

Abstract
Subjects with diabetes have increased cardiovascular disease risk compared to those without diabetes. Addressing residual cardiovascular disease risk in this disease, beyond blood pressure and LDL cholesterol control, remains important as the prevalence of diabetes increases worldwide. The accelerated atherosclerosis and cardiovascular disease in diabetes is likely multifactorial and there are numerous therapeutic approaches that can be considered. Results of mechanistic studies conducted in isolated cells, animals, or humans can provide important insights with potential to influence clinical management decisions and improve outcomes. In this review, we focus on three areas in which pathophysiologic considerations could be particularly informative in this regard; the roles of hyperglycemia, diabetic dyslipidemia (beyond LDL cholesterol level), and inflammation (including that in adipose tissue) for accelerating vascular injury and the rates of cardiovascular disease in Type 2 diabetes are outlined and evaluated.
INTRODUCTION
Multiple mechanisms likely contribute to the accelerated atherosclerosis and increased cardiovascular disease (CVD) risk observed in patients with Type 2 diabetes (T2DM). This review focuses on three areas in which basic mechanistic studies are most relevant to current clinical controversies for managing CVD risk in this disease. Pathophysiologic information relating hyperglycemia, diabetic dyslipidemia (beyond LDL cholesterol level), and inflammation to the accelerated vascular injury and CVD risk observed in T2DM will be evaluated and clinical considerations discussed.
HYPERGLYCEMIA AND THE VESSEL WALL
Although epidemiological studies show a consistent association between glycemic control and cardiovascular disease (1), the effect of tight glycemic control has been less convincing in clinical trials (2) The intensive glycemic control arm of the ACCORD (Action to Control Cardiovascular Risk in Diabetes) study was recently stopped due to increased cardiovascular deaths (3). A formal analysis of the study results has not yet been reported. The ADVANCE (Action in Diabetes and Vascular Disease) study will provide additional information regarding a potential CVD benefit of good glycemic control (4). Basic studies in vitro, in animal models, and in human subjects with diabetes suggest several mechanisms by which hyperglycemia might influence atherogenesis at the level of the artery wall (Fig 1).
Figure 1. Potential ways in which glucose and advanced glycation end-products (AGE-proteins) can affect atherogenesis in diabetes.

An early event in atherogenesis is adhesion of circulating monocytes to arterial endothelium. They enter the artery wall along a chemotactic gradient. Once inside the artery wall, monocytes can be activated and differentiate into macrophages. Atherogenic lipoproteins cross the endothelial barrier where they can be trapped by vascular proteoglycans and other matrix molecules such as collagen. Once retained by the matrix they can undergo modification by glycoxidation and other processes, which render the lipoproteins more toxic to vascular cells. They also can be taken up by macrophages, resulting in the formation of foam cells. Later, smooth muscle cell migrate from the media to the arterial intima. Glucose (*) and AGEs (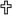 ) have been shown to affect various steps in these pathways as shown.
) have been shown to affect various steps in these pathways as shown.
Mechanisms by which hyperglycemia might affect vascular cells
Hyperglycemia could lead to vascular complications by several mechanisms. First, high glucose concentrations per se can activate nuclear factor κ-B (NFκB) (5;6), which in turn can increase the expression of a number of genes in endothelial cells, monocyte-macrophages and vascular smooth muscle cells. Advanced glycation end-products (AGEs) can be formed as a result of prolonged exposure of proteins and lipids to high concentration of glucose. AGEs include protein cross-links, fluorophors and other low molecular weight residues, which can generate reactive oxygen species. The ligation of AGEs to specific cell surface receptors can regulate gene expression in vessel wall cells.
Glucose also can increase oxidative stress, which in turn has several potential deleterious effects on the artery wall. For example, auto-oxidation of glucose leads to the formation of several reactive oxygen species such as the superoxide anion, and can facilitate LDL oxidation in vitro (7). Indirect evidence suggests that lipoprotein oxidation may be increased in diabetes in vivo (8) and is related to glycemic control (9). However, many of these studies relied on non-specific assays of oxidative stress. The absence of highly specific markers of oxidative stress in collagen (10) or in plasma or urine from subjects with diabetes (11) argues against a generalized increase in oxidative stress in diabetes. Thus, it has been suggested that glycoxidation reactions contribute to macrovascular disease in diabetes by damaging tissues in the local microenvironment of the arterial wall (11). Such pathways include the mitochondrial pathway for generation of superoxide, NADPH generation by monocyte-macrophages, or a redox-sensitive mechanism that generates hydroxyl radicals. The observation that products of hydroxyl radicals accumulate locally in arterial tissue of diabetic monkeys is consistent with the latter mechanism (12).
There has been a recent resurgence of interest in postprandial hyperglycemia as an important index of glycemic exposure and potential oxidative stress. 24-hour excretion of 8-iso prostaglandin F2α (8-iso PGF2α), an indicator of free radical production derived from arachidonic acid in cell membranes (13), was increased in patients with diabetes compared nondiabetic controls (14). Among those with diabetes, 8-iso PGF2α levels were highest in patients with the greatest glycemic variability. Moreover, glycemic variability was a strong predictor of total free radical production, whereas postprandial blood glucose levels were not. Indeed, swings in blood glucose levels accelerated atherosclerosis in apoE deficient mice (15). Additional studies are needed to determine the importance of oxidative stress that results from glycemic variability.
In the following sections we review experimental data of how glucose, AGEs and oxidative stress might influence atherogenesis. Available evidence will be reviewed for the three major types of vascular cells – endothelial cells, macrophages, and vascular smooth muscle cells (VSMC).
Glucose and the endothelium
An important initial event in the pathogenesis of atherosclerosis is the adhesion of monocytes to arterial endothelial cells, followed by their transmigration into the subendothelial space along a chemotactic gradient. Hyperglycemia enhances monocyte adhesion to cultured aortic endothelial cells (16) by affecting both cell types. One mechanism involves activation of NF-κB by hyperglycemia (5;6). NF-κB activation increases the expression of several inflammatory genes, including adhesion molecules that facilitate monocyte adhesion to endothelial cells (5).
Adhesion molecule expression may also result from impaired nitric oxide (NO) production, since agents that increase the production of NO reduce the expression of these adhesion molecules. Glucose- and AGE-mediated inhibition of NO production by endothelial cells also is associated with impaired endothelial dependent relaxation (17-19), an early marker of vascular injury. In addition to displaying marked impairment of endothelium-dependent relaxation, diabetic mice have evidence of increased peroxynitrite generation, nitrotyrosine expression, and lipid peroxidation in aortic tissue (20). Hyperglycemia and AGEs also stimulate the production of superoxide by endothelial cells in part by activating NADPH oxidase (6;21), thereby providing a link between hyperglycemia, AGEs and oxidative stress.
Glucose and monocyte-macrophages
Both high glucose conditions (22-24) and AGEs are associated with an increased state of activation of circulating monocytes in vitro and in vivo. Monocytes grown in high glucose conditions or isolated from subjects with poorly controlled diabetes are in an activated and inflammatory state, as evidenced for example by increased expression of the cytokines, interleukin-1 beta and interleukin-6 (22), and expression of CD36 and monocyte chemoattractant protein-1 (25). These inflammatory changes are associated with induction of protein kinase C, NF-κB activation and increased release of superoxide, which could play a role in the oxidative stress that occurs in the presence of hyperglycemia (26).
Monocytes entering the endothelial space in response to chemotactic factors, proliferate and differentiate into intimal macrophages, which accumulate in the artery wall in diabetes (26) . Although hyperglycemia is not sufficient to stimulate macrophage proliferation in lesions of atherosclerosis or in isolated murine macrophages, a combination of hyperglycemia and hyperlipidemia stimulates macrophage proliferation by a pathway that may involve glucose-dependent oxidation of LDL (27).
Arterial wall macrophages can accumulate lipid from modified forms of LDL, which are taken up by scavenger receptors. These include LDL that have become oxidized as a result of glucose-mediated oxidative stress (28), and AGE-modified LDL (29). In addition, AGE-modified albumin can inhibit the selective uptake of cholesteryl esters from HDL (30), a critical step in reverse cholesterol transport. Thus, modification of lipoproteins and other proteins that result from prolonged exposure to high glucose conditions can alter the delivery and removal of lipid from macrophages in a manner that is likely to promote atherosclerosis.
