

Abstract
Pompe disease results in the accumulation of lysosomal glycogen in multiple tissues due to a deficiency of acid alpha-glucosidase (GAA). Enzyme replacement therapy for Pompe disease was recently approved in Europe, the U.S., Canada and Japan using a recombinant human GAA (Myozyme, alglucosidase alfa) produced in CHO cells (CHO-GAA). During the development of alglucosidase alfa, we examined the in vitro and in vivo properties of CHO-cell derived rhGAA, an rhGAA purified from the milk of transgenic rabbits, as well as an experimental version of rhGAA containing additional mannose-6-phosphate intended to facilitate muscle targeting. Biochemical analyses identified differences in rhGAA N-termini, glycosylation types and binding properties to several carbohydrate receptors. In a mouse model of Pompe disease, glycogen was more efficiently removed from the heart than from skeletal muscle for all enzymes, and overall, the CHO-cell derived rhGAA reduced glycogen to a greater extent than that observed with the other enzymes. The results of these preclinical studies, combined with biochemical characterization data for the three molecules described within, led to the selection of the CHO-GAA for clinical development and registration as the first approved therapy for Pompe disease.
Keywords: Enzyme replacement therapy, Pompe disease, lysosomal storage disorder, recombinant proteins, glycoproteins, mannose-6-phosphate receptor, mannose receptor
INTRODUCTION
Pompe disease is a rare and fatal myopathy, inherited in an autosomal recessive manner, which results from a deficiency of the lysosomal enzyme acid alpha-glucosidase (GAA). The pathological hallmark of Pompe disease (also known as glycogen storage disease type II, acid maltase deficiency or glycogenosis type II) is lysosomal accumulation of glycogen in muscle tissues. Pompe disease has a wide range of clinical presentations and course. Initially described as an acutely life threatening infantile disease, the disorder has been historically classified with designations based on age at onset of symptoms (infantile, juvenile or adult), extent of organ involvement and rate of progression to death [1–3].
Prior to 2006, there were no approved products for the treatment of Pompe disease. The previous palliative and supportive care strategies used in patient management have been largely ineffective in preventing disease progression. This manuscript describes the head-to-head in vitro and in vivo comparison of three compositionally distinct recombinant human GAAs (rhGAA) that ultimately led to the approval of enzyme replacement therapy (ERT) in Europe, the U.S., Canada and Japan for the treatment of Pompe disease.
Several sources of GAA have been evaluated clinically for the treatment of patients with Pompe disease. Early attempts at ERT in infants using enzyme preparations purified from Aspergillis niger or human placenta were unsuccessful, presumably due to suboptimal dosing, disease stage and the lack of correct post-translational modifications necessary for muscle targeting [4, 5]. The first clinical studies using a rhGAA purified from the milk of transgenic rabbits (tgGAA) demonstrated that ERT could improve respiratory insufficiency and restore some muscle function in infants [6, 7]. Subsequent infantile trials using recombinant human GAA (rhGAA) from two different Chinese hamster ovary (CHO) cell lines have also been performed [8–10]. Collectively, these studies demonstrated that intravenous administration of highly purified tgGAA or CHO cell-derived rhGAA was safe and provided a beneficial effect on survival, cardiomyopathy, motor function and growth. Less clinical data are available for ERT in late-onset patients, however, some disease stabilization and functional improvement has been reported [11]. While the recent approval of ERT using rhGAA produced using a CHO cell based process represents a major milestone in the treatment of this devastating disorder, additional research on the pathophysiology of Pompe disease is necessary to further refine treatment strategies as a function of disease progression.
Correction of glycogen accumulation in the skeletal muscle of Pompe patients has proven to be a greater challenge than substrate removal in other lysosomal storage disorders (LSDs) that have been successfully treated by ERT. In Gaucher and Fabry diseases, the affected cells (macrophages and endothelial cells respectively) are typically found within or in close proximity to the confines of the capillary lumen or reticular endothelial system. Intravenously administered enzyme is therefore immediately accessible to many of the affected cells. Muscle cells, by contrast, are separated from the circulatory system by a blood-muscle barrier comprised of endothelial cells, basement membrane and other interstitial tissues. For Pompe disease, these structures represent physical and functional barriers, as well as non- productive sinks for the administered enzyme. Additionally, skeletal muscle represents roughly half the total body weight in healthy adults [12]; thus, clearance of lysosomal glycogen from this large tissue mass challenges the amount (dose) of enzyme required to effectively treat Pompe disease. Finally, treatment of the metabolic defect in skeletal muscle may be further complicated by the degree of glycogen accumulation and other biochemical differences between muscle fiber types. In humans, low levels of membrane bound glycogen, mild ultrastructural damage, and a high proportion of Type I muscle fibers were associated with a good histologic response [13]. Furthermore, in GAA knockout mice, it has been reported that rhGAA clears accumulated glycogen more efficiently from predominant fiber type I (oxidative) versus type IIb muscles [14]. Recent reports have determined that preferential accumulation of autophagosomes in type II fibers may represent a glycogen-containing compartment refractory to ERT [15]. These findings also suggest that, for optimal benefit, ERT should be initiated before significant secondary pathology develops. Taken together, these factors provide a plausible explanation why the enzyme levels required to treat Pompe disease are much higher (20–40 mg/kg) than the recommended doses for Gaucher and Fabry diseases (1.5 and 1.0 mg/kg respectively).
During the preclinical and clinical development of ERT for treatment of Pompe disease, we compared the biochemical and pharmacological properties of several different rhGAA preparations. These included a conventional CHO cell-produced form of the enzyme (CHO-GAA) and rhGAA produced in the milk of transgenic rabbits (tgGAA). The cation independent mannose-6-phosphate (M6P) receptor, which cycles to the cell surface, plays a key role in the trafficking of exogenous lysosomal enzymes containing this recognition marker [16]. Therefore, a carbohydrate engineered form of the enzyme containing high levels of mannose-6-phosphate produced in CHO cells using a stepwise enzymatic engineering approach was also included in this comparison (referred to as HP-GAA). The relative in vivo efficacy of these three different rhGAA preparations was evaluated in the GAA knockout mouse model of Pompe disease [17]. The combination of these preclinical studies with the clinical data cited above, and the detailed biochemical characterization of the three enzymes described below, ultimately led to the selection of the CHO-GAA candidate discussed within for development and registration of the current ERT for Pompe disease (Myozyme, alglucosidase alfa).
MATERIALS AND METHODS
Materials
Methods for the purification of the transgenically-produced rhGAA (tgGAA), the CHO-GAA, and the carbohydrate engineered form of rhGAA (HP-GAA) are described elsewhere [6, 18, 19]. The purity of the various forms of GAA used in these studies was ≥95% as judged by SDS gel electrophoresis.
