

Abstract
Purpose
To determine the inhibitory potency of letrozole and its main human metabolite, 4,4′-methanol-bisbenzonitrilee, on the activities of eight cytochrome P450 (CYP) enzymes.
Methods
Letrozole and its metabolite were incubated with human liver microsomes (HLMs) (or expressed CYP isoforms) and NADPH in the absence (control) and presence of the test inhibitor.
Results
Letrozole was a potent competitive inhibitor of CYP2A6 (Ki 4.6 ± 0.05 μM and 5.0 ± 2.4 μM in HLMs and CYP2A6, respectively) and a weak inhibitor of CYP2C19 (Ki 42.2 μM in HLMs and 33.3 μM in CYP2C19), while its metabolite showed moderate inhibition of CYP2C19 and CYP2B6. Letrozole or its metabolite had negligible effect on other CYPs.
Conclusions
Based on the in vitro Ki values, letrozole is predicted to be a weak inhibitor of CYP2A6 in vivo. Letrozole and its major human metabolite show inhibitory activity towards other CYPs, but clinically relevant drug interactions seem less likely as the Ki values are above the therapeutic plasma concentrations of letrozole.
Keywords: Letrozole, Metabolite, Cytochrome P450, Drug interaction, Aromatase inhibitor
Introduction
Estrogens have been strongly implicated in initiation and promotion of estrogen receptor (ER) positive breast cancer [1]. Thus, reducing the actions of estrogens in the breast through blockade of ERs (e.g. by tamoxifen) [2], down regulation of the ERs by fulvestrant [3], or depletion of plasma and tissue concentrations of estrogens in postmenopausal women by inhibiting aromatase [cytochrome P450 (CYP) 19] [4], a rate-limiting enzyme in the biosynthesis of estrogens [5], has become an effective strategy in the prevention and treatment of breast cancer.
The nonsteroidal triazole derivatives letrozole (4,4′-[(1H-1,2,4-triazol-1-yl) methylene] bis-benzonitrile) belong to the third generation potent and selective aromatase inhibitors (AIs) under wider clinical use for the treatment of breast cancer in postmenopausal women [6]; the other third generation AIs are anastrozole (a triazole) and exemestane (a steroid derivative). The use of letrozole and other AIs is on the increase because large randomized clinical trials have shown that these drugs are more effective than tamoxifen as adjuvant therapy of breast cancer [7–9]. The likelihood that AIs may be coadministered with other drugs is high, although information on the potential for drug–drug interactions with these drugs is very limited. As has been repeatedly emphasized in our recent work with tamoxifen [10, 11], the potential for adverse drug–drug interactions in breast cancer patients is of great concern.
Letrozole exhibits multiple interactions with the CYP enzyme system. First, of all the AIs, letrozole is the most potent inhibitor of aromatase by tightly binding to the hem iron of the enzyme complex [12], and it may do so the same with other CYPs involved in human drug metabolism [13]. Second, anastrozole, another nonsteroidal triazole AI, has been shown to inhibit CYP1A2, 2C9, and 3A in vitro, although the Ki values (8–10 μM) were much higher than the therapeutic plasma concentration of the drug and the nanomolar concentration needed to inhibit human aromatase (IC50 = ∼15 nM) [14]. Other triazole derivatives such as azole antifungal drugs are known to alter the pharmacokinetics of coadministered drugs through inhibition of CYPs [15]. Third, letrozole itself is a substrate of CYPs that include CYP2A6, CYP3A4, and CYP3A5 [13, 16, 17], and it is conceivable that it competitively inhibits CYPs. It is, therefore, possible that letrozole alters the pharmacokinetics of coadministered drugs through inhibition of CYPs. However, the limited in vitro information available [13, 17], has not been published in full to allow assessment of the relevance of these interactions and prediction of letrozole interactions in patients. In the present study, we determined the inhibitory effect of letrozole and its main human oxidative metabolite, 4,4′-methanol-bisbenzonitrile (Fig. 1) [18, 19], on the activities of eight drug-metabolizing CYPs and predicted in vivo relevance from in vitro data.
Fig. 1.
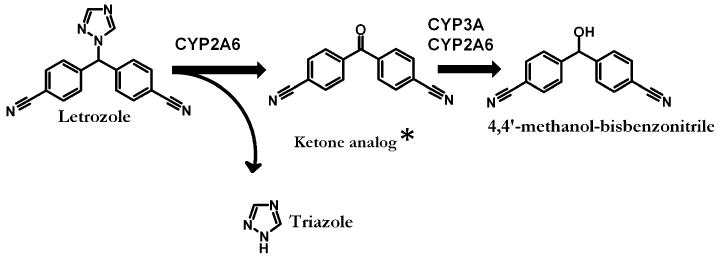
Proposed human metabolic pathways of letrozole and the specific cytochrome P450 (CYP) isoforms involved. There is no full publication describing in detail the precise metabolic pathways and CYPs involved in letrozole metabolism. Thus, the schematic presentation is based on information provided in the letrozole label [17] and our own data published in abstract form (16). *Represents the ketone analog of 4,4′-methanol-bisbenzonitrile
Materials and methods
Chemicals
Efavirenz, 7-hydroxycoumarin, 8-hydroxyefavirenz, letrozole ritonavir, midazolam, 1′-hydroxymidazolam, and desethylamodiaquine were purchased from Toronto Research Chemicals (North York, ON, Canada). Coumarin, glucose 6-phosphate, NADP, glucose-6-phosphate dehydrogenase, 8-methoxypsolaren, dextromethorphan, dextrorphan, chloroquine, desmethyldiazepam, phenacetin, and acetaminophen were purchased from Sigma-Aldrich (St Louis, MO). Amodiaquine and levallorphan were purchased from the United States Pharmacopeia (Rockville, MD). S-Mephenytoin was purchased from Biomol (Plymouth, PA). Letrozole metabolite 4,4′-methanol-bisbenzonitrilee was kindly provided by Novartis Pharma AG (Basel, Switzerland). All other chemicals and solvents which were all of HPLC grade were purchased from reliable commercial sources.
