

Abstract
Computational protein design relies on a number of approximations to efficiently search the huge sequence space available to proteins. The fixed backbone and rotamer approximations in particular are important for formulating protein design as a discrete combinatorial optimization problem. However, the resulting coarse-grained sampling of possible side-chain terminal positions is problematic for the design of protein function, which depends on precise positioning of side-chain atoms. Although backbone flexibility can greatly increase the conformation freedom of side-chain functional groups, it is not obvious which backbone movements will generate the critical constellation of atoms responsible for protein function. Here, we report an automated method for identifying protein backbone movements that can give rise to any specified set of desired side-chain atomic placements and interactions, using protein–DNA interfaces as a model system. We use a library of previously observed protein–DNA interactions (motifs) and a rotamer-based description of side-chain conformation freedom to identify placements for the protein backbone that can give rise to a favorable side-chain interaction with DNA. We describe a tree-search algorithm for identifying those combinations of interactions from the library that can be realized with minimal perturbation of the protein backbone. We compare the efficiency of this method with the alternative approach of building and screening alternate backbone conformations.
Keywords: computational protein design, protein–nucleic acid interactions
Introduction
The successes of computational protein design have been enabled by a set of algorithmic and representational choices that have rendered tractable the problem of selecting amino acid sequences that will fold to a stable conformation and carry out a desired function. It was recognized early on that the most general formulation of protein design presupposes a solution to the folding problem: to assess a given amino acid sequence, the structure must be first predicted and then screened for proper function in silico. To avoid this hopelessly unrealistic prescription, Pabo1 suggested instead that protein design should address the “inverse folding” problem. In this approach, the structure of the protein backbone is taken as a starting point, and an optimal amino acid sequence is determined to stabilize the assumed backbone conformation. This “fixed backbone” approximation was first used in practice by Ponder and Richards2 to identify amino acid combinations compatible with efficient packing of protein cores. Their computations required a further conformational simplification: amino acid side chains were restricted to a discrete set of rotational states that are frequently observed in experimentally determined protein structures and predicted to be energetically favorable based on small molecule analogs (the rotamer approximation). Despite numerous improvements in scoring functions and optimization techniques, this general protocol of protein design with a fixed backbone and a discrete palette of side-chain conformational options remains a core subtask in almost all computational design algorithms, and is typically the step at which sequence variation is introduced.
The simplifications due to the fixed backbone approximation extend beyond the computations involved in design. With a few notable exceptions,3,4 the assumed backbone conformation is taken from an experimentally determined structure. This ensures that the backbone is “designable,” because at least one sequence is capable of achieving the conformation. Furthermore, assuming the constancy of the backbone allows one to neglect the energetics of protein backbone movement when evaluating amino acid sequences. Overall, the fixed backbone approximation allows for the simplified recasting of protein design in its inverted form, ensures a reasonable end-state fold for the protein, and excludes the difficult balance between side-chain energetics and compensatory backbone motion from the scoring calculus. The ability of this approach to redesign native proteins, often with enhanced stability, has been demonstrated multiple times.5–8
Although the fixed backbone approximation is well-suited for designing stable proteins, it can hinder the design of proteins that perform specific functions. Protein function is usually mediated by side-chain atoms, and the combination of a rigid backbone and a discrete set of rotamers implies a limited ability to locate side-chain groups where they are required for function. The effect is most pronounced for amino acids with longer side chains, where the limitations of discretization in torsional space are amplified by a “lever arm” effect. Unfortunately, the charged and polar amino acids that typically serve as catalytic groups and mediate molecular recognition (e.g., of DNA) tend to fall within this group. This difficulty in sampling can be reduced (but not eliminated) by the use of larger rotamer libraries, with a corresponding increase in computational burden.
The design of the Top7 protein involved an unprecedented backbone topology, and as a result could not rely on the guarantees of designability conferred by the use of previously observed fixed backbones.4 In this case, backbone flexibility was included by coupling techniques from protein design and structure prediction: protein models were subjected to iterative rounds of sequence optimization followed by structural relaxation. However, no specific interactions were required, only that the final sequence-structure combination had very low energy. Because the design of functional proteins requires satisfaction of specific functional restraints in addition to energy optimization, the iterative design/relaxation model would require significant modification to address these restraints.
Although even subtle backbone motions can greatly expand the conformational freedom of side-chain functional groups, those motions which result in optimal placements of side-chain atoms for a given task cannot in general be determined until they have been tried and evaluated. In previous work, researchers have transplanted functional motifs from wild-type proteins onto redesigned scaffolds to confer these scaffolds with the analogous function. Typically, this involves finding a set of backbone locations on the scaffold whose mutual orientation approximates the wild-type context for the functional motif to be transplanted. For example, Pabo and coworkers9,10 identified two positions in the DNA-binding domain of lambda repressor suitably oriented to form a disulfide bond. Hellinga and coworkers11–16 have transplanted a number of metal-binding sites into protein scaffolds using chelating amino acid arrangements found in wild-type proteins. Finally, our group has developed efficient methods for placing sets of catalytic amino acids into arbitrary protein scaffolds, generating novel enzymatic activities.17,18 In each of these cases, the scaffold backbone is treated as fixed, and the functional residues are specified before hand.
In this report, we describe a new and general method for directing backbone movement in such a way as to incorporate functional amino acids into a scaffold protein. The method can be used to design any desired set of functional interactions; in this article, we use the recognition of specific DNA sequences as a concrete example. The functional interactions between protein and DNA are not specified, but are selected automatically from a library of previously observed possibilities as the algorithm proceeds. The algorithm is implemented in the Rosetta molecular modeling program, which provides efficient methods for backbone movement and includes a full accounting of side chain and backbone energetics. We also consider a “null model” method in which alternate backbones are generated using peptide fragment assembly and screened for the ability to incorporate functional motifs from the same library. We find that the motif-directed method is more efficient at generating minimally altered backbones incorporating one or more functional motifs than screening ensembles of rebuilt backbone conformations. Finally, we demonstrate that a motif-directed strategy for backbone relaxation can generate conformations suitable for homology modeling of DNA-binding proteins.