Biochemical and cell culture analyses
Enzymatic Activity
GAA activity was measured by determining the rate of GAA-catalyzed hydrolysis of a synthetic substrate, p-nitrophenyl-D-α-glucopyranoside, in 50 mM sodium acetate, 0.1% BSA, pH 4.3. The released chromophore, p-nitrophenol, was quantified spectrophotometrically at an alkaline pH (> 10.2) at 400 nm. One unit of GAA was defined as that amount of activity which resulted in the hydrolysis of 1 µmol of substrate per minute at 37°C under the assay conditions.
SDS-PAGE
Two hundred ng of each GAA were added to Laemmli sample buffer plus reductant (DTT) [20], boiled for 1 minute and loaded onto a 4–20% polyacrylamide gradient gel (Invitrogen). Invitrogen Mark 12 molecular weight markers were also loaded onto the gel. Electrophoresis was performed for approximately 1.5 hours at 150 volts and proteins visualized with PhastGel Blue R-350 stain.
N-Terminal Sequence Analysis
One hundred pmol of each GAA were spotted onto a Biobrene-conditioned filter and analyzed for 10 cycles using an ABI Procise N-terminal sequencer.
Isoelectric Focusing
Ten µg of each GAA were analyzed using an Invitrogen pH 3–7 IEF gel with Invitrogen running buffers. Proteins were focused for 1 hour at 100 volts, 1 hour at 200 volts and 30 minutes at 500 volts. The gel was stained using PhastGel Blue R-350 stain.
Mannose-6-Phosphate Content
Levels of mannose-6-phosphate (M6P) were determined using the method of Zhou et. al.[21]
GlcNAc-P-Man Phosphodiester Content
Fifty µg samples of each GAA were hydrolyzed in 1 M formic acid for 60 minutes at 80°C. Samples were dried and reconstituted in 200µL distilled water and filtered through 10 Kd filters. The filtered hydrolysate was then analyzed on a CarboPac PA1 column using high pH anion exchange chromatography and pulsed amperometric detection (HPAEC-PAD). The GlcNAc was eluted isocratically using 21 mM NaOH. Quantitation of the phosphodiester was calculated relative to authentic N-acetylglucosamine (GlcNAc) standards.
Sialic Acid Content
Fifty µg each of CHO-GAA and tgGAA were hydrolyzed in 0.5M formic acid for 60 minutes at 80°C. Samples were dried and reconstituted in 200 µL distilled water and filtered through 0.45 µm filters. The filtered hydrolysate was then analyzed on a 4 × 250mm CarboPac PA1 column using high pH anion exchange chromatography and pulsed amperometric detection (HPAEC-PAD). Buffer A was 100mM sodium hydroxide and Buffer B was 100mM sodium hydroxide/1M sodium acetate. Quantitation was performed with a standard curve constructed with N-acetylneuraminic acid standards.
Oligosaccharide Profiling
Five hundred µg samples of each GAA were sonicated in 20% 2-betamercaptoethanol for 25 minutes and then boiled for 1 minute at 100°C. The samples were cooled and 50mM sodium phosphate buffer pH 7.0 was added to a final volume of 300µL. After sonication for 25 minutes, samples were digested with peptide N-glycosidase F using a 1/30 enzyme/protein (wt/wt) ratio. The samples were filtered through 10,000 NMWL centrifugal filters, evaporated to dryness, reconstituted in 530µL distilled water and dialyzed against distilled water overnight. The dialyzed samples were again evaporated to dryness and then reconstituted in 200µL of deionized water. Fifty µL of sample were analyzed on a 4 × 250mm CarboPac PA100 column using high pH anion exchange chromatography and pulsed amperometric detection (HPAEC-PAD). Buffer A was 100mM sodium hydroxide and Buffer B was 100mM sodium hydroxide/500mM sodium acetate.
Glycoform Analysis
One hundred µg of each GAA were denatured with 300 µL 6M guanidine hydrochloride, reduced with 5µL 2M DTT and alkylated with 25 µL 10%4-vinylpyridine. The alkylation reaction was quenched with 25 µL 2M DTT. Reduced and alkylated samples of GAA were dialyzed against 50mM Tris, pH 8.5 and digested with trypsin using a 1/50 enzyme/protein (wt/wt) ratio. Digests were incubated overnight at 37°C and quenched by the addition of 0.1% TFA. The tryptic digests (2 µg) were analyzed by capillary LC/MS using a Bruker Esquire ion trap mass spectrometer. The peptide separations were performed using a capillary Vydac C18 column (Microtech Scientific, 320 µm × 15 cm) and a formic acid/acetonitrile solvent system with a flowrate of 4 µL/minute. The mass spectra were acquired in positive mode with a m/z range of 250–2000.
Binding to Mannose-6-Phosphate Receptor
The relative binding affinities of each GAA for the soluble cation independent mannose-6-phosphate receptor (sCIMPR), purified from bovine serum, were assessed by surface plasmon resonance using a Biacore 3000 instrument. sCIMPR was immobilized onto a CM5 chip using NHS/EDC chemistry. Samples were diluted to 5µg/mL using 10mM HEPES, pH 7.0, 150mM NaCl, 0.005% surfactant P20, 10mM CaCl2 and injected at a flowrate of 40µL/minute. After a three minute association and 2.5 minute dissociation phase, the receptor chip was regenerated with 10mM sodium acetate, 300mM NaCl. The binding signals were corrected by subtracting signals from a control mock flow cell and from buffer injections.
Binding to Mannose Receptor
Surface plasmon resonance was also used to evaluate binding of each GAA to the mannose receptor. The mouse carbohydrate recognition domain (CRD) 1–4 of the mannose receptor was expressed, purified and immobilized onto a CM5 chip using NHS/EDC chemistry. Samples were diluted to 5µg/mL using 10mM HEPES, pH 7.0, 150mM NaCl, 0.005% surfactant P20, 10mM CaCl2 and injected at a flowrate of 40µL/minute. After a three minute association and 2.5 minute dissociation phase, the receptor chip was regenerated using 5mM EDTA. The binding signals were corrected by subtracting signals from a control mock flow cell and from buffer injections.
Cell-based bioassay for GAA binding/uptake
Human Pompe fibroblasts (TR4192) were grown to confluence in a T-75 flask using MEM/FBS media. The cells were washed with PBS, trypsinized and plated at 1 × 106 cells/mL in a 96-well plate (100µL/well). Plates were incubated overnight at 37°C. Following incubation, samples of GAA (assayed in triplicate) were diluted in reduced serum media (MEM/1% FBS) and added to the cells. Following a 24 hour incubation at 37°C, cells were washed and lysed with the addition of PBS/1% Triton X-100 and freezing at −80°C. A protein determination (BCA) and an activity analysis (using 4-methylumbelliferyl-α-D-glucoside as the substrate) were performed on the cell lysates and the uptake of each GAA preparation was determined.