Human liver microsomal preparations
The Human liver microsomal preparations (HLMs) used were prepared from human liver tissues medically unsuitable for transplantation by ultracentrifugation using standard protocols. Protein concentrations were determined by the Bradford method [20], using bovine serum albumin as a standard. Additional HLMs were purchased from Cellzdirect (Pittsboro, NC). Baculovirus–insect cell–expressed human P450s (with oxidoreductase) were purchased from BD Biosciences (San Jose, CA). The microsomal pellets were suspended in a reaction buffer to a protein concentration of 10–20 mg/ml (stock). All microsomal preparations were stored at −80°C until used.
General incubation conditions
The inhibitory effects of letrozole and its metabolite (4,4′-methanol-bisbenzonitrile) on the activities of different CYP isoforms were studied in HLMs (and expressed CYPs when required) using reaction probes selective for each isoform. Using incubation conditions specific to each isoform that were linear for time, substrate and protein concentrations [21], isoform selective substrate probes were incubated in duplicate at 37°C with HLMs (or expressed enzymes when required), phosphate reaction buffer (pH 7.4) and NADPH-generating system (1.3 mM NADP, 3.3 mM glucose-6-phosphate, 3.3 mM MgCl2, and 0.4 U/ml glucose 6-phosphate dehydrogenase) (final incubation volume 250 μl) in the absence (control) or presence of varying concentrations of letrozole or its metabolite. The test inhibitors were dissolved and diluted in methanol to the required concentrations and any methanol was removed by drying in speed vacuum before the addition of the incubation components. Formation rate of acetaminophen from phenacetin (CYP1A2), of 7-hydroxycoumarin from coumarin (CYP2A6), of 8-hydroxyefavirenz from efavirenz (CYP2B6), of desethylamodiaquine from amodiaquine (CYP2C8), for 4-methyltolbutamide from tolbutamide (CYP2C9), of 4′-hydroxymephenytoin from S-mephenytoin (CYP2C19), of dextrorphan from dextromethorphan (CYP2D6), and 1′-hydroxymidazolam from midazolam (CYP3A) served as isoform-specific marker of activity [21].
The following published methods were adopted or slightly modified (see for details [21] to measure the activity of each isoform in the absence or presence of the test inhibitors. A method(s) described elsewhere were used to assay: CYP1A2 [22]; CYP2A6 [23–25]; CYP2B6 [26]; CYP2C8 [27, 28]; CYP2C9 [22]; CYP2C19 [22]; CYP2D6 [22]; CYP3A [29]. Human liver microsomal protein concentrations of 1 mg/ml (CYP1A2, CYP2C9 and CYP2CD6), 0.5 mg/ml (CYP2A6, CYP2B6, CYP2C19 and CYP3A) or 0.1 mg/ml (CYP2C8) were used. Expressed enzymes were used at concentration of 52 pmol/ml. The duration of incubation was 5 min (CYP3A), 10 min (CYP2B6), 15 min (CYP2A6 and CYP2C8), 30 min (CYP1A2 and CYP2D6), and 60 min (CYP2C9 and CYP2C19). The internal standards used were described elsewhere (809) and included 50 μl of 100 μM coumarin, 50 μl of 20 μg/ml 8-methoxypsolaren, 50 μl of 10 μg/ml ritonavir, 50 μl of 50 μM chloroquine, 50 μl of 10 μg/ml chlorpropamide, 50 μl of 5 μg/ml phenytoin, 40 μL of 16 μM levallorphan, and 50 μl of 5 μg/ml desmethyldiazepam, respectively, for CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A.
Unless specified, an incubation mixture that consists of the substrate probe, HLMs (or expressed CYP), and phosphate reaction buffer (pH 7.4) was prewarmed for 5 min at 37°C without (control) and with multiple concentrations of the test inhibitors. The reaction was initiated by addition of NADPH-generating system and allowed to proceed for time specific for each isoform and then terminated by placing tubes on ice and immediately adding appropriate stopping reagent as described elsewhere [21]. After the addition of an appropriate internal standard, the incubation mixture was vortex-mixed and centrifuged at 14,000 rpm for 5 min (Brinkmann Instruments, Westbury, NY). The supernatant layer was injected into an HPLC system directly or after it was extracted and reconstituted with mobile phase. The concentrations of the metabolites formed during incubation and internal standards were measured by HPLC with UV or fluorescent detection specific for each assay. The reagents to stop enzymatic reactions, sample processing, HPLC instrumentation, HPLC separation systems (mobile phase and columns), and the detection methods used in this study were the same as those described in our recent publication [21]. The ratio of the area under the curve for the metabolite to the area under the curve for each internal standard was calculated. Formation rate of metabolite from the respective probe substrate was quantified by using an appropriate standard curve. Intra- and inter-day coefficients of variation of the assays were less than 15%.
Kinetic analysis
For each enzyme assay, kinetic parameters for the metabolism of each probe substrate were determined by incubating increasing concentrations (6–10 points each) of probe substrates with HLMs and cofactors in the absence of the test inhibitors. The following probe concentrations were used: 5–500 μM (phenacetin, S-mephenytoin), 0.1–500 μM (efavirenz, tolbutamide), 0.1–50 μM (coumarin), 0.5–100 μM (amodiaquine), 1–200 μM (dextromethorphan), and 1-300 μM (midazolam). The kinetic parameters (Vmax and Km values) for each CYP isoform, as measured by selective probe substrate reaction in HLMs, have been published elsewhere [21], and the findings generally agree well with data published in the literature and served as a guide to select substrate probe concentrations for subsequent inhibition experiments.
