

Abstract
The assembly of major histocompatibility complex (MHC) class I molecules with peptides is orchestrated by several assembly factors including the transporter associated with antigen processing (TAP) and tapasin, the endoplasmic reticulum (ER) oxido-reductases ERp57 and protein disulfide isomerase (PDI), the lectin chaperones calnexin and calreticulin, and the ER aminopeptidase (ERAAP). Typically, MHC class I molecules present endogenous antigens to cytotoxic T lymphocytes (CTLs). However, the initiation of CD8+ T-cell responses against many pathogens and tumors also requires the presentation of exogenous antigens by MHC class I molecules. We discuss recent developments relating to interactions and mechanisms of function of the various assembly factors and pathways by which exogenous antigens access MHC class I molecules.
Introduction
The interaction between a T-cell receptor (TCR) on a cytotoxic T lymphocyte (CTL) and a major histocompatibility complex (MHC) class I peptide complex on an antigen presenting cell (APC) initiates many antiviral and antitumor immune responses. MHC class I molecules comprise a membrane-linked heavy chain, a soluble light chain [β2-microglobulin (β2m)] and a short peptide bound in a groove of the membrane-distal domains of the heavy chain. Complexes of MHC class I heavy chains, β2m and peptide are typically assembled in the endoplasmic reticulum (ER) of cells through a complex assembly pathway (reviewed in Ref. [1]). In the classical MHC class I assembly pathway, cytosol-derived proteins that are endogenous to cells are processed into peptides by the proteasome. The proteasome generates peptides optimized for MHC class I binding at the C terminus but extended at the N terminus relative to peptides bound to mature MHC class I molecules (reviewed in Ref. [2]). N-extended peptides are transported into the ER lumen by the transporter associated with antigen processing (TAP). In the ER, MHC class I heterodimers lacking peptides are recruited into complexes with the TAP transporter, in an interaction bridged by tapasin (Figure 1). ERp57, protein disulfide isomerase (PDI) and calreticulin also associate with the TAP-tapasin-MHC class I complex to form a large complex called the peptide loading complex (PLC) [1] (Figure 1; PDI not shown). Components of the PLC facilitate peptide binding to MHC class I molecules. ERAAP cleaves N-extended peptides at the N terminus, optimizing MHC class I peptide associations (reviewed in Ref. [2]). Peptide occupancy triggers dissociation of MHC class I from the PLC, exit from the ER and transit to the cell surface, where the MHC class I molecules become available for immune surveillance by T cells. This general pathway is relevant to the assembly of MHC class I peptide complexes in virally infected cells and tumor cells and to the subsequent immune recognition of such cells. Recent developments relating to this pathway will be reviewed in this article. Additionally, the initial priming of naïve CD8+ T cells into effector CTLs requires the processing of exogenous particulate and soluble antigens by dendritic cells (DCs) and presentation of such antigens via the MHC class I molecules of the DCs. This phenomenon is called cross-priming and is a key mechanistic step in the initial activation of CTL responses against tumor cells, viruses and other intracellular pathogens (reviewed in Ref. [3]). This review also summarizes recent information about cellular pathways used by antigen presenting cells (APCs) for the presentation of exogenous antigens to CD8+ T cells.
Figure 1.
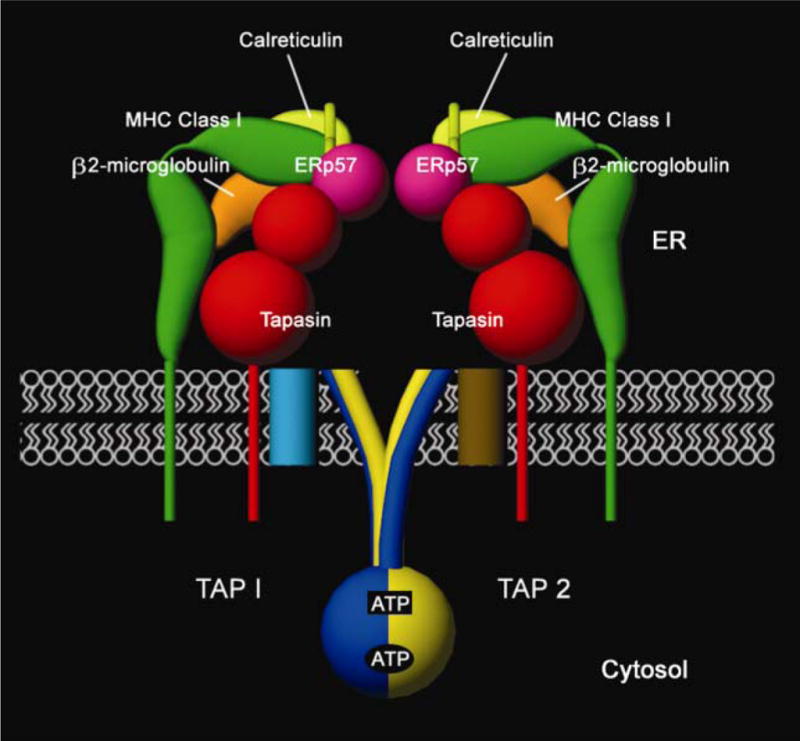
Components of the peptide-loading complex in the endoplasmic reticulum (ER). Major histocompatibility complex (MHC) class I heavy chains (green) and β2m (orange) are found in association with tapasin (red) and the TAP1-TAP2 (blue and yellow, respectively) complex. Many generic ER factors are also found in association with TAP, tapasin and MHC class I, including calreticulin (light green), ERp57 (purple) and PDI (not shown), which together constitute a complex called the PLC [1]. This complex forms an intricate molecular machine that recruits peptide-deficient heterodimers of heavy chains and β2m, transports peptide cargo from the cytosol to the ER, facilitates assembly of peptide with MHC class I heterodimers and ensures appropriate release of peptide-loaded MHC class I molecules. TAP1 and TAP2 each contain a cytosolic NBD and a transmembrane region with multiple membrane-spanning segments. The N-terminal region of the TAP transmembrane domain (light blue and brown cylinders in the membrane) forms a separate tapasin binding domain of the TAP complex [5–7]. The cytosolic face of TAP1-TAP2 complexes depicts the predicted closed-state NBD dimer [9,10]. Two ATP molecules are sandwiched at the NBD dimer interface; between the Walker A region of TAP1 NBD and the signature motif of TAP2 NBD (lower oval-shaped site, TAP1 site) and between the Walker A region of TAP2 NBD and the signature motif of TAP1 NBD (upper rectangular site, TAP2 site). The TAP2 site is the main catalytic site driving peptide transport [10–13]. The figure depicts a hypothetical scheme of interactions between the luminal domains of tapasin, ERp57, calreticulin and MHC class I, and the precise nature of the interactions remains to be defined. Only the tapasin-binding domain of ERp57 is depicted. ER, endoplasmic reticulum; ERp57, ER oxidoreductase of 57 KDa; NBD, nucleotide binding domain; PDI, protein disulfide isomerase; PLC, peptide loading complex; TAP, transporter associated with antigen processing.