Animal studies
Animal care and experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (US Department of Health and Human Services, NIH publication No. 86-23). Four to five month old GAA knockout mice [17] were administered 0 (vehicle only), 20, 60 or 100mg/kg of each enzyme preparation intravenously, by bolus injection, for four weekly doses (n=5 per group). Starting with the second dose (on day 8), mice were pre-treated with 5 mg/kg diphenhydramine (DPH) by intraperitoneal injection 20 minutes prior to enzyme administration to prevent anticipated hypersensitivity reactions. Despite the administration of diphenhydramine, one mouse was sacrificed following the fourth dose of 100 mg/kg tgGAA due to clinical signs consistent with a hypersensitivity reaction. Milder signs of hypersensitivity were observed in animals from all groups following drug administration. However, these reactions were transient and generally resolved within 1 hour of dose with no apparent long-term effects. Mice were sacrificed 24 hours after the last injection, and the heart, diaphragm, quadriceps, triceps and psoas muscle were collected for analysis.
Tissue glycogen content
Glycogen content was evaluated using high resolution light microscopy and computer assisted histomorphometry as previously described [22]. Briefly, representative samples of cardiac and skeletal muscle were fixed in 3% gluteraldehyde in 0.2mol/L sodium cacodylate buffer (Electron Microscopy Sciences, Fort Washington, PA) and embedded in epon-araldite. One micron sections were stained with Periodic acid-Schiff (PAS) reaction and counterstained with Richardson’s solution. This results in high quality tissue preservation in which glycogen is fully retained and appears purple against a blue counterstain of myocyte cytoplasm. One representative field from each slide was photographed with a Nikon DXM1200 digital camera (Nikon Inc, Instrument Group, Meville, NY) and analyzed using Metamorph Imaging Processing and Analysis software (version 4.6; Universal Imaging Corporation). For each image, glycogen load was expressed as a percentage of total tissue area.
Statistical Analysis
Statistical analysis was performed on the tissue glycogen content and GAA enzyme activity data using one-way analysis of variance followed by a Student-Newman Keuls test. A probability value of p<0.05 was considered statistically significant.
Measurement of GAA activity in tissue homogenates
GAA activity in tissue homogenates was assayed using 4-methylumbelliferyl-α-D-glucoside in a 96-well plate [23]. Briefly, the homogenate supernatant was diluted in 200 mM potassium acetate, 1.0 mg/ml BSA, 0.02% sodium azide, pH 4.0 and transferred in triplicate to 96 well black sample plates. An equal volume of 1.0 mM 4-methylumbelliferyl-α-d-glucoside in 200 mM potassium acetate, 0.02% sodium azide, pH 4.0 was added to the samples and the plate mixed on a micro-plate orbital shaker. A five point 4-methylumbelliferyl curve (10 – 0.16 nmol/mL) was included on each plate and the plate was incubated for 60 minutes at 37°C. The reaction was stopped by the addition of 125 µL of 1.0 M glycine, pH 10.5 to all wells. Fluorescence was measured and converted to nmol/ml/hr using the standard curve. The activity numbers were normalized to protein using the bicinchoninic acid (BCA) protein assay for a final value of nmol of 4MU released/mg of tissue/hour.
Measurement of GAA specific IgG antibodies
A sandwich ELISA was used to detect rhGAA specific antibodies. Plates were coated with rhGAA overnight and blocked with PBS/BSA. Samples, controls, and blanks were added and incubated for 1 hour at 37°C. HRP conjugated goat anti-mouse IgG was added and 3,3’,5,5’-Tetramethylbenzidine (TMB) was used as substrate. Plates were stopped with 1N HCL and a spectrophotometer reading was determined at 450nm. Sample titers were determined using a cutoff value of 0.200 OD.
Study Blinding
All aspects of this study, including sample labeling, characterization studies on the various enzymes, in-life animal studies, and all analyses were conducted in a blinded manner under the supervision of Genzyme’s R&D Quality Assurance group. Unblinding was only performed after all analyses, tables and statistical values had been finalized and the relative merits of the three enzymes had been agreed to by the study team.
RESULTS
Biochemical Analyses
A summary of the biochemical analyses performed on the various rhGAA preparations is shown in Table 1. The corresponding published biochemical properties of human placental-derived GAA are provided for comparison [24–27]. Each of the three recombinant GAA preparations was biochemically active towards the synthetic α-D-glucopyranoside substrate. However, SDS-PAGE analysis indicated that, unlike GAA purified from human placenta, the predominant species in each rhGAA preparation was the precursor or 110kd form of the molecule. The N-terminal sequence analyses indicated that the CHO-GAA and HP-GAA preparations contained chemically blocked N-termini. Further analysis of CHO-GAA indicated that the N-terminus was chemically blocked by a pyroglutamate residue at position 57 (data not shown). The N-terminus of tgGAA started at Asp-67. The isoelectric focusing (IEF) analysis, shown in Figure 1a, indicated that the CHO-GAA and tgGAA preparations focused as a heterogeneous mixture of isoforms between pIs 4.0 and 4.6. The HP-GAA was less heterogeneous and focused at a pI range of 4.6–4.9.
Table 1.
Summary of biochemical analyses performed on CHO cell-derived rhGAA, HP-GAA and tgGAA (n/a = not applicable; − = not present)
| GAA Source | Human Placental |
CHO-GAA | HP-GAA | tgGAA |
|---|---|---|---|---|
| Expression system | n/a | CHO cells | CHO cells | Transgenic rabbit milk |
|
Activity towards p-nitrophenyl-D- α-glucopyranoside substrate (U/mg) |
+ | 4.3 | 4.5 | 4.2 |
|
Molecular Weight by SDS-PAGE |
76kD, 70kD | 110kD | 110kD | 110kD |
|
N-terminal sequence |
Met-122 [18] Ala-204 |
Pyro-Gln-57 | Pyro-Gln-57 | Asp-67 |
| IEF (pI range) | 4.0 – 4.6 | 4.6 – 4.9 | 4.2 – 4.6 | |
|
Mannose-6- Phosphate (M/M) |
- | 1.2 | 3.5 | 1.3 |
|
GlcNAc-P-Man phosphodiester |
- | - | - | + |
| Sialic acid (M/M) | - | 6.2 | - | 4.6 |
|
Oligomannose structures |
- | + | +++ | ++ |
|
Phosphorylated oligomannose structures |
- | + | +++ | ++ |
|
Complex Structures |
- | + | - | + |
|
Binds to sCIMPr in vitro |
- | ++ | ++++ | + |
Figure 1.