Determination of IC50 values
Preliminary inhibition experiments were carried out to obtain initial information (IC50 values) on the inhibitory potency of letrozole and its metabolite. A single isoform-specific substrate concentration (around the Km value) was performed with HLMs and cofactors in the absence and presence of letrozole (or its metabolite) (0 to up to 100 μM). The concentration of each substrate probe used is detailed in legend for Fig. 2 and elsewhere [21]. In addition, positive control experiments were run in parallel by incubating probe substrates at the concentrations listed above in the absence (control) and presence of the following established isoform-specific inhibitors in HLMs at protein concentrations described below for each enzyme assay: furafylline (20 μM) for CYP1A2; pilocarpine (50 μM) for CYP2A6; thioTEPA (50 μM) and ticlopidine (5 μM) for CYP2B6; quercetin (10 μM) for CYP2C8; sulfaphenazole (25 μM) for CYP2C9; ticlopidine (5 μM) for CYP2C19; quinidine (1 μM) for CYP2D6; and ketoconazole (1 μM) for CYP3A. The specific conditions used with these inhibitors have been described in detail elsewhere [21, 26, 30–32]. As expected, the isoform specific inhibitor substantially decreased (by >50%) the activity of the respective isoform [809] and served as positive control.
Fig. 2.
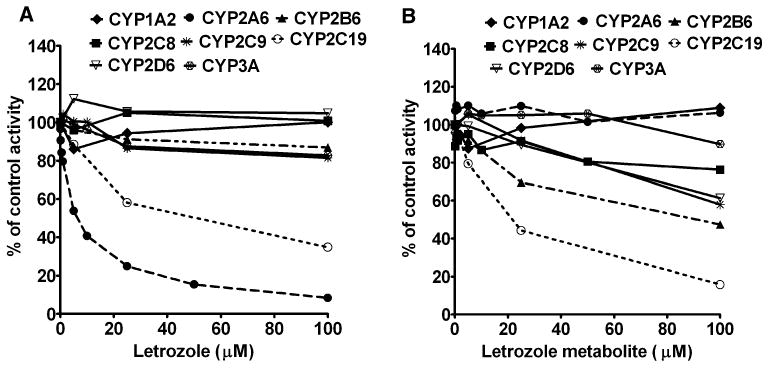
Inhibition of CYP isoforms by letrozole (a), and its metabolite 4,4′-methanol-bisbenzonitrile (b) in human liver microsomes. The activity of each enzyme was measured using substrate reaction probe selective for each isoform: phenacetin (50 μM) O-deethylation (CYP1A2); coumarin (10 μM) 7-hydroxylation (CYP2A6); efavirenz (10 μM) 8-hydroxylation (CYP2B6); amodiaquine (5 μM) desethylation (CYP2C8); tolbutamide (150 μM) 4-methyl-hydroxylation (CYP2C9); S-Mephenytoin (50 μM) 4′-hydroxylation (CYP2C19); for dextromethorphan (25 μM) O-demethylation (CYP2D6); and for midazolam (10 μM) 1′-hydroxylation (CYP3A). Each substrate probe was incubated in the absence (control) or presence of letrozole or its metabolite with HLMs and cofactors for time and protein concentrations specific for each reaction (see “Materials and methods” section). Inhibition of known CYP isoform-specific inhibitors served as positive control for inhibition. Each point represents average of duplicate incubations
Determination of Ki values
Whenever an inhibition was noted in the preliminary experiments, Dixon plots for the inhibition by letrozole and/or its metabolite were determined in both HLMs and expressed CYPs. Multiple concentrations of the substrate probe without or with multiple concentrations of the test inhibitor were incubated with HLMs (and expressed CYPs) and co-factors. Inhibitor and substrate concentrations used for constructing the Dixon plots are described in the corresponding figure legends. The inhibition data obtained from the pilot experiments (IC50 determination) were used as a guide to focus on those isoforms requiring further characterization and to generate appropriate probe substrate and test inhibitor concentrations for the determination of exact inhibition constants (Ki values).
Assessment of time-dependent inactivation
To test whether inhibition of CYP isoforms by letrozole or its metabolite involves mechanism-based inactivation, the test inhibitors were preincubated for 0 and 15 min with HLM and cofactors in the absence of a substrate. Probe substrate was then added at final concentration corresponding to Vmax and further incubated for time specific for each assay. Reaction was stopped and processed as described above for co-incubation.
Data analysis
Formation rates of metabolite versus substrate concentrations were fit to appropriate enzyme kinetic models using WinNonlin Version 5.0 (Pharsight, Mountain View, CA) to estimate apparent kinetic parameters (Km and Vmax). Data were best fit to a one site Michaelis–Menten equation; except for CYP1A2 which was fit to two-site enzyme kinetic equation (kinetic parameters for the high affinity component were used). The rates of metabolite formation from substrate probes in the presence of the test inhibitors or isoform-specific inhibitors were compared with those for controls in which the inhibitor was replaced with vehicle. IC50 values were determined by an analysis of the plot of the logarithm of inhibitor concentration versus percentage of activity remaining after inhibition using SigmaPlot, version 10.0 (Systat Software, Point Richmond, CA). The Ki values were determined by nonlinear least square regression analysis using WinNonlin Version 5.0 (Pharsight, Mountain View, CA). The inhibition data were fit to different models of enzyme inhibition (competitive, noncompetitive and uncompetitive). The equations for kinetic and inhibition analysis of the data by Winnonlin were written by ourselves. Before modeling the data using nonlinear models, initial information on the mode of inhibition was obtained by visual inspection of different plots (Lineweaver–Burk, Dixon, and Eadie–Scatchard plots). Final decision on the mechanisms of inhibition was made on model derived parameters such as the sum of squares of residuals, Akaike information criterion (AIC) and Schwartz criterion (SC) values.
