

Abstract
Sulfhydryl chemistry plays a vital role in normal biology and in defense of cells against oxidants, free radicals, and electrophiles. Modification of critical cysteine residues is an important mechanism of signal transduction, and perturbation of thiol–disulfide homeostasis is an important consequence of many diseases. A prevalent form of cysteine modification is reversible formation of protein mixed disulfides (protein–SSG) with glutathione (GSH). The abundance of GSH in cells and the ready conversion of sulfenic acids and S-nitroso derivatives to S-glutathione mixed disulfides suggests that reversible S-glutathionylation may be a common feature of redox signal transduction and regulation of the activities of redox sensitive thiol-proteins. The glutaredoxin enzyme has served as a focal point and important tool for evolution of this regulatory mechanism, because it is a specific and efficient catalyst of protein–SSG deglutathionylation. However, mechanisms of control of intracellular Grx activity in response to various stimuli are not well understood, and delineation of specific mechanisms and enzyme(s) involved in formation of protein–SSG intermediates requires further attention. A large number of proteins have been identified as potentially regulated by reversible S-glutathionylation, but only a few studies have documented glutathionylation-dependent changes in activity of specific proteins in a physiological context. Oxidative stress is a hallmark of many diseases which may interrupt or divert normal redox signaling and perturb protein–thiol homeostasis. Examples involving changes in S-glutathionylation of specific proteins are discussed in the context of diabetes, cardiovascular and lung diseases, cancer, and neurodegenerative diseases. Antioxid. Redox Signal, 10, 1941–1988.
Proteomics of Discovery of Potential Protein–SSG Intermediates
Deglutathionylation (Reversal) of Protein–SSG: Properties of the Glutaredoxin Enzymes
-
Diabetes and Implications of Changes in S-Glutathionylation Status
-
K+ channels: Grx regulated (Fig. 3, step 2a)
-
Ca2+ channels: SERCA-SSG and Grx-reversible RyR-SSG (Fig. 3, step 3a)
Insulin receptor: Grx-reversible PTP1B-SSG (Fig. 3, step 6b)
-
Signal transduction [Fig. 3, Ras-SSG (step 7b), MEKK-SSG (step 8b), c-Jun-SSG (step 9b), Akt-SSG (step 10b), IKK-SSG (step 11b), NF-κB(p50)-SSG (steps 5a and 12b), and PKC-SSG (step 4a)]
Summary and discussion: Grx as a therapeutic target in diabetic complications
-
Cardiovascular Diseases and Alterations in Protein-S-Glutathionylation Status
-
Implications of Reversible Protein S-Glutathionylation in Cancer
-
Implications of Protein S-Glutathionylation in Neurodegenerative Diseases
I. Introduction
Sulfhydryl chemistry plays a vital role in normal cell biology and in defense of cells against oxidants, free radicals, and electrophiles. Modulation of thiol–disulfide status of critical cysteines on enzymes, receptors, transport proteins, and transcription factors is recognized as an important mechanism of signal transduction and an important consequence of oxidative stress associated with aging, cardiovascular and neurodegenerative diseases, diabetes, and cancer. Within these contexts, a prevalent form of cysteine modification is reversible formation of protein mixed disulfides (protein-SSG) with glutathione (GSH), the major nonprotein thiol compound in cells. Protein glutathionylation increases globally during overt oxidative stress [e.g., cardiac ischemia–reperfusion (79)], but selective/local generation of reactive oxygen species (ROS) mediates physiological redox signaling (1, 19, 20, 317).
To facilitate interpretation of the growing literature on redox regulation via reversible glutathionylation, we have suggested five criteria for evaluating reported studies (Table 1). Briefly, S-glutathionylation must (a) be site-specific and functionally effective, (b) occur in a physiologically relevant context, (c) occur under physiologically relevant redox conditions, (d) occur via an efficient mechanism for protein-SSG formation, and (e) exhibit an efficient mechanism of reversal (i.e., deglutathionylation). A more complete discussion of the rationale for these criteria is presented in our previous review (273). In many reports, S-glutathionylation is characterized as inhibitory, for example, phospho-fructokinase (199, 331); carbonic anhydrase III (33); nuclear factor 1 (NF1) (18); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (175, 202); protein tyrosine phosphatase 1B (PTP1B-Cys215) (19, 20); protein kinase Cα (320); nuclear factor kappa B (NFκB) (237, 242); creatine kinase (249); actin-Cys374, (59, 61, 315, 317); protein phosphatase 2A (247); protein kinase A (124); tyrosine hydroxylase (28), mitochondrial complex I (293); IκB Kinase (IKK) (251). Likewise, there are many cases where S-glutathionylation represents an activation, for example, microsomal glutathione S-transferase (57); carbonic anhydrase III phosphatase-Cys186 (33); HIV-1 protease-Cys67 (64, 65); matrix metalloproteinase (220); hRas-Cys118 (1); sarco/endoplasmic reticulum calcium ATPase (SERCA) (2); and mitochondrial complex II (39). Although this list is not comprehensive, it reflects the breadth of protein activities that can be modulated up or down by S-glutathionylation.
Table 1.
Criteria for S-Glutathionylation as a Regulatory Mechanism
| 1. S-Glutathionylation occurs at a discrete site and changes the function of the modified protein. |
| 2. S-Glutathionylation occurs at relatively high GSH/GSSG ratio, i.e. physiological conditions. |
| 3. S-Glutathionylation occurs within intact cells in response to a physiological stimulus, and elicits a physiological response. |
| 4. There is a rapid and efficient mechanism for formation of specific proteins-SSG |
| 5. There is a rapid and efficient mechanism for reversing the S-Glutathionylation reaction. |
The table lists criteria for evaluating reports of reversible S-glutathionylation of specific proteins. The criteria serve as a guide for interpreting data on protein-SSG formation as a potential mechanism of regulation of the cellular functions of the glutathionylated proteins. (See text for further explanation).
A large number of proteins have been identified as potentially regulated by reversible S-glutathionylation; however, studies of only a few effectively fulfill most of the criteria of Table 1. These examples include the protein tyrosine phosphatases (PTPs) (19, 20, 142), hRas (1), and actin (315, 317). These three well-characterized cases, however, represent a spectrum of complexity. First, the PTPs are the most straightforward. In response to an extracellular signal (e.g., growth factor) ROS is generated as the second messenger. This oxidative impulse mediates oxidation of the low-pKa active site cysteine residues of the PTPs [i.e., mixed disulfide formation (PTP-SSG)], thereby leading to inactivation of the enzymes. The process is reversed by the specific deglutathionylase enzyme glutaredoxin, which reactivates the PTPs. Second, hRas is regulated analogously, but S-glutathionylation in this case occurs at an allosteric site and leads to activation of hRas. Third, actin regulation is the most complex. Glutathionylation of the normal-pKa cysteine residue near the C-terminus of actin pre-exists at steady state (actin-Cys374-SSG). In response to growth factor stimulation, ROS is produced intracellularly, but this oxidative signal leads to deglutathionylation of actin-SSG (i.e., reduction). This paradoxical sequence of events indicates at least an oxidant-induced conformational change in the actin to expose the disulfide moiety to Glutaredoxin (Grx), but other explanations are plausible. Thus, much remains to be learned about regulation via reversible glutathionylation, especially regarding the intracellular organization of the signaling cascades (e.g., ref. 294), and the events that occur between production of the activated second messengers [ROS and/or reactive nitrogen species (RNS)], and the changes in protein–glutathionylation status that result from various physiological or pathophysiological stimuli with various types of cells.