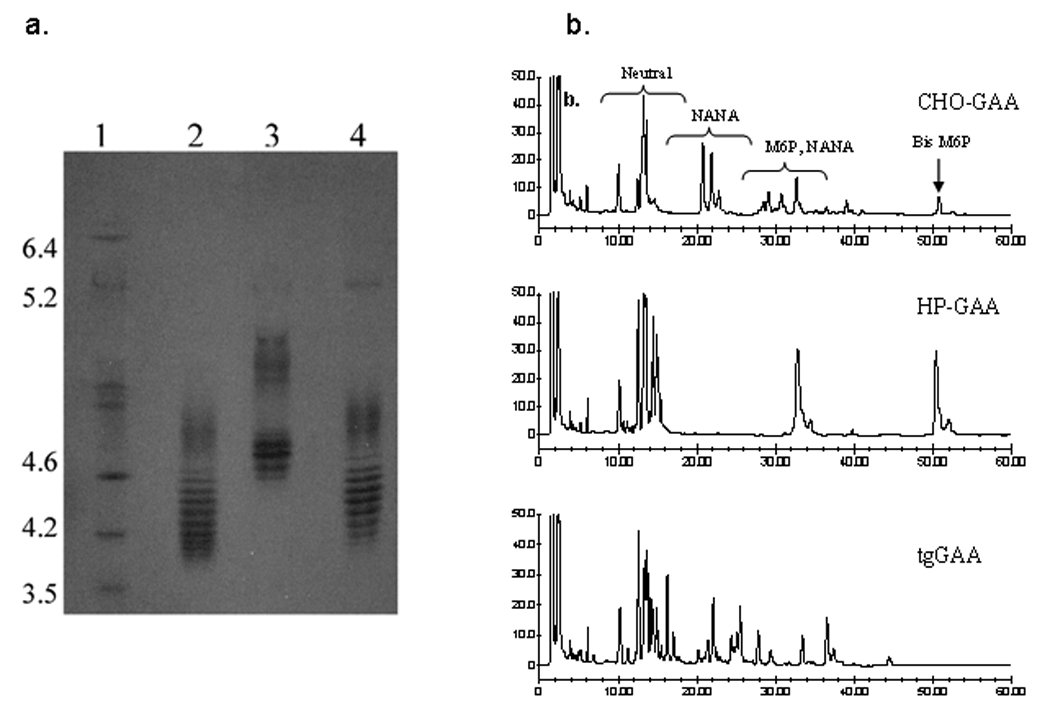
IEF and oligosaccharide profiling analyses of recombinant acid α-glucosidase preparations. (a) IEF analysis of CHO-GAA (lane 2), HP-GAA (lane 3) and tgGAA (lane 4). Isoelectric point markers are shown in lane 1. (b) Oligosaccharide profiles of CHO-GAA, HP-GAA and tgGAA. Regions of the chromatogram containing neutral, sialylated (NANA), mannose-6-phosphate-containing (M6P) and bis mannose-6-phosphate-containing (Bis M6P) glycans are noted.
Oligosaccharide Analyses
As shown in Table 1, all rhGAA preparations contained mannose-6-phosphate. As expected, the carbohydrate engineered HP-GAA contained a higher level of mannose-6-phosphate (M6P) than the CHO-GAA or tgGAA preparations. The M6P groups of HP-GAA and CHO-GAA contained exclusively phosphomonoesters while the tg-GAA glycans contained a combination of phosphomonoesters and phosphodiesters with N-acetylglucosamine (GlcNAc). Oligosaccharide profile analysis by high pH anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD), confirmed the presence of additional bis-mannose-6-phosphate (biphosphorylated oligomannose structures) on HP-GAA relative to the other enzymes (Fig. 1b). Glycoform analysis by LCMS (Table 2) revealed that the seven N-linked glycosylation sites of CHO-GAA and tgGAA were comprised of a mixture of oligomannose, phosphorylated oligomannose and complex oligosaccharide structures. For tgGAA, the phosphorylated oligomannse structures were found to be covered by GlcNAc [28]. The seven N-linked sites of HP-GAA were comprised of oligomannose and phosphorylated oligomannose structures.
Table 2.
Site-specific glycoform analysis of rhGAA, HP-GAA and tgGAA. The predominant glycoforms were determined by LCMS analysis of tryptic GAA peptides. Data are shown for each of the seven GAA asparagine-linked sites of glycosylation
| Location of Asn-linked glycosylation |
CHO-GAA | HP-GAA | tgGAA |
|---|---|---|---|
| Asn-140 | Complex + sialic acid Oligomannose + 2P |
Oligomannose + 2P Oligomannose + P |
Hybrid + P + GlcNAc Oligomannose + 2P + 2GlcNAc |
| Asn-233 | Oligomannose Hybrid |
Oligomannose Oligomannose + P |
Oligomannose Oligomannose + P + GlcNAc |
| Asn-390 | Complex + sialic acid Complex |
Oligomannose + P Oligomannose |
Oligomannose |
| Asn-470 | Complex + sialic acid Oligomannose + P |
Oligomannose + 2P Oligomannose + P |
Oligomannose + P + GlcNAc Oligomannose |
| Asn-652 | Complex + sialic acid Complex |
Oligomannose Oligomannose + P |
Hybrid + sialic acid Oligomannose |
| Asn-882 | Complex + sialic acid Complex |
Oligomannose + P Oligomannose |
Complex + sialic acid |
| Asn-925 | Complex + sialic acid | Oligomannose Oligomannose + P |
Complex + sialic acid |
Binding to Mannose-6-Phosphate and Mannose Receptor
The relative affinities of the various rhGAA preparations for the soluble cation independent mannose-6-phosphate receptor (sCIMPr) and the mannose receptor were determined by surface plasmon resonance (Biacore). As shown in Figure 2a, HP-GAA had the highest relative affinity for the immobilized sCIMPr versus tgGAA with the lowest. Although the CHO-GAA and tgGAA had similar quantities of M6P (as noted above), the M6P residues found on tgGAA were covered by GlcNAc and were therefore less accessible to the immobilized sCIMPr. The relative affinities of the GAA preparations for the mouse carbohydrate recognition domain (CRD) 1 through 4 of the mannose receptor were evaluated. The data (Fig. 2b) indicate a relative binding rank of HP-GAA > tgGAA > CHO-GAA. As noted in Table 2, the HP-GAA molecule contains significant quantities of oligomannose structures which likely facilitated binding to the mannose receptor construct used in these experiments. The tgGAA preparation also contained oligomannose structures at the N-linked site found at position 390 which may contribute to the enhanced binding of this molecule to the mannose receptor, compared to the CHO-GAA.
Figure 2.