Prediction of in vivo changes from in vitro inhibition constants
Anticipated in vivo drug interaction from the in vitro competitive or noncompetitive inhibition data was estimated using the following equation:
where AUCI/AUCUI is the ratio of AUC of the substrate after inhibition (AUCI) to that of AUC uninhibited (AUCUI), [I] is the concentration of inhibitor and Ki is the in vitro inhibition constant. The average letrozole Cmax concentration of 0.5 μM was considered for this calculation [18]. In addition, 1 μM letrozole was also included in the calculation to capture higher-than-average concentrations in some individuals. The fraction unbound was estimated assuming a plasma protein binding of 60% [33].
Results
The present study was performed simultaneously with another similar interaction study (voriconazole interaction study) [21]. The kinetic parameters (Vmax and Km) derived from incubation of probe substrates without the inhibitor as well as inhibition by isoform-specific inhibitors have been published recently in the voriconazole study [21]. Thus the same values (Kms) were used to guide further experiments and to serve as positive controls for inhibition.
The effect of multiple concentrations of letrozole (0–100 μM) and letrozole metabolite (0–100 μM) on the activity of each CYP isoform was determined in HLMs (Fig. 2) [and in expressed CYP2A6, CYP2C19 and CYP2B6 (data not shown)]. A single concentration of the respective phenotyping probe around the Km value was used (see legend to Fig. 2). Letrozole inhibited CYP2A6 activity in HLMs (HL-SD109) (Fig. 2a) and expressed CYP2A6, with IC50 value of 5.90 and 12.52 μM respectively. Letrozole also showed modest inhibition of CYP2C19 in HLMs (IC50 = 24.8 μM) (Fig. 2a) and expressed CYP2C19 (IC50 = 62.9 μM). The inhibitory effect of letrozole on the other isoforms tested (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2D6 and CYP3A) was negligible (by less than 20% at 100 μM letrozole) (Fig. 2). The degree of inhibition of CYPs by letrozole was not modified during preincubation experiments (data not shown); suggesting that letrozole inhibition of CYPs is not time dependent.
The inhibitory effect of letrozole metabolite on the activity of different CYPs in HLMs is summarized in Fig. 2b. Modest inhibition of CYP2C19 and CYP2B6 was observed, with IC50 values of 19.5 and 33.1 μM, respectively. A week inhibition of CYP2D6 (IC50 = 158.0 μM) and CYP2C9 (IC50 = 133.9 μM) was also noted. The effect of letrozole metabolite on the activity of other CYPs was negligible.
The preliminary inhibition data generated using a single probe substrate reaction were then used to simulate appropriate range of substrate and inhibitor concentrations to construct Dixon plots for the inhibition of CYP isoforms by letrozole and its metabolites in HLMs and/or expressed enzymes from which inhibition constants (Ki values) were estimated.
In Fig. 3, the inhibition of CYP2A6 by letrozole in HLMs and expressed CYP2A6 are shown. Visual inspection of the Dixon plots and further analysis of the parameters of the enzyme inhibition models suggested that the mode of inhibition competitive. The Ki values (±SD) estimated for competitive enzyme inhibition is summarized in Table 1. The Ki values in HLMs (4.6 ± 0.05 μM) were close to those derived in expressed CYP2A6 (5.0 ± 2.4 μM), suggesting that the same enzyme is inhibited in both microsomal systems.
Fig. 3.
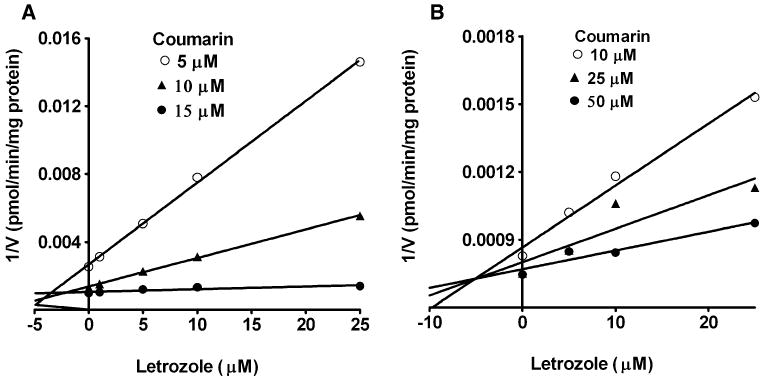
Dixon plots for the inhibition of CYP2A6-catalyzed coumarin 7-hydroxylation by letrozole in HLMs (a), and expressed CYP2A6 (b). Coumarin (5–50 μM) was incubated without or with letrozole (5–25 μM) with HLMs (0.5 mg/ml, SD-109) and expressed CYP2A6 (52 pmol/ml) with cofactors at 37°C for 15 min. Each point represents mean of duplicate incubations
Table 1.