Transport of peptides for MHC class I antigen presentation
The role of TAP is to transport peptides from the cytosol into the ER. Two subunits, TAP1 and TAP2 (also known as ABCB2 and ABCB3, respectively; Figure 1), are required for peptide transport. TAP1 and TAP2 belong to the ATP binding cassette (ABC) family, transmembrane proteins that function in the ATP-dependent transport of various substrates across cellular membranes. TAP1 and TAP2 each contain a cytosolic nucleotide binding domain (NBD) and a transmembrane region with several predicted membrane-spanning segments (reviewed in Ref. [4]). Compared with other ABC transporters, TAP1 and TAP2 contain additional N-terminal transmembrane segments (Figure 1) that form the accessory domains for tapasin binding and MHC class I bridging [5–7]. Peptide translocation by TAP is preceded by peptide binding to TAP, and cytosol to ER translocation of peptides requires nucleotide hydrolysis (reviewed in Ref. [8]). Crystal structures of TAP1 NBD and of homologous ABC transporters have predicted that, at some point during the transport cycle, two ATP molecules are bound at the interface between TAP1 and TAP2 NBD dimers, [9,10], generating a ‘closed conformation‘ of the NBDs. The TAP1 nucleotide binding site contains naturally occurring modifications that attenuate its ATPase activity, and the TAP2 site is the main catalytically active site driving peptide transport [10–13].
There is emerging consensus that substrate transport by ABC transporters might involve nucleotide-dependent reorganizations between two main conformational states [14–16], as depicted in Figure 2 for peptide transport by TAP. The question of what molecular events initiate the catalytic cycle of ABC transporters and determine transitions between these two conformational states is of major interest to the field. The catalytic cycle of TAP is a target for immune evasion by different viruses. In addition to the well-known TAP inhibitors ICP47 and US6 encoded by herpes simplex viruses and human cytomegalovirus, respectively (reviewed in Ref. [4]), recent studies have revealed that the UL49.5 proteins of bovine herpesvirus 1, pseudorabies virus and equine herpesviruses 1 and 4 target different steps of the TAP catalytic cycle [17]. Additionally, the BNFL2a protein of Epstein Barr virus and homologous proteins of other viruses are also inhibitors of peptide and nucleotide binding by TAP [18]. A better understanding of the TAP catalytic cycle will allow further insights into mechanisms that viruses use to suppress the antigen shuttle pathway, and conversely, viral inhibitors of TAP may be useful reagents to further elucidate steps of the TAP catalytic cycle.
Figure 2.

Model for the transporter associated with antigen processing (TAP) catalytic cycle deduced from structural and functional studies [9–16]. TAP1 is indicated in blue (dark), TAP2 in yellow (light), peptide in red, and the membrane in white. TAP is predicted to alternate between two major conformational states, induced by the binding and hydrolysis of ATP. The conformations would correspond to a cytosol-facing high-affinity peptide binding site (substrate-trapping conformation, also referred to as the ‘inward-facing conformation’, or open conformation) (a and d) and an endoplasmic reticulum (ER)-facing low-affinity peptide-binding site (substrate-release conformation, also referred to as the ‘outward-facing conformation’ or closed conformation) (b and c). In the resting state of TAP complexes (a), the nucleotide binding domains (NBDs) of TAP1 and TAP2 are loosely engaged, and a high-affinity peptide binding site is oriented toward the cytosol. Peptide binding promotes ATP binding to the TAP2 site and conformational changes that facilitate tight TAP1 NBD–TAP2 NBD interactions (b). This step may be followed by a reorientation of the peptide binding site toward an opening in the membrane, accompanied by a reduction in the peptide binding affinity, which results in peptide release into the ER lumen (step 1). ATP hydrolysis occurs at the TAP2 site (upper rectangular site, step 2). Hydrolysis induces further conformational rearrangements at the TAP1/TAP2 NBD interface, which are transmitted to the transmembrane domains to restore a peptide binding site on the cytosolic surface and reinitiate a new cycle (c->d, step 3).