Results
Inverse rotamers for protein–DNA interactions
We constructed a library of interactions (termed “motifs”) between amino acids and bases in duplex DNA (see Fig. 1). The interactions were drawn from several sources. Interactions implicated in conferring binding specificity have been previously reported,20 and databases containing exhaustive sets of polar interactions have been compiled.21 We augmented these with hydrophobic (packing) interactions culled from protein–DNA complexes in the protein databank.22 The vast majority of the motifs come from the AANT database of polar interactions.21 Although many interactions in this database are redundant, we erred on the side of completeness and did not attempt to eliminate similar interactions. Two coordinate systems were defined for the base and the amino acid atoms involved in the interaction (denoted by the expanded atoms in Figure 1, in many cases hydrogen bond donors and acceptors), and the geometric relationship between the two coordinate systems was expressed as a translation vector and a set of Euler angles.
Figure 1.

Examples of interaction motifs. Each panel depicts a single interaction motif. The three coordinate system-defining atoms in both the base (or bases) and the amino acid used to describe the motif are rendered as larger spheres. The sticks representing the amino acid backbone atoms are rendered with a decreased radius. A: A bidentate hydrogen-bond interaction between an arginine and a guanine. B: An interaction between an asparagine and two adjacent stacked bases. The noninteracting bases paired with the stacked bases are colored in solid grey. In this motif, the coordinate system on the DNA side of the interface is defined using atoms from both of the interacting bases. C: An interaction between a lysine and two bases that are diagonally related—the bases are on opposite strands but in adjacent base pairs. The noninteracting bases paired with the stacked bases are colored in solid grey. D: A hydrophobic interaction between a phenylalanine ring and a thymine methyl group. This and all other figures were generated using PyMOL.19
The motif library was used to build inverse rotamers,23,24 which are free-floating residues in the correct orientation to make the interaction described by the motif. As in the more common use of rotamers to describe side chain conformational freedom given a fixed backbone, the inverse rotamers generated for a single motif comprise a discrete set of conformational possibilities, and differ in the values of their side chain bond torsions. They are “inverted” in the sense that the residues are superimposed on the three atoms that define the coordinate system for the amino acid functional group, rather than on the backbone atoms (see Fig. 2). As a result, a set of inverse rotamers describes the main chain spatial locations that can give rise to the interaction described by the motif.
Figure 2.
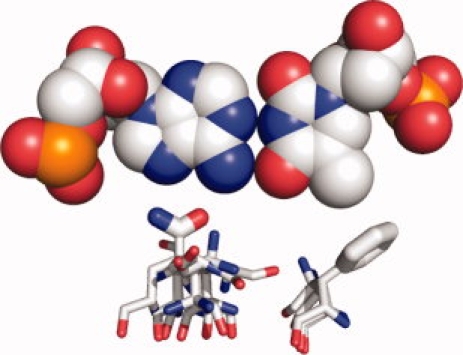
Backbone positions for interaction motifs. For an example A-T base pair, inverse rotamers are shown for two interaction motifs: a commonly observed bidentate hydrogen bond interaction between a glutamine and an adenine base, and a packing interaction between a phenylalanine side chain and a thymine methyl group. Although the coordinates for the terminal atoms contacting the DNA are specified by the motif geometry, the location of possible backbone atoms are determined by building the amino acid backward from side chain to main chain using torsional values taken from a rotamer library. A full set of inverse rotamers for this base pair would include more motifs, each with multiple amino acid conformations capable of realizing the interaction encoded in the motif.
Computational model system for protein–DNA interfaces
We choose for our test case the I-AniI homing endonuclease. Homing endonucleases recognize extended DNA sequences, and are commonly composed of two domains.25 We focus here on the N-terminal domain of I-AniI. The wild-type recognition site for I-AniI differs by five base pairs from a site in the IL-2Rγ gene in a mouse model of severe combined immunodeficiency disease (SCID) [Fig. 3(A)]. The IL-2Rγ locus is frequently mutated in human patients with X-linked SCID,26 and repair of this gene by homologous recombination from an exogenous DNA template stimulated by endonuclease-targeted double-strand breaks is a possible therapeutic strategy. We modeled the central three base pair mutations from the mouse SCID site into the experimentally determined structure of the I-AniI-DNA complex.27 Inverse rotamers were constructed for a stretch of four base pairs in the DNA (positions −9 through −6 in the recognition site), spanning the three mutated base pairs and one wild-type base pair that was retained but falls within the mutated region. A 14-residue region of the protein backbone (positions 19–32) was allowed to move in an attempt to incorporate interaction motifs into the protein [shown in magenta in Fig. 3(B)]. When all applicable motifs are used to generate inverse rotamer libraries for the eight selected bases, 37,924 inverse rotamers are found which do not clash with the DNA or the fixed elements of the protein. These are further filtered to exclude those whose backbone atoms are sufficiently far from any backbone position that incorporation is impossible. This results in 19,208 inverse rotamers to consider.
Figure 3.
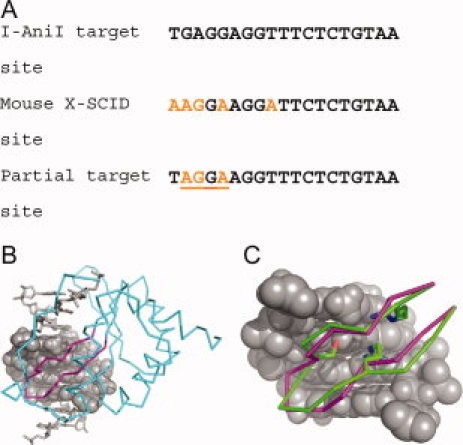
Test case for motif-directed relaxation. A: The wild-type recognition sequence for the homing endonuclease I-AniI is similar to a site found in exon six of the IL-2Rγ gene of a mouse model for SCID (severe combined immunodeficiency disease). To evaluate our algorithm, we used motif-directed relaxation to generate altered backbone conformations that could make interactions from our library with a target site incorporating three of the mutations required to change the I-AniI target site to the mouse SCID target site. B: The N-terminal domain of I-AniI and its DNA target half-site are shown with the three base pair changes mutated in silico. Inverse rotamers were built for the mutated base pairs and the intervening wild-type base pair (rendered in space-fill). The protein backbone region that was allowed to move to incorporate interaction motifs is colored magenta, with fixed regions of the protein colored cyan. C: The result of a three-motif loop relaxation (shown in green, with the incorporated motif side chains Arg20, Ser24, and Arg29 rendered as sticks) is overlaid on the native backbone conformation (in magenta).