Prediction of in vivo letrozole drug interactions from in vitro data
| CYP isoforms | Microsomes | Ki values (mean ± SE or SD)a | AUCI/AUCUI ratio expected in vivob | |
|---|---|---|---|---|
| Letrozole (μM) | Metabolite (μM)c | Letrozole plasma concentrationd 0.5–1 μM (fu 0.2–0.4)e | ||
| CYP2A6 | HLMs (SD-109) | 4.6 ± 0.1 | NA | 1.11–1.22 (free 1.04–1.09) |
| Expressed 2A6 | 5.0 ± 2.4 | |||
| CYP2B6 | HLMs (HL-091499) | NA | 12.9 ± 3.8 | |
| Expressed CYP2B6 | 40.4 ± 14.6 | |||
| CYP2C19 | HLMs (SD-101) | 42.2 ± 3.8 | 19.5 ± 0.5 | 1.01–1.02 (free 1.004–1.009) |
| Expressed 2C19 | 33.3 ± 7.6 | 4.5 ± 0.7 | ||
Values derived from our in vitro experiments (μM). For all Ki values, data are presented as mean ± SE (standard error of parameter estimates from the nonlinear computer model), except for the values for CYP2A6 in HLMs and expressed CYP2A6 which was mean ± SD (standard deviation)
The ratio of in vivo area under the concentration–time curve with the inhibitor (AUCI) and without the inhibitor (AUCUI) was predicted from the in vitro data (AUCI/AUCUI = 1 + [I[/Ki), where I is steady state letrozole concentration (μM); only Ki values from HLMs were used for this calculation
AUCI/AUCUI ratio was calculated for letrozole since systemic exposure of the metabolite (4′,4-methanol-bisbenzonitrile) is unknown after the administration of 2.5 mg/day letrozole
The average maximum plasma concentration (Cmax) of letrozole (2.5 mg/day) at steady state that was estimated at 0.5 μM was used as total I, while 1 μM letrozole was included in the calculation to capture higher-than-average concentrations in some individuals [6, 18]
fu (fraction unbound) was estimated to be 0.2–0.4 assuming a plasma protein binding of 60% of letrozole [33]
The next isoform that was inhibited by letrozole was CYP2C19. Dixon plots for the inhibition of CYP2C19 by letrozole in HLMs and expressed CYP2C19 are shown in Fig. 4. Letrozole was a competitive inhibitor of CYP2C19 in HLMs (Fig. 4a) (Ki = 42.2 μM) and expressed CYP2C19 (Fig. 4b) (Ki = 33.34 μM). Of note, the Ki value for the inhibition of CYP2C19 in HLMs was over ninefold higher than that derived for CYP2A6 in HLMs. Similarly, letrozole metabolite was a competitive inhibitor of CYP2C19 activity in HLMs with a Ki of 19.5 μM (Fig. 4c) and expressed CYP2C19 (Ki = 4.5 μM) (Fig. 4d; Table 1).
Fig. 4.
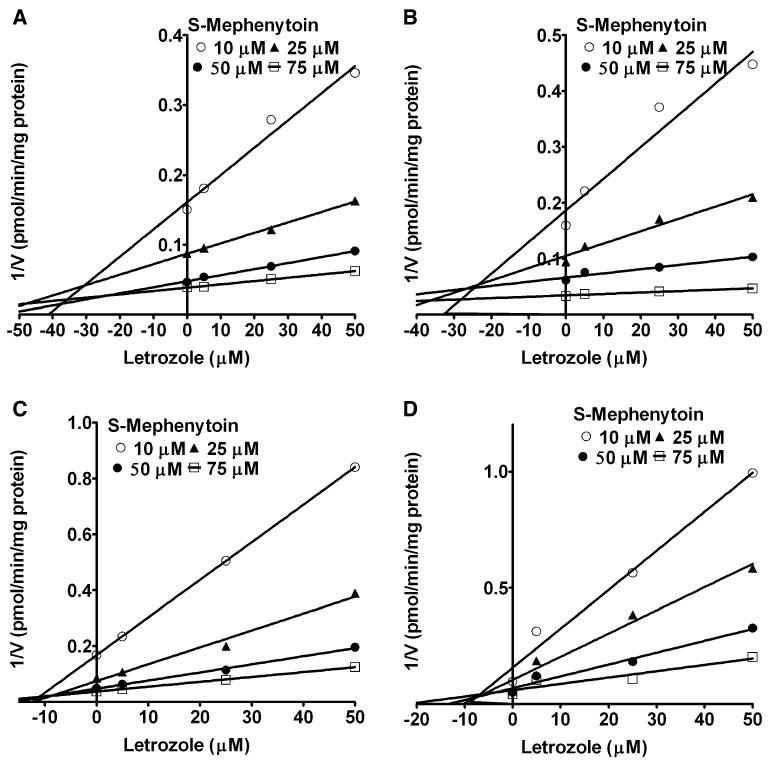
Dixon plots for the inhibition of CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation by letrozole and letrozole metabolite in HLMs and expressed CYP2C19. S-Mephenytoin (10–75 μM) was incubated without (control) or with 5–50 μM letrozole or letrozole metabolite with HLMs (0.5 mg/ml, SD-101) and expressed CYP2C19 (52 pmol/ml) and cofactors at 37°C for 60 min. Inhibition by letrozole in HLMs (a) and in expressed CYP2C19 (b), and by letrozole metabolite in HLMs (c) and expressed CYP2C19 (d) is shown. Each point represents the mean of duplicate measurements
The inhibitory potency of letrozole metabolite towards CYP2B6 was determined in HLMs (Fig. 5a) and expressed CYP2B6 (Fig. 5b). The data were best fit to competitive inhibition equation. Letrozole metabolite was relatively more potent inhibitor of CYP2B6 in HLMs (Ki = 12.9 μM) than in expressed CYP2B6 (40.4 μM).
Fig. 5.