Stages of MHC class I assembly and formation of the PLC
A model for the assembly of MHC class I molecules involves the sequential interactions of (i) class I heavy chains with calnexin, (ii) class I heterodimers with calreticulin and (iii) class I heterodimers-calreticulin with TAP-tapasin-ERp57 (reviewed in Ref. [1]). However, the precise sequence of the MHC class I or the PLC assembly pathway is difficult to dissect unambiguously, because each component involved in MHC class I assembly and PLC formation can mediate multiple interactions. Tapasin uses its transmembrane and/or cytosolic domains for efficient TAP binding, and its ER luminal domains are able to interact with MHC class I and ERp57 [19,20]. Tapasin and ERp57 interact within the PLC via a disulfide bond between C95 of tapasin and C57 of ERp57 [1,20]. ERp57 can interact with both calreticulin and calnexin, and these complexes can bind and promote folding and assembly of MHC class I, tapasin and other partially folded glycoproteins via N-glycan and polypeptide-based binding interactions (reviewed in Ref. [21]). Finally, tapasin and peptide-receptive MHC class I molecules interact in the absence of other PLC components [22–24].
Recent knockout mice–based experiments have been important to further dissect the role of ERp57 and calreticulin in PLC formation and function. In ERp57-deficient B cells, MHC class I molecules are recruited into complexes with TAP and tapasin but dissociate more rapidly [25]. Furthermore, the C95A mutant of tapasin that is unable to bind ERp57 is able to recruit MHC class I molecules into a complex with TAP when expressed in the human tapasin-deficient 721.220 cells [26]. Interactions between TAP, tapasin and MHC class I molecules are also observable in fibroblasts derived from calreticulin-deficient mouse embryos [27,28]. Thus, neither ERp57 nor calreticulin is essential for the TAP-tapasin-MHC class I interaction, although interactions within the PLC might be stabilized by ERp57 and/or calreticulin. However, MHC class I assembly and cell surface expression are negatively impacted in cells deficient in ERp57 or calreticulin, indicating that both factors are important for optimal function of the PLC [25,27,28]. Studies with mutant PLC components may provide further insights into the dynamics of the functional interactions within the PLC.
Redox chemistry and mechanisms of tapasin-assisted peptide loading
Tapasin stabilizes TAP and enhances peptide transport by TAP [19], a function shown to be crucial for enhancing peptide loading onto MHC class I molecules of tapasin-deficient mouse cells [29]. In the tapasin-deficient human melanoma cells (M553), interferon-γ (IFN-γ) increases TAP protein levels dramatically but has a small effect on induction of MHC class I surface expression in the absence of exogenous tapasin [30]. Thus, in some cells, tapasin-mediated enhancement in TAP activity may be insufficient to significantly induce cell surface MHC class I expression at least for some MHC class I allotypes. By what additional mechanisms could tapasin facilitate assembly and cell surface expression of MHC class I molecules? Early studies showed that soluble tapasin that was unable to bind or stabilize TAP was partially able to induce MHC class I cell surface expression [19]. Using purified soluble versions of MHC class I and tapasin, tapasin alone has no effect or only a small effect in enhancing peptide binding to MHC class I molecules [24,31], suggesting that the requirement for tapasin relates to the specifics of peptide loading in an intracellular environment. In studies that measured peptide binding by MHC class I molecules in lysates of tapasin-deficient 721.220 cells, reconstitution with soluble tapasin-ERp57 conjugates, but not soluble tapasin alone, significantly enhanced peptide loading of MHC class I molecules [32], coincident with enhanced stabilization of the heavy chain. The peptide binding groove of MHC class I heavy chains has a disulfide bond within its α2 domain (Figure 3). This disulfide bond is sensitive to reduction until the peptide binding groove becomes stably occupied with peptide [33], and reduction of the disulfide bond is expected to be unfavorable for peptide binding. Blockage of tapasin-ERp57 conjugate formation by the tapasin C95A mutation enhanced reduction of HLA-B*4402, coincident with enhanced ERp57 binding to HLA-B*4402, which led to the suggestion that the formation of tapasin-ERp57 conjugates might sequester the reductase activity of ERp57 away from the α2 domain disulfide bond [26]. Based on these results, one mechanism for tapasin-assisted peptide loading could involve the maintenance of the α2 domain disulfide bond in an oxidized form favorable for peptide loading [26]. The primary function of PDI was also suggested to relate to the maintenance of the α2 domain disulfide bond, based on analyses of MHC class I heavy chain oxidation state and cell surface expression in PDI-depleted cells [33]. Thus, important questions to be addressed include whether the functions of PDI and tapasin-ERp57 are linked or represent two independent mechanisms for maintenance of the α2 domain disulfide bond. It is alternatively possible that enhanced reduction of the HLA-B*4402 heavy chain that was observed in the presence of the tapasin C95A mutant [26] is secondary to the diminished functional activity of the tapasin C95A mutant in promoting MHC class I peptide loading. Further studies are needed to clarify mechanisms of ERp57 and PDI functions within the PLC and to better understand the effects of ERp57 on tapasin structure and functional activities.
Figure 3.