None of the studies described above elucidated the mechanisms of formation of protein-SSG in vivo [i.e., criterion 4 (92); and see below]. In contrast, the primary mechanism of deglutathionylation has been characterized well and attributed to the enzyme glutaredoxin (41, 140). This review considers the status of knowledge of mechanisms of formation and reversal of protein–SSG in mammalian systems pertinent to human health and disease; then particular sections are devoted to assessing current understanding of perturbations of regulation by S-glutathionylation in a variety of disease contexts.
II. Potential Mechanisms of Protein–SSG Formation
Unlike the history of phosphorylation as a regulatory mechanism, where specific kinases and their substrates were characterized before phosphatases were discovered, regulation via reversible S-glutathionylation has gained momentum through characterization of the deglutathionylation mechanism (see below). Thus, S-glutathionylation is a prevalent protein modification, but the mechanisms of protein–SSG formation are not resolved. Figure 1 depicts potential mechanisms of protein–SSG formation that may occur spontaneously or be catalyzed by enzymes that are yet to be identified. These mechanisms, described briefly here, are discussed in more detail in our previous review (92).
FIG. 1.

Potential mechanisms of protein S-glutathionylation. This figure depicts various biochemical mechanisms by which protein thiol moieties could be converted to protein–SSG mixed disulide adducts. (1) via thiol-disulfide exchange; (2) via sulfenic acid intermediates; (3) via sulfenyamide intermediates; (4) via thiyl radical intermediates; (5) via thiosulfinate intermediates; (6) via S-nitrosyl intermediates. (See text for further explanation).
A. Thiol-disulfide exchange
The glutathionylation status of a protein-SH depends on the GSH/GSSG ratio (ca. 100/1 in nonstressed cells) and the specific oxidation potential for formation of the mixed disulfide (protein–SSG), termed “Kmix;” typically Kmix ∼1 (99). This means for most protein–thiols the intracellular GSH/GSSG ratio would have to decline markedly (i.e., from 100/1 to 1/1) to drive 50% conversion of protein-SH to protein–SSG (98). Hence thiol–disulfide exchange with GSSG is an unlikely mechanism of protein–SSG formation. However, there may be exceptions. For example, c-Jun, whose DNA binding activity is inhibited by S-glutathionylation, displays an unusually high thiol redox potential [Kmix ∼ 13 (156)], rendering it susceptible to S-glutathionylation by exchange with GSSG at relatively high GSH/GSSG ratios (i.e., 50% protein-SSG at GSH/GSSG ∼ 13). In general, mechanisms that involve intermediate formation of reactive thiol derivatives (described below) are more likely to mediate protein-SSG formation in cells.
B. Sulfenic acid intermediates
Protein- and glutathione-sulfenic acids are expected to form by reaction of the cysteine moieties with endogenously produced ROS and/or RNS (19, 123, 239). Exposed sulfenic acids are highly unstable (155, 239), rapidly undergoing further oxidation to sulfinic (-SO2H) or sulfonic (-SO3H) acids, or reacting with neighboring thiols (vicinal disulfide formation), or with GSH to form protein–SSG (Fig. 1, #2). Usually protein sulfenic acids are thought to be short-lived species in cells, although there are well-characterized enzymes that utilize uniquely stabilized, active-site cysteine-sulfenic acids (Cys-SOH) or selenocysteine-selenenic acids (Cys-Se-OH) as intermediates in their catalytic mechanisms (89, 239, 252). In the context of redox regulation, many proteins have been identified as candidates for modulation by sulfenic acid formation (e.g., c-Jun, Fos, bovine papillomavirus E2 protein, nuclear factor 1, NFκB-p50, GAPDH) (45, 237, 331). In most cases, however, sulfenic acid formation was either not documented directly, or shown to occur only under artificial oxidative conditions in the absence of GSH, which would react quickly with most protein-SOH to form protein–SSG. Studies on mammalian protein tyrosine phosphatases support the likelihood of rapid conversion of protein-SOH to protein–SSG by GSH (19, 20).
C. Sulfenylamide intermediates
Sulfenylamide formation is a unique post-translational modification described for protein tyrosine phosphatase 1B (PTP1B) after treatment with H2O2 or with 2-phenyl-isoxazolidine-3,5-dione, and interpreted to proceed from an initial sulfenic acid species (261, 309). The sulfenylamide moiety was reducible to PTP1B-SH by GSH, suggesting interconversion with PTP1B–SSG (261). Whether sulfenylamide formation represents a more generalizable redox signaling intermediate or a side reaction requires further study, including characterization of the kinetics of its formation and breakdown in the presence of GSH (92).
D. Thiyl radical intermediates
Production of thiyl radicals has been reported under various conditions in vitro and in vivo, including exposure to ROS and RNS, potentially representative of redox signaling conditions (139, 145, 161, 188). Thiyl radicals (RS·) are among the shortest-lived sulfhydryl derivatives (269, 321, 326), readily mediating formation of protein–SSG via radical recombination, or reaction with a thiolate followed by reaction with O2 (Fig. 1, #4). Several proteins whose functions are redox-sensitive have been shown to undergo S-glutathionylation in the presence of GS-radical generating systems in vitro, and these reactions can be catalyzed by glutaredoxin (287) (see below).
E. Thiosulfinate intermediates
The thiosulfinate derivative of GSH (GS(O)SG) has been detected by HPLC analysis in aqueous solutions containing decomposed GSNO, and in tissues treated with the O2·−/H2O2-producing system xanthine/xanthine oxidase (X/XO) (169). Like sulfenic acids, thiosulfinates are reported to be highly reactive, particularly with thiols, forming disulfide and water almost exclusively (123). This reactivity suggests that glutathionyl-thiosulfinate would be chemically and kinetically competent to glutathionylate proteins by reaction with protein cysteines (Fig. 1 #5). However, it is difficult to distinguish whether GS(O)SG would be the primary mediator of protein glutathionylation, or whether a different activated species generated under conditions of the X/XO or decomposed GSNO experiments (e.g., GS· or GSSG) would play a more prominent role. It is also feasible that protein thiosulfinates could react with GSH to form protein–SSG and GSOH (Fig. 1 #5), however definite evidence to support this concept has not been reported as yet (92).