Glucose and vascular smooth muscle cells
As lesions progress, smooth muscle cells migrate from the media to the intima, where they proliferate, generate growth factors, and participate in the formation of a fibrous cap. High glucose concentrations can stimulate the proliferation of VSMC in vitro (31). Similar finding are observed with exposure of cell to AGEs (32) and high insulin concentrations (33), which often accompany hyperglycemia in T2DM.
VSMC generate several matrix molecules that are involved in atherogenesis. Vascular proteoglycans bind atherogenic lipoproteins, leading to their retention in the subendothelial space (34). An increase in chondroitin sulfate and dermatan sulfate proteoglycans, and a decrease in heparan sulfate proteoglycans in atherosclerotic lesions at autopsy from subjects with diabetes compared to lesions from nondiabetic subjects (35). The increase in chondroitin and dermatan sulfate proteoglycans in diabetes may contribute to the increased atherosclerosis in diabetes by increasing LDL retention in the artery wall (34). Diabetes also is associated with a loss of intimal elastin content and increased elastin fragmentation in both a rat (36) and pig (37) model of diabetes. Lower intimal elastin content, whether through decreased elastin production or increased elastin breakdown, appears to promote atherosclerosis by mechanisms that are not clear (37). Therefore, it is possible that elastin fragmentation is another mechanism by which hyperglycemia increases atherosclerosis in diabetes. Finally, collagen, which is made by VSMC, also accumulates in atherosclerosis. Advanced nonenzymatic glycation of collagen increases LDL binding, which might lead to increased lipoprotein retention by the artery wall in diabetes (38).
DIABETIC DYSLIPIDEMIA AND THE VESSEL WALL
Diabetic dyslipidemia is strongly related to atherosclerosis in human studies. Even though diabetic subjects may not have significantly elevated LDL cholesterol levels compared to matched subjects without diabetes, a cornerstone of managing CVD risk in diabetes rests on the use of LDL cholesterol-lowering statin drugs. Statin therapy generally reduces CVD events by 25-50% (39;40)but even among statin-treated subjects there appears to be excess residual CVD risk among those with diabetes compared to those without (41). Some of this residual risk could be attributed to lipoprotein abnormalities in diabetes that are not adequately addressed by statin therapy in T2DM. T2DM is characterized by decreased concentration of HDL cholesterol, increased concentration of triglyceride-rich lipoproteins, and abnormalities in the composition of HDL, LDL, and triglyceride-rich lipoprotein particles (Table 1) (42;43). In this section we will evaluate mechanisms relating diabetic dyslipidemia to CVD risk. Available evidence will be reviewed organized by lipoprotein parameters, as these highlight the therapeutic questions that confront the clinician for managing cardiovascular risk in patients with diabetes.
Table 1.
Lipoprotein Alterations in T2DM
| Triglyceride-rich lipoproteins |
| Increased particle number |
| Increased post-prandial level |
| Triglyceride and cholesterol enriched particles |
| LDL |
| Increased particle number |
| Small dense particles |
| HDL |
| Decreased particle number |
| Numerous changes in particle composition |
Patients with T2DM display characteristic abnormalities in lipoprotein level and composition.
Triglyceride-rich lipoproteins in diabetes
The triglyceride-rich lipoproteins in human diabetes are VLDL and its metabolites, and chylomicron remnants, and these can be elevated in the fasting or post-prandial state. The role of triglyceride-rich lipoproteins in human diabetic atherosclerosis remains controversial. Triglyceride levels vary inversely with HDL cholesterol levels, confounding interpretations relating elevations of triglyceride-rich lipoproteins to atherosclerosis (44). An interesting recent observation suggests that post-prandial triglyceride level may be a better predictor of CVD events than fasting triglyceride level independent of HDL cholesterol level (45;46). There is substantial in vitro support for a pro-atherogenic influence of triglyceride-rich lipoproteins in the vessel wall (Fig 2). Triglyceride-rich lipoproteins enhance a pro-inflammatory phenotype in endothelial cells and macrophages and produce apoptosis in endothelial cells (47). They increase tumor necrosis factor α (TNFα) expression in macrophages and increase expression of adhesion receptors resulting in increased adherence of monocytes and monocyte-derived macrophages to endothelial cells (48). ApoCIII, a component of triglyceride-rich lipoproteins and an inhibitor of lipoprotein lipase activity, increases adhesion of monocytic cells to endothelial cells (49). Chylomicron remnants and triglyceride-rich lipoproteins also produce lipid accumulation in macrophages (50). Uptake of larger lipid-rich VLDL particles is favored in macrophages, promoting lipid accumulation (51). Disruption of the VLDL receptor in macrophages reduces atherosclerosis in cholesterol-fed mice, while VLDL receptor expression in VLDL receptor-deficient recipient mice increases atherosclerosis (52). Lowering triglyceride-rich lipoproteins in a mouse model of diabetes and hyperlipidemia prevented disruption of atherosclerotic plaques (53). Elevated levels of post-prandial remnant lipoprotein particles have been shown to contribute to impaired arterial compliance (54).
Figure 2. Diabetic dyslipidemia and the vessel wall.

Insulin resistance and relative insulin deficiency in T2DM leads to abnormal lipoprotein particle number, size and composition. These lipoprotein changes impact gene expression and lipid flux in macrophages, endothelial cells, and arterial smooth cells to favor increased lipoprotein retention, sterol content, and inflammatory response in the vessel wall. These changes in the vessel wall favor growth of the atherosclerotic plaque and may predispose to instability and plaque rupture.
The fatty acid composition of chylomicron remnants influences their uptake, and the induction of lipid accumulation in macrophages (50). The ability of triglyceride-rich lipoprotein particles to induce an inflammatory phenotype in macrophages may be enhanced by lipolytic release of fatty acids from VLDL (50;55). Elevated free fatty acid levels are also a component of diabetic dyslipidemia and accompany elevated triglyceride level. Fatty acids can directly lead to changes in the composition of extracellular matrix produced by arterial smooth muscle cells in a manner that favors increased immobilization and retention of lipoproteins (56). Excess free fatty acid delivery to peripheral tissues can worsen insulin resistance and may play a role in activating inflammatory processes through activation of toll-like receptors (57). Free fatty acids have also been shown to impair endothelium-dependent vasodilatation (58), and to disrupt the function of cellular sterol transporters important for reverse cholesterol transport (59), as further discussed below. On the other hand, there is data suggesting that in certain circumstances physiologic lipolysis of triglyceride-rich lipoproteins may have beneficial anti-inflammatory effects. In some model systems, the lipolytic release of fatty acids can provide a ligand for nuclear hormone receptors involved in repressing inflammation such as PPARγ (60;61). Taken together, these data suggest that inappropriate generation and/or handling of fatty acids may represent a fundamental abnormality in diabetes leading to accelerated atherosclerosis.
