured state energetics and thereby the protein stability have been altered by altering the bias specific interactions in the denatured states.16,45,49 The observation that the residual structure in denatured state has profound influence on the stability is well established.2,7,9 In Ribonuclease H from thermophile and mesophiles, RNAse Sa, the importance of the residual structure and its perturbation by mutations in altering stability and kinetics of folding have been well documented.44,50
In conclusion, mutants identified in this study by a combination of in vitro evolution methods and site-saturation mutagenesis reveals a mechanism by which thermal aggregation could be prevented in proteins. Predictions of such mutations are very difficult because it requires structural information of the denatured state and folding pathways. Our study highlights the importance of in vitro methods for isolating such mutations. The ability to prevent aggregation by the introduction of a proline or a charge indicates the importance of intramolecular interactions in denatured ensemble in unfolding and refolding pathways.
Methods
Reagents
Taq DNA polymerase and dNTPs were obtained from Invitrogen. Restriction enzymes were from New England Biolabs (Beverley,MA). Fast link™ DNA Ligation kit was from EPICENTRE Biotechnologies (Madison, WI). QIAquick® gel extraction kit and QIAprep® spin Miniprep kits were from Qiagen (Hilden, Germany). Phenyl Sepharose 6 Fast Flow (High Sub) was from Amersham Biosciences. Bis-ANS, PNPA, PNPB and Triton-X 100 were from Sigma Chemical Co. GdmCl was purchased from SERVA Electrophoresis GmbH (Heidelberg, Germany). Oligonucleotides were purchased from BioServe (Hyderabad, India). All other reagents were of analytical grade or higher.
Construction of vectors used for screening and protein purification
For efficient screening, mutant lipase gene was cloned in pET22b expression vector (Novagen), under pelB signal sequence for periplasmic expression. The structural gene, encoding 4D3 mutant lipase, cloned in pET21b,26 was PCR amplified using primers NcoFOR (5′-GAAGGAGATATACCCATGGCAGAACACA-3′) and T7-terminator (5-GCTAGTTATTGCTCAGCGG-3′) (NcoI site underlined). An internal NcoI site within the structural gene was knocked down by silent mutagenesis using two internal primers DNCOF (5′-GAAACGTTCAAATTCATGGCGTTGGA-3′) and DNCOR (5′-GTCCAACGCCATGAATTTGAACGTTTCTA-3′). The amplified product was digested with NcoI and HindIII followed by gel extraction. The digested and gel-purified product was ligated with similarly digested pET22b vector to get pET22-4D3. This construct was used for screening of thermostable mutants upon transformation of E. coli BL21 (DE3), which directs the expression of protein in periplasm upon induction with IPTG.
For protein over-expression and purification, structural genes of lipase mutants cloned in pET22b, were PCR amplified using primers PET22NDE (5′-GCCGGCGATGCATATGGCAGAACACA-3′) and T7-terminator (NdeI site underlined). The amplified products were digested with NdeI and HindIII followed by gel extraction and ligated with similarly digested pET21b. The forward primer also introduced start codon ATG, which adds an extra methionine residue, before the N-terminal alanine of mature protein, upon expression in E. coli.
Site-saturation mutagenesis
Site-saturation mutagenesis at selected positions, in 4D3 mutant lipase gene, was performed using a pair of oligonucleotide primers. The target amino acid position was coded by NNK (sense strand) and MNN (antisense strand), where N = A, G, C or T, K = G or T and M = A or C. The reaction was carried out by the overlap extension method using two vector specific primers T7-promoter and T7-terminator (Novagen), along with position specific mutagenic primers, using pET22-4D3 as template. The amplified products were digested with NcoI and HindIII followed by gel purification. Digested and purified products were ligated with similarly digested pET22b and used for transformation of E. coli BL21 (DE3) for gene expression.
Screening
The thermostability of mutants was assessed by following a two tier screening protocol as described earlier with some modifications.26 Initial screening was performed using a 96-well plate assay format followed by rigorous assessment of positives in tube assays.