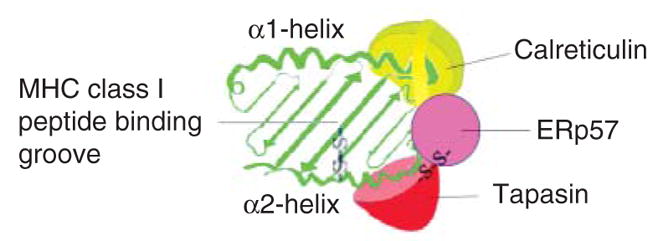
Two crucial disulfide bonds in the peptide loading of major histocompatibility complex (MHC) class I molecules. Schematic representation of the peptide binding groove of a peptide-receptive MHC class I heavy chain (green), as viewed from the top, and indicated components of the peptide loading complex (PLC). Tapasin (red) forms a disulfide bond with ER oxidoreductase of 57 Kda (ERp57; purple) and binding of tapasin-ERp57 to an empty MHC class I molecule could promote peptide binding by stabilization of the peptide-binding groove. Calreticulin (light green) interactions could also contribute to enhancing the stability of the peptide binding groove. A disulfide bond (C101–C164) connects the MHC class I α2 domain α-helix to the floor of the peptide binding groove. This disulfide bond is predicted to be surface accessible in the groove of an MHC class I molecule lacking peptide, and maintenance of the disulfide bond may be important for efficient peptide loading. Tapasin-ERp57 has been suggested to promote peptide binding by inhibiting the reductase activity of ERp57 toward the α2 domain disulfide bond [26]. Protein disulfide isomerase (PDI) has also been proposed to the important for the maintenance of the α2 domain disulfide bond [33]. In the interactions scheme shown here, tapasin is depicted as interacting with a region of the heavy chain that includes residue T134 (in the vicinity of the N terminus of the α2 domain α-helix) that has been suggested to be important for MHC class I-tapasin binding (reviewed in Ref. [1]). Calreticulin is depicted as interacting with a region of the heavy chain that includes residue N86 (in the vicinity of the C terminus of the a1 domain a-helix), a site for N-linked glycosylation suggested to be important for calreticulin-MHC class I binding (reviewed in Ref. [1]). ERp57, which interacts independently with both tapasin [20] and calreticulin (reviewed in Ref. [21]), is shown as partially bridging a tapasin-calreticulin–MHC class I interaction. The depicted modes of interaction between the different components remain to be verified. PDI is not shown, and the mode of PDI interaction with the PLC remains to be defined.
Tapasin and ERAAP as editors of the MHC class I peptide repertoire
Some studies suggest that tapasin edits the MHC class I peptide repertoire toward the binding of high-affinity peptides [32,34], although cellular evidence for a tapasin-induced MHC class I peptide repertoire that is high affinity or high stability has not been obtained [35,36]. The tapasin–MHC class I interaction is intrinsically peptide sensitive and dissociated by peptide binding [22,24,32]. More effective disengagement of tapasin–MHC class I complexes is induced by high-affinity peptides [24], and by this mechanism, tapasin could establish an affinity threshold for exit of peptide–MHC class I complexes from the ER. More direct editing might also occur; for instance, tethering of tapasin to MHC class I molecules via a fos-jun linkage enhanced dissociation rates of some peptide–MHC class I complexes [31]. In these studies, however, only a subset of the tested peptides was sensitive to tapasin during the dissociation reactions, and the magnitude of tapasin’s effect could not be predicted from the intrinsic dissociation rates of the peptide–MHC class I complexes, suggesting that other parameters are relevant to tapasin-mediated peptide selection [31]. ERAAP is also a key editor of the MHC class I peptide repertoire, and its peptidase activity generates optimal peptides from suboptimal N-extended sequences (reviewed in Ref. [2]). In ERAAP-deficient cells, despite the presence of tapasin, MHC class I molecules present many unstable and N-extended peptides to CD8+ T cells [37]. Thus, tapasin’s editing abilities might at best be restricted to some peptides or perhaps linked to the presence of ERAAP. Further studies are needed to address these possibilities.
Mechanisms and possible consequences of tapasin-dependent and independent MHC class I assembly
Recent studies have shown that HLA class I allotypes can differ dramatically in their intracellular assembly requirements [38–40]. For example, HLA-B*4402 is highly tapasin dependent for its assembly, whereas HLA-B*4405 is relatively tapasin independent [39,40]. Mechanisms that govern these assembly differences and the functional effects of such differences are of considerable interest, because the assembly profile could impact the efficiency of the immune response to tumors and intracellular infections [41]. Kienast et al. [26] observed that, compared with HLA-B*4405, HLA-B*4402 was more susceptible to reduction of its α2 domain disulfide bond in the absence of tapasin and suggested that tapasin dependency reflected susceptibility to reduction of the α2 domain disulfide bond. However, as noted above, peptide occupancy influences the extent of reduction of the α2 domain disulfide bond [33], and we noted that the intrinsic peptide loading efficiency of HLA-B*4402 was lower than HLA-B*4405 when comparing binding of different peptide libraries [23]. Thus, it remains to be established whether reduction of the α2 domain disulfide is the cause or the effect of reduced peptide loading of HLA-B*4402 compared with HLA-B*4405 in the absence of tapasin.
Various genetic studies have elucidated the fact that the presence of particular HLA alleles can profoundly impact infectious disease and cancer outcomes. Not much is known about the impact of HLA-B*4402 versus HLA-B*4405 in disease outcomes, because HLA-B*4405 is a rare allele in population groups in which genetic studies were undertaken. However, other HLA class I associations with disease are well established. For example, HLA-B*57 and HLA-B*27 alleles are associated with slow AIDS progression, whereas certain HLA-B*35 alleles are associated with rapid AIDS progression (reviewed in Ref. [42]). A major cause of the strong influence of HLA allotypes on disease outcomes probably relates to differences in peptides presented by each HLA class I molecule. However, it is possible that another level of influence arises from the assembly phenotype of a HLA class I molecule. It has been shown that, compared with tapasin-independent allotypes, tapasin-dependent allotypes are more susceptible to inhibition by herpesviral factors that target TAP/tapasin [40,43]. Thus, in the context of infections that target ER assembly factors of the MHC class I pathway, a tapasin-dependent assembly profile may be disadvantageous for the CTL response. However, a tapasin-dependent assembly profile could benefit the immune response in other situations. For example, tapasin-dependent assembly, which seems to correlate with slow rates of ER-medial Golgi trafficking [23], could result in more consistent loading of antigen epitopes during infections with viruses that do not interfere with MHC class I assembly in the ER. More studies are needed to better understand the impact of MHC class I assembly characteristics on immune responses and disease outcomes in different infection models.