LDL in diabetes
Subjects with T2DM may not have significantly higher LDL cholesterol levels than matched subjects without diabetes, but for any LDL cholesterol level, those with diabetes generally have more LDL particles (42;44). This is because in T2DM, small dense lipid-poor LDL particles accumulate in the circulation. Because each LDL particle contains one apoB molecule, for any level of LDL cholesterol, subjects with T2DM will also have a higher level of apoB. An increased number of LDL particles, either directly measured or as indicated by apoB levels, may contribute to atherogenesis and CVD risk (62-64). Increased LDL particle number in diabetes can be therapeutically addressed by statin therapy. A separate issue has arisen, however, whether or not small dense LDL particles are inherently more atherogenic on a per particle basis, compared to larger buoyant particles. Evidence for increased atherogenicity of small dense LDL particles has been obtained from in vitro studies showing that small LDL particles rapidly enter the arterial wall, can be more toxic to endothelial cells, cause greater production of pro-coagulant factors, are oxidized more readily, and are more readily immobilized by proteoglycans present in the arterial wall (65). These particles have also been shown to bind less well to the LDL receptor, which may lead to impaired clearance by the liver (65). It remains unclear, however, how these in vitro results translate to the in vivo milieu. There is no completely satisfactory in vivo model for testing atherogenicity of small dense LDL particles on a per particle basis compared to larger particles and all apoB-containing lipoproteins with the exception of the largest chylomicrons can enter the subendothelial space and accumulate in the arterial wall. In non-human primates fed fat-modified diets, LDL particle size per se was not found to be independently atherogenic (66). Recent studies in humans found that both large and small LDL particles are related to atherosclerosis and CVD events (67;68)
HDL in diabetes
Humans with T2DM have a decreased HDL cholesterol level and a decrease in the circulating concentration of apolipoprotein A1, the major apolipoprotein found in HDL (69). Abnormalities in size and composition of the HDL particle have also been noted (42;44;70;71). HDL and apoA1 remove excess cholesterol from atherosclerotic plaque cells, and their reduced level in diabetes would be expected to have a detrimental effect on vessel wall cholesterol content (Fig 2). The cell type of most interest here is monocyte-derived macrophages as cholesterol ester engorged macrophages (i.e. foam cells) are hallmarks of the atherosclerotic plaque. Removal of cholesterol from macrophages has been considered an important first step in the process of reverse cholesterol transport, and may be important for preventing progression or producing regression of atherosclerotic plaques (72). The HDL particle and its apoA1 component may act via distinct cellular sterol transporters for removing cholesterol from cells. The HDL particle appears to rely mainly on ATP binding cassette -G1 (ABCG1) to facilitate sterol efflux, and expression of ABCG1 in cells can be suppressed by exposure to glycosylated proteins (73). In addition, glycosylation of apoA1, which primarily acts via ABCA1, has been shown to suppress its ability to remove cholesterol from cells (74). HDL also has anti-inflammatory and anti-oxidant properties in cells of the vessel wall (75;76) Monocyte-derived macrophages isolated from subjects with low HDL cholesterol manifest a pro-inflammatory phenotype (77).
In addition to changes to HDL cholesterol and apoA1 level, subjects with diabetes have altered HDL composition. HDL is perhaps the most heterogeneous and complex of all lipoprotein particles, and changes in composition may importantly impact HDL atheroprotective properties (78) (Fig 2). In isolated cells, HDL particles of different size and composition display different abilities to remove cholesterol from cells (79). Changes in content of numerous proteins associated with HDL, for example paroxonase, may impact its atheroprotective properties (80). Compositional abnormalities of HDL isolated from human diabetic subjects have been linked to impaired anti-atherogenic properties (70). The failure of cholesterol ester transfer protein inhibition with torcetrapib to protect against CVD events also underscores the notion that HDL particle composition may be more important than HDL cholesterol level for reducing CVD risk (81).
Mice with absent apoA1 and very low HDL cholesterol levels have more atherosclerosis because of both reduced cholesterol transport and increased inflammation (82). Conversely, increased expression of apoA1 with higher HDL cholesterol levels produces less atherosclerosis in the apoE-/- mouse – a model of accelerated and progressive atherosclerosis (83). An increase in HDL cholesterol in diabetic subjects has been linked to reduced carotid atherosclerosis (84;85). HDL has also been shown to improve mobilization and function of endothelial precursor cells (86); and to protect the myocardium from ischemia/re-perfusion injury (87).
Glycemia vs hyperlipidemia in the pathogenesis of atherosclerosis in diabetes
The relative roles of hyperglycemia and hyperlipidemia in atherogenesis have been difficult to separate in animal models of diabetes. Hyperlipidemia is usually exacerbated by the onset of hyperglycemia in mouse models such as LDL receptor and apoE deficient mice, thereby confounding the effect of hyperglycemia. However, two animal models suggest that hyperglycemia plays an independent role. First, fat-fed diabetic swine had more atherosclerosis than equally dyslipidemic fat-fed animals without diabetes (88). Second, consumption of a cholesterol-free diet by LDL receptor-deficient mice with a novel form of diabetes induced by a beta cell-directed viral antigen resulted in hyperglycemia without changes in lipids and lipoproteins (89). Hyperglycemia per se was associated with lesion initiation. Addition of increasing amounts of dietary cholesterol led to dyslipidemia, which was the major factor in atherosclerosis progression, independent of hyperglycemia (89).
CHRONIC SUB-CLINICAL INFLAMMATION AND THE VESSEL WALL
Extensive evidence ranging from human pathologic studies to in vivo mouse models establishes the role of inflammatory cells, like macrophages and T lymphocytes, and inflammatory mechanisms, like cytokine release in the pathogenesis of atherosclerosis (90). Several issues frame any consideration of the extensive pre-clinical data implicating inflammation in diabetic atherosclerosis. T2DM and atherosclerosis are both chronic conditions arising over decades, a timeline that makes discerning cause and effect a challenge (Fig. 3). Inflammation is implicated in the pathogenesis of both T2DM and atherosclerosis (91;92). Since T2DM itself promotes atherosclerosis and increases cardiovascular events, a distinction may exist between inflammation that fosters T2DM versus inflammation that occurs subsequent to T2DM and that promotes atherosclerosis directly (Fig. 3). Most of the inflammatory mechanisms under discussion here also appear involved in the atherosclerosis seen in pre-diabetic as well as non-diabetic states. Here the focus is on those inflammatory mechanisms in diabetes that promote atherosclerosis and its complications, whether based in a given cell type or part of a more general pathway. Such an approach is required given that, although the evidence implicating inflammation in atherosclerosis and T2DM is extensive, no one specific mechanism, nor a single integrated framework, has emerged to explain precisely why patients with diabetes are more prone to inflammation or atherosclerosis.
Figure 3. The intersection of inflammation, T2DM and atherosclerosis.

Important relationships among inflammation, atherosclerosis, and key characteristics of T2DM are shown. Sorting out mechanistic relationships in humans is challenging given the chronicity of these problems, including relatively long pre-clinical phases and common, overlapping antecedents, like increased insulin resistance. For example, the constellation of abnormalities associated with T2DM, and pre-diabetic states, are also associated with cardiovascular risk. One common theme in these connections is inflammation.
Inflammation in Diabetic Atherosclerosis: Cellular Players
The endothelium, as the cellular interface between the circulation and the hyperglycemia and dyslipidemia that characterizes T2DM, responds to such stimuli by exhibiting an inflammatory response (93). Most of the responses induced in atherosclerosis are common to both diabetic and non-diabetic atherosclerosis. Classic pro-atherosclerotic endothelial responses – adhesion molecule expression, secretion of chemokines and coagulation proteins (plasminogen activator inhibitor 1, total plasminogen activator, tissue factor), release of vasoactive mediators (endothelial NO and bradykinin) – are induced or regulated by inflammatory stimuli in diabetes models in vitro and/or in vivo (94;95) as discussed earlier.
Lymphocytes, which provide critical pro-inflammatory signals to monocyte/macrophages, and VSMC, are activated by metabolic stimuli (96-98). Macrophages also directly respond to common abnormalities found in T2DM like glucose, free fatty acids and hypertriglyceridemia, by augmenting inflammatory responses (90;99). Multiple stimuli and cellular pathways are involved in these macrophage effects (Fig. 4), including increased foam cell formation, release of matrix metalloproteinases and secretion of growth factors and cytokines (92;100). Macrophages also highlight the important connections between insulin resistance, inflammation and atherosclerosis. When bone marrow from insulin receptor deficient mice was transplanted into LDL receptor deficient mice, more complex lesions were noted (101). Macrophage specific deficiency of the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) in mice alters insulin resistance (102;103), suggesting that the presence of this ligand-activated transcription factor in macrophages regulates insulin sensitivity, an intriguing finding given PPARγ’s role in repressing inflammation (104). Similar issues apply to retinoid signaling through the retinoid X receptor, the required partner for PPARγ and many other nuclear receptors (100;105).
Figure 4. Macrophage biology in T2DM and atherosclerosis.