The mutant library obtained after site-saturation mutagenesis was used to transform E. coli BL21 (DE3). Cells were plated on LB (1% tryptone, 0.5% yeast extract and 1% NaCl) agar plates having 100 μg/mL ampicillin and incubated at 37°C for 12 h. Transformants obtained were inoculated in individual wells of 96-well plates containing 200 μL of LB medium having 100 μg/mL ampicillin. Plates were incubated at 37°C with shaking at 200 rpm for 6 h. Grown cultures were used to inoculate fresh medium in identical plates and were incubated at 37°C with shaking at 200 rpm After 6 h, cultures were induced by adding IPTG in each well to a final concentration of 0.5 mM and incubated further for 12 h. Cells were harvested by spinning the plates at 4000 rpm for 30 min at 4°C in an Eppendorf centrifuge, 5810 R. Supernatant (100 μL) was mixed 1:1 with 100 mM sodium phosphate buffer (pH 7.2) and splitted into two identical 96-well PCR plates (Axygen Scientific, Union City, CA). One plate was incubated at 70°C for 20 min, cooled at 4°C for 20 min followed by equilibration at 25°C for 15 min in a PCR thermal cycler (GeneAmp 9700, Applied Biosystems, Foster City, CA). The other plate was incubated under identical conditions, except incubation at high temperature. Activity measurements were performed by mixing 80 μL of diluted supernatant with 80 μL of 2× PNPB- Triton X-100 substrate solution (4 mM PNPB micellized in 40 mM Triton X-100). Increase in absorbance at 405 nm with time was monitored using Spectramax 190 plate reader (Molecular Devices, Sunnyvale, CA) while data was analyzed by using SoftMaxPro 4.7.1 software provided with the instrument. The ratio of activity of each clone after incubation at higher temperature versus without incubation was taken as residual activity and was used to identify the positives.
Positives were further confirmed by performing tube assays. Ten milliliters of cell culture in LB medium, having 100 μg/mL ampicillin, was inoculated with 1% of overnight grown culture, followed by incubation at 37°C with vigorous shaking (250 rpm). After 6 h, culture was induced with 0.5 mM IPTG and incubated further for 12 h after which cells were separated from culture supernatant by centrifugation at 18,000g for 10 min at 4°C. Culture supernatant was diluted 1:1 with 100 mM sodium phosphate buffer (pH 7.2) and incubated (100 μL) at various temperatures for 20 min followed by cooling at 4°C for 20 min and final equilibration at 25°C for 15 min in a PCR thermal cycler. Activity was measured by adding 80 μL of diluted supernatant to 920 μL of 50 mM sodium phosphate buffer (pH 7.2) containing 2 mM PNPB micellized in 20 mM Triton X-100 as substrate. Increase in absorbance at 405 nm with time was measured with a spectrophotometer (U-2000, Hitachi, Japan). Residual activity corresponding to each temperature of incubation was determined by taking the ratio of activities obtained upon incubation with that of without incubation. The temperature at which enzyme loses 50% of its activity after incubation for 20 min (T50) was taken as the thermostability index for the selection of mutants. Plasmids were purified from mutants displaying higher T50 values with respect to parent and genes were sequenced to identify the mutation occurred.
DNA sequencing
Plasmid DNA was isolated from E. coli DH5α or BL21 (DE3), using QIAprep® spin Miniprep kit according to manufacturer's instructions. DNA sequencing was performed using ABI Prism BigDye™ v3.0 reaction kit according to manufacturer's instructions using ABI 3730 sequence analyzer (Applied Biosystems).