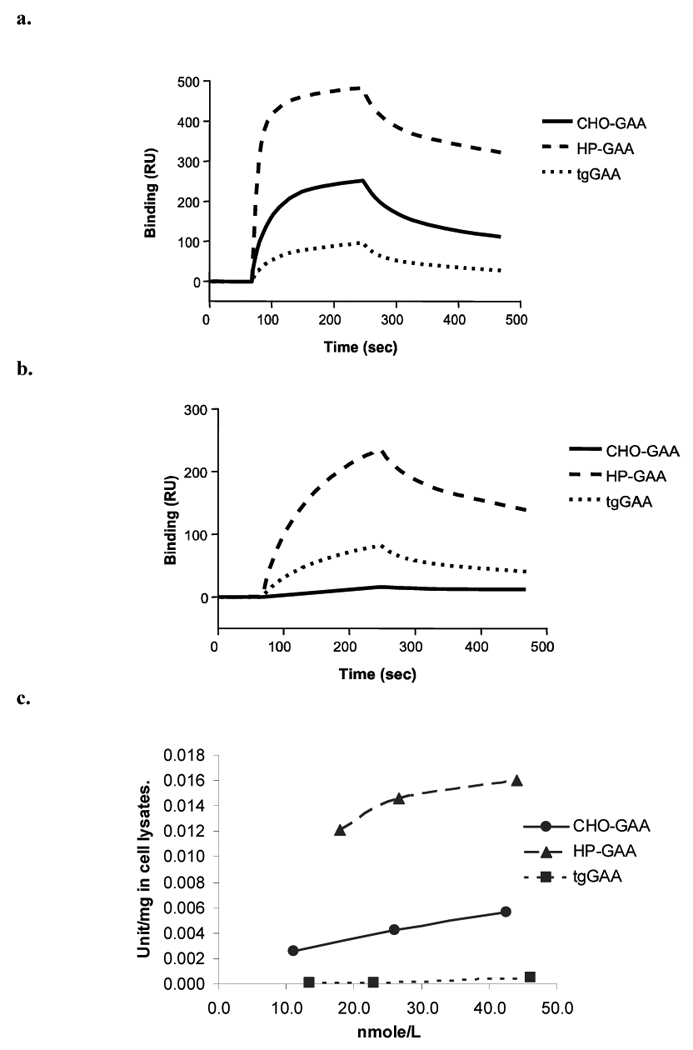
Receptor binding and fibroblast cell uptake analyses of CHO-GAA, HP-GAA and tgGAA. (a and b) The relative binding affinities of CHO-GAA, HP-GAA and tgGAA for the mannose-6-phosphate receptor (panel a) and the mannose receptor (panel b) were evaluated by surface plasmon resonance (BIAcore). (c) Cellular uptake of these recombinant acid α-glucosidase preparations was determined using human Pompe fibroblasts.
Binding and uptake of various rhGAA preparations by cells in vitro
Uptake of the rhGAA preparations into Pompe fibroblasts is shown in Figure 2c. HP-GAA has the highest cellular uptake followed by CHO-GAA, with tgGAA showing little cellular uptake. These results are in good agreement with the relative sCIMPr binding data obtained using surface plasmon resonance and are consistent with a M6P receptor dependent uptake mechanism.
Tissue glycogen clearance
Pharmacodynamic efficacy of the different enzymes was determined in the GAA knockout mice using a histomorphometric evaluation of muscle glycogen content. The results of this analysis in two representative tissues (heart and quadriceps muscle) are shown in Figure 3. Of note, biochemical glycogen analysis was also performed on the GAA knockout mouse muscle samples. Highly similar results were observed following the biochemical analysis as compared to the histomorphometric evaluation (data not shown). In general, a dose dependent reduction in the tissue glycogen content was observed with each of the enzyme preparations. In the heart, the CHO-GAA preparation reduced glycogen content more effectively than HP-GAA or tgGAA. Specifically, 100mg/kg of CHO-GAA cleared greater than 90% of accumulated glycogen when compared to the vehicle control; whereas only 76% clearance was observed with 100mg/kg HP-GAA, and less than 40% reduction was observed with the tgGAA. In the skeletal muscles, glycogen clearance was less effective for all enzymes. However, as illustrated in the quadriceps, the CHO-GAA appeared more efficacious than either the HP-GAA or the tgGAA, particularly at the lower doses.
Figure 3.
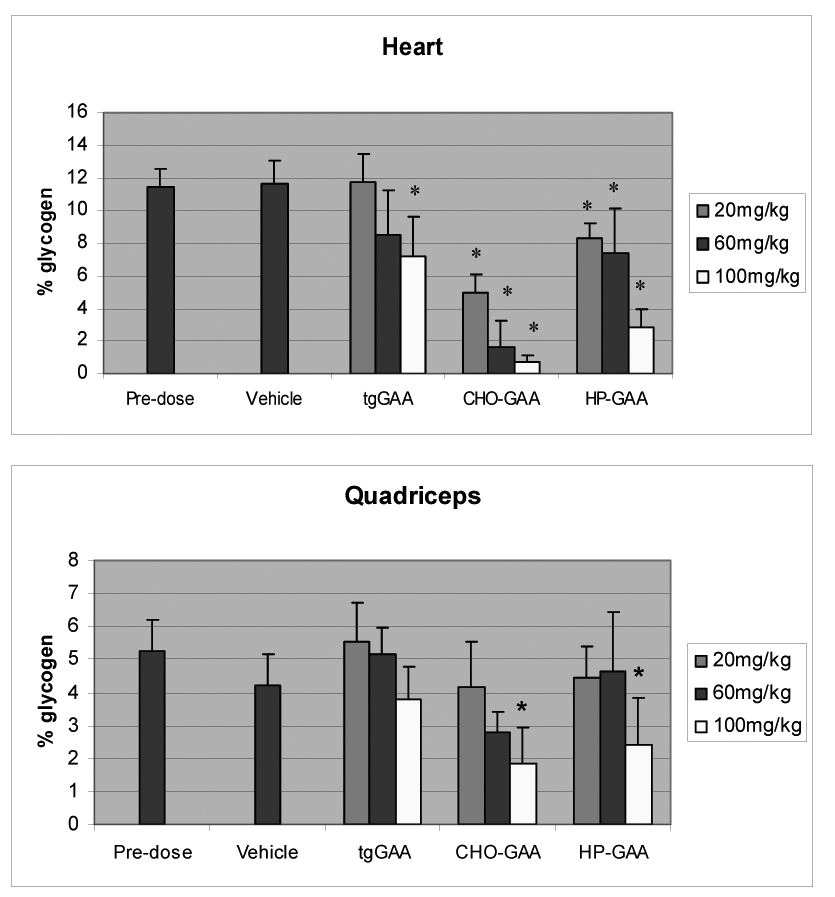
Metamorph glycogen analysis of heart and quadriceps tissue from GAA knockout mice administered CHO-GAA, tgGAA and HP-GAA for 4 weekly doses of 20, 60 and 100 mg/kg (mean ± SD). * indicates p<0.05 as compared to vehicle.
Tissue GAA activity levels
Higher levels of GAA activity were detected in both the heart and quadriceps muscles of GAA knockout mice treated with tgGAA, when compared to those treated with CHO-GAA (Fig. 4). Higher levels of GAA activity were also observed in the hearts of the mice treated with HP-GAA, although similar levels of enzyme activity were detected in the quadriceps when compared to CHO-GAA. These observations are of particular interest since the amount of GAA activity present in the tissues does not appear to correlate with the extent of glycogen clearance. For example, significantly higher enzyme activity levels were observed in the heart following administration of 100 mg/kg tgGAA or HP-GAA as compared to 100 mg/kg CHO-GAA; however, in the same tissue, at the same dose, CHO-GAA cleared greater than 90% of the accumulated glycogen which is notably higher than the reduction observed following either tgGAA or HP-GAA administration (less than 40% and 76% respectively). A similar trend was also observed with 100 mg/kg tgGAA in the quadriceps muscle when compared to both CHO-GAA and HP-GAA. Western blot analysis was conducted to establish whether each of the enzyme preparations was processed correctly upon uptake into the target tissues. The results demonstrated that all molecules were processed appropriately in the tissue homogenates examined (data not shown), suggesting that each of the enzymes was targeted appropriately to the lysosomes.