The macrophage is a key player in atherosclerosis and may play an important role in the accelerated atherosclerosis of diabetes. Various factors commonly encountered in T2DM – hyperglycemia, elevated circulating cytokokines, increased free fatty acids (FFA), LDL and its modified forms, AGEs and cellular debris in the arterial wall (e.g. apoptotic bodies) can incite multiple responses in macrophages, including ER stress, generation of reactive oxygen species (ROS), and increased ceramide levels. These and other stressors can impinge on downstream inflammatory signaling pathways, such as JNK/AP1 and NFκB, further amplifying expression of a pro-inflammatory macrophage phenotype. Other mechanisms, like PPARγ activation, may balance some of these inflammatory responses.
Inflammation in Diabetic Atherosclerosis: Mechanisms
The available data suggests cellular responses to injury, inflammation and metabolism may converge on control points that are important in atherosclerosis. A central regulator of inflammation is NF-κB (106), a transcriptional complex activated by various stimuli including cytokines, oxidized LDL, lipopolysaccharide and oxidant stress (Fig. 4) (92). NF-κB reportedly regulates LDL oxidative modification, chemokine and cytokine expression, macrophage growth and differentiation, apoptosis, and VSMC proliferation. NF-κB, its regulatory proteins like inhibitor κB (IkB), and distal targets like the c-Jun n-terminal kinase (JNK) have all been strongly implicated in insulin sensitivity as well as atherosclerosis (92;100;107;108) (Fig. 4). As such, NF-kB may serve as a final common pathway linking many inputs that are activated in T2DM to atherosclerotic responses. NF-κB is activated by factors commonly abnormal in T2DM including fatty acids, glucose itself, AGE pathways, and certain toll like receptors (TLRs), a family of pattern recognition receptors found in various inflammatory cells (109). The number of NF-κB regulated targets involved in diabetic atherosclerosis are extensive including TNF-α, which increases insulin resistance, TLRs, and resistin to name a few. In mice, inhibiting NF-κB activation can improve insulin sensitivity and decrease atherosclerosis, a concept under study in humans through several approaches, including high dose salicylates (92). PPARγ’s anti-inflammatory and anti-atherosclerotic effects evident in vitro and in mice may also involve NF-κB inhibition (Fig 4) (104).
Several mechanistic pathways have been proposed for how glucose induces cellular injury and subsequent inflammation. A broad theme in this work suggests that cells lacking the ability to counter elevated intracellular glucose levels activate pathways of cellular injury and inflammation (see above) (110). These mechanisms include activation of protein kinase C, the formation of polyols, which promotes intracellular oxidative stress, and increased hexosamine activation, with subsequent increases in reactive oxidant species and mitochondrial stress (111-113). Although much of this evidence was linked to diabetic microvascular disease, increased flux of free fatty acids into the endothelium may influence macrovascular disease through similar pathways, inducing inflammation
All secretory and membrane proteins, many nutrients, and many pathogens pass through the endoplasmic reticulum (ER). Several lines of study implicate ER stress in promoting inflammation (114). Hypoxia, hyperglycemia, and increased fatty acids can all induce ER stress and a specific cellular process known as the unfolded protein response (UPR) (115), which is a homeostatic mechanism that restores normal ER function. ER stress, present in liver and in adipose tissue, can activate pro-oxidant and pro-inflammatory pathways and has been implicated in both diabetes and atherosclerosis (116).
Adipose tissue inflammation: A core mechanism in diabetic atherosclerosis?
Adipose tissue is now recognized as a biologically active endocrine and paracrine organ. This new view of fat has many implications for the intersection of inflammation, atherosclerosis and T2DM, especially given clinical data placing adiposity as a core defect in the metabolic abnormalities found before and during diabetes (94). Inflammation in adipose tissue itself may contribute to abnormal metabolism and atherosclerosis in T2DM.
Many of the pathways described above – oxidant stress, ER stress, NF-κB activation – also operate in adipocytes (94;100). Oxidative stress and inflammation in adipose tissue can be amplified by hyperglycemia (117). Fatty acids released from adipose tissue may signal to macrophages via pathways that involve toll-like receptors, leading to NF-κB activation. Interestingly, many of the same pathways involved in recruiting leukocytes to the arterial wall also recruit inflammatory cells to fat, including monocyte chemoattractant protein-1 (118). Indeed, mice lacking C chemokine receptor-2 (CCR2), the receptor for monocyte chemoattractant protein -1 (MCP-1), enjoy some degree of protection from diet-induced insulin resistance and induction of inflammatory programs (119). Excess lipid accumulation in other tissues, like skeletal muscle and the liver, may also modulate inflammation, contributing to insulin resistance and atherosclerosis (120;121).
Increased levels of inflammatory cytokines released from visceral fat in diabetes and obesity can act directly on the liver to increase the circulating levels of pro-inflammatory molecules such as C reactive protein (CRP) and serum amyloid A (SAA) (122). The former may directly amplify injury at the vessel wall and the latter unfavorably modifies the composition and function of HDL. The expression of adipose tissue apolipoproteins which impact adipocyte lipid metabolism, is also modified by inflammatory cytokines (123;124). Several adipocyte-specific mediators have been implicated in the inflammation contributing to insulin resistance and atherosclerosis. Leptin is an adipocyte-specific signal that appears to exert systemic pro-inflammatory effects (125). Leptin produces pro-inflammatory changes in endothelial cells and macrophages, and administration of leptin to apoE-deficient mice promotes atherosclerosis (122). Adiponectin, which circulates in the plasma in various multimeric forms, limits inflammatory and atherosclerotic responses (126;127). Adiponectin levels are lower in obesity and diabetes (122;126) and the treatment of apoE-deficient mice with an adiponectin-expressing adenovirus has been shown to reduce atherosclerotic plaque formation (122). Adiponectin is present at higher levels in adipocytes from subcutaneous fat than visceral fat, one of many examples that suggest both depot-specific differences in fat and increased pathogenicity from visceral fat (122).
SUMMARY
Even in the statin era of lower LDL levels, hyperglycemia, lipoprotein abnormalities, and inflammation all appear intertwined in accelerating atherosclerosis and CVD risk in patients with diabetes. It is obvious from the data summarized here that important and complex links between hyperglycemia, lipoprotein abnormalities and systemic and vessel wall inflammation need to be considered when evaluating how each of these impact vessel wall health in diabetes. It is equally apparent that despite intensive study, persistent questions regarding these interactions require further investigation. Changes in glycemia impact lipoprotein levels, composition and metabolism, and can also directly impact inflammatory pathways, like NF-κB. Altered lipoprotein and lipid flux can modulate insulin sensitivity, glucose disposal, and thereby influence circulating glucose levels, as well as directly impact inflammation. Inflammation, both systemically and in adipose tissue, may play an important pathophysiologic role in overall insulin sensitivity and alter carbohydrate metabolism, lipid metabolism and inflammatory pathways in the liver, and directly add to vessel wall injury.
While pathophysiologic relationships do not always translate into high-value therapeutic targets, the abundance of pathophysiologic and mechanistic information from in vitro and animal models provides important insights for the design and interpretation of human clinical studies. These insights are also essential for understanding the implications of equivocal or negative results in randomized clinical trials. The recent failures of the intensive glucose control arm in the ACCORD trial (3), and of HDL cholesterol raising by the CETP inhibitor torcetrapib (81), to reduce cardiovascular death must be viewed in terms of the pathophysiologic and animal data related to these therapeutic areas. For example, data from in vitro and animal models would support the argument that the failure of intensive glycemic control in ACCORD should not eliminate the future consideration of hyperglycemia as an important therapeutic target for reducing CVD in diabetes, but may relate to an unfavorable benefit/risk ratio of currently available glucose-lowering therapies in the specific populations recruited for that trial. This elderly at-risk population may have been more susceptible to the adverse effects of hypoglycemia, off target drug effects, or the risk imposed by ectopic fat deposition that usually accompany intensification of glucose lowering therapy (128). With respect to the failure of torcetrapib, understanding the complexity of HDL metabolism and composition, and the potent atheroprotective effect HDL can have in animal models, argues that other methods of modifying HDL metabolism warrant evaluation. The lipid arm of ACCORD will provide information regarding the value of adding a fibrate (which can raise HDL cholesterol) to statin therapy in diabetic subjects (129). While therapeutic interventions in humans can never be as targeted or specific as the experimental manipulations achievable in isolated cells or in animal models, pathophysiologic and mechanistic information from these models remain key sources of insight for designing and evaluating new therapeutic options to reduce CVD in diabetes.