Protein purification
Lipase mutant proteins, genes of which cloned in pET21b, were purified upon expression in E. coli BL21 (DE3) as described earlier with some modifications.26 All mutant proteins bind to phenyl Sepharose and were eluted under conditions similar to that of 4D3 mutant. In the subsequent ion-exchange chromatography step, all the mutants bind to anion exchanger Mono-Q (Amersham Biosciences) in 20 mM glycine-NaOH buffer (pH 10.5). Elution of bound protein was performed by using a linear gradient of NaCl, from 0 to 1M, in binding buffer. Active fractions were pooled, dialyzed overnight against 2 mM glycine-NaOH buffer (pH 10) at 4°C and stored at −20°C. Purity was checked by running SDS-PAGE and was found to be more than 95%. Protein quantitation was done by the modified Lowry method.51
Thermal inactivation and unfolding
Heat treatment of purified protein was carried out by incubating the protein in 0.2 mL PCR tubes in a programmable thermal cycler for precise temperature control. Proteins (25 μL of 0.05 mg/mL in 50 mM sodium phosphate buffer, pH 7.2) were heated at different temperatures for required time, cooled at 4°C for 20 min followed by equilibration at 25°C for 15 min. Samples were centrifuged to get rid off any aggregated protein before assaying for enzymatic activity. Thermal inactivation profiles were plotted by incubating the enzymes at various temperatures (from 60 to 100°C) for 20 min followed by residual activity measurement at room temperature. Kinetics of thermal inactivation was monitored by incubating the proteins at 75 and 85°C. At various time intervals, an aliquot was removed followed by residual activity measurement at room temperature. Typically, inactivation was followed until >80% of the activity was lost. The heat-treated protein sample (20 μL) was added to 1 mL of 50 mM sodium phosphate buffer (pH 7.2) having 2 mM PNPA as substrate. Enzymatic activity was monitored by measuring the rate of increase in absorbance at 405 nm as described earlier.26
Thermal unfolding of lipase mutants was monitored by circular dichroism spectroscopy in a JASCO J-815 spectropolarimeter fitted with Jasco Peltier-type temperature controller (CDF-426S/15). The protein concentration used was 0.05 mg/mL in 50 mM sodium phosphate buffer (pH 7.2) with path length of 1 cm. Temperature dependent unfolding profiles were obtained by heating protein at a constant rate of 1°C per minute from 25 to 95°C and measuring the change in ellipticity at 222 nm.
Circular dichroism
Far-UV CD spectra were recorded in the 250–200 nm range using JASCO J-815 spectropolarimeter fitted with Jasco Peltier-type temperature controller (CDF-426S/15). All spectra reported are the average of three accumulations. Scan speed of 100 nm/minute, responsetime of 2 sec, bandwidth of 2 nm and data pitch of 0.2 nm was used for measurement. All spectra were corrected for buffer baseline by subtracting the respective blank spectra recorded identically without protein.
Fluorescence
Fluorescence measurements were performed using Hitachi F-4500 fluorimeter. Protein concentration of 0.05 mg/mL, in 50 mM sodium phosphate buffer (pH 7.2) was used. Intrinsic tryptophan fluorescence was recorded between 310 and 380 nm by exciting the sample at 295 nm. The excitation and emission band passes were set at 5 nm while all spectra were recorded in corrected spectrum mode. All spectra were corrected for buffer baseline by subtracting the respective blank spectra, without protein, recorded under identical conditions.
Bis-ANS fluorescence at elevated temperature was monitored using Fluorolog 3-22 fluorimeter, fitted with Peltier based cuvette holder, controlled by LFI-3751 temperature controller (Jobin Yvon, USA). Sample composition and measurement parameters were same as described above. Samples were excited at 390 nm while emission was recorded in the range of 400–600 nm. Both excitation and emission band passes were set at 2 nm. Protein was added to preheated buffer at desired temperature and incubated for 5 min for temperature equilibration. Bis-ANS was added to a final concentration of 10 μM from methanolic stock and spectra were recorded after an additional incubation of 1 min.