Mechanisms and pathways of antigen cross-presentation
Uptake of antigen
It has been well established that both soluble and cell-associated or particulate exogenous antigens can be taken up by APCs by the general mechanisms of endocytosis and phagocytosis and further processed for presentation by MHC class I molecules (reviewed in Refs. [3,44]). Additionally, some exogenous soluble proteins can directly traffic to the ER of DC during cross-presentation [45].
The physiologically relevant form of cross-presented antigen used for activation of a CD8 T-cell response is vigorously debated, and evidence supports a role for cellular proteins that are proteasome substrates [46,47] or preprocessed peptides [48], in particular those sequestered in complex with different heat-shock proteins (HSPs) [49]. Evidence suggests that high antigen stability might favor priming of CD8 T cells [46,47,50], probably by promoting steady-state antigen accumulation and persistence in multiple intracellular and extracellular environments encountered during cross-presentation. However, as with endogenous presentation (for example, Ref. [51]), interactions of less stable or partially degraded antigens with specific cellular chaperones and HSPs could enhance stability, inhibit premature degradation and promote cross-presentation. Additionally, HSP associations could assist with antigen uptake into APC via specific HSP receptors as previously suggested [49] and with antigen targeting into intracellular compartments favorable for cross-presentation. Although several HSP receptors have been defined (reviewed in Ref. [52]), the role of particular HSP receptors in actively promoting cross-presentation remains to be demonstrated.
Features of receptors and APCs used in cross-presentation
Use of specific receptor and/or adaptor systems during antigen uptake can indeed strongly dictate the intracellular localization of antigen [53] and the efficiency of cross-presentation [54]. Cross-presentation of soluble ovalbumin by mouse APCs was dependent on mannose receptor–mediated endocytosis, which directed ovalbumin into a stable early endosomal compartment. In the absence of the mannose receptor, scavenger receptor endocytosed or pinocytosed ovalbumin was rapidly transported to lysosomes, which resulted in efficient MHC class II presentation but severely compromised cross-presentation [54]. For phagocytosed antigens, variations in phagosome properties among different APCs can impact the cross-presentation efficiency of an APC. The increased efficiency of cross-presentation by mouse bone marrow–derived dendritic cells (BMDCs) compared with macrophages [55] was attributed to some unique DC traits. Compared with macrophages, the neutral pH and low proteolytic activity in BMDC phagosomes in the first few hours after phagocytosis is thought to favor cross-presentation by BMDCs [56]. The NADPH oxidase NOX2, which is significantly recruited to the BMDC phagosome, is thought to be important for maintaining a neutral pH in the BMDC phagosomal lumen and for inhibiting rapid degradation of antigens, which could destroy potential epitopes for cross-presentation [56].
Intracellular pathways of cross-presentation
Proposed pathways for cross-presentation fall into two main categories: those dependent on the TAP and proteasomal processing and those relatively independent of these factors. The latter, also termed the vacuolar pathway (Figure 4a, reviewed in [3,44]), involves proteolysis of endocytic antigens by cysteine proteases such as cathepsin S [57] and loading of recycling MHC class I within the same compartments. Because peptide loading in the vacuolar pathway is generally proteasome and TAP independent, the ability to use the vacuolar pathway might be restricted to a few select antigens that are appropriately cleavable within endosomes. A recent study showed that human plasmacytoid DCs (pDCs) displayed unusual accumulation of MHC class I molecules within recycling endosomes. Presentation of a matrix protein–derived peptide in the context of HLA-A2 occurred in a cathepsin-dependent and proteasome-independent manner [58], invoking a vacuolar pathway, as depicted in Figure 4a. However, other studies indicate that this mode of antigen presentation by pDCs might depend on particular antigens and/or MHC class I allotypes rather than being a general feature of pDC [59].
Figure 4.

Proposed cellular pathways for antigen cross-presentation. (a) The transporter associated with antigen processing (TAP)-independent vacuolar pathway; endocytosed antigens are proteolytically processed by cysteine proteases such as cathepsin S. Peptide is loaded onto recycling major histocompatibility complex (MHC) class I molecules within the endosome, and the newly formed MHC class I/peptide complex traffics back to the plasma membrane. (b) The retrograde translocation model proposes that some soluble antigens are directly targeted to the endoplasmic reticulum (ER), following retro-trafficking through the trans-Golgi network (TGN) and Golgi [45]. Once in the ER, the antigen is retro-translocated into the cytosol by ERAD machinery (Sec61) and processed similarly to an endogenous protein for MHC class I presentation [60,61]. (c) The TAP-dependent phagosomal pathway relies on the presence of ER components on phagosomes [67–69]. Phagocytosed antigens use the Sec61 channel to egress out of the phagosome, are proteasomally processed and are reimported into the phagosome for loading onto MHC class I molecules within the phagosome. Once the MHC class I molecules are loaded, they traffic to the plasma membrane. Phagocytosed antigens that have egressed into the cytoplasm could also follow the classical ER-routed MHC class I pathway for endogenous proteins. (d) The newly described TAP-dependent endosomal pathway [63] proposes that TAP is recruited to an early endosome through TLR4/MyD88 signaling. Antigen egresses from the endosome by an unknown transporter and after proteasomal proteolysis, processed peptides are shuttled back into endosomes by recruited TAP and loaded onto recycling MHC class I molecules. This figure is adapted from Ref. [74] and redrawn.