Motif-directed backbone relaxation
As described in detail in the methods, we have developed a computational protocol that uses inverse rotamers to constrain backbone conformational sampling in an automated way (see Fig. 4). The coordinate rmsds between the Cβ, Cα, and C atoms of each inverse rotamer and the closest position in the protein are evaluated. Those within a cutoff value (1.2 Å was used for this study) are selected for incorporation [Fig. 4(A)], which is attempted in two steps. First, harmonic constraints are introduced between the Cβ, Cα, and C atoms of the inverse rotamer and the closest position in the backbone. The flexible region of the protein backbone is subjected to backrub conformational sampling28,29 under the Rosetta energy function augmented by the harmonic constraints. If the constraint score after conformational sampling is below a threshold, the inverse rotamer is placed onto the protein backbone position, otherwise the incorporation attempt is aborted [Fig. 4(B)]. However, after the backbone movement the constrained atoms will not overlay perfectly with the corresponding atoms in the inverse rotamer. As a result, the side chain atoms directly involved in the interaction will differ between the original inverse rotamer and the copy superimposed on the protein backbone [Fig. 4(C)]. A second round of conformational sampling is then performed in which the backbone constraints are removed, and side-chain constraints are imposed between the positions of the three motif-defining side-chain atoms before and after transfer onto the protein backbone. In essence, the constraints “pull” the side-chain atoms to their original, interacting locations [Fig. 4(D)]. Conformational optimization is again performed, and if the constraint score is below a threshold, the incorporation is considered successful.
Figure 4.
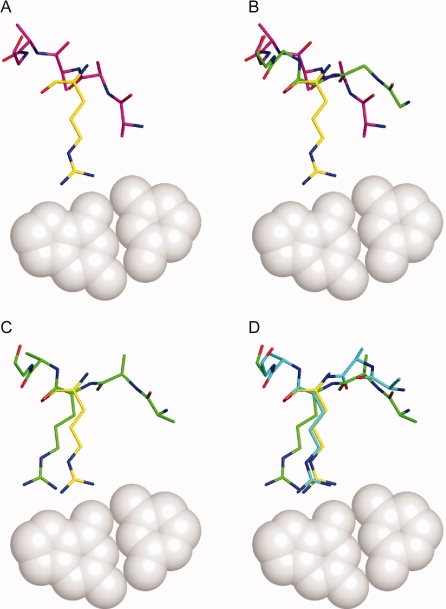
Single motif incorporation. A: An inverse rotamer (shown in yellow) is selected for incorporation if a subset of its backbone atoms (see Methods section) are approximately super imposable with any residue in the protein (starting conformation shown in magenta). B: Backbone conformational relaxation under a potential augmented with constraints to force the coincidence of inverse rotamer and protein backbone atoms yields an altered protein backbone (shown in green). The incorporation attempt is terminated if the final rmsd between the two sets of backbone atoms is above a threshold value. C: The inverse rotamer is superimposed onto the protein backbone (shown in green). Small differences in backbone atom positions (amplified by a lever arm effect along the side chain) result in the displacement of the side chain functional atoms from the original interacting positions. D: A second round of relaxation is performed with constraints between the functional atoms of the original and superimposed inverse rotamers to restore the desired interaction (final conformation show in cyan).
Successful incorporation of an interaction motif yields a structure with an altered backbone in the flexible region of the protein, and a mutated amino acid making a contact with the DNA. This structure is then used as the starting point for a further round of motif incorporation (see Fig. 5). Because the backbone has been altered, the set of inverse rotamers that will be considered for incorporation will be different from that used during previous relaxation attempts. The number of motifs to incorporate is typically limited to three, as the calculation time required to explore all combinations of rotamers increases significantly with the number of desired motifs.
Figure 5.
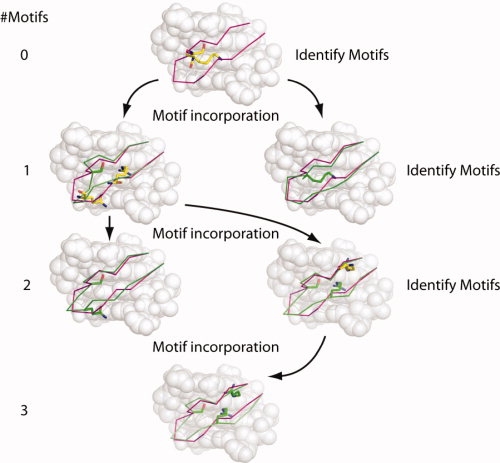
Incorporation of multiple motifs. The starting (wild-type) backbone conformation is shown throughout in magenta, and altered backbones and incorporated motifs are shown in green. Unincorporated motifs that are identified as close to the current backbone are shown with yellow carbon atoms. The number of motifs incorporated at each level is indicated on the left side of the figure. Each round of incorporation begins by identifying those inverse rotamers whose backbone atoms may be made to coincide with corresponding atoms in the flexible protein region with small perturbations of the protein backbone. For each of these inverse rotamers, the backbone relaxation protocol is applied in an attempt to “thread” the protein backbone through the main chain atoms of the inverse rotamer. If successful, the rotamer is transplanted onto the protein backbone (and that position disallowed from downstream incorporation), and the altered conformation serves as a starting point for the next round of incorporation. As the algorithm proceeds, inverse rotamers considered too far from the initial conformation may be identified as close enough to altered backbones to attempt incorporation in later rounds. The procedure takes the form of a tree, in which the starting conformation is the root, and each successful incorporation of an inverse rotamer begins a new branch. Along each branch of the tree, the procedure terminates when no inverse rotamers are found to attempt another round (first right-hand branch), or when a specified number of motifs have been incorporated (final branch with three motifs).