F. S-Nitrosylated intermediates
Cysteine sulfhydryls on proteins and GSH undergo nitrosylation in vivo under a variety of normal and pathological conditions (32, 82, 121, 139, 187), forming protein-SNO, and GSNO, respectively. GSNO is the major nitrosothiol in cells, detected in micromolar concentrations in nonstressed tissues, with increased amounts in certain disease states (97), whereas protein-SNO is more prevalent in extracellular spaces (197, 286). Both GSNO and protein-SNO are relatively stable sulfhydryl derivatives, with half lives on the order of hours in aqueous solution (13, 229, 278). Nevertheless, biochemical studies support the potential role of GSNO to promote protein S-glutathionylation. Thus, GSNO reacts rapidly with a variety of isolated proteins (papain, creatine phosphokinase, GAPDH), forming protein-SSG within minutes of treatment (101, 202, 324); and many other proteins have been reported to be glutathionylated by treatment of the isolated proteins or cultured cells with GSNO, but reaction rates were not determined systematically (37, 134, 157, 169, 202). Alternatively, reaction of GSNO with protein-SH may result in transnitrosation (i.e., protein-SNO) with the propensity to glutathionylation vs. nitrosylation by GSNO likely being influenced by the microenvironment of the modified Cys residues (101). On the other hand, the possibility that certain protein-SNO intermediates might serve as precursors to protein–SSG via reaction with GSH has not been as thoroughly studied as the converse (Fig. 1, #6) (92).
III. Potential Catalysis of Protein Glutathionylation
A. GSTπ
It is conceivable that a glutathione-S-transferase enzyme could catalyze conjugation of GSH to an activated cysteine residue, made electrophilic by oxidation (e.g., Cys-SOH). Indeed evidence for such a mechanism has been described for GSTπ interacting with 1-cysteine peroxiredoxin sulfenic acid (1CysPRx-SOH) (186). However, it is not known whether this type of catalysis is peculiar to this one example, or a more general phenomenon (92, 295).
B. Grx
Oxidized derivatives of GSH increase during oxidative stress, including GS·, GSNO, and GSSG (155, 296), and they are proposed to contribute to formation of protein–SSG, as described above. Based on the low pKa of the active site-Cys22 of Grx1 (199), and the increased stability of disulfide-anion radicals compared to thiyl radicals (269, 321), we anticipated Grx1 might catalyze protein glutathionylation via stabilization of the GS· thiyl radical as an enzyme disulfide anion radical intermediate (Grx1-SSG·−), facilitating GS-radical recombination with a protein thiyl radical (287). Indeed, Grx1 promoted glutathionylation of isolated GAPDH, PTP1B, and actin in the presence of a GS· radical generating system, but protein–SSG formation was competitively inhibited by reaction of O2 with the Grx1-SSG·− intermediate. These observations suggest that the mode of catalysis by Grx1 could depend upon the redox environment of the cell; for example, transiently acting as a glutathionylating enzyme under an oxidative stimulus, and as a deglutathionylase when the oxidative signal or stress subsides. In a recent study of hypoxia/N-acetyl cysteine-induced apoptosis of pancreatic cancer cells, the data implicated Grx1 as the mediator of NFκB inactivation via S-glutathionylation of p65 (242).
C. Flavoprotein sulfhydryl oxidase (QSOX)
Sulfhydryl oxidase enzymes, ubiquitous in multicellular organisms but absent in prokaryotes and yeast, utilize metals (metalloenzyme family) or flavins (flavoenzyme family) to catalyze disulfide bond formation from diverse thiol substrates with concomitant reduction of O2 to H2O2 (297). Based on the ability of the QSOX enzyme to utilize both low molecular weight and protein thiols as substrates (52, 223), we anticipated QSOX might catalyze protein-SSG formation. Initial studies showed QSOX- and GSH-dependent inhibition of PTP1B in a time-dependent manner, suggesting glutathionylation of the active site Cys215 of PTP1B (92).
D. Other potential mechanisms of catalysis/control of protein S-glutathionylation
By analogy to their typical catalytic reactions, other enzymes may be implicated in catalysis of S-glutathionylation. For example, a glutathione peroxidase-like mechanism could apply if protein-SH is substituted for one of the two GSH molecules in its typical reaction. Similarly, glutathionylation could proceed via a monooxygenase-like mechanism (utilizing either a heme-enzyme or a flavo-enzyme). To the best of our knowledge, glutathionylation activity of well-known peroxidases and monooxygenases has not been reported. In general, it remains uncertain whether protein glutathionylation may be an important function of known or as yet undiscovered enzymes. Alternatively, the specificity of S-glutathionylation reactions pertinent to redox signal transduction may be governed instead by the organization of receptors and ROS/RNS-producing enzymes with their specific protein substrates on scaffolds that provide localized control of the concentrations of the activated species and spatial orientation of the signaling intermediates.
IV. Proteomics of Discovery of Potential Protein–SSG Intermediates
Efficient methodology continues to emerge that can detect proteins with oxidant–sensitive cysteine residues, the oxidative insult being produced in various ways, for example, diamide, menadione, hydrogen peroxide, ischemia/reperfusion, and TNF-α. However, many of the current approaches do not distinguish protein–SSG formation from other possible types of oxidized cysteine modifications, and there are shortcomings that preclude definitive and quantitative assessment of the role of the identified proteins in actual signal transduction situations. Further studies are needed to evolve this methodology into an efficient and accurate detection for S-glutathionylated proteins within cell signaling pathways both in the absence and presence of an oxidative signal (reviewed in refs. 60, 272, and 273).
V. Deglutathionylation (Reversal) of Protein–SSG: Properties of the Glutaredoxin Enzymes
The net reaction catalyzed by glutaredoxin is appropriately depicted as a thiol–disulfide exchange reaction involving nucleophilic displacement reactions rather than single electron transfer reactions that would involve radical intermediates. Accordingly, the original name “transhydrogenase” which was applied to the enzyme activity from rat liver (244) was replaced by the name “thioltransferase,” because it more accurately represents the nature of the reaction that is catalyzed (16). In another context, Holmgren discovered an enzyme that catalyzed GSH-dependent turnover of oxidized ribonucleotide reductase in a mutant of Escherichia coli that lacked thioredoxin, and he named it “glutaredoxin” (122). Subsequent to those earlier studies, “thioltransferase” and “glutaredoxin” enzymes from a variety of organisms and mammalian tissues were isolated and characterized, and a high degree of structure–function congruence has led to the widely accepted supposition that “thioltransferase” and “glutaredoxin” simply represent alternative names for the same family of enzymes. Although “thioltransferase” better depicts the reaction catalyzed by these enzymes, the name “glutaredoxin” has been adopted as the most commonly used name internationally. Recently, other enzymes (e.g., sulfiredoxin and PDI) (87a, 233a) have been reported to exhibit deglutathionylating activity; however, it is uncertain whether they contribute significantly to intracellular protein de–glutathionylation (reviewed in ref. 92).