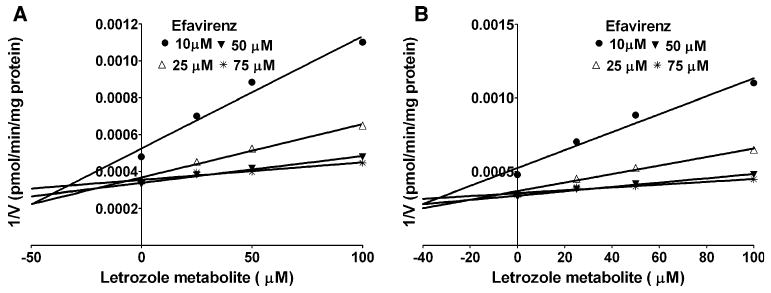
Dixon plots for the inhibition of efavirenz 8-hydroxylation by letrozole metabolite 4,4′-methanol-bisbenzonitrile in HLMs (a), and expressed CYP2B6 (b). Efavirenz (10–75 μM) was incubated without or with letrozole metabolite (5–100 μM) with HLMs (0.5 mg/ml) (or expressed CYP2B6, 52 pmol/ml) and NADPH generating system at 37°C for 10 min. Each point represents the mean of duplicate measurements
Discussion
In the present study, we assessed the ability of letrozole and its principal human metabolite 4,4′-methanol-bis-benzonitrile to inhibit eight major drug-metabolizing CYP enzymes in vitro. We found that letrozole is a relatively potent competitive inhibitor of CYP2A6 and a modest inhibitor of CYP2C19, while its metabolite is a weak inhibitor of CYP2B6 and CYP2C19. Letrozole or its metabolite has minimal inhibitory effect on the other isoforms tested.
Of the CYP isoforms studied, CYP2A6 was the most susceptible to letrozole (but not to its metabolite) inhibition. CYP2A6 metabolizes only few clinically used drugs, but it is an important enzyme in the metabolism of nicotine and a number of other environmental chemicals [34]. From the in vitro Ki values, we attempted to predict AUC changes of CYP2A6 substrates during coadministration with letrozole. To do so, it was informative to evaluate the in vitro Ki values derived in the context of steady state concentrations of letrozole or its metabolite in humans. In women with breast cancer, the therapeutic dose of letrozole is 2.5 mg/day and the average maximum plasma concentration of letrozole at steady state is ∼0.5 μM, with relatively high intersubject variability [18]. In certain cases, higher plasma concentrations of letrozole may be expected due to its nonlinear pharmacokinetics [6] due to an auto-inhibition or saturation of oxidative metabolism [18] or due to coadministration of letrozole with drugs that inhibit its elimination. To represent higher therapeutic concentration, a 1 μM letrozole concentration was also examined in the predictive model. Assuming letrozole's complete absolute bioavailability (99.9%) [35], a plasma protein binding of ∼60% [33], and competitive in vitro inhibition (Table 1), the in vivo change in AUC ratios for a drug primarily cleared by CYP2A6 was estimated to be 1.11–1.22 at total letrozole concentrations (bound + unbound) of 0.5 and 1 μM, respectively (Table 1), although the AUC ratio was close to unity when the fraction unbound was used. On the basis of this prediction, letrozole may be categorized as a weak inhibitor of CYP2A6 in vivo. Indeed, letrozole exposure at steady state has been reported to be 28%, higher than that observed at a single dose, suggesting the possibility that letrozole may alter its own elimination through inhibition of CYP2A6 [18]. Together, our data suggest that letrozole might slightly alter the pharmacokinetics of CYP2A6 substrates, but the clinical relevance remains to be tested. Another implication of our findings is the observation that letrozole is a relatively selective inhibitor of CYP2A6 (over ninefold difference in Ki value between CYP2A6 and CYP2C19), with no meaningful effect on other isoforms, suggesting that letrozole may be utilized as an inhibitor probe for CYP2A6 to dissect its contribution to human drug and chemical metabolism in vitro. In addition, the relatively higher affinity of letrozole to CYP2A6 provides further evidence that CYP2A6 is involved in the metabolism of letrozole.
The next enzyme that was inhibited in vitro by letrozole was CYP2C19. However, given that the Ki value for the inhibition of CYP2C19 by letrozole was over 40-fold higher than the therapeutic plasma concentrations of letrozole, no in vivo interaction with CYP2C19 substrates is expected.
We also assessed the potential contribution of 4,4′-methanol-bisbenzonitrile, the main human metabolite that accounts for over 60% of letrozole dose [18], and found a modest inhibitory effect on the activities of CYP2B6 and CYP2C19. These in vitro data do not support any meaningful inhibition of these enzymes in vivo in humans. Although data on plasma exposure of 4,4′-methanol-bis-benzonitrile after letrozole administration is lacking, its systemic concentrations is likely to be low [18] relative to the in vitro Ki values for the inhibition of CYP2C19 and CYP2B6 (Table 1). Approximately 82% of the radioactivity in plasma following 14C-labeled letrozole (2.5 mg) has been accounted by unchanged drug [17]. Once formed, this metabolite seems to undergo rapid glucuronidation and renal excretion [17, 18]. Nevertheless, the inhibition data of letrozole metabolite are useful in that they provide important information about the structural requirements for inhibition of CYPs by letrozole. The lack of any effect of letrozole metabolite on CYP2A6 activity, the fact that CYP2C19 was inhibited by both letrozole and it metabolite, and the metabolite (but hardly letrozole) inhibited CYP2B6 may suggest that (1) the triazole ring is crucial for binding of letrozole with CYP2A6, (2) the bisbenzonitrile moiety of letrozole is important for the interaction with CYP2C19 and probably CYP2B6. These structural activity relationships might form the basis for synthesizing letrozole-based chemical inhibitor of CYPs that could be exploited as inhibitor probes and to study active site of CYPs.
