

Abstract
Purpose
The study demonstrates the functional candidate gene analysis in a cataract family of German descent.
Methods
We screened a German family, clinically documented to have congenital cataracts, for mutation in the candidate genes CRYG (A to D) and CRYBB2 through polymerase chain reaction analyses and sequencing.
Results
Congenital cataract was first observed in a daughter of healthy parents. Her two children (a boy and a girl) also suffer from congenital cataracts and have been operated within the first weeks of birth. Morphologically, the cataract is characterized as nuclear with an additional ring-shaped cortical opacity. Molecular analysis revealed no causative mutation in any of the CRYG genes. However, sequencing of the exons of the CRYBB2 gene identified a sequence variation in exon 5 (383 A>T) with a substitution of Asp to Val at position 128. All three affected family members revealed this change but it was not observed in any of the unaffected persons of the family. The putative mutation creates a restriction site for the enzyme TaiI. This mutation was checked for in controls of randomly selected DNA samples from ophthalmologically normal individuals from the population-based KORA S4 study (n=96) and no mutation was observed. Moreover, the Asp at position 128 is within a stretch of 12 amino acids, which are highly conserved throughout the animal kingdom. For the mutant protein, the isoelectric point is raised from pH 6.50 to 6.75. Additionally, the random coil structure of the protein between the amino acids 126-139 is interrupted by a short extended strand structure. In addition, this region becomes hydrophobic (from neutral to +1) and the electrostatic potential in the region surrounding the exchanged amino acid alters from a mainly negative potential to an enlarged positive potential.
Conclusions
The D128V mutation segregates only in affected family members and is not seen in representative controls. It represents the first mutation outside exon 6 of the human CRYBB2 gene.
Introduction
Several different cataract mutations have been characterized in man and mouse over the last ten years. They demonstrate a broad spectrum of genetic and phenotypical variety; however, the analysis of congenital (mainly dominant) cataracts revealed a high number of mutations in genes coding for γ-crystallins and to a slightly less extent in genes coding for β-crystallins (for a recent review see [1]). Furthermore, structural proteins in the lens represent also membrane proteins. The most prominent membrane proteins are the main intrinsic protein (MIP; which belongs to the family of aquaporins) and gap-junction proteins (which belong to the connexin family). Mutations in the corresponding genes, MIP/Mip, GJA3/Gja3, and GJA8/Gja8 (encoding the gap junction membrane channel protein alpha 3 and alpha 8, respectively), have been shown to underlie some congenital, dominant cataracts in mouse and man (for a review see [1]). Additional causes for congenital, hereditary cataracts are mutations affecting enzymes of sugar metabolism (e.g. GALK1 [2]) or other metabolic disorders like hyperferritinemia [3].
Among the crystallins, the βB2-crystallin was, for a long time, also referred to as the "basic principle" crystallin because of its high expression level in the lens. Therefore, it is not surprising that mutations in that gene correspond frequently to hereditary cataracts. Because of its expression pattern it is also consistent with the observation that it is mainly involved in congenital cataracts. Crystallins, the βB2-crystallin in particular, are considered to act mainly as structural proteins of the lens; however, some of them have also been detected in other ocular tissues and other organs [4-6]. Recently, it was shown that the mouse Crybb2 is also involved in elongation of axons during regeneration of retinal ganglion cells [7]. Moreover, mice harboring a mutation in the Crybb2 gene exhibit subfertility in the Swiss Webster genetic background (but not in C57BL/6 background) based upon its expression in the sperm [8].
The present paper demonstrates clinical data for a dominant congenital lens opacity in three affected patients of a German family. Since the family size did not permit linkage analysis, a functional candidate gene approach was attempted towards the identification of the underlying molecular lesion. This resulted in the identification of a new cataract-causing allele of the CRYBB2 gene (encoding βB2-crystallin); it is the first one outside exon 6 of CRYBB2.
Methods
Congenital cataract was observed in three members of a German three-generation family, a mother and her two children (the grandparents were not affected). Informed consent was obtained from the family.