Mechanisms of TAP and proteasome-dependent antigen cross-presentation
For TAP and proteasome-dependent presentation, antigens must reach the cytosol. Recent studies provide evidence for retro-transport of some antigens into the cytosol after their direct uptake into the ER of DCs [45]. The ER-associated degradation (ERAD) machinery, including Sec61, a putative translocon channel, is thought to function in ER to cytosol retro-transport [60,61] (Figure 4b). Antigen transport to the cytosol can also occur from phagocytic [62], endocytic [54,63] or macropinocytic [55,64,65] compartments. After a report that the phagosomal membranes are formed in part from the ER membrane [66], three laboratories described the observation of ERAD factors and components of the MHC class I peptide loading machinery within phagosomal membranes [67–69] (Figure 4c). The presence of ERAD components on phagosomes provides an attractive mechanism for the translocation of phagosomal antigens to the cytosol for processing. However, the findings of ER components in phagosomal membranes were refuted in a subsequent report that described an inability to detect ER contribution to phagosome formation, based on various quantitative approaches [70]. Thus, the presence of ER components within phagosomal membranes remains controversial.
In related findings, a recent report suggests that TAP can be recruited to early endosomes in a TLR (Toll-like receptor) 4-MyD88–dependent manner [63]. A transferrin receptor–conjugated inhibitor of TAP, which localized to recycling endosomes, was found to inhibit cross-presentation of exogenous ovalbumin but not the presentation of endogenously expressed ovalbumin [63]. These results support a pathway in which soluble exogenous antigens are retro-translocated from endosomal compartments into the cytosol, followed by re-import of proteasomally processed peptides back into endosomes via endosomal TAP, where peptide loading of recycling MHC class I occurs (Figure 4d). Related mechanisms may determine endosomal and phagosomal localization of TAP, and thus, further understanding of the mechanisms by which TAP becomes endosomally localized, as well as additional quantitative demonstrations of the presence of other PLC and ER components in TAP-bearing endosomes may help resolve some of the current controversy surrounding the presence of ER components in phagosomal membranes. In this regard, it is also noteworthy that previous studies by Lizee et al. [71] have suggested the existence of an endo-lysosomal compartment for peptide loading of MHC class I molecules during cross-presentation and shown that a tyrosine-based targeting signal within the MHC class I cytoplasmic domain is important for routing MHC class I molecules through the endo-lysosomal compartments [71].
Conclusions
Thus, in many cases, the cytosol seems to be the main antigen processing site for both the endogenous and cross-presentation pathways, and TAP is a common crucial component to both pathways. Many aspects relating to TAP localization during cross-presentation remain to be clarified or further elucidated. Mechanisms by which phagosomal or endosomal antigens access the cytosol during cross-presentation remain an important area for further investigations, and why the cytosolic products of endosomally derived antigens might be preferentially retargeted into the endosome rather than the ER [63] (as may be predicted for a freely diffusing antigen) also remains to be addressed. Receptor systems that allow the best manipulations of cross-priming efficiencies remain to be fully defined and will be important toward vaccine studies. Structural and biochemical studies of PLC components and complexes will yield better insights into the specific functions of individual components of the PLC. Finally, research into novel pathogen-encoded mechanisms for evasion of the CTL responses will undoubtedly reveal new and unexpected details of the physiological assembly, trafficking and degradation pathways that are exploited by pathogens (see Refs. [17,33,72,73] for some recent examples).
Acknowledgments
The authors thank all our colleagues for numerous contributions that ensure that the field is thriving and generating debate and discussion. The authors also thank Yesung Park and Ben Schwartz for the artwork. Our contributions summarized here were supported by NIH Grants AI044115 and A1066131 (to M.R.).