In summary, we have shown that letrozole appreciably inhibits CYP2A6 in vitro, which was predicted to result in a small increase in exposure of CYP2A6 substrates (maximum 11–22%). In rat liver tissues, letrozole concentrations are much higher (>sevenfold) than in plasma [36]. If the same relationship exists in humans, the interaction of letrozole with CYP2A6 is likely to be significant and therefore needs clinical testing. Letrozole is unlikely to alter the pharmacokinetics of drugs metabolized by the other CYPs tested, consistent with the lack of letrozole's effect on the pharmacokinetics of known CYP substrates including warfarin, diazepam, and tamoxifen [17, 37]. However, letrozole drug interactions caused by induction or down regulation of CYPs in vivo or due to interaction with drug transporters cannot be ruled out. In addition, as many genes including those encoding drug-metabolizing enzymes are known to be regulated by estrogens, it is conceivable that depletion of tissue and plasma estrogen resulting from inhibition of the aromatase (CYP19) in postmenopausal women could influence rate of drug metabolism.
Acknowledgments
This study was supported in part by a Pharmacogenetics Research Network Grant (U-01 GM61373), which supports the Consortium on Breast Cancer Pharmacogenomics (COBRA), a Clinical Pharmacology training Grant (T32GM008425) and an RO1 grant (1R01GM078501-01A1) from the National Institute of General Medical Sciences, National Institutes of Health (Bethesda, MD), and by a fellowship award to Dr. Jeong from the Research Institute of Medical Science of Chonnam National University, Gwangju, S. Korea.
Abbreviations
- CYPs
Cytochrome P450s
- HLMs
Human liver microsomes
- HPLC
High-performance liquid chromatography
Footnotes
Conflict of interest statement The authors have no conflict of interest.
Contributor Information
Seongwook Jeong, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, 1001 West 10th Street, WD Myers Bldg., W7123, Indianapolis, IN 46202, USA.
Margaret M. Woo, Novartis Pharmaceutical Corporation, Florham Park, NJ, USA
David A. Flockhart, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, 1001 West 10th Street, WD Myers Bldg., W7123, Indianapolis, IN 46202, USA
Zeruesenay Desta, Email: zdesta@iupui.edu, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, 1001 West 10th Street, WD Myers Bldg., W7123, Indianapolis, IN 46202, USA.
References
- 1.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 2.Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339:1609–1618. doi: 10.1056/NEJM199811263392207. [DOI] [PubMed] [Google Scholar]
- 3.Buzdar AU, Robertson JF. Fulvestrant: pharmacologic profile versus existing endocrine agents for the treatment of breast cancer. Ann Pharmacother. 2006;40:1572–1583. doi: 10.1345/aph.1G401. [DOI] [PubMed] [Google Scholar]
- 4.Smith IE, Dowsett M. Aromatase inhibitors in breast cancer. N Engl J Med. 2003;348:2431–2442. doi: 10.1056/NEJMra023246. [DOI] [PubMed] [Google Scholar]
- 5.Ryan KJ. Biological aromatization of steroids. J Biol Chem. 1959;234:268–272. [PubMed] [Google Scholar]
- 6.Scott LJ, Keam SJ. Letrozole: in postmenopausal hormone-responsive early-stage breast cancer. Drugs. 2006;66:353–362. doi: 10.2165/00003495-200666030-00010. [DOI] [PubMed] [Google Scholar]
- 7.Thurlimann B, Keshaviah A, Coates AS, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. 2005;353:2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 8.Coombes RC, Hall E, Gibson LJ, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med. 2004;350:1081–1092. doi: 10.1056/NEJMoa040331. [DOI] [PubMed] [Google Scholar]
- 9.Buzdar AU, Coombes RC, Goss PE, Winer EP. Summary of aromatase inhibitor clinical trials in postmenopausal women with early breast cancer. Cancer. 2008;112:700–709. doi: 10.1002/cncr.23193. [DOI] [PubMed] [Google Scholar]
- 10.Jin Y, Desta Z, Stearns V, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97:30–39. doi: 10.1093/jnci/dji005. [DOI] [PubMed] [Google Scholar]
- 11.Borges S, Desta Z, Li L, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006;80:61–74. doi: 10.1016/j.clpt.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Bhatnagar AS. The discovery and mechanism of action of letrozole. Breast Cancer Res Treat. 2007;105(Suppl 1):7–17. doi: 10.1007/s10549-007-9696-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wrz B, Valles B, Parkinson A, Madan A, Probst A, Zimmerlin A, Gut J. CYP3A4 and CYP2A6 are involved in the biotransformation of letrozole. ISSX Proc. 1996;10:359. Abstract. [Google Scholar]
- 14.Grimm SW, Dyroff MC. Inhibition of human drug metabolizing cytochromes P450 by anastrozole, a potent and selective inhibitor of aromatase. Drug Metab Dispos. 1997;25:598–602. [PubMed] [Google Scholar]
- 15.Venkatakrishnan K, Von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111–180. doi: 10.2165/00003088-200038020-00002. [DOI] [PubMed] [Google Scholar]