Blood samples (5-10 ml) were collected from all available family members (affected and unaffected). Genomic DNA was isolated according to standard procedures [9] and amplified by polymerase chain reaction (PCR) for the exons (and their flanking regions) of the CRYGC, CRYGD [10], and CRYBB2 genes [11]. PCR products were checked in 1.5% agarose gels and purified through Nucleospin columns (Macherey and Nagel, Düren, Germany). Sequencing was done either commercially (SequiServe, Vaterstetten, Germany) or at the Genome Analysis Center of the GSF (ABI3730; Applied Biosystems, Darmstadt, Germany) according to standard procedures.
The mutation was confirmed by the presence of the cleavage site for the restriction enzyme TaiI. For control, we analyzed 96 randomly chosen ophthalmologically normal individuals of the KORA Survey 4 (Cooperative Health Research in the Augsburg Region), which studied a population-based sample of 4,261 subjects aged 25-74 years old during the years 1999-2001 [12].
Biophysical predictions of the altered protein were analyzed using the Bioinformatics tool of the Expasy Proteomics server. To predict the secondary structure we used the GOR4 prediction method [13]. For tertiary structure predictions, we used EsyPred3D [14] and DEEP VIEW/SWISS-Pdb viewer [15] for automated homology protein modeling and visualization of resulting protein database files.
Results
Case history
The index patient (II.2) was operated on at the age of two years because of bilateral cataract. Her two children, a girl (III.2) and a boy (III.1), suffered also from congenital, bilateral cataract and have been operated within the first few weeks of birth. Figure 1A shows the eyes of the boy before operation and Figure 1B after operation in the right eye (the left eye is still cataractous). The figures demonstrate a central nuclear cataract with a surrounding cortical opacity. The pedigree of the family (Figure 2) suggests an autosomal dominant mode of inheritance. The parents of the index patient are not affected.
Figure 1.
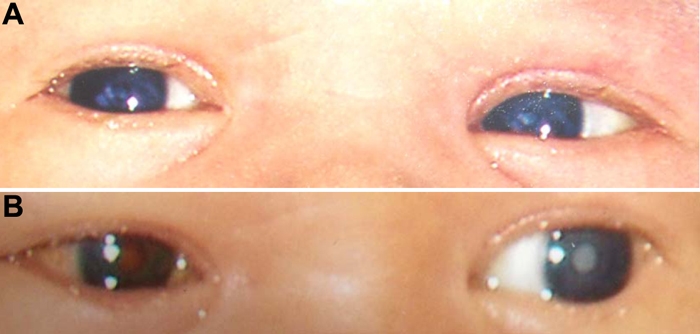
Clinical features of the cataract. A: The photograph of the affected boy demonstrates a nuclear cataract in both eyes surrounded by a ring-shaped opacity in both eyes. B: After operation in the right eye, the lens is clear and the "red dot" effect can be observed after flash-light photography. The left eye remains opaque. An observation of the increased dense nuclear opacity indicates slight progression of the cataractous process. The photographs are private, with permission from the parents.
Figure 2.
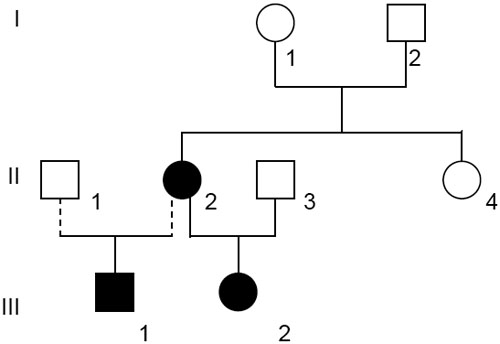
Pedigree of the affected family. The family history demonstrated that the cataract was present in two generations. The dark symbols represent the affected members of the family, while the clear symbols indicate the healthy ones. Circles are for female and squares for male family members.
Molecular genetics
Because of the small family size, we performed a functional candidate gene analysis instead of linkage analysis. Since most hereditary cataracts are caused by mutations in genes of the CRYG-cluster, these four genes (CRYGA-CRYGD) have been checked first. In the index patient, no alteration was observed in the CRYGA-CRYGC genes as compared to the database. However, in CRYGD, two changes have been found (in intron 2: IVS2+30; 517 T>C and in exon 3: 303/304 AG>GA, V101M). Both sites have been identified previously as being polymorphic and occurred in nine out of nine tested samples, which suggests either a sequencing error or a rare single nucleotice polymorphism (SNP) in the database [11].