GdmCl unfolding
Equilibrium unfolding in the presence of GdmCl was monitored by using CD and Fluorescence spectroscopy. Protein (0.5 mg/mL in 50 mM sodium phosphate buffer, pH 7.2), was incubated in varying concentrations of GdmCl for 12 h at room temperature prior to measurements. Far-UV CD spectra were recorded in 250 to 210 nm range using 1 mm path length cuvette as described above. The same protein sample was diluted to 0.1 mg/mL concentration in equimolar denaturant solution and intrinsic tryptophan fluorescence measurements were performed, as described above. Unfolding profiles were determined by monitoring the change in ellipticity at 222 nm and shift in tryptophan emission maxima (λmax) with increasing concentration of GdmCl. The concentration of stock GdmCl solution was determined by refractive index measurements. Data was analyzed as described by Pace et al.27
Reversibility of thermal unfolding
Thermal unfolding of lipase mutants were monitored using CD by heating purified protein (0.05 mg/mL in 50 mM sodium phosphate buffer, pH 7.2) from 25 to 95°C at a constant rate of 1°C/minute in 1 cm path length cuvette. Refolding was monitored by reversing the scan from 95 to 25°C with identical rate of cooling. Change in ellipticity at 222 nm with temperature was observed to monitor unfolding and refolding transitions. Reversibility of thermal unfolding of lipase mutants, upon incubation at elevated temperatures for 20 min, was probed by Far-UV CD, by estimating residual activity and the amount of protein left in the supernatant after removal of aggregated fraction. Three milliliters of protein solution (0.05 mg/mL) in 50 mM sodium phosphate buffer (pH 7.2), was incubated at elevated temperatures for 20 min followed by rapid cooling and equilibration at 25°C for 20 min in a 1 cm path length cuvette within the cuvette holder of CD spectropolarimeter. Far-UV CD spectra of native and heat treated protein was recorded as described above. Rest of the sample was centrifuged at 20,000g for five minutes followed by passing through Millex-GV, 0.22 μm filter (Millipore, Japan), to remove any aggregated protein. Enzymatic assay and protein estimation was performed to determine residual activity and the amount of protein left in the supernatant.
Static light scattering
Static light scattering was performed on Fluorolog 3-22 fluorimeter, fitted with Peltier based cuvette holder, controlled by LFI-3751 temperature controller (Jobin Yvon, USA). Both monochromators were set at 360 nm, while slit-widths at 2 nm each. Protein sample of 0.05 mg/mL in 50 mM sodium phosphate buffer (pH 7.2), was rapidly heated to desired temperature followed by incubation at elevated temperature for 10 min. Data acquisition was started as soon as the temperature of sample reaches the fixed temperature.
Activity measurements
All enzyme kinetic parameters were determined at 25°C by measurements performed on thermostatted spectrophotometer (Lamda-35 attached with PTP-1 Peltier temperature programmer, Perkin Elmer) using PNPA substrate as described earlier.25
Acknowledgments
The authors thank Virender Kumar for purifying wt-M137P mutant protein.
Glossary
Abbreviations:
- bis-ANS
1, 1′-bis-(4 anilino) naphthalene-5, 5′-disulfonic acid
- CD
circular dichroism
- GdmCl
guanidinium chloride
- PNPA
p-nitrophenyl acetate
- PNPB
p-nitrophenyl butyrate.
References
- 1.Pace CN, Shirley BA, McNutt M, Gajiwala K. Forces contributing to the conformational stability of proteins. FASEB J. 1996;10:75–83. doi: 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- 2.Shortle D. The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 1996;10:27–34. doi: 10.1096/fasebj.10.1.8566543. [DOI] [PubMed] [Google Scholar]
- 3.Shortle D, Wang Y, Gillespie JR, Wrabl JO. Protein folding for realists: a timeless phenomenon. Protein Sci. 1996;5:991–1000. doi: 10.1002/pro.5560050602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rees DC, Robertson AD. Some thermodynamic implications for the thermostability of proteins. Protein Sci. 2001;10:1187–1194. doi: 10.1110/ps.180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 6.Razvi A, Scholtz JM. Lessons in stability from thermophilic proteins. Protein Sci. 2006;15:1569–1578. doi: 10.1110/ps.062130306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dill KA, Shortle D. Denatured states of proteins. Annu Rev Biochem. 1991;60:795–825. doi: 10.1146/annurev.bi.60.070191.004051. [DOI] [PubMed] [Google Scholar]