Relative efficiencies of motif-directed relaxation and undirected backbone rebuilding
The relaxation procedure used to move the protein backbone to coincide with the inverse rotamers involves a nontrivial amount of computation. To ensure that such effort was warranted, we compared motif-directed backbone relaxation with three alternative methods for generating a diverse set of backbone conformations that are not motif-directed. First, we constructed models of the I-AniI homing endonuclease with the flexible backbone region (positions 19–32) replaced with the structurally homologous regions from other homing endonuclease structures. The homologous positions were: 151–167 Z from I-AniI (pdb code: 2qoj27), 25–41 A from I-CreI (pdb code: 1g9y30), 71–87 A from I-CeuI (pdb code: 2ex531), 24–38 A from I-DmoI (pdb code: 2vs732), and 27–44 A from I-MsoI (pdb code: 1m5x33). Second, we created models where the flexible region was replaced with extended fragments of similar length taken from the protein database with start and end residues that could be oriented to overlay simultaneously on the original residues. Because these methods generated a relatively small number of alternate backbones and were rarely compatible with the motifs in our library, we also used an algorithm for rebuilding backbone segments from peptide fragments to generate a much larger set of backbone conformations.34 All of these methods result in reasonable backbone conformations that have not undergone any selection for the ability to make favorable interactions with DNA. As a result, performance for all three approaches was similar, and we present only the data for the rebuilt backbone conformations, for which we have by far the largest test set.
The same backbone region subjected to motif-directed relaxation above was rebuilt 2000 times using peptide fragments and energy-minimized using the all-atom potential.35 A comparison of the number of motifs found to be compatible with each of these loops is shown in Table I. The number of single motifs found to be compatible with the rebuilt loops gives an estimate of how often loops selected or constructed without regard for the inverse rotamers will be suitable for incorporating a motif. Approximately one in every six loops can accommodate a motif from our library. If only one motif is required, the directed approach is marginally more efficient: more motifs are found per unit time than for undirected loop rebuilding. However, the chance of simultaneously accommodating two motifs in a rebuilt loop drops off sharply—this occurs only once every 286 loops. Per unit time, the directed approach generates ∼48 times as many two-motif loop conformations as the undirected approach. We did not observe any rebuilt backbones incorporating three interactions. Although exploring all possible three-motif combinations is more time consuming (taking four times as long as exploring all two-motif combinations), our method readily identifies 134 triple motif-incorporating backbones.
Table I.
Comparison of Backbone Rebuilding and Motif-Directed Backbone Relaxation for Incorporating Inverse Rotamers
| Method for generating backbone diversity |
||
|---|---|---|
| No. of motifs incorporated | Motif-directed | Backbone rebuilding |
| 1 | 10 (4.5 h)a | 341 |
| 2 | 58 (45 h)a | 7 |
| 3 | 134 | 0 |
| Run time | 173 h | 260 h |
Times in parentheses show the runtimes if the motif-directed search is terminated at less than three motifs. Thus, all single motifs are found in 4.5 h.
The motif-directed approach also yields backbones with smaller Cα rmsd to the starting structure (data not shown) which could prove advantageous for further modeling or design applications, as it minimizes the impact of the introduced perturbation on the rest of the protein structure. The Cα rmsd values between the starting and ending conformations range from 0.66 to 2.6 Å (average of 1.34 Å). Although some of the more perturbed loops may be initially incompatible with the scaffold protein, we anticipate that downstream steps in design protocols will entail further rounds of iterative relaxation and design, and we prefer not to filter candidate backbones before further refinements and corrections may be applied. In all the altered loops, motifs were incorporated into eight different positions, with a total of 20 distinct amino acid-position combinations found.
Motif-directed modeling of homologous loop regions
To assess the motif-based protocol's suitability for homology modeling, we modeled the structural response of the I-CeuI protein–DNA complex31 when the +5 to +12 base pairs and the residues in positions 69–89 A were mutated to the corresponding bases and amino acids in the I-CreI protein–DNA complex30 (base pairs +5 to +12 and amino acid positions 23–43A; the base pairs differ at four of eight positions in this range). These structures were chosen because they have the same length throughout the flexible region and have similar conformations in the turn between the beta strands. Thus, even though significant lateral movement is required to superimpose one on the other (the Cα rmsd between the two is 2.6 Å over the 21 residues, with some corresponding Cα atoms differing by ∼3.2 Å), the I-CeuI loop serves as a reasonable starting template for comparative modeling. Furthermore, the I-CreI protein makes several commonly occurring direct interactions to DNA that, if recognized, can guide conformational relaxation [Fig. 6(A)].
Figure 6.

Transition between homologous loop conformations. A: The N-terminal half-site of the I-CeuI crystal structure31 was used as the starting point for a test of loop homology modeling. The flexible protein backbone region is shown in magenta. The sequence of DNA was computationally altered to match the recognition sequence for the I-CreI homing endonuclease30 (rendered in grey spheres). When the I-CeuI and I-CreI structures are structurally aligned using only the atoms of the phosphate backbone and deoxyribose rings, it is seen that the strand-turn-strand regions for the two enzymes adopt different orientations (Cα rmsd of 2.6 Å over the 21 residue loop region) with respect to the major groove (I-CreI loop shown in blue). Three direct contacts between the I-CreI homing endonuclease and the DNA are rendered as sticks (Lys 28A, Asn 30A, and Gln 38A). B: After motif-driven backbone relaxation was performed using the I-CeuI backbone conformation as the starting point and the I-CreI recognition sequence as the target, a number of altered backbone loops incorporating motifs consistent with the I-CreI protein sequence were generated. The loop with the smallest Cα rmsd to the I-CreI backbone (1.5 Å) is shown in green. Three inverse rotamers were incorporated during the process, shown in blue sticks for comparison with the experimentally determined I-CreI side chains. C: Close-up view with the native I-CreI and incorporated motif interactions shown in blue and green, respectively. D: Backbone traces for the I-CeuI (magenta), I-CreI (blue), and the altered loop regions (green) are shown for comparison.