Figure 4.
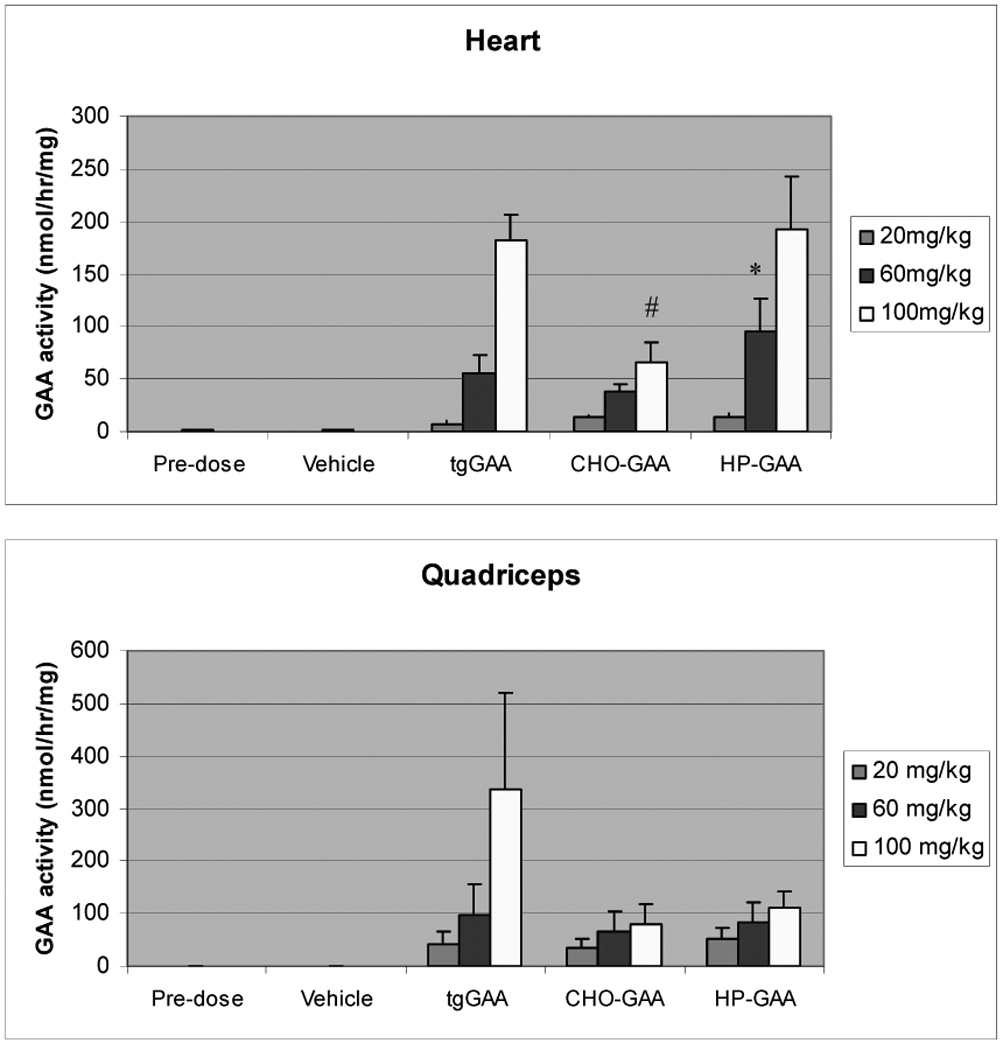
Enzyme activity analysis of heart and quadriceps from GAA knockout mice administered CHO-GAA, tgGAA and HP-GAA for 4 weekly doses of 20, 60 and 100 mg/kg (mean ± SD). * indicates p<0.05 as compared to tgGAA and CHO-GAA; # indicates p<0.05 as compared to tgGAA and HP-GAA.
Development of anti-GAA specific antibodies
Anti-GAA specific antibodies to rhGAA were also detected in the serum of all animals treated with enzyme. This was not unexpected given the human sequence of the recombinant protein. In the 100mg/kg treatment groups, titers of 625 – 3125 were detected for both CHO-GAA and tgGAA. The antibody titers observed after administration of 100mg/kg HP-GAA were slightly higher than those for the other enzymes since a titer of 15625 was observed in all animals.
DISCUSSION
The development and regulatory approval of Myozyme (alglucosidase alfa) for the treatment of Pompe disease represents the first major scientific and clinical breakthrough in the treatment of a life-threatening human myopathy. Several different forms of rhGAA were evaluated prior to the selection of CHO-GAA (as described within) for final clinical development and commercialization. In addition to CHO-GAA, these included rhGAA produced in milk of transgenic rabbits (tgGAA) and a carbohydrate engineered form of rhGAA containing high levels of M6P (HP-GAA) also produced in CHO cells. The HP-GAA, prepared by enzymatic addition of M6P to mannose residues, was of particular interest because effective targeting of GAA to the lysosomal compartment of cardiac and skeletal muscle depends upon M6P receptor mediated uptake [29–31].
During trafficking to the lysosome, GAA undergoes complex processing during which glycans are remodeled and peptide segments are excised from the parent polypeptide to yield a multi-subunit complex [18]. Previous studies have shown that the processed forms of GAA lack the M6P targeting signal and are not effectively taken up by human fibroblasts [26, 32]. Accordingly, the rhGAAs used in this study were restricted to the precursor form of the enzyme containing varying levels of M6P to ensure more effective targeting of exogenous enzyme to the lysosomal compartment of skeletal muscle.
The N-terminus of the CHO and HP-GAAs were located at residue 57, which was confirmed to be a pyroglutamic acid residue. The N-terminus of tgGAA was located at aspartic acid 67. Heterogeneity at the N-terminus of other GAA preparations either expressed in CHO cells or purified from human urine and placenta has been previously described and does not appear to impact enzyme function [24, 26, 33]. Biochemical analyses of the various rhGAA preparations revealed that the oligosaccharides of HP-GAA, unlike those of CHO-GAA or tgGAA, were predominantly oligomannose and phosphorylated oligomannose structures. M6P analysis of these different preparations indicated levels of ~1.2 mol/mol for CHO-GAA, ~1.3 mol/mol for tgGAA and ~3.5 mol/mol for HP-GAA. Although the M6P levels of CHO and tgGAAs were comparable, the M6P residues of tgGAA were partially covered by N-acetylglucosamine (GlcNAc). Previous studies have shown that oligosaccharides containing GlcNAc-covered M6P structures have low affinity for the M6P receptor [34]. The relative binding of the various rhGAAs to the immobilized sCIMPR, as judged by surface plasmon resonance analyses, was consistent with the level of exposed M6P (HP-GAA > CHO-GAA > tg-GAA). Likewise, the relative binding and uptake of the various forms of rhGAA by cells in vitro demonstrated a strong correlation with the level of exposed M6P, with HP-GAA exhibiting an enhanced cellular uptake compared to either CHO-GAA or tgGAA. However, binding of HP-GAA to the mannose receptor was significantly higher than the binding of either tgGAA or CHO-GAA. The enhanced affinity of HP-GAA for the mannose receptor can be attributed to the predominance of oligomannose structures or glycan isoforms found at each of the seven N-linked sites of HP-GAA.