- 8.Smith LJ, Fiebig KM, Schwalbe H, Dobson CM. The concept of a random coil. Residual structure in peptides and denatured proteins. Fold Des. 1996;1:R95–R106. doi: 10.1016/S1359-0278(96)00046-6. [DOI] [PubMed] [Google Scholar]
- 9.Millett IS, Doniach S, Plaxco KW. Toward a taxonomy of the denatured state: small angle scattering studies of unfolded proteins. Adv Protein Chem. 2002;62:241–262. doi: 10.1016/s0065-3233(02)62009-1. [DOI] [PubMed] [Google Scholar]
- 10.Mittag T, Forman-Kay JD. Atomic-level characterization of disordered protein ensembles. Curr Opin Struct Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Shortle D. The expanded denatured state: an ensemble of conformations trapped in a locally encoded topological space. Adv Protein Chem. 2002;62:1–23. doi: 10.1016/s0065-3233(02)62003-0. [DOI] [PubMed] [Google Scholar]
- 12.Robic S, Guzman-Casado M, Sanchez-Ruiz JM, Marqusee S. Role of residual structure in the unfolded state of a thermophilic protein. Proc Natl Acad Sci USA. 2003;100:11345–11349. doi: 10.1073/pnas.1635051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jahn TR, Radford SE. Folding versus aggregation: polypeptide conformations on competing pathways. Arch Biochem Biophys. 2008;469:100–117. doi: 10.1016/j.abb.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pace CN, Alston RW, Shaw KL. Charge-charge interactions influence the denatured state ensemble and contribute to protein stability. Protein Sci. 2000;9:1395–1398. doi: 10.1110/ps.9.7.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho JH, Sato S, Raleigh DP. Thermodynamics and kinetics of non-native interactions in protein folding: a single point mutant significantly stabilizes the N-terminal domain of L9 by modulating non-native interactions in the denatured state. J Mol Biol. 2004;338:827–837. doi: 10.1016/j.jmb.2004.02.073. [DOI] [PubMed] [Google Scholar]
- 16.Cho JH, Raleigh DP. Mutational analysis demonstrates that specific electrostatic interactions can play a key role in the denatured state ensemble of proteins. J Mol Biol. 2005;353:174–185. doi: 10.1016/j.jmb.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 17.Bershtein S, Tawfik DS. Advances in laboratory evolution of enzymes. Curr Opin Chem Biol. 2008;12:151–158. doi: 10.1016/j.cbpa.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 18.Jackel C, Kast P, Hilvert D. Protein design by directed evolution. Annu Rev Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- 19.Tracewell CA, Arnold FH. Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr Opin Chem Biol. 2009;13:1–7. doi: 10.1016/j.cbpa.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reetz MT, Kahakeaw D, Sanchis J. Shedding light on the efficacy of laboratory evolution based on iterative saturation mutagenesis. Mol Biosyst. 2009;5:115–122. doi: 10.1039/b814862g. [DOI] [PubMed] [Google Scholar]
- 21.Lutz S, Bornscheurer UT. Protein engineering handbook. Weinheim: Wiley-VCH; 2009. [Google Scholar]
- 22.Wintrode PL, Arnold FH. Temperature adaptation of enzymes: lessons from laboratory evolution. Adv Protein Chem. 2000;55:161–225. doi: 10.1016/s0065-3233(01)55004-4. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Nussinov R. How do thermophilic proteins deal with heat? Cell Mol Life Sci. 2001;58:1216–1233. doi: 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eijsink VG, Gaseidnes S, Borchert TV, van den BB. Directed evolution of enzyme stability. Biomol Eng. 2005;22:21–30. doi: 10.1016/j.bioeng.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Acharya P, Rajakumara E, Sankaranarayanan R, Rao NM. Structural basis of selection and thermostability of laboratory evolved Bacillus subtilis lipase. J Mol Biol. 2004;341:1271–1281. doi: 10.1016/j.jmb.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 26.Ahmad S, Kamal MZ, Sankaranarayanan R, Rao NM. Thermostable Bacillus subtilis lipases: in vitro evolution and structural insight. J Mol Biol. 2008;381:324–340. doi: 10.1016/j.jmb.2008.05.063. [DOI] [PubMed] [Google Scholar]
- 27.Pace CN, Shirley BA, Thompson JA. Mechanism of protein folding. In: Creighton ED, editor. Protein structure: a practical approach. Oxford: Oxford Press; 1998. pp. 311–330. [Google Scholar]
- 28.Kelly SM, Price NC. The use of circular dichroism in the investigation of protein structure and function. Curr Protein Pept Sci. 2000;1:349–384. doi: 10.2174/1389203003381315. [DOI] [PubMed] [Google Scholar]
- 29.Jaenicke R, Lilie H. Folding and association of oligomeric and multimeric proteins. Adv Protein Chem. 2000;53:329–401. doi: 10.1016/s0065-3233(00)53007-1. [DOI] [PubMed] [Google Scholar]