We assumed that the sequence alignment of the I-Ceul protein into the I-Crel protein and the I-Ceul binding site onto the I-Crel binding site are both known. Thus, although inverse rotamers for all appropriate motifs were constructed at each base of the binding, backbone relaxation and motif incorporation were only attempted when the amino acid identity of the motif matched the I-CreI sequence at the corresponding closest backbone position. To prevent the remainder of the I-CeuI template from limiting backbone conformational freedom, the helical secondary structure elements that pack against the flexible region (positions 129–211A) were omitted from energy calculations during the relaxation protocol. Shown in Figure 6(B) is a resulting altered loop that has incorporated three inverse rotamers that make I-CreI-like interactions with the DNA (in green) overlaid with the analogous interactions and backbone from the I-CreI crystal structure (in blue). All three incorporated motifs are derived from the AANT database. A closer view of the two sets of interactions is shown in Figure 6(C). The movement of the backbone is substantial: the Cα rmsd between the I-CeuI and I-CreI backbones [shown in magenta and blue, respectively, in Fig. 6(D)] is 2.6 Å, whereas the rmsd between the altered loop [shown in green in Fig. 6(D)] and the I-CreI backbone is 1.5 Å. However, the protocol generates many loop conformations and incorporates some inverse rotamers that make contacts unlike those found in I-CreI. To become a practical tool for homology modeling, it will be necessary not only to generate native-like backbone orientations, but also to identify them with our scoring function. This will involve predicting and evaluating the conformations of the remaining side chains in the flexible region as well as reintroducing the surrounding secondary structural elements computationally removed to maximize the conformational freedom of the loop region. The construction of a more complete homology modeling computational pipeline is currently under development.
Discussion
Motif-based interactions for protein–DNA modeling and design
Interactions between specific amino acids and bases were predicted even before atomic resolution structures of protein–DNA complexes were available.36 This led to hope that a purely sequence-based recognition code for protein–DNA interactions might be possible. However, structural work has demonstrated that each of the bases in DNA may be recognized by different amino acids, and furthermore may interact in more than one mode with the same amino acid.37 Pabo and Nekludova38 analyzed the geometric relationship between bases and the backbone atoms of amino acid residues to determine which DNA bases may be productively contacted by a given position in a protein, given the relative orientation of the backbone to the DNA. The interaction motif description we utilize here specifies instead the relative orientation between the base and the side-chain atoms with which it interacts. The backbone positions capable of making these interactions are determined by assuming that the amino acid conformations are consistent with a rotamer library. Because our method retains the precise orientation between directly interacting residues but allows for multiple main chain locations consistent with the interaction, it can be viewed as generalization of the geometric description of interactions developed by Pabo and Nekludova.
Comparison with other approaches to backbone diversity
When redesigning a protein–DNA interface, an experimentally determined structure of a protein–DNA complex is used as a template to model a different DNA sequence, and mutations on the protein side of the interface are introduced to recover complementarity across the interface. Unfortunately, the interactions that provide this complementarity require geometric precision not achievable with a fixed protein backbone. As a result, backbone flexibility is required to realize favorable interactions. When the only optimization goal is to minimize the system energy, high-resolution refinement using the energy function as a guide is an excellent choice, and efficient methods may be appropriated from structure prediction protocols. When additional functional constraints are present, beneficial changes in backbone conformation are in general unknown until evaluated for some desired quality.
In the case of protein–DNA interactions, the ability to make one or more interactions previously observed in native complexes can serve as a metric for whether a backbone conformation is useful for recognizing a given DNA sequence. We have found that the geometric precision involved in the exquisite interactions between proteins and DNA is such that simply screening large ensembles of backbone conformations is inefficient when a single interaction is required, and becomes more so for multiple interactions. In contrast, utilizing high-resolution structural relaxation techniques to adapt a starting backbone conformation to incorporate a library of interaction motifs can generate minimally perturbed backbones capable of realizing several interactions with DNA.
Of course, there is no guarantee that a given starting backbone conformation can be adapted to bind specifically to arbitrary DNA sequences. Our method can be extended by diversifying the starting backbones available for a given protein scaffold. In the case of the I-AniI homing endonuclease, structurally homologous regions from other homing endonucleases can be grafted on the protein. Because different homing endonucleases recognize different DNA sequences, using the conformation of a homologous region from a different homing endonuclease may permit the transfer of its sequence preferences. By judiciously selecting recognition elements most similar to a desired target site, the amount of perturbation required for a protein conformation to conform to a new DNA sequence can be minimized.
Although we intentionally selected a search algorithm (backrub relaxation29) that samples backbone conformations relatively close to the starting conformation, we note that the amount of backbone flexibility can be increased by other algorithms for generating diversity. The alternate loop conformations generated by the de novo fragment-based construction algorithm can be used as starting points for the inverse rotamer selection and optimization protocol described in the Methods section, biasing the resulting loop regions toward non-native conformations. Alternatively, the constraints that are specified by the inverse rotamer construction can be incorporated into the fragment-based loop generation algorithm itself, providing for another route toward non-native but motif-incorporating backbone conformations.