ACKNOWLEDGEMENTS
TM is supported by grants DK71711 and HL39653 from the National Institutes of Health; AC is supported by grants HL30086 and HL19645 from the National Institutes of Health; JP is supported by grants HL071745 and HL48743 from the National Institutes of Health. The authors thank Stephanie Thompson for assistance with manuscript preparation.
Role of Funding Source No funding source was directly involved in the development of this review.
Footnotes
Conflict of Interest Statement The authors disclose the following relationships. TM has received research support from Novartis, Takeda, and has received honoraria from Amylin, GlaxoSmithKline, Merck, Takeda, and; AC has consulted with Merck, Pfizer, Novo Nordisk, Novartis, Astra-Zeneca, Takeda; JP has consulted with Astra-Zeneca, Dainippon, GlaxoSmithKline, Merck, Novo Nordisk, Pfizer, Sanofi-Aventis. No other potential conflicts.
Search Strategy Publicly available databases (PubMed, Medline) were searched. References were selected from peer-reviewed journals based on importance, novelty and relevance to the subject of the review.
Reference List
- (1).Balkau B, Hu G, Qiao Q, Tuomilehto J, Borch-Johnsen K, Pyorala K. Prediction of the risk of cardiovascular mortality using a score that includes glucose as a risk factor: The DECODE Study. Diabetologia. 2004;47:2118–28. doi: 10.1007/s00125-004-1574-5. [DOI] [PubMed] [Google Scholar]
- (2).UK Prospective Diabetes Study (UKPDS) Group Intensive blood-glucose control with sulphonylureas or insluin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–53. [PubMed] [Google Scholar]
- (3).For Safety, NHLBI Changes Intensive Blood Sugar Treatment Strategy in Clinical Trial of Diabetes and Cardiovascular Disease [news release] National Heart, Lung, and Blood Institute Communication Office; [Accessed March 7, 2006]. http://public.nhlbi.nih.gov/newsroom/home/GetPressRelease.aspx?id=2551. 2-3-2006. Ref Type: Internet Communication. [Google Scholar]
- (4).ADVANCE Management Committee Study rationale and design of ADVANCE: action in diabetes and vascular disease-preterax and diamicron MR controlled evaluation. Diabetologia. 2008;44:1118–20. doi: 10.1007/s001250100612. [DOI] [PubMed] [Google Scholar]
- (5).Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T. Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis. 2007;193:328–34. doi: 10.1016/j.atherosclerosis.2006.09.016. [DOI] [PubMed] [Google Scholar]
- (6).Yan SD, Schmidt AM, Anderson GM, Zhang JH, Brett J, Zou YS, et al. Enhanced Cellular Oxidant Stress by the Interaction of Advanced Glycation End-Products with Their Receptors Binding-Proteins. J Biol Chem. 1994;269:9889–97. [PubMed] [Google Scholar]
- (7).Kawamura M, Heinecke JW, Chait A. Pathophysiological Concentrations of Glucose Promote Oxidative Modification of Low-Density-Lipoprotein by A Superoxide-Dependent Pathway. J Clin Invest. 1994;94:771–8. doi: 10.1172/JCI117396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McSorley PT, Young IS, McEneny J, Fee H, McCance DR. Susceptibility of low-density lipoprotein to oxidation and circulating cell adhesion molecules in young healthy adult offspring of parents with type 2 diabetes. Metabolism. 2004;53:755–9. doi: 10.1016/j.metabol.2003.11.026. [DOI] [PubMed] [Google Scholar]
- (9).Hussein OA, Gefen Y, Zidan JM, Karochero EY, Luder AS, Assy NN, et al. LDL oxidation is associated with increased blood hemoglobin A1c levels in diabetic patients. Clinica Chimica Acta. 2007;377:114–8. doi: 10.1016/j.cca.2006.09.002. [DOI] [PubMed] [Google Scholar]
- (10).Wells-Knecht MC, Lyons TJ, McCance DR, Thorpe SR, Baynes JW. Age-dependent increase in ortho-tyrosine and methionine sulfoxide in human skin collagen is not accelerated in diabetes - Evidence against a generalized increase in oxidative stress in diabetes. J Clin Invest. 1997;100:839–46. doi: 10.1172/JCI119599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Pennathur S, Heinecke JW. Mechanisms for oxidative stress in diabetic cardiovascular disease. Antiox Redox Sig. 2007;9:955–69. doi: 10.1089/ars.2007.1595. [DOI] [PubMed] [Google Scholar]
- (12).Pennathur S, Wagner JD, Leeuwenburgh C, Litwak KN, Heinecke JW. A hydroxyl radical-like species oxidizes cynomolgus monkey artery wall proteins in early diabetic vascular disease. J Clin Invest. 2001;107:853–60. doi: 10.1172/JCI11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dogne JM, Hanson J, Pratico D. Thromboxane, prostacyclin and isoprostanes: therapeutic targets in atherogenesis. Trends Pharmacol Sci. 2005;26:639–44. doi: 10.1016/j.tips.2005.10.001. [DOI] [PubMed] [Google Scholar]
- (14).Davi G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, et al. In vivo formation of 8-iso-prostaglandin F-2 alpha and platelet activation in diabetes mellitus - Effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99:224–9. doi: 10.1161/01.cir.99.2.224. [DOI] [PubMed] [Google Scholar]
- (15).Mita T, Otsuka A, Azuma K, Uchida T, Ogihara T, Fujitani Y, et al. Swings in blood glucose levels accelerate atherogenesis in apolipoprotein E-deficient mice. Biochem Biophys Res Comm. 2007;358:679–85. doi: 10.1016/j.bbrc.2007.04.118. [DOI] [PubMed] [Google Scholar]
- (16).Otsuka A, Azuma K, Iesaki T, Sato F, Hirose T, Shimizu T, et al. Temporary hyperglycaemia provokes monocyte adhesion to endothelial cells in rat thoracic aorta. Diabetologia. 2005;48:2667–74. doi: 10.1007/s00125-005-0005-6. [DOI] [PubMed] [Google Scholar]
- (17).Cohen RA. Role of nitric oxide in diabetic complications. Am J Ther. 2005;12:499–502. doi: 10.1097/01.mjt.0000178776.77267.19. [DOI] [PubMed] [Google Scholar]
- (18).Bucala R, Tracey KJ, Cerami A. Advanced Glycosylation Products Quench Nitric-Oxide and Mediate Defective Endothelium-Dependent Vasodilatation in Experimental Diabetes. J Clin Invest. 1991;87:432–8. doi: 10.1172/JCI115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Decaterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, et al. Nitric-Oxide Decreases Cytokine-Induced Endothelial Activation - Nitric-Oxide Selectively Reduces Endothelial Expression of Adhesion Molecules and Proinflammatory Cytokines. J Clin Invest. 1995;96:60–8. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, et al. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci. 2007;104:5217–22. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, Zuodar G, et al. Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: The distinct role of protein kinase C and mitochondrial. superoxide production. Atherosclerosis. 2005;183:259–67. doi: 10.1016/j.atherosclerosis.2005.03.015. [DOI] [PubMed] [Google Scholar]
- (22).Dasu MR, Devaraj S, Jialal I. High glucose induces IL-1 beta expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab. 2007;293:E337–E346. doi: 10.1152/ajpendo.00718.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased TLR2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a pro-inflammatory state. J Clin Endocrinol Metab. 2007;93:578–83. doi: 10.1210/jc.2007-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. 2006;55:774–9. doi: 10.2337/diabetes.55.03.06.db05-1417. [DOI] [PubMed] [Google Scholar]
- (25).Cipolletta C, Ryan KE, Hanna EV, Trimble ER. Activation of peripheral blood CD14(+) monocytes occurs in diabetes. Diabetes. 2005;54:2779–86. doi: 10.2337/diabetes.54.9.2779. [DOI] [PubMed] [Google Scholar]