The relative efficacy of these different rhGAAs to reduce accumulated muscle glycogen in vivo was assessed in a GAA knockout mouse model of Pompe disease [17]. Systemic administration of all three forms of rhGAA resulted in a dose-dependent reduction of glycogen content in the cardiac and skeletal muscles of these animals as judged by histological analyses [22]. Glycogen clearance from the cardiac muscle was significantly greater than that observed in skeletal muscle following administration of all three enzymes. Both CHO-GAA and HP-GAA were significantly more effective in clearing glycogen from the heart and skeletal muscle than was tgGAA. Furthermore, CHO-GAA appeared to be more effective, on a dose-dependent basis, than HP-GAA in clearing accumulated glycogen from the myocardium. Infantile clinical studies have shown that both CHO-GAA and tgGAA were effective in treating the myocardium [6–8, 10, 35], and overall, our results suggest that all three recombinant enzymes are relatively effective in this tissue. The response in skeletal muscle is more difficult to interpret, partly because histopathologic findings from infantile muscle indicate significantly greater pathology than in GAA knockout mice [13]. Comparison of these findings with human data is complicated by the relatively low glycogen load in Pompe mouse quadriceps (≈2–5%). Glycogen content in the quadriceps muscle of Pompe infants pre-treatment, measured by the same histomorphometric method, varied from ≈10–65% [13]. Furthermore, infant muscle displays serious destructive intracellular pathology in correlation with glycogen load to the point of almost total obliteration of recognizable intracellular morphology in the worst cases, and this adds an additional factor with regards to therapeutic response. Our skeletal muscle data from the mouse clearly separated the in vivo pharmacodynamic behavior of the three recombinant enzymes tested; however, translation of these results to human skeletal muscle is complicated by the considerable difference in the pathology of GAA deficiency in the two species.
Interestingly, heart and skeletal muscle tissue levels of the various rhGAA preparations did not correlate with efficacy in terms of glycogen clearance since the tissue levels of both tgGAA and HP-GAA were significantly higher than was observed for CHO-GAA. Western blot analysis indicated that all three enzymes were properly processed in the lysosomes of homogenized tissue. These data suggest that the higher levels of tgGAA and HP-GAA, compared to CHO-GAA, in target tissues may be a consequence of unproductive uptake of enzyme by resident endothelial cells and/or fibroblasts rather than glycogen-containing myocytes. This is particularly likely in the case of HP-GAA in which higher levels of exposed mannose may result in unproductive targeting of the enzyme to endothelial cells via the mannose receptor. Of note, recent studies demonstrated that chemically attaching synthetic bis-M6P containing glycans to oxidized sialic acid residues of CHO-GAA (neo-rhGAA) resulted in improved glycogen reduction from the skeletal muscles of GAA knockout mice [30, 31]. Since neo-rhGAA contains significantly less exposed mannose residues as compared to HP-GAA, these data demonstrate that if non-productive targets can be avoided, the concept of further exploiting uptake via the cation independent M6P receptor may offer a path to an improved therapeutic.
Finally, all of the enzyme preparations tested induced anti-GAA specific antibodies in the serum of the treated mice. The observation was not unexpected given that the mice were repeatedly administered high doses of a human recombinant protein to mice. Development of anti-GAA specific antibodies has also been observed in Pompe patients treated with ERT [10]. However, it has been possible to clinically manage the immune response to ERT in both Pompe patients and GAA knockout mice in a manner that decreases infusion associated adverse reactions and in a large number of patients appears to provide for glycogen clearance from the muscle even in the presence of circulating antibodies.
Taken together, our in vitro and in vivo studies provided clear evidence that neither cell binding/uptake experiments nor the level of enzyme activity in whole tissues were predictive of efficacy in terms of clearance of accumulated muscle glycogen in the myocytes of GAA knockout mice. These results serve to emphasize the risks and uncertainties associated with using in vitro or tissue enzyme activity endpoints to predict efficacy in animal models of ERT for treatment of lysosomal storage disorders. Finally, the data presented in this report were instrumental in selecting the CHO-GAA candidate for further development and registration, leading to the first approved therapy for Pompe disease (Myozyme, alglucosidase alfa).
ACKNOWLEDGEMENTS
We thank members of Genzyme’s Department of Pathology who helped support this work, as well as Misty Troutt, Susan Boucher, Lorraine Copertino, Josephine Kyazike, Karen Albee, Denise Honey, Qun Zhou, Joseph Serriello, Jason DelCarpini and Nikkol Atwell for their technical support. This work was funded by Genzyme Corporation. AMW, KLL, HQ, XJ, HD, RG, BLT, CR, MOC, WC, LA and RJM are employees of Genzyme Corporation. NR has received research/grant support from Genzyme Corporation. We are also grateful to Edward S. Cole, Y.T. Chen, Priya Kishnani, Arnold Reuser and Ans van der Ploeg for numerous helpful discussions during the development of alglucosidase alfa.