Applications beyond protein–DNA design
In many respects, the design of DNA-binding proteins is an ideal application for a strategy involving a library of previously observed interactions. Nucleic acid duplexes present the atoms in their bases in a relatively uniform context, suggesting that modes of interaction may be transferable. Furthermore, bases are observed to interact with different amino acids in different interfaces. This makes an approach based on a library of interactions desirable, as multiple interactions and combinations of interactions can be evaluated as potential solutions to a design problem. Also important is the availability of numerous interaction motifs in structural databases and previous literature.20,21
Another application for this method is the homology modeling of protein–DNA complexes. Even subtle differences between homologous structures can be problematic when attempting to use one as a template for generating models of the other.39,40 Relaxation of structural models is often necessary to improve on models generated from a homologous template alone. Our results demonstrate that motif-directed relaxation can generate alternate backbones from a starting template that incorporate plausible interactions with altered DNA. This ability to identify potential interactions made by residues that differ between the template and target structures by analogy to previously observed motifs can greatly improve the efficiency of these methods by providing a constraint on the conformational degrees of freedom and effectively reducing the search space.
A third application that could benefit from this approach is enzyme and protein–protein interaction specificity redesign and optimization. In these applications, the analogs to previously observed protein–DNA interactions are either interactions between catalytic residues and substrates or amino acid-amino acid interactions between interacting proteins. Given a starting complex of an enzyme or protein–protein interface, specific side-chain interactions can be made with a disembodied inverse rotamer library on the target substrate or protein, and the approach developed here used to mold the backbone of the enzyme or protein partner to realize the desired functional side-chain interactions.
Our algorithm generates altered backbones that can incorporate interactions taken from a predefined library. An implicit assumption in this method is that any interaction is assumed to be as desirable as another. However, in certain design problems, a particular interaction may be required. A complementary algorithm that devotes its computational resources towards the focused incorporation of a required interaction has recently been developed in our group (P.M. Murphy, J.M. Bolduc, J.L. Gallagher, B.L. Stoddard, and D. Baker, in press).
Future work
We anticipate that, similar to rotamer-based side chain placement, motif-directed backbone placement will be an important subtask of more complex protocols. We are currently working to improve the efficiency our method by assembling a more compact and nonredundant motif library from a fresh analysis of currently available protein–DNA complexes, and extending the scope of interactions to include those mediated by water. We are also developing and evaluating different protocols for incorporating motif-directed loop placement into protein–DNA homology modeling and design.
Our algorithm produces multiple alternate backbone conformations with a small number of incorporated interactions. Each of these represents the starting point for a complete design calculation. We are currently exploring design protocols in which each of these starting structures is subjected to multiple (∼10) rounds of redesign and refinement, during which the amino acid identities of the incorporated motifs are held fixed. In most cases, this will yield too many final protein sequences to test experimentally. One resolution to this problem is to select a small number of sequences to evaluate based on our scoring function (ranking by predicted affinity perhaps in combination with predicted specificity). Another approach under investigation is to use the full set of designed sequences to generate an initial library for directed evolution.
Methods
Assembly of motif library
Interactions between amino acid side chains and the bases of nucleic acids in the major groove were collected from several sources. First, bidentate interactions involving two hydrogen bonds between an amino acid and one or two bases were taken from the set identified by Thornton and coworkers.20 Second, polar interactions between single amino acids and bases were taken from the AANT database.21 Finally, hydrophobic interactions were identified by visual inspection of all protein–DNA complexes involving regulatory and enzymatic proteins. For each interaction, three atoms on both the amino acid and the base were selected to define a coordinate system. The translation vector and Euler angles relating the coordinate systems were determined. Each interaction motif is defined by the identities of the amino acid and the base(s), the coordinate system defining atoms from the amino acid and base(s), the components of the translation vector, and the Euler angles. For some motifs involving rotatable hydroxyl groups, the polar hydrogen is included as one of the coordinate system defining atoms, and in these cases, the hydrogen are placed using the Rosetta molecular modeling program before the geometric parameters are calculated. The complete set of motifs used is given in the Supporting Information data.
Construction of an inverse rotamer library
To determine whether a protein backbone in a complex with DNA is capable of realizing a given interaction between a base and an amino acid described in the motif library, all possible torsional states of the amino acid must be constructed and properly oriented relative to the base, and the resulting backbone fragments compared with the backbone of the protein. For a specified subset of base pairs in a protein–DNA complex, we construct a set of isolated amino acids for which the side-chain atoms realize an interaction in the motif library and the main chain atoms are colocalized with corresponding atoms in the pre-existing protein backbone. For simplicity and efficiency, we assume that the possible conformations of the amino acids involved in the motif interaction are well described by a rotamer library. In most applications, rotamer libraries are used to describe the side chain conformations that are possible given a fixed backbone location. Here, we “invert” the library such that all members are superimposed on the three atoms that define the motif interaction. When this set of structurally aligned singleton residues is placed opposite the corresponding motif-defining atoms of a nucleic acid base, the backbone atoms of the members of the set describe (within the rotamer approximation) the possible backbone locations capable of realizing the interaction. For each specified base pair in the complex, inverse rotamer libraries are constructed for each applicable motif. Inverse rotamers that clash with the DNA or the fixed regions of the protein (outside the designated flexible loop), or that have main chain atoms too far from any protein residue (rmsd > 3.6 Å over the Cβ, Cα, and C atoms for all protein positions) are discarded.
Backbone assembly from fragments
Alternate backbone conformations were generated by rebuilding specified backbone segments using protein fragment insertion.41 The protocol was developed for high-resolution refinement of protein models for structure prediction, and has been described in detail elsewhere.34 Before full-atom backbone rebuilding, all residues in the rebuilt region were mutated to alanine, except positions occupied by glycine and proline, which were retained. Fragments for the native I-AniI sequence were selected as for protein structure prediction, and random 9-mer, 5-mer, or 1-mer fragments were inserted into the specified backbone segment. The integrity of the protein backbone was enforced using a cyclic coordinate descent loop closure algorithm.42 Acceptance of each attempted insertion was determined using the Metropolis criterion.43 Fragment insertion proceeded through 10 rounds, with the penalty for chain breaks in the backbone gradually increasing with subsequent rounds.