- 16.Desta Z, Ward BA, Flockhart DA. In vitro letrozole N-dealkylation is mainly catalyzed by human cytochrome P450 (CYP) 3A. Clin Pharmacol Ther. 2005;77:79. Abstract. [Google Scholar]
- 17.Product information. Novartis Pharmaceutical Canada Inc. Prescribing information femara1 (letrozole) http://www.pharma.ca.novartis.com/downloads/e/femara_scrip_e.pdf (online 23 April 2007)
- 18.Pfister CU, Martoni A, Zamagni C, et al. Effect of age and single versus multiple dose pharmacokinetics of letrozole (Femara) in breast cancer patients. Biopharm Drug Dispos. 2001;22:191–197. doi: 10.1002/bdd.273. [DOI] [PubMed] [Google Scholar]
- 19.Tao X, Piao H, Canney DJ, Borenstein MR, Nnane IP. Biotransformation of letrozole in rat liver microsomes: effects of gender and tamoxifen. J Pharm Biomed Anal. 2007;43:1078–1085. doi: 10.1016/j.jpba.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Jeong S, Nguyen PD, Desta Z. Comprehensive in vitro inhibition analysis of 8 cytochrome P450 (CYP) enzymes by voriconazole: major effect on CYPs 2B6, 2C9, 2C19 and 3. A Antimicrob Agents Chemothe. 2009;53:541–51. doi: 10.1128/AAC.01123-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko JW, Sukhova N, Thacker D, Chen P, Flockhart DA. Evaluation of omeprazole and lansoprazole as inhibitors of cytochrome P450 isoforms. Drug Metab Dispos. 1997;25:853–862. [PubMed] [Google Scholar]
- 23.Greenlee WF, Poland A. An improved assay of 7-ethoxycoumarin O-deethylase activity: induction of hepatic enzyme activity in C57BL/6J and DBA/2J mice by phenobarbital, 3-methylcholanthrene and 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. J Pharmacol Exp Ther. 1978;205:596–605. [PubMed] [Google Scholar]
- 24.Pearce R, Greenway D, Parkinson A. Species differences and interindividual variation in liver microsomal cytochrome P450 2A enzymes: effects on coumarin, dicumarol, and testosterone oxidation. Arch Biochem Biophys. 1992;298:211–225. doi: 10.1016/0003-9861(92)90115-d. [DOI] [PubMed] [Google Scholar]
- 25.Draper AJ, Madan A, Parkinson A. Inhibition of coumarin 7-hydroxylase activity in human liver microsomes. Arch Biochem Biophys. 1997;341:47–61. doi: 10.1006/abbi.1997.9964. [DOI] [PubMed] [Google Scholar]
- 26.Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. The cytochrome P4502B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: Implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther. 2003;306:287–300. doi: 10.1124/jpet.103.049601. [DOI] [PubMed] [Google Scholar]
- 27.Li XQ, Bjorkman A, Andersson TB, Ridderstrom M, Masimirembwa CM. Amodiaquine clearance and its metabolism to N-desethylamodiaquine is mediated by CYP2C8: a new high affinity and turnover enzyme-specific probe substrate. J Pharmacol Exp Ther. 2002;300:399–407. doi: 10.1124/jpet.300.2.399. [DOI] [PubMed] [Google Scholar]
- 28.Parikh S, Ouedraogo JB, Goldstein JA, Rosenthal PJ, Kroetz DL. Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther. 2007;82:197–203. doi: 10.1038/sj.clpt.6100122. [DOI] [PubMed] [Google Scholar]
- 29.Wang YH, Jones DR, Hall SD. Differential mechanism-based inhibition of CYP3A4 and CYP3A5 by verapamil. Drug Metab Dispos. 2005;33:664–671. doi: 10.1124/dmd.104.001834. [DOI] [PubMed] [Google Scholar]
- 30.Desta Z, Kerbusch T, Soukhova N, Richard E, Ko JW, Flockhart DA. Identification and characterization of human cytochrome P450 isoforms interacting with pimozide. J Pharmacol Exp Ther. 1998;285:428–437. [PubMed] [Google Scholar]
- 31.Desta Z, Soukhova N, Mahal SK, Flockhart DA. Interaction of cisapride with the human cytochrome P450 system: metabolism and inhibition studies. Drug Metab Dispos. 2000;28:789–800. [PubMed] [Google Scholar]
- 32.Bourrie M, Meunier V, Berger Y, Fabre G. Cytochrome P450 isoform inhibitors as a tool for the investigation of metabolic reactions catalyzed by human liver microsomes. J Pharmacol Exp Ther. 1996;277:321–332. [PubMed] [Google Scholar]
- 33.Colussi DM, Parisot CY, Lefevre GY. Plasma protein binding of letrozole, a new nonsteroidal aromatase enzyme inhibitor. J Clin Pharmacol. 1998;38:727–735. doi: 10.1002/j.1552-4604.1998.tb04813.x. [DOI] [PubMed] [Google Scholar]
- 34.Xu C, Goodz S, Sellers EM, Tyndale RF. CYP2A6 genetic variation and potential consequences. Adv Drug Deliv Rev. 2002;54:1245–1256. doi: 10.1016/s0169-409x(02)00065-0. [DOI] [PubMed] [Google Scholar]
- 35.Sioufi A, Gauducheau N, Pineau V, et al. Absolute bioavailability of letrozole in healthy postmenopausal women. Biopharm Drug Dispos. 1997;18:779–789. doi: 10.1002/(sici)1099-081x(199712)18:9<779::aid-bdd64>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Liu XD, Xie L, Zhong Y, Li CX. Gender difference in letrozole pharmacokinetics in rats. Acta Pharmacol Sin. 2000;21:680–684. [PubMed] [Google Scholar]
- 37.Ingle JN, Suman VJ, Johnson PA, et al. Evaluation of tamoxifen plus letrozole with assessment of pharmacokinetic interaction in postmenopausal women with metastatic breast cancer. Clin Cancer Res. 1999;5:1642–1649. [PubMed] [Google Scholar]