References
- 1.Cresswell P, et al. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev. 2005;207:145–157. doi: 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 2.Blanchard N, Shastri N. Coping with loss of perfection in the MHC class I peptide repertoire. Curr Opin Immunol. 2008;20:82–88. doi: 10.1016/j.coi.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen L, Rock KL. Priming of T cells by exogenous antigen cross-presented on MHC class I molecules. Curr Opin Immunol. 2006;18:85–91. doi: 10.1016/j.coi.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Scholz C, Tampe R. The intracellular antigen transport machinery TAP in adaptive immunity and virus escape mechanisms. J Bioenerg Biomembr. 2005;37:509–515. doi: 10.1007/s10863-005-9500-1. [DOI] [PubMed] [Google Scholar]
- 5.Koch J, et al. Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP) J Biol Chem. 2004;279:10142–10147. doi: 10.1074/jbc.M312816200. [DOI] [PubMed] [Google Scholar]
- 6.Procko E, et al. Identification of domain boundaries within the N-termini of TAP1 and TAP2 and their importance in tapasin binding and tapasin-mediated increase in peptide loading of MHC class I. Immunol Cell Biol. 2005;83:475–482. doi: 10.1111/j.1440-1711.2005.01354.x. [DOI] [PubMed] [Google Scholar]
- 7.Leonhardt RM, et al. Critical role for the tapasin-docking site of TAP2 in the functional integrity of the MHC class I-peptide-loading complex. J Immunol. 2005;175:5104–5114. doi: 10.4049/jimmunol.175.8.5104. [DOI] [PubMed] [Google Scholar]
- 8.van Endert PM, et al. Powering the peptide pump: TAP crosstalk with energetic nucleotides. Trends Biochem Sci. 2002;27:454–461. doi: 10.1016/s0968-0004(02)02090-x. [DOI] [PubMed] [Google Scholar]
- 9.Smith PC, et al. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Procko E, et al. Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol Cell. 2006;24:51–62. doi: 10.1016/j.molcel.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, et al. Functional non-equivalence of ATP-binding cassette signature motifs in the transporter associated with antigen processing (TAP) J Biol Chem. 2004;279:46073–46081. doi: 10.1074/jbc.M404042200. [DOI] [PubMed] [Google Scholar]
- 12.Ernst R, et al. Engineering ATPase activity in the isolated ABC-cassette of human TAP1. J Biol Chem. 2006;281:27471–27480. doi: 10.1074/jbc.M601131200. [DOI] [PubMed] [Google Scholar]
- 13.Perria CL, et al. Catalytic site modifications of TAP1 and TAP2 and their functional consequences. J Biol Chem. 2006;281:39839–39851. doi: 10.1074/jbc.M605492200. [DOI] [PubMed] [Google Scholar]
- 14.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 15.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 16.Ward A, et al. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppers-Lalic D, et al. Varicellovirus UL 49.5 proteins differentially affect the function of the transporter associated with antigen processing, TAP. PLoS Pathog. 2008;4:e1000080. doi: 10.1371/journal.ppat.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hislop AD, et al. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J Exp Med. 2007;204:1863–1873. doi: 10.1084/jem.20070256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehner PJ, et al. Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line. 220. Immunity. 1998;8:221–231. doi: 10.1016/s1074-7613(00)80474-4. [DOI] [PubMed] [Google Scholar]
- 20.Dick TP, et al. Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity. 2002;16:87–98. doi: 10.1016/s1074-7613(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 21.Williams DB. Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J Cell Sci. 2006;119:615–623. doi: 10.1242/jcs.02856. [DOI] [PubMed] [Google Scholar]
- 22.Schoenhals GJ, et al. Retention of empty MHC class I molecules by tapasin is essential to reconstitute antigen presentation in invertebrate cells. EMBO J. 1999;18:743–753. doi: 10.1093/emboj/18.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thammavongsa V, et al. HLA-B44 polymorphisms at position 116 of the heavy chain influence TAP complex binding via an effect on peptide occupancy. J Immunol. 2006;177:3150–3161. doi: 10.4049/jimmunol.177.5.3150. [DOI] [PubMed] [Google Scholar]
- 24.Rizvi SM, Raghavan M. Direct peptide regulatable interactions between tapasin and MHC class I molecules. Proc Natl Acad Sci U S A. 2006;103:18220–18225. doi: 10.1073/pnas.0605131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garbi N, et al. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol. 2006;7:93–102. doi: 10.1038/ni1288. [DOI] [PubMed] [Google Scholar]
- 26.Kienast A, et al. Redox regulation of peptide receptivity of major histocompatibility complex class I molecules by ERp57 and tapasin. Nat Immunol. 2007;8:864–872. doi: 10.1038/ni1483. [DOI] [PubMed] [Google Scholar]
- 27.Gao B, et al. Assembly and antigen-presenting function of MHC class I molecules in cells lacking the ER chaperone calreticulin. Immunity. 2002;16:99–109. doi: 10.1016/s1074-7613(01)00260-6. [DOI] [PubMed] [Google Scholar]
- 28.Ireland BS, et al. Lectin-deficient calreticulin retains full functionality as a chaperone for class I histocompatibility molecules. Mol Biol Cell. 2008;19:2413–2423. doi: 10.1091/mbc.E07-10-1055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Garbi N, et al. A major role for tapasin as a stabilizer of the TAP peptide transporter and consequences for MHC class I expression. Eur J Immunol. 2003;33:264–273. doi: 10.1002/immu.200390029. [DOI] [PubMed] [Google Scholar]
- 30.Belicha-Villanueva A, et al. Differential contribution of TAP and tapasin to HLA class I antigen expression. Immunology. 2008;124:112–120. doi: 10.1111/j.1365-2567.2007.02746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen M, Bouvier M. Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. EMBO J. 2007;26:1681–1690. doi: 10.1038/sj.emboj.7601624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wearsch PA, Cresswell P. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat Immunol. 2007;8:873–881. doi: 10.1038/ni1485. [DOI] [PubMed] [Google Scholar]
- 33.Park B, et al. Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell. 2006;127:369–382. doi: 10.1016/j.cell.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 34.Howarth M, et al. Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc Natl Acad Sci U S A. 2004;101:11737–11742. doi: 10.1073/pnas.0306294101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zarling AL, et al. Tapasin is a facilitator, not an editor, of class I MHC peptide binding. J Immunol. 2003;171:5287–5295. doi: 10.4049/jimmunol.171.10.5287. [DOI] [PubMed] [Google Scholar]