Motif-directed backbone movement
Backbone segments were modified to overlap with the backbone atoms of target motifs in the inverse rotamer library in two steps. First, the backbone conformation was optimized to minimize the Rosetta energy function with additional harmonic constraints between the Cβ, Cα, and C atoms of a target motif and the corresponding atoms in the residue in the protein backbone region initially closest to the target motif. A spring constant of 20.0 kcal/(mol Å2) was used for the constraint energy term. If no backbone position in the flexible region had an initial rmsd below a cutoff value for a given inverse rotamer (1.2 Å for the calculations summarized in Table I), incorporation was not attempted. Monte Carlo minimization using “backrub” moves was used to sample backbone conformations.29 If the final value for the constraint energy is above a given cut-off, corresponding to an rmsd of 0.5 Å over the three atoms, incorporation of the inverse rotamer is abandoned. Otherwise, a copy of the inverse rotamer is superimposed onto the backbone atoms at the position, and the protein is modified to adopt the chemical identity and conformation of the inverse rotamer at that position [see Fig. 4(C)]. Because the match between the backbone atoms of the protein and the motif will not be exact, a second round of minimization is performed to overlay the side chain atoms that define the interaction with the DNA for the motif. For this second round, the constraints on the backbone atoms are removed, and harmonic constraints are applied between the motif-defining side chain atoms in the motif and on the newly placed residue in the protein. If, after this second round of backrub minimization, the rmsd between the side chain atoms are below the cut-off, structural data for the complex, including the mutated position and the perturbed backbone coordinates are written to a file for further modeling or design.
The structure that results from incorporation of a motif into a protein structure also serves as an initial structure for further attempts to incorporate more motifs. When used in this way, the possible combinations of motifs that may be incorporated into an existing protein backbone takes the form of a tree (see Fig. 5), with each node of the tree corresponding to either the starting structure or a structure with one or more incorporated motifs. Edges branch off from these nodes for each additional motif that may be incorporated. This tree is searched in a depth-first manner, and branches in the tree that correspond to motifs that cannot be incorporated are pruned. Typically the depth of the tree structure that is explored (corresponding to the maximum number of motifs to be incorporated) is limited, as the number of combinations increases rapidly at each level.
Retrieval of I-CreI loop conformation in I-CeuI scaffold
Chain A from the I-CeuI crystal structure (pdb code: 2ex531) was used as the starting point for this calculation. The DNA sequence from bases +5 to +12 in the I-CreI structure (pdb code: 1g9y)30 was modeled into the corresponding positions in the I-CeuI structure. To remove any steric hindrance from surrounding residues, positions 129–211A were removed from the model during the calculation. Inverse rotamers were constructed, and loop relaxation and motif incorporation were performed as above with two modifications. First, because the displacements between corresponding Cα atoms in the I-CeuI and I-CreI structures are know to be large [≥3.0 Å in some cases; see Fig. 6(A)], the rmsd cutoff beyond which inverse rotamer incorporation is not attempted was increased to 3.5 Å. Second, motif incorporation was only attempted when the amino acid in the motif matched that of I-CreI at the corresponding position.
Conclusions
The fixed backbone approximation used to facilitate side chain repacking and protein design calculations carries with it certain limitations. The combination of a fixed backbone with a limited set of rotamers for describing side chain conformational freedom does not provide adequate sampling for applications where geometrically precise interactions such as hydrogen bonding dominate. This is the case for modeling protein–DNA interfaces, where slight errors in backbone positions can be amplified through long side chains, making the identification of correct contacts or the selection of optimal amino acids for altered target bases very difficult. We have presented a method for identifying potential interactions between protein and DNA by analogy to observed interactions, and for generating minimally altered protein backbones capable of making these interactions. The method is more efficient at generating modified backbones capable of realizing multiple interactions than the alternative of generating ensembles of backbones from peptide fragments and screening these for close matches to motifs. We expect that this approach of automated enforcement of constraints by analogy to pre-existing systems will find broad use beyond protein–DNA modeling alone.
The algorithm described has been implemented in the C++ language as part of the Rosetta++ molecular modeling suite (version 2.3.0). The source code is available free of charge to academic users.
Acknowledgments
The authors thank Colin Smith and Tanja Kortemme for sharing code for backrub minimization prior to publication, Barry Stoddard for sharing the 2QOJ crystal structure prior to publication, and Justin Ashworth and John Karanicolas for helpful discussions.