There are two forms of Grx that have been characterized in mammals, Grx1 and Grx2. Five forms of Grx (Grx1-5) have been identified in E. coli and yeast, and the gene for a mammalian form of Grx5 has been reported. However, it is not clear whether the mammalian Grx5 exhibits deglutathionylase activity [reviewed in (92)]. Grx1 is the better characterized isoform in mammalian systems. It has been reported to catalyze deglutathionylation of diverse protein substrates in vitro and in situ, (e.g., hemoglobin, HIV-1-protease, nuclear factor-1, PTP1B, actin, Ras, IκB kinase, procaspase 3, and IRF-3) (1, 18–20, 65, 199, 227, 241, 251, 317), and it is far more efficient than other thiol–disulfide oxidoreductase enzymes at catalyzing protein deglutathionylation in vitro (41, 296); for example, Grx1 displays a 5,000-fold greater kcat/KM for Cys-SSG as substrate vs. thioredoxin (41). The deglutathionylating activity of Grx has been implicated in regulation of many vital functions, including actin polymerization, vasodilation, cellular hypertrophy, transcription factor activation, and propagation of apoptosis (1, 2, 227, 251, 315, 317).
Primarily localized to the cytosol, Grx1 recently was documented to exist also in the intermembrane space of mitochondria (226); however, the specific functions of Grx1 in the mitochondria have yet to be elucidated. Localization of Grx1 in the nucleus of particular types of cells has been reported (179, 232, 258, 259, 288). However, neither confocal microscopy of immunostained cells nor isolation of Grx1 from purified nuclei has been performed to document Grx1 in the nucleus. These studies will need to be extended to include analysis of endogenous Grx1 to determine if Grx1 exists normally in the nucleus, constitutively or via translocation in response to stimuli.
Grx2 displays only ∼30% sequence homology to Grx1. To date, two human clones (Grx2a and Grx2b) have been discovered with distinct N-terminal sequences, and a third splice variant (Grx2c) has been described (102, 172, 176, 178). Grx2a contains a mitochondrial localization sequence which is cleaved upon entry into the mitochondria. Confirmation of mitochondrial localization was performed with a GFP fusion protein (102, 178), and via analysis of isolated mitochondria in which matrix localization of Grx2 was documented (226). Within mitochondria, Grx2a is reported to exist in part as an inactive dimer associated with a 2Fe–2S cluster (24, 135, 171, 178) involving coordination by four Cys residues, two from the active sites of each Grx2 enzyme, and two from coordinated GSH molecules.
Both mammalian Grx isoforms have been implicated in regulation of mitochondrial complex I via reversible glutathionylation. In the case of Grx1, knockdown of the enzyme in mice was accompanied by a decrease in complex I activity, suggesting Grx1 plays a role in maintenance of complex I activity (72, 150). The situation for Grx2 is less straightforward, because the enzyme was reported to catalyze both deglutathionylation and glutathionylation of complex I under different conditions. This is consistent with the Grx catalytic mechanism, which is bidirectional depending on the relative concentrations of protein–SSG, protein-SH, GSH, and GSSG (Fig. 2). However, the deglutathionylase activity of Grx2 in situ may be limited by the amount of enzyme that exists as active monomer rather than inactive dimer (178). Whether Grx2-mediated glutathionylation of complex I by GSSG is physiologically meaningful is also called into question, because the high concentrations of GSSG that were used may not be representative of the mitochondrial milieu (23).
FIG. 2.

Glutaredoxin catalytic mechanism. This figure depicts glutaredoxin-catalyzed deglutathionylation of protein-SSG mixed disulfides. The central portion shows Grx catalysis proceeding via a monothiol mechanism involving a selective double displacement reaction. The glutathionylated sulfur moiety of the protein–SSG is attacked by the thiolate anion of the enzyme (Grx-S−), forming the covalent enzyme intermediate (GRx-SSG) and releasing the reduced protein-SH as the first product. The second rate-determining step involves reduction of the Grx-SSG by GSH to produce glutathione disulfide (GSSG) as the second product, recycling the reduced enzyme (Grx-S−). The left side depicts a dithiol catalytic mechanism which has also been proposed; however the monothiol mechanism is more prevalent and favored by a preponderance of evidence (see text). The right side depicts the side reaction involving formation of an intramolecular disulfide at the active site of the enzyme (C22-SS-C25) which detracts from catalysis (see text).
To explore the potential for Grx2 in the nucleus, the Grx2b variant was tagged with GFP and thereby found localized to the perinuclear region (102); a nuclear localization sequence has been proposed, but not confirmed (178). More recently, splice variants Grx2b and Grx2c were reported to be expressed only in testes among normal tissues, but they were also expressed in several cancer cell lines (176). Localization of Grx2b,c or translocation of Grx1 to the nucleus would have multiple functional implications, especially regarding regulation of transcription factors. However, localization and functional studies have yet to be performed on isolated nuclei to confirm localization and functional activity within the nucleus.
VI. Glutaredoxin Mechanism of Action
Grx functions with GSH as the co-substrate to reduce protein–SSG mixed disulfides as shown in Fig. 2. Grx catalysis proceeds via a monothiol mechanism (central portion of Fig. 2) through a selective double displacement reaction in which the nucleophilic attack is performed by the N-terminal active site Cys of the CPYC motif, which exists as a thiolate anion due to its unusually low pKa of 3.5 (199, 200, 284). The glutathionylated sulfur moiety of the protein-SSG is attacked by the thiolate anion of the enzyme (Grx-S−), forming the covalent enzyme intermediate (Grx-SSG) and releasing the reduced protein-SH as the first product. The second rate-determining step involves reduction of the Grx-SSG by GSH to produce glutathione disulfide (GSSG) as the second product, and recycle the reduced enzyme (Grx-S−) (108, 131, 284, 327). GSSG is subsequently reduced to GSH by GSSG reductase (GRase) and NADPH. Also, in systematic kinetics studies Grx was shown to be specific for glutathione-containing disulfides as the first substrate (41, 108, 198, 327), and GSH is the preferred second substrate (284) for the two-step reaction. The reaction mechanism (Fig. 2, center) is documented by so-called ping-pong kinetics, giving a characteristic parallel line pattern for the 1/V vs. 1/S plots at several fixed concentrations of the co-substrate. This kinetic behavior has been documented for both isozymes of mammalian glutaredoxin (Grx1 and Grx2) (93, 108). If a nonglutathionylated precursor is tested as the first substrate, then the two-substrate kinetics pattern changes to an ordered mechanism with a double reciprocal plot displaying converging lines at the same point on the x-axis (i.e., identical apparent KM values), reflecting the requirement of the initial reaction of the precursor with GSH to form the actual glutathionylated substrate for the enzyme [(108, 327) details reviewed in (198)].
Both a monothiol mechanism (i.e., requiring only one active site Cys) and a dithiol mechanism (i.e., requiring both active site Cys; see Fig. 2, left side) have been proposed for Grx activity; however the monothiol mechanism is prevalent. Mutagenesis studies that replace the second Cys at the active site (distal from the C-terminus) have supported the monothiol mechanism. When this Cys is replaced (e.g., C25S mutation of human Grx1), the mutant enzyme retains its normal catalytic function, and in fact becomes a better catalyst than the natural enzyme (327). This observation documents that the side reaction involving formation of the intramolecular disulfide form of the enzyme (C22-SS-C25) detracts from catalysis (Fig. 2, right side).