- 36.Everett MW, Edidin M. Tapasin increases efficiency of MHC I assembly in the endoplasmic reticulum but does not affect MHC I stability at the cell surface. J Immunol. 2007;179:7646–7652. doi: 10.4049/jimmunol.179.11.7646. [DOI] [PubMed] [Google Scholar]
- 37.Hammer GE, et al. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol. 2007;8:101–108. doi: 10.1038/ni1409. [DOI] [PubMed] [Google Scholar]
- 38.Peh CA, et al. HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity. 1998;8:531–542. doi: 10.1016/s1074-7613(00)80558-0. [DOI] [PubMed] [Google Scholar]
- 39.Williams AP, et al. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity. 2002;16:509–520. doi: 10.1016/s1074-7613(02)00304-7. [DOI] [PubMed] [Google Scholar]
- 40.Zernich D, et al. Natural HLA class I polymorphism controls the pathway of antigen presentation and susceptibility to viral evasion. J Exp Med. 2004;200:13–24. doi: 10.1084/jem.20031680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khanna R, et al. Hierarchy of Epstein-Barr virus-specific cytotoxic T-cell responses in individuals carrying different subtypes of an HLA allele: implications for epitope-based antiviral vaccines. J Virol. 1997;71:7429–7435. doi: 10.1128/jvi.71.10.7429-7435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin MP, Carrington M. Immunogenetics of viral infections. Curr Opin Immunol. 2005;17:510–516. doi: 10.1016/j.coi.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 43.Park B, et al. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity. 2004;20:71–85. doi: 10.1016/s1074-7613(03)00355-8. [DOI] [PubMed] [Google Scholar]
- 44.Groothuis TA, Neefjes J. The many roads to cross-presentation. J Exp Med. 2005;202:1313–1318. doi: 10.1084/jem.20051379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ackerman AL, et al. Access of soluble antigens to the endoplasmic reticulum can explain cross-presentation by dendritic cells. Nat Immunol. 2005;6:107–113. doi: 10.1038/ni1147. [DOI] [PubMed] [Google Scholar]
- 46.Norbury CC, et al. CD8+ T cell cross-priming via transfer of proteasome substrates. Science. 2004;304:1318–1321. doi: 10.1126/science.1096378. [DOI] [PubMed] [Google Scholar]
- 47.Shen L, Rock KL. Cellular protein is the source of cross-priming antigen in vivo. Proc Natl Acad Sci U S A. 2004;101:3035–3040. doi: 10.1073/pnas.0308345101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blachere NE, et al. Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol. 2005;3:e185. doi: 10.1371/journal.pbio.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binder RJ, Srivastava PK. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol. 2005;6:593–599. doi: 10.1038/ni1201. [DOI] [PubMed] [Google Scholar]
- 50.Bins AD, et al. In vivo antigen stability affects DNA vaccine immunogenicity. J Immunol. 2007;179:2126–2133. doi: 10.4049/jimmunol.179.4.2126. [DOI] [PubMed] [Google Scholar]
- 51.Kunisawa J, Shastri N. Hsp90alpha chaperones large C-terminally extended proteolytic intermediates in the MHC class I antigen processing pathway. Immunity. 2006;24:523–534. doi: 10.1016/j.immuni.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 52.Calderwood SK, et al. Extracellular heat shock proteins in cell signaling and immunity. Ann N Y Acad Sci. 2007;1113:28–39. doi: 10.1196/annals.1391.019. [DOI] [PubMed] [Google Scholar]
- 53.Lakadamyali M, et al. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124:997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burgdorf S, et al. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316:612–616. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez A, et al. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol. 1999;1:362–368. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- 56.Savina A, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 57.Shen L, et al. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity. 2004;21:155–165. doi: 10.1016/j.immuni.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 58.Di Pucchio T, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol. 2008;9:551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoeffel G, et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity. 2007;27:481–492. doi: 10.1016/j.immuni.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 60.Ackerman AL, et al. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity. 2006;25:607–617. doi: 10.1016/j.immuni.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 61.Imai J, et al. Exogenous antigens are processed through the endoplasmic reticulum-associated degradation (ERAD) in cross-presentation by dendritic cells. Int Immunol. 2005;17:45–53. doi: 10.1093/intimm/dxh184. [DOI] [PubMed] [Google Scholar]
- 62.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 63.Burgdorf S, et al. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol. 2008;9:558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- 64.Brossart P, Bevan MJ. Presentation of exogenous protein antigens on major histocompatibility complex class I molecules by dendritic cells: pathway of presentation and regulation by cytokines. Blood. 1997;90:1594–1599. [PMC free article] [PubMed] [Google Scholar]
- 65.Norbury CC, et al. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity. 1995;3:783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 66.Gagnon E, et al. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110:119–131. doi: 10.1016/s0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 67.Ackerman AL, et al. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci U S A. 2003;100:12889–12894. doi: 10.1073/pnas.1735556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guermonprez P, et al. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 2003;425:397–402. doi: 10.1038/nature01911. [DOI] [PubMed] [Google Scholar]
- 69.Houde M, et al. Phagosomes are competent organelles for antigen cross-presentation. Nature. 2003;425:402–406. doi: 10.1038/nature01912. [DOI] [PubMed] [Google Scholar]
- 70.Touret N, et al. Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell. 2005;123:157–170. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 71.Lizee G, et al. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol. 2003;4:1065–1073. doi: 10.1038/ni989. [DOI] [PubMed] [Google Scholar]
- 72.Loureiro J, Ploegh HL. Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv Immunol. 2006;92:225–305. doi: 10.1016/S0065-2776(06)92006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Byun M, et al. Cowpox virus exploits the endoplasmic reticulum retention pathway to inhibit MHC class I transport to the cell surface. Cell Host Microbe. 2007;2:306–315. doi: 10.1016/j.chom.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 74.Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. doi: 10.1016/j.coi.2007.12.002. [DOI] [PubMed] [Google Scholar]