- (26).Venugopal SK, Devaraj S, Yang T, Jialal I. Alpha-tocopherol decreases superoxide anion release in human monocytes under hyperglycemic conditions via inhibition of protein kinase C-alpha. Diabetes. 2002;51:3049–54. doi: 10.2337/diabetes.51.10.3049. [DOI] [PubMed] [Google Scholar]
- (27).Burke AP, Kolodgie FD, Zieske A, Fowler DR, Weber DK, Varghese PJ, et al. Morphologic findings of coronary atherosclerotic plaques in diabetics - A postmortem study. Arterioscler Thromb Vasc Biol. 2004;24:1266–71. doi: 10.1161/01.ATV.0000131783.74034.97. [DOI] [PubMed] [Google Scholar]
- (28).Kawamura M, Heinecke JW, Chait A. Increased uptake of alpha-hydroxy aldehyde-modified low density lipoprotein by macrophage scavenger receptors. J Lipid Res. 2000;41:1054–9. [PubMed] [Google Scholar]
- (29).Miyazaki A, Nakayama H, Horiuchi S. Scavenger receptors that recognize advanced glycation end products. Trends Cardiovasc Med. 2002;12:258–62. doi: 10.1016/s1050-1738(02)00171-8. [DOI] [PubMed] [Google Scholar]
- (30).Ohgami N, Miyazaki A, Sakai M, Kuniyasu A, Nakayama H, Horiuchi S. Advanced glycation end products (AGE) inhibits a new crossroad of AGE to cholesterol metabolism. J Atheroscler Thromb. 2003;10:1–6. doi: 10.5551/jat.10.1. [DOI] [PubMed] [Google Scholar]
- (31).Carmody BJ, Arora S, Wakefield MC, Weber M, Fox CJ, Sidawy AN. Progesterone inhibits human infragenicular arterial smooth muscle cell proliferation induced by high glucose and insulin concentrations. J Vasc Surg. 2002;36:833–8. [PubMed] [Google Scholar]
- (32).Seki N, Hashimoto N, Sano H, Horiuchi S, Yagui K, Makino H, et al. Mechanisms involved in the stimulatory effect of advance glycation end products on growth of rat aortic smooth muscle cells. Metabolism. 2003;52:1558–63. doi: 10.1016/j.metabol.2003.07.010. [DOI] [PubMed] [Google Scholar]
- (33).Pfeifle B, Ditschuneil H. The effect of insulin and insulin-like growth factors on cell proliferation of human smooth muscle cells. Artery. 1980;8:336–41. [PubMed] [Google Scholar]
- (34).Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–44. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- (35).Heickendorff L, Ledet T, Rasmussen LM. Glycosaminoglycans in the Human Aorta in Diabetes-Mellitus - A Study of Tunica Media from Areas with and Without Atherosclerotic Plaque. Diabetologia. 1994;37:286–92. doi: 10.1007/BF00398056. [DOI] [PubMed] [Google Scholar]
- (36).Kwan CY, Wang RRJ, Beazley JS, Lee RMKW. Alterations of Elastin and Elastase-Like Activities in Aortae of Diabetic Rats. Biochimica et Biophysica Acta. 1988;967:322–5. doi: 10.1016/0304-4165(88)90027-x. [DOI] [PubMed] [Google Scholar]
- (37).McDonald TO, Gerrity RG, Jen C, Chen HJ, Wark K, Wight TN, et al. Diabetes and arterial extracellular matrix changes in a porcine model of atherosclerosis. J Histochem Cytochem. 2007;55:1149–57. doi: 10.1369/jhc.7A7221.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Brownlee M, Vlassara H, Cerami A. Nonenzymatic Glycosylation Products on Collagen Covalently Trap Low-Density Lipoprotein. Diabetes. 1985;34:938–41. doi: 10.2337/diab.34.9.938. [DOI] [PubMed] [Google Scholar]
- (39).Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–16. doi: 10.1016/s0140-6736(03)13636-7. [DOI] [PubMed] [Google Scholar]
- (40).Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364:685–96. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- (41).Costa J, Borges M, David C, Vaz CA. Efficacy of lipid lowering drug treatment for diabetic and non-diabetic patients: meta-analysis of randomised controlled trials. BMJ. 2006;332:1115–24. doi: 10.1136/bmj.38793.468449.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Garvey WT, Kwon S, Zheng D, Shaughnessy S, Wallace P, Hutto A, et al. Effects of insulin resistance and type 2 diabetes on lipoprotein subclass particle size and concentration determined by nuclear magnetic resonance. Diabetes. 2003;52:453–62. doi: 10.2337/diabetes.52.2.453. [DOI] [PubMed] [Google Scholar]
- (43).Deeg MA, Buse JB, Goldberg RB, Kendall DM, Zagar AJ, Jacober SJ, et al. Pioglitazone and rosiglitazone have different effects on serum lipoprotein particle concentrations and sizes in patients with type 2 diabetes and dyslipidemia. Diabetes Care. 2007;30:2458–64. doi: 10.2337/dc06-1903. [DOI] [PubMed] [Google Scholar]
- (44).Goldberg IJ. Clinical review 124: Diabetic dyslipidemia: causes and consequences. J Clin Endocrinol Metab. 2001;86:965–71. doi: 10.1210/jcem.86.3.7304. [DOI] [PubMed] [Google Scholar]
- (45).Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298:309–16. doi: 10.1001/jama.298.3.309. [DOI] [PubMed] [Google Scholar]
- (46).Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- (47).Shin HK, Kim YK, Kim KY, Lee JH, Hong KW. Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase-mediated production of superoxide and cytokines via lectin-like oxidized low-density lipoprotein receptor-1 activation - Prevention by cilostazol. Circulation. 2004;109:1022–8. doi: 10.1161/01.CIR.0000117403.64398.53. [DOI] [PubMed] [Google Scholar]
- (48).Ting HJ, Stice JP, Schaff UY, Hui DY, Rutledge JC, Knowlton AA, et al. Triglyceride-rich lipoproteins prime aortic endothelium for an enhanced inflammatory response to tumor necrosis factor-alpha. Circ Res. 2007;100(3):381–90. doi: 10.1161/01.RES.0000258023.76515.a3. [DOI] [PubMed] [Google Scholar]
- (49).Kawakami A, Aikawa M, Libby P, Sacks FM. Apolipoprotein C-III in ApoB lipoproteins enhances the adhesion of monocytes to endothelial cells by increasing the active form of beta-1 integrin. Arterioscler Thromb Vasc Biol. 2005;25(5):E53. [Google Scholar]
- (50).De Pascale C, Avella M, Perona JS, Ruiz-Gutierrez V, Wheeler-Jones CPD, Botham KM. Fatty acid composition of chylomicron remnant-like particles influences their uptake and induction of lipid accumulation in macrophages. Febs J. 2006;273:5632–40. doi: 10.1111/j.1742-4658.2006.05552.x. [DOI] [PubMed] [Google Scholar]
- (51).Palmer AM, Nova E, Anil E, Jackson K, Bateman P, Wolstencroft E, et al. Differential uptake of subfractions of triglyceride-rich lipoproteins by THP-1 macrophages. Atherosclerosis. 2005;180:233–44. doi: 10.1016/j.atherosclerosis.2004.12.038. [DOI] [PubMed] [Google Scholar]
- (52).Van Eck M, Oost J, Goudriaan JR, Hoekstra M, Hildebrand RB, Bos IST, et al. Role of the macrophage very-low-density lipoprotein receptor in atherosclerotic lesion development. Atherosclerosis. 2005;183:230–7. doi: 10.1016/j.atherosclerosis.2005.03.045. [DOI] [PubMed] [Google Scholar]
- (53).Johansson F, Kramer F, Barnhart S, Kanter JE, Vaisar T, Merrill RD, et al. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc Natl Acad Sci. 2008;105:2082–7. doi: 10.1073/pnas.0709958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Nestel PJ, Shige H, Pomeroy S, Cehun M, Chin-Dusting J. Post-prandial remnant lipids impair arterial compliance. J Am Coll Cardiol. 2001;37:1929–35. doi: 10.1016/s0735-1097(01)01251-7. [DOI] [PubMed] [Google Scholar]
- (55).Saraswathi V, Hasty AH. The role of lipolysis in mediating the proinflammatory effects of very low density lipoproteins in mouse peritoneal macrophages. J Lipid Res. 2006;47:1406–15. doi: 10.1194/jlr.M600159-JLR200. [DOI] [PubMed] [Google Scholar]