Analogous to Grx1, Grx2 exhibits deglutathionylating activity for peptide and protein substrates, but its activity is ∼10-fold lower than that of Grx1 (93, 136, 178). Mutating the active site of Grx2 (CSYC) to mimic that of Grx1 (CPYC) partially enhances the Grx2 activity but still remains less active than Grx1 (136), indicating that other features of the two proteins contribute the distinction in activity. Remarkably, Grx2 was reported to be as an enzyme with “high affinity” for the glutathione moiety, but this interpretation is problematic, because it was based on limited kinetic analysis (136). Instead, comparison of Grx1 and Grx2 with the prototype substrate Cys-SSG, which represents the common feature of all protein–SSG substrates and avoids steric constraints, shows little difference in apparent KM for Cys-SSG for the two isoforms under the same assay conditions with a fixed concentration of GSH. Furthermore, two-substrate kinetic analysis of Grx2 indicates a ping-pong mechanism analogous to Grx1, whereby rapid covalent reactions (i.e., high commitment to catalysis) supersede reversible binding of the protein–SSG or GSH substrates (93). Hence, the typical interpretation of “substrate affinity” does not apply to the Grx enzymes.
Overexpression studies have suggested a role for Grx2 in protection of cells and preservation of mitochondrial integrity after treatment with exogenous oxidants (85, 87, 171); however, it is not clear whether these effects are due to the deglutathionylation activity of Grx2 (92). The role of Grx2 activity within the cell may be limited due to its sequestration as dimers with a bridging Fe–S cluster masking the active site (24, 135, 171, 178). It has recently been reported that S-nitroglutathione treatment causes the release of Grx2 from these Fe–S clusters in vitro allowing for activity to be reestablished (114). Hence, oxidative stimuli may lead to release of Grx2 from Fe–S clusters as a protective response to oxidative stress, thereby providing for thiol–disulfide homeostasis within the mitochondria.
VII. Modulation of Grx Expression
Various natural and synthetic compounds, including oxidatively labile diphenols and organic hydroperoxides, are known to protect cells against chemical and radiation-induced carcinogenesis by elevating phase II detoxification enzymes [e.g., γ-glutamylcysteine synthetase, glutathione-S-transferase, and glutathione peroxidase (31, 240)]. The inducers are believed to elicit cellular signals that activate gene transcription via the antioxidant response element (ARE) or electrophile response element (EpRE). In the mouse, the critical DNA regions that respond to antioxidant inducers are comparable to activator protein 1 (AP-1) sites and thus linked to the c-fos and c-Jun transduction pathway (91). Radical scavengers like tert-butylated hydroxyanisole (BHA) are among the agents characterized as inducers of the antioxidant response, and BHA has been reported to induce glutathione-S-transferase and glutaredoxin in mice (71). In the mammalian system, the human glutaredoxin (glrx) gene has been cloned and reported to have an AP-1 site in its promoter region, implying regulation by oxidants as well as other factors that induce at AP-1 sites, including epidermal growth factor, TGFβ, cyclic AMP, and retinoic acid (228). Thus, in some situations, induction of glutaredoxin may be part of a pleiotropic response to stimulation of the antioxidant response element. Grx levels were reported to be elevated in cells resistant to the anti-cancer agent adriamycin that is a generator of oxyradicals (323). In addition, Grx content is elevated in rat brain in response to oxidative stress injury (149), and H2O2 was reported to stimulate expression of Grx in a time- and dose-dependent manner in cultured human coronary artery smooth muscle cells (221). Grx content has been reported to increase in various other oxidative stress contexts in mammalian cells (83, 160). It was recently proposed that 17β-estradiol protects H9c2 cardiomyocytes from oxidant induced cell death via transcriptional upregulation of both Grx and GSH (307). Evidence implicating estradiol in the regulation of Grx1 was provided previously by the observation that pretreatment of bovine aortic endothelial cells with 17β estradiol results in an increased Grx protein content as well as resistance to oxidative stress (83). A potential mechanism by which estrogen may regulate Grx1 expression is via selective binding to an EpRE-1 site contained within the glrx gene promoter (307). UVB radiation is another oxidative stimulant to cause upregulation of transcription of Grx in rat keritinocytes potentially through activation of the AP-1 site (257). Understanding of transcriptional regulation of Grx is still rudimentary, leaving much to be discovered. Also, there is evidence that in some situations glutaredoxin activity may be enhanced in response to an oxidative stimulus without an increase in Grx protein expression (227), however the mechanism of such Grx activation remains to be discovered.
The remainder of this review is focused on perturbations in glutaredoxin content and activity and/or alterations in protein-SSG status in various disease conditions.
VIII. Diabetes and Implications of Changes in S-Glutathionylation Status
A. Mechanism of hyperglycemic damage and ROS
Diabetes arises from chronic elevations in blood glucose, and has been characterized as a multifaceted disease of oxidative stress that can lead to complications through multiple mechanisms. Four primary pathways that mediate glucose damage are (a) increased advanced glycation end products (AGEs); (b) polyol pathway activation; (c) protein kinase C (PKC) activation; and (d) hexosamine pathway activation (30). All four pathways coincide with increased superoxide from the mitochondrial electron-transport chain, and the diabetes-induced activation of three of these pathways (a–c) can be blocked by inhibition of the overproduction of mitochondrial superoxide (30, 77). ROS from the mitochondria are a primary source of oxidative stress/signaling in diabetes, though NADPH oxidases also contribute (54). The significance of ROS involvement is reflected by entire reviews dedicated to ROS in specific tissues. For example, Coughlan et al. (54) devote eleven pages to the role of RAGE (receptor for AGE) and ROS in the microvasculature of the diabetic kidney.
1. Insulin–glucose dynamics and diabetic complications
Glucose metabolism leads to insulin secretion from pancreatic β-cells via exocytosis, and Fig. 3 depicts the well-characterized insulin exocytosis “triggering pathway” modeled primarily from two other reviews on insulin release (15, 117). Insulin is essential in facilitating cellular glucose uptake, and glucose is the major energy source of most cells. However, excess glucose metabolism can have deleterious effects within the cell. For example, hyperglycermia has been associated with poly (ADP-ribose) polymerase (PARP)-mediated inhibition of GAPDH and overproduction of superoxide by mitochondria(78). Additionally, chronic exposure to high glucose can lead to insulin resistance (i.e., cells no longer take up glucose in response to insulin). When insulin levels are insufficient, glucose is not taken up by the cells and starvation ensues. Devastating complications of diabetes include cardiovascular disease, stroke, retinopathy, nephropathy, neuropathy, depression, and other problems associated with poor circulation.
FIG. 3.