- 30.Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res. 2008;25:1487–1499. doi: 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 32.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 33.Brockwell DJ, Radford SE. Intermediates: ubiquitous species on folding energy landscapes? Curr Opin Struct Biol. 2007;17:30–37. doi: 10.1016/j.sbi.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nabuurs SM, Westphal AH, van Mierlo CP. Extensive formation of off-pathway species during folding of an alpha-beta parallel protein is due to docking of (non)native structure elements in unfolded molecules. J Am Chem Soc. 2008;130:16914–16920. doi: 10.1021/ja803841n. [DOI] [PubMed] [Google Scholar]
- 35.Matthews BW, Nicholson H, Becktel WJ. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc Natl Acad Sci USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim PS, Baldwin RL. Intermediates in the folding reactions of small proteins. Annu Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 37.Chen BL, Baase WA, Nicholson H, Schellman JA. Folding kinetics of T4 lysozyme and nine mutants at 12 degrees C. Biochemistry. 1992;31:1464–1476. doi: 10.1021/bi00120a025. [DOI] [PubMed] [Google Scholar]
- 38.Osvath S, Gruebele M. Proline can have opposite effects on fast and slow protein folding phases. Biophys J. 2003;85:1215–1222. doi: 10.1016/S0006-3495(03)74557-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmid FX. Prolyl isomerase: enzymatic catalysis of slow protein-folding reactions. Annu Rev Biophys Biomol Struct. 1993;22:123–142. doi: 10.1146/annurev.bb.22.060193.001011. [DOI] [PubMed] [Google Scholar]
- 40.Jaenicke R, Bohm G. The stability of proteins in extreme environments. Curr Opin Struct Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 41.Ballano G, Zanuy D, Jimenez AI, Cativiela C, Nussinov R, Aleman C. Structural analysis of a β-helical protein motif stabilized by targeted replacements with conformationally constrained amino acids. J Phys Chem B. 2008;112:13101–13115. doi: 10.1021/jp8032116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flores-Ortega A, Casanovas J, Nussinov R, Aleman C. Conformational preferences of β- and γ-aminated proline analogues. J Phys Chem B. 2008;112:14045–14055. doi: 10.1021/jp807638p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flores-Ortega A, Jimenez AI, Cativiela C, Nussinov R, Aleman C, Casanovas J. Conformational preferences of alpha-substituted proline analogues. J Org Chem. 2008;73:3418–3427. doi: 10.1021/jo702710x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trefethen JM, Pace CN, Scholtz JM, Brems DN. Charge-charge interactions in the denatured state influence the folding kinetics of ribonuclease Sa. Protein Sci. 2005;14:1934–1938. doi: 10.1110/ps.051401905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cho JH, Raleigh DP. Electrostatic interactions in the denatured state and in the transition state for protein folding: effects of denatured state interactions on the analysis of transition state structure. J Mol Biol. 2006;359:1437–1446. doi: 10.1016/j.jmb.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 46.Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed Engl. 2006;45:7745–7751. doi: 10.1002/anie.200602795. [DOI] [PubMed] [Google Scholar]
- 47.Reetz MT, Carballeira JD. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc. 2007;2:891–903. doi: 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- 48.Rajakumara E, Acharya P, Ahmad S, Sankaranaryanan R, Rao NM. Structural basis for the remarkable stability of Bacillus subtilis lipase (Lip A) at low pH. Biochim Biophys Acta. 2007;1784:302–311. doi: 10.1016/j.bbapap.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 49.Cho JH, Sato S, Horng JC, Anil B, Raleigh DP. Electrostatic interactions in the denatured state ensemble: their effect upon protein folding and protein stability. Arch Biochem Biophys. 2008;469:20–28. doi: 10.1016/j.abb.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hollien J, Marqusee S. A thermodynamic comparison of mesophilic and thermophilic ribonucleases H. Biochemistry. 1999;38:3831–3836. doi: 10.1021/bi982684h. [DOI] [PubMed] [Google Scholar]
- 51.Markwell MAK, Hass SM, Tolbert NE, Bieber LL. Protein determination in membrane and lipoprotein samples: Manual and automated procedures. Methods Enzymol. 1981;72:296–303. doi: 10.1016/s0076-6879(81)72018-4. [DOI] [PubMed] [Google Scholar]