- (56).Rodriguez-Lee M, Bondjers G, Camejo G. Fatty acid-induced atherogenic changes in extracellular matrix proteoglycans. Curr Opin Lipidology. 2007;18:546–53. doi: 10.1097/MOL.0b013e3282ef534f. [DOI] [PubMed] [Google Scholar]
- (57).Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Steinberg HO, Tarshoby M, Monestel R, Hook G, Cronin J, Johnson A, et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J Clin Invest. 1997;100:1230–9. doi: 10.1172/JCI119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wang YT, Oram JF. Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a protein kinase C delta pathway. J Lipid Res. 2007;48:1062–8. doi: 10.1194/jlr.M600437-JLR200. [DOI] [PubMed] [Google Scholar]
- (60).Ziouzenkova O, Plutzky J. Lipolytic PPAR activation: new insights into the intersection of triglycerides and inflammation? Curr Opin Clin Nutr Metab Care. 2004;7:369–75. doi: 10.1097/01.mco.0000134358.46159.61. [DOI] [PubMed] [Google Scholar]
- (61).Ziouzenkova O, Perrey S, Asatryan L, Hwang J, MacNaul KL, Moller DE, et al. Lipolysis of triglyceride-rich lipoproteins limits inflammatory responses through PPAR-alpha activation. Proc Natl Acad Sci USA. 2003;100:2730–2735. doi: 10.1073/pnas.0538015100. Ref Type: Journal (Full) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jiang R, Schulze MB, Li TC, Rifal N, Stampfer MJ, Rimm EB, et al. Non-HDL cholesterol and apolipoprotein B predict cardiovascular disease events among men with type 2 diabetes. Diabetes Care. 2004;27(8):1991–7. doi: 10.2337/diacare.27.8.1991. [DOI] [PubMed] [Google Scholar]
- (63).Kuller L, Arnold A, Tracy R, Otvos J, Burke G, Psaty B, et al. Nuclear magnetic resonance spectroscopy of lipoproteins and risk of coronary heart disease in the Cardiovascular Health Study. Arteriosclerosis Thrombosis and Vascular Biology. 2002;22:1175–80. doi: 10.1161/01.atv.0000022015.97341.3a. [DOI] [PubMed] [Google Scholar]
- (64).El Harchaoui K, van der Steeg WA, Stroes ES, Kuivenhoven JA, Otvos JD, Wareham NJ, et al. Value of low-density lipoprotein particle number and size as predictors of coronary artery disease in apparently healthy men and women: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol. 2007;49:547–53. doi: 10.1016/j.jacc.2006.09.043. [DOI] [PubMed] [Google Scholar]
- (65).Sniderman AD, Scantlebury T, Cianflone K. Hypertriglyceridemic hyperapoB: The unappreciated atherogenic dyslipoproteinemia in type 2 diabetes mellitus. Ann Intern Med. 2001;135:447–59. doi: 10.7326/0003-4819-135-6-200109180-00014. [DOI] [PubMed] [Google Scholar]
- (66).Rudel LL, Johnson FL, Sawyer JK, Wilson MS, Parks JS. Dietary Polyunsaturated Fat Modifies Low-Density Lipoproteins and Reduces Atherosclerosis of Nonhuman-Primates with High and Low Diet Responsiveness. Am J Clin Nutr. 1995;62:S463–S470. doi: 10.1093/ajcn/62.2.463S. [DOI] [PubMed] [Google Scholar]
- (67).Mora S, Szklo M, Otvos JD, Greenland P, Psaty BM, Goff DC, et al. LDL particle subclasses, LDL particle size, and carotid atherosclerosis in the Multi-Ethnic Study of Atherosclerosis (MESA) Atherosclerosis. 2007;192:211–7. doi: 10.1016/j.atherosclerosis.2006.05.007. [DOI] [PubMed] [Google Scholar]
- (68).Otvos JD, Collins D, Freedman DS, Shalaurova I, Schaefer EJ, McNamara JR, et al. Low-density lipoprotein and high-density lipoprotein particle subclasses predict coronary events and are favorably changed by gemfibrozil therapy in the Veterans Affairs High-Density Lipoprotein Intervention Trial. Circulation. 2006;113:1556–63. doi: 10.1161/CIRCULATIONAHA.105.565135. [DOI] [PubMed] [Google Scholar]
- (69).Mooradian AD, Haas MJ, Wong NCW. Transcriptional control of apolipoprotein A-I gene expression in diabetes. Diabetes. 2004;53:513–20. doi: 10.2337/diabetes.53.3.513. [DOI] [PubMed] [Google Scholar]
- (70).Gowri MS, Van der Westhuyzen DR, Bridges SR, Anderson JW. Decreased protection by HDL from poorly controlled type 2 diabetic subjects against LDL oxidation may be due to the abnormal composition of HDL. Arterioscler Thromb Vasc Biol. 1999;19:2226–33. doi: 10.1161/01.atv.19.9.2226. [DOI] [PubMed] [Google Scholar]
- (71).Chahil TJ, Ginsberg HN. Diabetic dyslipidemia. Endocrinol Metab Clin North Am. 2006;35:491–510. doi: 10.1016/j.ecl.2006.06.002. [DOI] [PubMed] [Google Scholar]
- (72).Cuchel M, Rader DJ. Macrophage reverse cholesterol transport - Key to the regression of atherosclerosis? Circulation. 2006;113:2548–55. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- (73).Isoda K, Folco EJ, Shimizu K, Libby P. AGE-BSA decreases ABCG1 expression and reduces macrophage cholesterol efflux to HDL. Atheroscler. 2007;192:298–304. doi: 10.1016/j.atherosclerosis.2006.07.023. [DOI] [PubMed] [Google Scholar]
- (74).Hoang A, Murphy AJ, Coughlan MT, Thomas MC, Forbes JM, O’Brien R, et al. Advanced glycation of apolipoprotein A-I impairs its anti-atherogenic properties. Diabetologia. 2007;50:1770–9. doi: 10.1007/s00125-007-0718-9. [DOI] [PubMed] [Google Scholar]
- (75).Gharavi NM, Gargalovic PS, Chang I, Araujo JA, Clark MJ, Szeto WL, et al. High-density lipoprotein modulates oxidized phospholipid signaling in human endothelial cells from proinflammatory to anti-inflammatory. Arterioscler Thromb Vasc Biol. 2007;27:1346–53. doi: 10.1161/ATVBAHA.107.141283. [DOI] [PubMed] [Google Scholar]
- (76).Norata GD, Callegari E, Marchesi M, Chiesa G, Eriksson P, Catapano AL. High-density lipoproteins induce transforming growth factor-beta(2) expression in endothelial cells. Circulation. 2005;111:2805–11. doi: 10.1161/CIRCULATIONAHA.104.472886. [DOI] [PubMed] [Google Scholar]
- (77).Sarov-Blat L, Kiss RS, Haidar B, Kavaslar N, Jaye M, Bertiaux M, et al. Predominance of a proinflammatory phenotype in monocyte-derived macrophages from subjects with low plasma HDL-cholesterol. Arterioscler Thromb Vasc Biol. 2007;27:1115–22. doi: 10.1161/ATVBAHA.106.138990. [DOI] [PubMed] [Google Scholar]
- (78).Mazzone T. HDL cholesterol and atherosclerosis. Lancet. 2007;370:107–8. doi: 10.1016/S0140-6736(07)61063-0. [DOI] [PubMed] [Google Scholar]
- (79).Asztalos BF, Llera-Moya M, Dallal GE, Horvath KV, Schaefer EJ, Rothblat GH. Differential effects of HDL subpopulations on cellular ABCA1- and SR-BI-mediated cholesterol efflux. J Lipid Res. 2005;46:2246–53. doi: 10.1194/jlr.M500187-JLR200. [DOI] [PubMed] [Google Scholar]
- (80).Ng CJ, Bourquard N, Hama SY, Shih D, Grijalva VR, Navab M, et al. Adenovirus-mediated expression of human paraoxonase 3 protects against the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:1368–74. doi: 10.1161/ATVBAHA.106.134189. [DOI] [PubMed] [Google Scholar]
- (81).Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- (82).Moore RE, Navab M, Millar JS, Zimetti F, Hama S, Rothblat GH, et al. Increased atherosclerosis in mice lacking apolipoprotein A-I attributable to both impaired reverse cholesterol transport and increased inflammation. Circ Res. 2005;97:763–71. doi: 10.1161/01.RES.0000185320.82962.F7. [DOI] [PubMed] [Google Scholar]
- (83).Rong JX, Li J, Reis ED, Choudhury RP, Dansky HM, Elmalem VI, et al. Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation. 2001;104:2447–52. doi: 10.1161/hc4501.098952. [DOI] [PubMed] [Google Scholar]