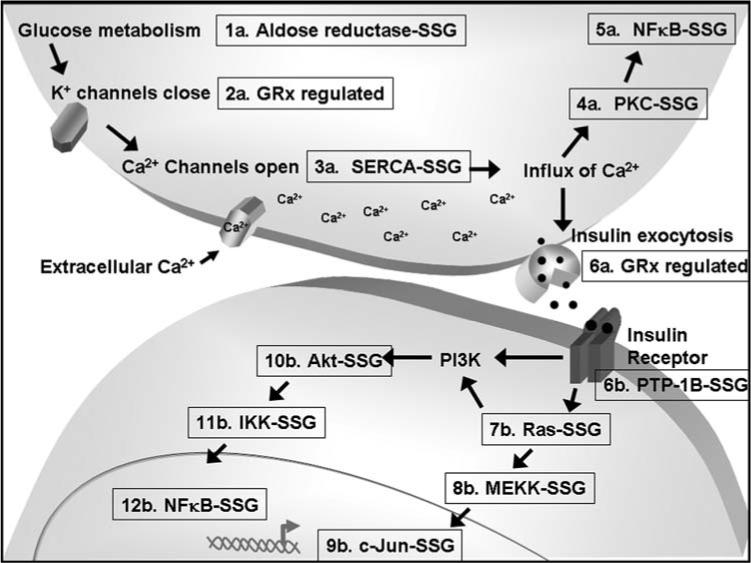
S-glutathionylation and GRx-regulation of proteins involved in insulin secretion (1a–6a) and insulin signaling (6b–12b). S-glutathionylation has been implicated in regulation of aldose reductase (1a), SERCA calcium channels (3a), PKC (4a), NF-κB (5a and 12b), PTP-1B (6b), Ras (7b), MEKK (8b), c-Jun (9b), Akt (10b), and IKK (11b). Grx has been reported to be involved in potassium channel gating (2a) and insulin secretion (6a).
As described above, S-glutathionylation of proteins is the primary mechanism of thiol redox signaling, and therefore likely has significant impact on the pathogenesis of diabetes. Few studies have analytically evaluated glutathionlyation for the criteria of a regulatory mechanism (Table 1), and even fewer of these studies have been conducted in the context of diabetes. We evaluate here key events in insulin secretion (Fig. 3, #1a–#6a) and classical signaling transduction pathways (Fig. 3, #6b–#12b) that have been implicated for regulation by glutathionylated proteins or Grx.
B. Glucose metabolism: aldose reductase-SSG (Fig. 3, step 1a)
During normal metabolism, glucose is phosphorylated by hexokinase and enters into the glycolysis pathway. Elevated glucose concentrations saturate hexokinase and trigger a second metabolic pathway called the polyol (sorbitol) pathway. The first step in this pathway depends on aldose reductase to convert glucose to sorbitol, and serves as a ‘backup’ system since hexokinase has a lower KM for glucose than does aldose reductase (AR) (154, 298). Mechanisms of sorbitol-related complications include sorbitol accumulation leading to osmotic swelling and cataract formation in diabetes (225, 285), and nonosmotic decreases in vascular and neuronal Na+K+ ATPase activity (154). The polyol pathway has been long known for generating ROS and most notably, antioxidants have been found to prevent cataract formation in rats in the presence of elevated polyol levels (285).
Inhibition of AR in diabetes has been a popular therapeutic goal for many years. Cys298 is in the active site of AR, and thiol modifications to this residue regulate substrate binding (285). Specifically, S-glutathionylation of AR at Cys298 inhibits its activity in the presence of normal glucose concentrations (285), suggesting basal glutathionylation analogous to that observed with actin (315, 317). Inhibitors of AR thus far have proven more promiscuous than effective (285), but since AR is a regulatory target for Grx, inhibition of Grx may provide additional means for therapeutic intervention in diabetes. Grx has been reported to be increased in the diabetic heart and retina of rats (170, 274), and decreased in platelets of diabetic patients (70). AR is glutathionylated and inhibited under basal conditions, suggesting limited Grx activity within the cellular microdomain of AR, possibly due to a high KM for AR-SSG or sequestration of the glutathionyl moiety on AR-SSG. Whatever the mechanism, AR is more active in diabetes, consistent with a decrease in glutathionylation of AR (↓AR-SSG, ↑AR-SH) corresponding to an increase in Grx activity.
C. K+ channels: Grx regulated (Fig. 3, step 2a)
Insulin secretion can occur via KATP channel-independent and -dependent processes, but the mechanism has been established only for the latter (117). Regulation of many potassium channels has widespread significance throughout physiology, but this discussion will be limited to two specific types that current information indicates are likely candidates for regulation by S-glutathionylation and/or Grx in diabetes; namely, (a) the ATP-sensitive channels containing inward rectifier potassium ion channels that signal insulin secretion in the pancreatic β-cell, and (b) the voltage-gated potassium channels (Kto) that regulate the transient outward potassium current (IKto) in the initial depolarization phase of an action potential in cardiac cells.
1. ATP-sensitive potassium channels
The ATP-sensitive potassium channels (KATP) mediate glucose-stimulated insulin secretion, and gene mutations lead to diabetic complications (15). Increased glucose metabolism leads to the closing of KATP channels via increased ATP concentrations (Fig. 3) (15, 117). The subsequent decrease in potassium efflux causes a depolarization in the membrane potential of pancreatic β-cells and triggers an influx of calcium ions via the voltage-gated calcium ion channels (15, 117). Elevated intracellular calcium is essential for initiating insulin release (Fig. 3, step 4a) (15, 117).
KATP are comprised of four regulatory sulphonylurea receptors (SUR) of the ATP-binding cassette (ABC) class of transporter proteins and four Kir6.2 inward rectifier potassium channel subunits (15, 117). SUR1 subunits are found in pancreatic β cells and SUR2 (SUR2A) is specific for cardiac muscle cells, unlike Kir6.2 which is found in both types of cells (305). Thiol-modifying agents inhibit KATP channel activity at critical cysteine residues. KATP channels from ventricular myocytes of guinea pigs exhibited DTT-reversible p-chloromercuriphenylsulphonate (pCMPS) inhibition, spontaneously reversible 5,5′dithio-bis-(2-nitrobenzioc acid) (DTNB) inhibition, DTT reversible thimerosal inhibition of rundown channels, and decreased ATP sensitivity to thimerosal (50). Glutathione disulfide (GSSG, 3 mM) did not inhibit channel activity. The authors speculated that insufficient exposure times due to time limitations of channel rundown may have prevented channel inhibition by GSSG; however, this does not preclude inhibition by glutathionylation via another mechanism (see Fig. 1, above). KATP channel activity was also shown to be inhibited by N-ethylmaleimide (NEM), DTNB, o-iodobenzoate, chloramine-T, and hydrogen peroxide in either toe muscle fibers of mice or CRI-G1 insulin-secreting cells (163, 322). pCMPS inhibition occurs specifically via Cys42 of Kir6.2, and activity is restored by DTT (304). Likewise, NEM inhibits ATP binding to SUR1 in the nucleotide binding fold (NBF) via Cys717 (191). Taken together, these reports suggest that the activity of KATP channels can be modulated by thiol and redox sensitive cysteines, suggestive of potential regulation by S-glutathionylation. Sulfonylurea drugs increase insulin release by closing KATP channels (15), and similarly, if glutathionylation leads to inhibition and downstream insulin release, inhibition of Grx would have therapeutic benefits for diabetic patients.