In contrast, a causative change has been found in the CRYBB2 gene encoding the βB2-crystallin, which was examined next because of its frequent involvement in hereditary cataracts. In exon 5 of the CRYBB2 gene, a heterozygous mutation was found in the index patient (383 A>T; Figure 3A,B). The mutation was confirmed by a TaiI digest of the PCR amplified exon 5 of the CRYBB2 gene. The same alteration has been found in the two other family members who were also affected (both children). This was confirmed by sequencing with the reverse primer. In contrast, the mutation was not seen in healthy family members (Figure 3C). This is particularly true for the parents of the index patient. Therefore, the mutation most likely occurred spontaneously in one of the germ lines of the parents. Finally, this TaiI restriction site could not be found in 96 healthy persons collected in the Augsburg region (Germany; data not shown). DNA translation programs predict an exchange of the highly conserved Asp at amino acid position 128 to Val (D128V). The Asp at position 128 lies within a stretch of 12 amino acids, which are identical in rat, mouse, cow, dog, human, chimpanzee, chick, and zebrafish (both isoforms).
Figure 3.

CRYBB2 mutation cosegregates with the disease in the family. A: A partial fragment of the fifth exon of the CRYBB2 gene is given: at sequence position 178 (equal to position 383 in CRYBB2 exon 5, counting the A of the ATG start codon as number 1), the heterozygous situation of the index patient is obvious (red arrow). B: A partial fragment of the corresponding genomic sequence of exon 5 of the CRYBB2 gene is given. The mutated sequence is shown below. The underlined bases (ACGT) define a TaiI restriction site, which is created by the mutation. The amino acid sequence is given below the DNA sequence. The wild-type base and amino acid are given in green and the mutated forms in red. C: A schematic overview of the PCR fragment (706 bp) including exon 5 of the CRYBB2 gene is given. The positions of the TaiI restriction sites are indicated for the wild type and the mutant allele. In the wild-type form there are two major fragments of 275 and 417 bp. In the disease form, the 275 bp fragment is cut again resulting in two smaller fragments of 207 and 68 bp. These smaller fragments (indicated by asterisks) can only be observed in the three affected family members (C, unaffected control; "+", digested fragment; "-", undigested fragment).
In-silico protein analysis
An initial attempt to understand the underlying molecular mechanisms was derived from the computer-assisted analysis of the structure of the mutated D128V βB2-crystallin. Using the proteomics program of the Expasy Proteomics server, we compared several features between the wild-type and the mutant protein. The isoelectric point is shifted from pH 6.50 in the wild-type protein up to pH 6.75 in the mutant βB2-crystallin due to the exchange of the acidic and hydrophilic Asp to the neutral, nonpolar and hydrophobic Val. It is very likely that the impact of this alteration in the immediate neighborhood of the exchanged amino acid might be much stronger. Thus, it is not surprising that changes in the secondary structure are predicted as follows: the Arg-Val exchange presumably has an effect on H-bond building and causes a loss of an H-bond from Val to Arg at position 191; and depending on the orientation of the side chain of Val, a new H-bond can be formed between Val and Tyr at position 159. This might explain the predicted interruption of the random coil structure by a shortly extended strand structure in this area.
Finally, an even more striking consequence is the modification of the electrostatic potential in this area. The negative electrostatic potential surrounding the Asp disappears and is replaced by a remarkably enlarged positive potential in this region (Figure 4). This might be the electrostatic explanation for the predicted change of the hydrophobic nature from neutral to +1 in this region.
Figure 4.