REFERENCES
- 1.Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid-α-glucosidase (acid maltase) deficiency. In: Scriver CR, editor. The metabolic and molecular basis of inherited disease. 2001. pp. 3389–3420. [Google Scholar]
- 2.Engel AG, Gomez MR, Seybold ME, Lambert EH. The spectrum and diagnosis of acid maltase deficiency. Neurology. 1973;23:95–106. doi: 10.1212/wnl.23.1.95. [DOI] [PubMed] [Google Scholar]
- 3.Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II Pompe disease) Curr. Mol. Med. 2002;2:145–166. doi: 10.2174/1566524024605789. [DOI] [PubMed] [Google Scholar]
- 4.Lauer RM, Mascarinas T, Racela AS, Diehl AM, Brown BI. Administration of a mixture of fungal glucosidases to a patient with type II glycogenosis (Pompe's disease) Pediatrics. 1968;42:672–676. [PubMed] [Google Scholar]
- 5.de Barsy T, Jacquemin P, Van Hoof F, Hers HG. Enzyme replacement in Pompe disease: an attempt with purified human acid alpha-glucosidase. Birth Defects Orig. Artic. Ser. 1973;9:184–190. [PubMed] [Google Scholar]
- 6.Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356:397–398. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- 7.Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, Van Hirtum H, Van Diggelen OP, Boer M, Kroos MA, Van Doorn PA, Van der Voort E, Sibbles B, Van Corven EJ, Brakenhoff JP, Van Hove J, Smeitink JA, de Jong G, Reuser AJ, Van der Ploeg AT. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–e457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 8.Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet. Med. 2001;3:132–138. [PubMed] [Google Scholar]
- 9.Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A, Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J. Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, De la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 11.Winkel LPF, Van den Hout JMP, Kamphoven JHJ, Disseldorp JAM, Remmerswaal M, Arts WFM, Loonen MCB, Vulto AG, Van Doorn PA, De Jong G, Hop W, Smit GPA, Shapira SK, Boer MA, van Diggelen OP, Reuser AJJ, Van der Ploeg AT. Enzyme replacement therapy in late-onset Pompe's disease: A three-year follow-up. Ann. Neurol. 2004;55:495–502. doi: 10.1002/ana.20019. [DOI] [PubMed] [Google Scholar]
- 12.Wackerhage H, Rennie MJ. How nutrition and exercise maintain the human musculoskeletal mass. J. Anat. 2006;208:451–458. doi: 10.1111/j.1469-7580.2006.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thurberg BL, Lynch Maloney C, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, O'Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for pompe disease. Lab. Invest. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- 14.Raben N, Fukuda T, Gilbert AL, de Jong D, Thurberg BL, Mattaliano RJ, Meikle P, Hopwood JJ, Nagashima K, Nagaraju K, Plotz PH. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent but neither completely clears glycogen from type II muscle fibers. Mol Ther. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol. Ther. 2006;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat. Rev. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 17.Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L, LaMarca M, King C, Ward J, Sauer B, Plotz P. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 18.Moreland RJ, Jin X, Zhang XK, Decker RW, Albee KL, Lee KL, Cauthron RD, Brewer K, Edmunds T, Canfield WM. Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J. Biol. Chem. 2005;280:6780–6791. doi: 10.1074/jbc.M404008200. [DOI] [PubMed] [Google Scholar]
- 19.Chavez CA, Bohnsack RN, Kudo M, Gotschall RR, Canfield WM, Dahms NM. Domain 5 of the cation-independent mannose 6-phosphate receptor preferentially binds phosphodiesters (mannose 6-phosphate N-acetylglucosamine ester) Biochem. 2007;46:12604–12617. doi: 10.1021/bi7011806. [DOI] [PubMed] [Google Scholar]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Q, Kyazike J, Edmunds T, Higgins E. Mannose 6-phosphate quantitation in glycoproteins using high-pH anion-exchange chromatography with pulsed amperometric detection. Anal. Biochem. 2002;306:163–170. doi: 10.1006/abio.2002.5703. [DOI] [PubMed] [Google Scholar]
- 22.Lynch CM, Johnson J, Vaccaro C, Thurberg BL. High-resolution light microscopy (HRLM) and digital analysis of Pompe disease pathology. J. Histochem. Cytochem. 2005;53:63–73. doi: 10.1177/002215540505300108. [DOI] [PubMed] [Google Scholar]
- 23.Reuser AJ, Koster JF, Hoogeveen A, Galjaard H. Biochemical immunological and cell genetic studies in Glycogenosis type II. Am. J. Hum. Genet. 1978;30:132–143. [PMC free article] [PubMed] [Google Scholar]
- 24.Wisselaar HA, Kroos MA, Hermans MM, van Beeumen J, Reuser AJ. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J. Biol. Chem. 1993;268:2223–2231. [PubMed] [Google Scholar]
- 25.Chadalavada DM, Sivakami S. Purification and biochemical characterisation of human placental acid alpha-glucosidase. Bio. Mol. Biol. Int. 1997;42:1051–1061. doi: 10.1080/15216549700203511. [DOI] [PubMed] [Google Scholar]
- 26.Fuller M, Van der Ploeg A, Reuser AJ, Anson DS, Hopwood JJ. Isolation and characterisation of a recombinant precursor form of lysosomal acid alpha-glucosidase. Eur. J. Biochem. 1995;234:903–909. doi: 10.1111/j.1432-1033.1995.903_a.x. [DOI] [PubMed] [Google Scholar]
- 27.Oude Elferink RP, Strijland A, Surya I, Brouwer-Kelder EM, Kroos M, Hilkens J, Hilgers J, Reuser AJ, Tager JM. Use of a monoclonal antibody to distinguish between precursor and mature forms of human lysosomal alpha-glucosidase. Eur. J. Biochem. 1984;139:497–502. doi: 10.1111/j.1432-1033.1984.tb08033.x. [DOI] [PubMed] [Google Scholar]
- 28.Jongen SP, Gerwig GJ, Leeflang BR, Koles K, Mannesse ML, van Berkel PH, Pieper FR, Kroos MA, Reuser AJ, Zhou Q, Jin X, Zhang K, Edmunds T, Kamerling JP. N-glycans of recombinant human acid {alpha}-glucosidase expressed in the milk of transgenic rabbits. Glycobiology. 2007;17:600–619. doi: 10.1093/glycob/cwm015. [DOI] [PubMed] [Google Scholar]
- 29.Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL, Mattaliano RJ, Nagaraju K, Plotz PH. Enzyme replacement therapy in the mouse model of Pompe disease. Mol. Genet. Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Y, Li X, Kyazike J, Zhou Q, Thurberg BL, Raben N, Mattaliano RJ, Cheng SH. Conjugation of mannose 6-phosphate-containing oligosaccharides to acid alpha-glucosidase improves the clearance of glycogen in Pompe mice. J. Biol. Chem. 2004;279:50336–50341. doi: 10.1074/jbc.M409676200. [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, Li X, McVie-Wylie A, Jiang C, Thurberg BL, Raben N, Mattaliano RJ, Cheng SH. Carbohydrate-remodeled acid alpha-glucosidase with higher affinity for the cation-independent mannose 6-phosphate receptor demonstrates improved delivery to muscles of Pompe mice. Biochem. J. 2005 doi: 10.1042/BJ20050364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang HW, Kikuchi T, Hagiwara Y, Mizutani M, Chen YT, Van Hove JL. Recombinant human acid alpha-glucosidase corrects acid alpha-glucosidase-deficient human fibroblasts quail fibroblasts and quail myoblasts. Pediatr. Res. 1998;43:374–380. doi: 10.1203/00006450-199803000-00011. [DOI] [PubMed] [Google Scholar]
- 33.Bijvoet AG, Kroos MA, Pieper FR, Van der Vliet M, De Boer HA, Van der Ploeg AT, Verbeet MP, Reuser AJ. Recombinant human acid alpha-glucosidase : high level production in mouse milk biochemical characteristics correction of enzyme deficiency in GSDII KO mice. Hum. Mol. Genet. 1998;7:1815–1824. doi: 10.1093/hmg/7.11.1815. [DOI] [PubMed] [Google Scholar]
- 34.Varki A, Kornfeld S. The spectrum of anionic oligosaccharides released by endo-beta-N-acetylglucosaminidase H from glycoproteins. Structural studies and interactions with the phosphomannosyl receptor. J. Biol. Chem. 1983;258:2808–2818. [PubMed] [Google Scholar]
- 35.Kishnani PS, Howell RR. Pompe disease in infants and children. J. Pediatr. 2004;144:S35–S43. doi: 10.1016/j.jpeds.2004.01.053. [DOI] [PubMed] [Google Scholar]