2. Voltage-gated potassium channels
In cardiac cells, in response to action potentials special potassium channels open, accounting for the initial phase of depolarization. A decrease in calcium-independent transient outward K+ current contributes to the extended action potential duration observed in hearts of diabetic rats (170). Decreased IKto in ventricular myocytes isolated from 3–5 week diabetic rats was correlated to a 1.7-fold increase in thioredoxin, a 2.5-fold increase in glutaredoxin, and a decrease in their corresponding reductase enzymes to similar extents (170). Grx activity was reported by the nonspecific hydroxyethyl disulfide reduction assay (HEDS), which reflects total cellular reducing capacity (i.e., the combined reduction of HEDS disulfide by Trx and HEDS-SSG mixed disulfide reduction by Grx) (25). Nevertheless, the induction of Grx activity in the diabetic rat ventricle is apparent since Trx was independently assayed via the insulin disulfide reduction assay and the increase in Trx alone does not account for the increase in HEDS reduction. This study proposes a protective role of Trx and Grx systems in regulation of cardiac potassium ion channels. Recent advances in Grx and Trx using genetic modeling provide critical tools for elucidating molecular mechanisms and subsequent functional roles of the thiol disulfide oxididoreductases (TDORs) in the heart and in the context of diabetes (120, 183, 190). These studies have already suggested a protective role for Grx against ischemia/reperfusion-induced cardiovascular damage in mice, although further characterization of Grx activity and protein change-of-function in these models is needed (120, 183) [see further discussion under the Cardiovascular Disease section, below]. Unlike the beneficial effects of potassium channel inhibition on insulin release in β-cells (see above), inhibition of these cardiac channels have undesirable consequences. The association of increased Grx with channel inhibition promotes Grx as a therapeutic target in diabetes. Further studies are needed to determine whether channel deglutathionylation is the mechanism of action of Grx in these systems.
D. Ca+2 channels: SERCA-SSG and Grx-reversible RyR-SSG (Fig. 3, step 3a)
Elevated extracellular calcium (Ca2+) is a critical step for insulin secretion from pancreatic β-cells (Fig. 3) (15, 117). Calcium levels are largely regulated by cellular transport systems in the endoplasmic reticulum (ER) (Ca2+ uptake system and Ca2+ release channels), mitochondria, and plasma membrane (Ca2+ entry channels and Ca2+ extrusion system) (40). Within the context of glutathionylation and diabetes, we focus here on the ER calcium release channel, ryanodine receptor (RyR), and the ER uptake system, sarco/endoplasmic reticulum calcium ATPase (SERCA) pump.
1. RyR-SSG
Ryanodine receptors release Ca2+ from the endoplasmic reticulum into the cytosol. All three known isoforms (RyR1, RyR2, and RyR3) have been found in pancreatic β-cells (76), whereas RyR2 is the predominant isoform in the heart (212). RyR-regulated Ca2+ release is a critical step in β-cell survival via a presenilin-HIF pathway (76), and regulates glucose-independent β-cell insulin secretion (137). Whether protein expression of RyR is decreased in diabetes is contentious (26, 328), but a decrease in function of the RyR in hearts of diabetic rats is generally accepted.
RyR1 has 100 cysteine residues, and glutathionylation of some of these has been related to magnesium-inhibitable calcium release from sarcoplasmic reticulum vesicles (SRV) from rabbit muscle (10). Ten cysteines were documented for glutathionylation in tryptic peptides from GSNO-treated SRV, and twelve glutathionylated cysteines were found in response to H202 plus GSH (11). Five specific cysteines could undergo glutathionylation or nitrosylation, three could be oxidized to disulfides or glutathionylated cysteines, and two were glutathionylation specific. However, the only cysteine (Cys3635) thus far known to be involved in RyR-regulated calcium release is also the only cysteine that can be glutathionylated, nitrosylated, or disulfide-linked, but it is not critical for H202 channel activation (11). The same study reports Grx-dependent reduction with subsequent alkylation with fluorescently labeled C2-maleimide. These data were interpreted to suggest that Grx could reduce both protein–SSG and protein–SNO; however, a more likely explanation would be conversion of protein–SNO to protein–SSG before Grx action. Regulation of RyR via glutathionylation will continue to be an important subject of study with many pathological implications, including diabetes (26, 76, 137, 328), nondiabetic cardiovascular complications (see Cardiovascular Section), and Alzheimer's disease (AD) (76, 148).
2. SERCA-SSG
In direct contrast to the function of the RyRs, SERCA pumps actively transport cytosolic Ca2+ into the sarcoplasmic reticulum, quenching cytoplasmic Ca2+ signals and regulating calcium oscillations in response to glucose (14). Each of the three isoforms of SERCA (SERCA1, SERCA2, and SERCA3) has highly spliced tissue-dependent variants (86). Nearly all forms of SERCA are found in the muscle, and SERCA2a has the most profound impact on cardiac calcium regulation (86). SERCA2b is predominant in smooth muscle cells, and SERCA2b and SERCA3 have implications in insulin secretion in pancreatic β-cells (14, 22).
Nitric oxide (NO)-activated calcium uptake into the SER is reported to be mediated by increases in SERCA2 activation via SERCA2-SSG at Cys674 (2). Additional cysteines were glutathionylated basally in pig carotid arteries (Cys498-SSG), in response to bradykinin in pig carotid arteries (Cys268-SSG, Cys528-SSG, Cys560-SSG, Cys669-SSG, Cys674-SSG), and after stimulation with NO in rabbit aorta (Cys498-SSG, Cys524-SSG, Cys613-SSG, Cys674-SSG) but a single serine mutation at Cys674 accounted for changes in SERCA activity (2). Observations like these reinforce the need to distinguish S-glutathionylation events that are responsible for change in protein function and physiological outcome from inconsequential protein–SSG events (re: Table 1).
Production of nitric oxide with either IL-1β or S-nitro-N-acetylpenicillamine (SNAP) was recently shown to induce activation of SERCA2b via Cys674-SSG in vascular smooth muscle cells (VSMC), and this induction inhibited the elevated cytosolic Ca2+ and VSMC migration under normal glucose conditions (299). High glucose prevented NO-induced inhibition of VSMC migration, and the inhibition could be circumvented by overexpression of wild-type SERCA, over-expression of SOD, or treatment with the SOD mimetic, Tempol (299). A serine mutant at Cys674 of SERCA did not allow for VSMC migration under either normal or high glucose conditions (299). Furthermore, high glucose decreased the amount of label associated with SERCA-Cys674-IAM-biotin and NO-induced SERCA-Cys674-SSG-biotin, and also led to sulfonic acid formation in VSMC (299). These results suggest a scenario involving increased Grx deglutathionylation activity producing more reduced SERCA-Cys674-SH that is then susceptible to irreversible oxidation to the sulfonic acid. Such a scenario is consistent with reports that high glucose and streptozotocin-induced diabetes lead to upregulation of Grx content and activity in the heart and the retina (170, 274).