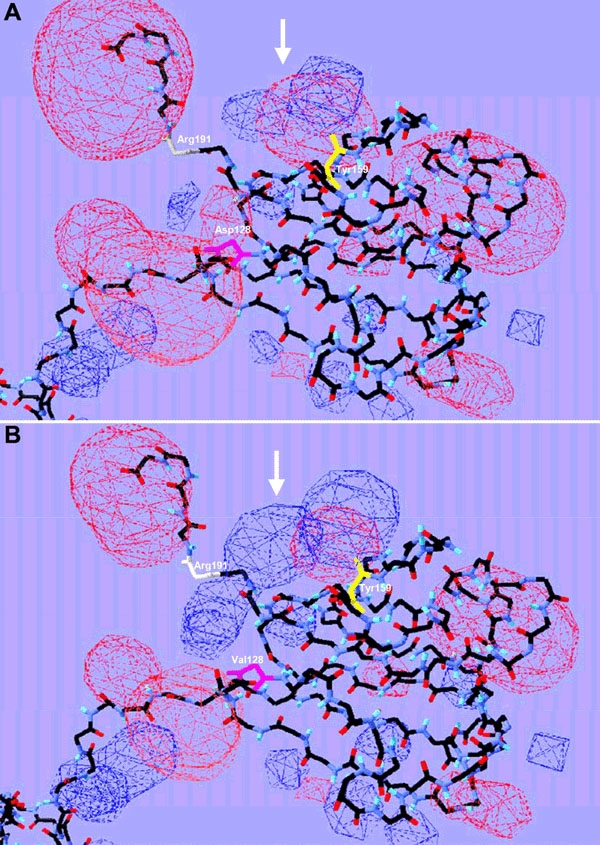
Three-dimensional protein structure. Schematic view of predicted three-dimensional protein structures for CRYBB2WT (A) and CRYBB2D128V (B), without side chains. The electrostatic potential is displayed in red (negative potential) and blue (positive potential) clouds. The alteration from a negative to a positive potential is indicated by white arrows. Exchanged amino acids, Asp and Val, at position 128 are marked in pink. Amino acids involved into H-bond building are shown in white (Arg, 191) and yellow (Tyr, 159).
Discussion
We describe a mother and her two children suffering from a dominant, congenital cataract. Her parents and sister are healthy with normal vision. The affected family members had no other ocular defects. Because of the small size of the family, linkage analysis could not be attempted. Therefore, we molecularly analyzed functional candidate genes and identified a mutation in the fifth exon of the CRYBB2 gene (D128V). Since this mutation segregates perfectly within this family and could not be found in 96 healthy subjects from the region of Augsburg/Germany, we consider this new allele as probable causative molecular lesion for the observed clinical findings.
Several other cataract mutations have been reported both in mouse and human to affect the CRYBB2 gene, which is considered to be one of the most important genes for lens transparency. The CRYBB2 gene product was earlier referred to as the "basic principle β-crystallin" because of its abundance in water soluble lens extracts [16]. Presently, this protein is referred to as βB2-crystallin.
The first mutation identified in the CRYBB2 gene was shown to be causative for a cerulean cataract (CCA2: congenital cataract of cerulean type 2) featuring peripheral bluish and white opacities in concentric layers with occasional central lesions arranged radially. Litt et al. [17] mapped this particular type of cataract to a region of the human chromosome 22 containing the cluster encoding four CRYB genes. Sequence analysis revealed that a chain-termination mutation at the beginning of exon 6 of the CRYBB2 gene (C475T; Gln155X) is associated with this particular type of cataract. Surprisingly, the same mutation was also found in a family suffering from a Coppock-like cataract [18] and in a five-generation Indian family with suture cataract and cerulean opacities [19]. The authors explain the identity of the three mutations by a gene-conversion mechanism between the CRYBB2 gene and its flanking pseudogene. The diversity of the phenotypes might be caused by variations in the promoter region, possibly influencing the expression of CRYBB2 in the lens or other crystallin genes as modifiers from surrounding loci. Two additional cases of this mutation were identified recently in a Chinese and a Chilean family. The phenotype in these families is highly variable [20,21].