References
- 1.Pabo C. Molecular technology-designing proteins and peptides. Nature. 1983;301:200–200. doi: 10.1038/301200a0. [DOI] [PubMed] [Google Scholar]
- 2.Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 3.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 4.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 5.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 6.Malakauskas SM, Mayo SL. Design, structure, and stability of a hyperthermophilic protein variant. Nat Struct Biol. 1998;5:470–475. doi: 10.1038/nsb0698-470. [DOI] [PubMed] [Google Scholar]
- 7.Dantas G, Kuhlman B, Callender D, Wong M, Baker D. A large scale test of computational protein design: folding and stability of nine completely redesigned globular proteins. J Mol Biol. 2003;332:449–460. doi: 10.1016/s0022-2836(03)00888-x. [DOI] [PubMed] [Google Scholar]
- 8.Shah PS, Hom GK, Ross SA, Lassila JK, Crowhurst KA, Mayo SL. Full-sequence computational design and solution structure of a thermostable protein variant. J Mol Biol. 2007;372:1–6. doi: 10.1016/j.jmb.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 9.Pabo CO, Suchanek EG. Computer-aided model-building strategies for protein design. Biochemistry. 1986;25:5987–5991. doi: 10.1021/bi00368a023. [DOI] [PubMed] [Google Scholar]
- 10.Sauer RT, Hehir K, Stearman RS, Weiss MA, Jeitler-Nilsson A, Suchanek EG, Pabo CO. An engineered intersubunit disulfide enhances the stability and DNA binding of the N-terminal domain of lambda repressor. Biochemistry. 1986;25:5992–5998. doi: 10.1021/bi00368a024. [DOI] [PubMed] [Google Scholar]
- 11.Hellinga HW, Caradonna JP, Richards FM. Construction of new ligand binding sites in proteins of known structure. II. Grafting of a buried transition metal binding site into Escherichia coli thioredoxin. J Mol Biol. 1991;222:787–803. doi: 10.1016/0022-2836(91)90511-4. [DOI] [PubMed] [Google Scholar]
- 12.Coldren CD, Hellinga HW, Caradonna JP. The rational design and construction of a cuboidal iron-sulfur protein. Proc Natl Acad Sci USA. 1997;94:6635–6640. doi: 10.1073/pnas.94.13.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinto AL, Hellinga HW, Caradonna JP. Construction of a catalytically active iron superoxide dismutase by rational protein design. Proc Natl Acad Sci USA. 1997;94:5562–5567. doi: 10.1073/pnas.94.11.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benson DE, Wisz MS, Liu W, Hellinga HW. Construction of a novel redox protein by rational design: conversion of a disulfide bridge into a mononuclear iron-sulfur center. Biochemistry. 1998;37:7070–7076. doi: 10.1021/bi980583d. [DOI] [PubMed] [Google Scholar]
- 15.Wisz MS, Garrett CZ, Hellinga HW. Construction of a family of Cys2His2 zinc binding sites in the hydrophobic core of thioredoxin by structure-based design. Biochemistry. 1998;37:8269–8277. doi: 10.1021/bi980718f. [DOI] [PubMed] [Google Scholar]
- 16.Benson DE, Haddy AE, Hellinga HW. Converting a maltose receptor into a nascent binuclear copper oxygenase by computational design. Biochemistry. 2002;41:3262–3269. doi: 10.1021/bi011359i. [DOI] [PubMed] [Google Scholar]
- 17.Jiang L, Kuhlman B, Kortemme TA, Baker D. A “solvated rotamer” approach to modeling water-mediated hydrogen bonds at protein-protein interfaces. Proteins. 2005;58:893–904. doi: 10.1002/prot.20347. [DOI] [PubMed] [Google Scholar]
- 18.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 19.Delano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. Available at: http:/www.pymol.org. accessed June 2008. [Google Scholar]
- 20.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman MM, Khrapov MA, Cox JC, Yao J, Tong L, Ellington AD. AANT: the amino acid-nucleotide interaction database. Nucleic Acids Res. 2004;32:D174–D181. doi: 10.1093/nar/gkh128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, Rothlisberger D, Baker D. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15:2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, III, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29:3757–3774. doi: 10.1093/nar/29.18.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer A. Severe combined immunodeficiencies (SCID) Clin Exp Immunol. 2000;122:143–149. doi: 10.1046/j.1365-2249.2000.01359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scalley-Kim M, McConnell-Smith A, Stoddard BL. Coevolution of a homing endonuclease and its host target sequence. J Mol Biol. 2007;372:1305–1319. doi: 10.1016/j.jmb.2007.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis IW, Arendall WB, III, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Smith CA, Kortemme T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J Mol Biol. 2008;380:742–756. doi: 10.1016/j.jmb.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chevalier BS, Monnat RJ, Jr, Stoddard BL. The homing endonuclease I-CreI uses three metals, one of which is shared between the two active sites. Nat Struct Biol. 2001;8:312–316. doi: 10.1038/86181. [DOI] [PubMed] [Google Scholar]
- 31.Spiegel PC, Chevalier B, Sussman D, Turmel M, Lemieux C, Stoddard BL. The structure of I-CeuI homing endonuclease: evolving asymmetric DNA recognition from a symmetric protein scaffold. Structure. 2006;14:869–880. doi: 10.1016/j.str.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Marcaida MJ, Prieto J, Redondo P, Nadra AD, Alibes A, Serrano L, Grizot S, Duchateau P, Paques F, Blanco FJ, Montoya G. Crystal structure of I-DmoI in complex with its target DNA provides new insights into meganuclease engineering. Proc Natl Acad Sci USA. 2008;105:16888–16893. doi: 10.1073/pnas.0804795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chevalier B, Turmel M, Lemieux C, Monnat RJ, Jr, Stoddard BL. Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J Mol Biol. 2003;329:253–269. doi: 10.1016/s0022-2836(03)00447-9. [DOI] [PubMed] [Google Scholar]
- 34.Qian B, Raman S, Das R, Bradley P, McCoy AJ, Read RJ, Baker D. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–264. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das R, Qian B, Raman S, Vernon R, Thompson J, Bradley P, Khare S, Tyka MD, Bhat D, Chivian D, Kim DE, Sheffler WH, Malmstrom L, Wollacott AM, Wang C, Andre I, Baker D. Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home. Proteins. 2007;8(69 Suppl):118–128. doi: 10.1002/prot.21636. [DOI] [PubMed] [Google Scholar]
- 36.Seeman NC, Rosenberg JM, Rich A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc Natl Acad Sci USA. 1976;73:804–808. doi: 10.1073/pnas.73.3.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthews BW. Protein-DNA interaction. No code for recognition. Nature. 1988;335:294–295. doi: 10.1038/335294a0. [DOI] [PubMed] [Google Scholar]
- 38.Pabo CO, Nekludova L. Geometric analysis and comparison of protein-DNA interfaces: why is there no simple code for recognition? J Mol Biol. 2000;301:597–624. doi: 10.1006/jmbi.2000.3918. [DOI] [PubMed] [Google Scholar]
- 39.Havranek JJ, Duarte CM, Baker D. A simple physical model for the prediction and design of protein-DNA interactions. J Mol Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 40.Morozov AV, Havranek JJ, Baker D, Siggia ED. Protein-DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 42.Canutescu AA, Dunbrack RL. Cyclic coordinate descent: a robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Metropolis N, Rosembluth A, Rosembluth M, Teller A. Equation of state calculations by fast computing machines. J Chem Phys. 1953;21:1087–1092. [Google Scholar]