- (84).Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D’Agostino RB, et al. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes - A randomized trial. JAMA. 2006;296:2572–81. doi: 10.1001/jama.296.21.joc60158. [DOI] [PubMed] [Google Scholar]
- (85).Davidson M, Meyer P, Haffner S, Feinstein S, D’Agostino R, Kondos G, et al. Increased HDL-C predicts the pioglitazone-mediated reduction of carotid intima-media thickness progression in patients with Type 2 diabetes. Circulation. 2008 doi: 10.1161/CIRCULATIONAHA.107.746610. in press. [DOI] [PubMed] [Google Scholar]
- (86).Tso C, Martinic G, Fan WH, Rogers C, Rye KA, Barter PJ. High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler Thromb Vasc Biol. 2006;26:1144–9. doi: 10.1161/01.ATV.0000216600.37436.cf. [DOI] [PubMed] [Google Scholar]
- (87).Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P(3) lysophospholipid receptor. Circulation. 2006;114:1403–9. doi: 10.1161/CIRCULATIONAHA.105.607135. [DOI] [PubMed] [Google Scholar]
- (88).Gerrity RG, Natarajan R, Nadler JL, Kimsey T. Diabetes-induced accelerated atherosclerosis in swine. Diabetes. 2001;50:1654–65. doi: 10.2337/diabetes.50.7.1654. [DOI] [PubMed] [Google Scholar]
- (89).Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114:659–68. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- (91).Biddinger SB, Kahn CR. From mice to men: Insights into the insulin resistance syndromes. Ann Rev Physiol. 2006;68:123–58. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- (92).Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Rask-Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- (94).Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- (95).Wu KK, Huan YM. Diabetic atherosclerosis mouse models. Atheroscler. 2007;191:241–9. doi: 10.1016/j.atherosclerosis.2006.08.030. [DOI] [PubMed] [Google Scholar]
- (96).Back M, Sultan A, Ovchinnikova O, Hansson GK. 5-lipoxygenase-activating protein - A potential link between innate and adaptive immunity in atherosclerosis and adipose tissue inflammation. Circ Res. 2007;100:946–9. doi: 10.1161/01.RES.0000264498.60702.0d. [DOI] [PubMed] [Google Scholar]
- (97).Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nulcear factor-kappa B by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:2910–20. doi: 10.2337/diabetes.53.11.2910. [DOI] [PubMed] [Google Scholar]
- (98).Wu HZ, Ghosh S, Perrard XD, Feng LL, Garcia GE, Perrard JL, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted Upregulation in adipose tissue in obesity. Circulation. 2007;115:1029–38. doi: 10.1161/CIRCULATIONAHA.106.638379. [DOI] [PubMed] [Google Scholar]
- (99).Liang CP, Han S, Senokuchi T, Tall AR. The macrophage at the crossroads of insulin resistance and atherosclerosis. Circ Res. 2007;100:1546–55. doi: 10.1161/CIRCRESAHA.107.152165. [DOI] [PubMed] [Google Scholar]
- (100).Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- (101).Han S, Liang CP, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, et al. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–66. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- (102).Hevener AL, Olefsky JM, Reichart D, Nguyen MTA, Bandyopadyhay G, Leung HY, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117:1658–69. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPAR gamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115:518–33. doi: 10.1161/CIRCULATIONAHA.104.475673. [DOI] [PubMed] [Google Scholar]
- (105).Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Hoffmann A, Baltimore D. Circuitry of nuclear factor kappa B signaling. Immunol Rev. 2006;210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- (107).de Winther MPJ, Kanters E, Kraal G, Hofker MH. Nuclear factor kappa B signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–14. doi: 10.1161/01.ATV.0000160340.72641.87. [DOI] [PubMed] [Google Scholar]
- (108).Hirosumi J, Tuncman G, Chang LF, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- (109).Carmody RJ, Chen YH. Nuclear Factor-kappa B: Activation and regulation during toll-like receptor signaling. Cell Mol Immunol. 2007;4:31–41. [PubMed] [Google Scholar]
- (110).Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- (111).Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005;2005:13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- (112).Rask-Madsen C, King GL. Proatherosclerotic mechanisms involving protein kinase C in diabetes and insulin resistance. Arterioscler Thromb Vasc Biol. 2005;25:487–96. doi: 10.1161/01.ATV.0000155325.41507.e0. [DOI] [PubMed] [Google Scholar]
- (113).Stark RRM. ESCI Award 2006. Mitochondrial function and endocrine disease. Eur J Clin Invest. 2007;37:236–48. doi: 10.1111/j.1365-2362.2007.01773.x. [DOI] [PubMed] [Google Scholar]
- (114).Gregor MF, Hotamisligil GS. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48:1905–14. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- (115).Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- (116).Gargalovic PS, Gharavi NM, Clark MJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–6. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- (117).Lin Y, Berg AH, Iyangar P, Lam TKT, Giacca A, Combs TP, et al. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J Biol Chem. 2005;280:4617–26. doi: 10.1074/jbc.M411863200. [DOI] [PubMed] [Google Scholar]
- (118).Sell H, Eckel J. Monocyte chemotactic proten-1 and its vole in insulin resistance. Curr Opin Lipidol. 2007;18:258–62. doi: 10.1097/MOL.0b013e3281338546. [DOI] [PubMed] [Google Scholar]
- (119).Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–24. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose Expression of Tumor-Necrosis-Factor-Alpha - Direct Role in Obesity-Linked Insulin Resistance. Science. 1993 January 1;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- (121).Neels JG, Olefsky JM. Inflamed fat: what starts the fire? J Clin Invest. 2006;116:33–5. doi: 10.1172/JCI27280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis - exploring the connection. Arterioscler Thromb Vasc Biol. 2007;27:996–1003. doi: 10.1161/ATVBAHA.106.131755. [DOI] [PubMed] [Google Scholar]
- (123).Yue L, Rassouli N, Ranganathan G, Kern PA, Mazzone T. Divergent effects of PPAR agonists and TNF on adipocyte apoE expression. J Biol Chem. 2004;279:47626–32. doi: 10.1074/jbc.M408461200. [DOI] [PubMed] [Google Scholar]
- (124).Huang ZH, Reardon CA, Mazzone T. Endogenous apoE expression modulates adipocyte triglyceride content and turnover. Diabetes. 2006;55:3394–402. doi: 10.2337/db06-0354. [DOI] [PubMed] [Google Scholar]
- (125).Friedman JM. The function of leptin in nutrition; Weight, and physiology. Nutr Rev. 2002;60:S1–S14. doi: 10.1301/002966402320634878. [DOI] [PubMed] [Google Scholar]
- (126).Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–49. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- (127).Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–92. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Unger RH. Reinventing type 2 diabetes: pathogenesis, treatment, and prevention. JAMA. 2008;299:1185–7. doi: 10.1001/jama.299.10.1185. [DOI] [PubMed] [Google Scholar]
- (129).Buse JB, Bigger JT, Byington RP, Cooper LS, Cushman WC, Friedewald WT, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol. 2007;99:21i–33i. doi: 10.1016/j.amjcard.2007.03.003. [DOI] [PubMed] [Google Scholar]