The first human mutation in CRYBB2, which did not fit within the gene conversion mechanism, was identified by Santhiya et al. [11] who described a W151C mutation affecting the sixth exon of the CRYBB2 gene. The clinical findings of both mutations are similar in that the cataracts were present at birth, the children needed an operation within a few weeks of birth, and the morphology was described as a central nuclear cataract. However, the actual mutation reported here is the first that does not affect exon 6 of the CRYBB2 gene. Since the PCR primers used for amplification of this particular exon are located fairly far in the flanking introns, no contamination with the corresponding pseudogene is observed. This pseudogene is different from the active CRYBB2 gene in this particular exon only in three bases and these distinct bases are located in the 3'-end of the exon.
So far, in mice, two mutant lines have been reported to involve the Crybb2 gene, the Philly mouse [22] and the Aey2 mutant line [23]. Both cataracts are progressive and recognizable from the second week of birth as an anterior suture and as a subcapsular opacity. In the Philly mouse, the ultimate phenotype is characterized as a strong opacity of the lens nucleus and of the anterior suture six to seven weeks after birth [24]. In Aey2 mutants, gradual opacification of the whole lens was completed at the age of 11 weeks. Therefore, the mouse mutants represent a less severe phenotype than most of the human cases.
Nevertheless, all mutants described in mouse and in man affect the COOH-terminal part of the βB2-crystallin. Recently, in a systematic analysis, Liu and Liang [25] investigated the influence of several mutations along the βB2-crystallin gene with respect to the possibility of the protein forming dimers. They found that mutations, which affect the formation of β-strands in the COOH-terminal part of the protein, have reduced protein interactions. The β-strand 11, which belongs to the third Greek key motif, is affected in the new allele reported here and is the most anterior mutation reported so far for the CRYBB2 gene. Since the negative electrostatic potential surrounding the Asp disappears and is replaced by a remarkably enlarged positive potential in this region, protein-protein interactions are most likely changed. Nevertheless, further physico-chemical experiments are necessary to demonstrate the reduced ability to form dimers in this novel CRYBB2 allele.
Acknowledgements
We thank the family members for their participation. The excellent technical assistance of Erika Bürkle and Monika Stadler (GSF Neuherberg) is gratefully acknowledged. Oligonucleotides were synthesized by Utz Linzner, GSF-Institute of Experimental Genetics.
References
- 1.Graw J. The genetic and molecular basis of congenital eye defects. Nat Rev Genet. 2003;4:876–88. doi: 10.1038/nrg1202. [DOI] [PubMed] [Google Scholar]
- 2.Sangiuolo F, Magnani M, Stambolian D, Novelli G. Biochemical characterization of two GALK1 mutations in patients with galactokinase deficiency. Hum Mutat. 2004;23:396. doi: 10.1002/humu.9223. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar-Martinez P, Schved JF, Brissot P. The evaluation of hyperferritinemia: an updated strategy based on advances in detecting genetic abnormalities. Am J Gastroenterol. 2005;100:1185–94. doi: 10.1111/j.1572-0241.2005.40998.x. [DOI] [PubMed] [Google Scholar]
- 4.Magabo KS, Horwitz J, Piatigorsky J, Kantorow M. Expression of betaB(2)-crystallin mRNA and protein in retina, brain, and testis. Invest Ophthalmol Vis Sci. 2000;41:3056–60. [PMC free article] [PubMed] [Google Scholar]
- 5.Runkle S, Hill J, Kantorow M, Horwitz J, Posner M. Sequence and spatial expression of zebrafish (Danio rerio) alphaA-crystallin. Mol Vis. 2002;8:45–50. http://www.molvis.org/molvis/v8/a6/ [PMC free article] [PubMed] [Google Scholar]
- 6.Horwitz J. Alpha-crystallin. Exp Eye Res. 2003;76:145–53. doi: 10.1016/s0014-4835(02)00278-6. [DOI] [PubMed] [Google Scholar]
- 7.Liedtke T, Schwamborn JC, Schroer U, Thanos S. Elongation of Axons during Regeneration Involves Retinal Crystallin beta b2 (crybb2). Mol Cell Proteomics. 2007;6:895–907. doi: 10.1074/mcp.M600245-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Duprey KM, Robinson KM, Wang Y, Taube JR, Duncan MK. Subfertility in mice harboring a mutation in betaB2-crystallin. Mol Vis. 2007;13:366–73. http://www.molvis.org/molvis/v13/a40/ [PMC free article] [PubMed] [Google Scholar]
- 9.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santhiya ST, Shyam Manohar M, Rawlley D, Vijayalakshmi P, Namperumalsamy P, Gopinath PM, Loster J, Graw J. Novel mutations in the gamma-crystallin genes cause autosomal dominant congenital cataracts. J Med Genet. 2002;39:352–8. doi: 10.1136/jmg.39.5.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santhiya ST, Manisastry SM, Rawlley D, Malathi R, Anishetty S, Gopinath PM, Vijayalakshmi P, Namperumalsamy P, Adamski J, Graw J. Mutation analysis of congenital cataracts in Indian families: identification of SNPS and a new causative allele in CRYBB2 gene. Invest Ophthalmol Vis Sci. 2004;45:3599–607. doi: 10.1167/iovs.04-0207. [DOI] [PubMed] [Google Scholar]
- 12.Illig T, Bongardt F, Schopfer A, Holle R, Muller S, Rathmann W, Koenig W, Meisinger C, Wichmann HE, Kolb H, KORA Study Group. The endotoxin receptor TLR4 polymorphism is not associated with diabetes or components of the metabolic syndrome. Diabetes. 2003;52:2861–4. doi: 10.2337/diabetes.52.11.2861. [DOI] [PubMed] [Google Scholar]
- 13.Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: network protein sequence analysis. Trends Biochem Sci. 2000;25:147–50. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 14.Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–6. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 15.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 16.Herbrink P, Bloemendal H. Studies on β-crystallin. 1. Isolation and partial characterization of the principal polypeptide chain. Biochim Biophys Acta. 1974;336:370–82. [Google Scholar]
- 17.Litt M, Carrero-Valenzuela R, LaMorticella DM, Schultz DW, Mitchell TN, Kramer P, Maumenee IH. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet. 1997;6:665–8. doi: 10.1093/hmg/6.5.665. [DOI] [PubMed] [Google Scholar]
- 18.Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Heon E. Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci. 2000;41:159–65. [PubMed] [Google Scholar]
- 19.Vanita, Sarhadi V, Reis A, Jung M, Singh D, Sperling K, Singh JR, Burger J. A unique form of autosomal dominant cataract explained by gene conversion between beta-crystallin B2 and its pseudogene. J Med Genet. 2001;38:392–6. doi: 10.1136/jmg.38.6.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao K, Tang X, Shentu X, Wang K, Rao H, Xia K. Progressive polymorphic congenital cataract caused by a CRYBB2 mutation in a Chinese family. Mol Vis. 2005;11:758–63. http://www.molvis.org/molvis/v11/a91/ [PubMed] [Google Scholar]
- 21.Bateman JB, von-Bischhoffshaunsen FR, Richter L, Flodman P, Burch D, Spence MA. Gene conversion mutation in crystallin, beta-B2 (CRYBB2) in a Chilean family with autosomal dominant cataract. Ophthalmology. 2007;114:425–32. doi: 10.1016/j.ophtha.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Chambers C, Russell P. Deletion mutation in an eye lens beta-crystallin. An animal model for inherited cataracts. J Biol Chem. 1991;266:6742–6. [PubMed] [Google Scholar]
- 23.Graw J, Loster J, Soewarto D, Fuchs H, Reis A, Wolf E, Balling R, Hrabe de Angelis M. Aey2, a new mutation in the betaB2-crystallin-encoding gene of the mouse. Invest Ophthalmol Vis Sci. 2001;42:1574–80. [PubMed] [Google Scholar]
- 24.Kador PF, Fukui HN, Fukushi S, Jernigan HM, Jr, Kinoshita JH. Philly mouse: a new model of hereditary cataract. Exp Eye Res. 1980;30:59–68. doi: 10.1016/0014-4835(80)90124-4. [DOI] [PubMed] [Google Scholar]
- 25.Liu BF, Liang JJ. Domain interaction sites of human lens betaB2-crystallin. J Biol Chem. 2006;281:2624–30. doi: 10.1074/jbc.M509017200. [DOI] [PubMed] [Google Scholar]