In an earlier study, peroxynitrite (0.4 mM) and GSH (5 mM) induced glutathionylation of SERCA1 from rabbit skeletal muscle at Cys344, Cys349, Cys364, Cys498, Cys525, and Cys614 (311). The difference in glutathionylated residues reported by Viner et al. (311) and Adachi et al. (2) could be due to the specific stimuli and conditions of the experiments or to intrinsic characteristics of the isoforms. The flanking residues of Cys674 of SERCA1 are “RRAC674C675FARVEP” and contain an additional Cys675 residue distinct from the amino acid sequence surrounding Cys674 of SERCA2 “NARC674FARVEP”. It remains to be discovered whether Cys675 of SERCA3 (RTARC675FARVEP) is regulated by glutathionylation. Since SERCA3 has implications in diabetes and SERCA3-deficiency leads to increased islet cell response (14), inhibition of SERCA by deglutathionylation due to increased Grx may serve as a focus for intervention in diabetic complications. However, comparative analysis of the catalytic efficiency of Grx for each of the SERCA isoforms would be necessary for assessing the utility of Grx as a therapeutic target, since both decreased SERCA3 in islet cells and increased SERCA2b in VSMCs appear to be beneficial in diabetic complications.
E. Insulin exocytosis: Grx regulated (Fig. 3, step 6a)
Glucose-induced insulin exocytosis occurs via a calcium-dependent mechanism (15, 117). Glucose metabolism generates NADPH, which elicits a similar extent of membrane depolarization as that associated with calcium-mediated exocytosis in mouse β-cells, suggesting that NAPDH mediates glucose-induced exocytosis of insulin (129). The authors rationalized that, since the Grx and Trx systems accept electrons from NAPDH, they would be mediators in NAPDH-induced capacitance. However, Trx inhibited NADPH-induced capacitance whereas Grx and GSH potentiated it. Whether the effect of Grx involves regulation via reversible S-glutathionylation of specific proteins remains to be discovered, and the basis for the different effect of Trx is unknown.
F. Insulin receptor: Grx-reversible PTP1B-SSG (Fig. 3, step 6b)
Phosphorylation of the insulin receptor is at the core of insulin signaling, and relies on protein tyrosine phosphatases (PTPs) for deactivation once insulin is no longer present (105). Specifically, PTP-1B is the predominant form that inhibits insulin signaling (105). Genetic manipulations and inhibition of PTP-1B increase insulin signaling (105, 300), and PTP-1B has become a prime candidate for therapeutic intervention in diabetes and obesity (104, 231, 300, 333).
Goldstein et al. (105) review in detail the roles of ROS in potentiating insulin signaling, paradoxical to the deleterious effects of oxidative stress well known in the complications of diabetes. Primarily, ROS have been shown to mediate insulin signaling and inhibit PTP1B (105). Isolated PTP-1B can be inactivated by glutathionylation in a Grx-reversible fashion, and it is converted to PTP-1B-SSG in situ when A431 cells respond to EGF, generating ROS intracellularly (19, 20). Since PTP-1B inhibition is beneficial in diabetes, inhibition of Grx would provide an alternative therapeutic approach, leading to increased glutathionylation of PTP-1B and its concomitant inactivation.
G. Signal transduction [Fig. 3, Ras-SSG (step 7b), MEKK-SSG (step 8b), c-Jun-SSG (step 9b), Akt-SSG (step 10b), IKK-SSG (step 11b), NF-κB(p50)-SSG (steps 5a and 12b), and PKC-SSG (step 4a)]
We review here only a subset of signaling mediators that are linked to diabetes and have been implicated for regulation by glutathionylation. These signaling proteins are grouped here because they are inter-related pathways, they reflect signal transduction downstream of the insulin receptor, and most have been reviewed previously in another context (273).
1. Ras–SSG
The Ras superfamily of small GTPases activate signaling in the GTP-bound state but are not transducive in the GDP-bound state, and the role of Ras in diabetes is complex and cell-type specific. For example, insulin signaling activates Ras and PI3K, and Ras activates PIK3 (Fig. 3), but Ras is not essential in insulin activation of PI3K in adipocytes (103, 308). Furthermore, Ras is an insufficient trigger for insulin-induced glucose uptake in adipocytes (308). In male diabetic mice but not female, Ras is involved in destruction of β-cells (80). Glucose activates H-Ras in retinal capillary endothelial cells, and the data suggest that superoxide plays an important role in this event (158).
Glutathionylation activates Ras in VSMC, and is decreased in Grx-overexpressing cells (1). Moreover, glutathionylation of Cys118 and activation of Ras leads to endothelial insulin resistance, and insulin signaling was recovered with Grx over-expression, implicating a role for Grx treatment in diabetes (48).
2. MEKK-SSG
The MAPK/ERK kinase kinae (MEKK) mediates Ras-Raf signal transduction to JNK and c-Jun (312), and p21ras is reported to act both downstream (308) and upstream (48) of insulin receptor substrate-1 (IRS-1) in the insulin signaling cascade. S-glutathionylation inhibits MEKK1 in menadione-treated lymph node carcinoma prostrate cells (55), and glutaredoxin enhances NFκB activation through MEKK in HEK293 cells (118). By analogy, reversible glutathionylation may play a role in regulating MEKK-dependent insulin signaling.
3. c-Jun-SSG
c-Jun is phosphorylated by c-Jun NH2-terminal kinase (JNK), and JNK has implications in the pathology of β-cells via IL-1β or specifically in mediating IRS-1-insulin receptor interactions in response to TNFα or anisomycin in Chinese hamster ovary cells (CHO) (3, 182). Perturbation of the redox-regulation of the JNK signaling cascade is important in diabetes, and inhibition of JNK signaling leads to beneficial effects in type 1 and type 2 diabetic mice (143). In this regard, c-Jun has been shown to undergo glutathionylation in vitro (156). Furthermore, c-Jun activation in MCF-1/ADR cells is reported to be hindered by Grx binding to ASK-1 (ASK:Grx), a mechanism of regulation distinct from the typical deglutathionylation activity of Grx (282). Hence, interference with these modes of regulation by alteration in Grx content may be important also in understanding the complications of diabetes.
4. Akt-SSG
Akt is a downstream effector of PI3K, a signaling caspase important in many cellular processes including glucose metabolism, and cell death and survival. Decreased Akt activity in response to peroxynitrite or high glucose has been reported in diabetic rats and human umbilical vein endothelial cells (HUVEC) (283, 332). Many reports suggest a strong link between ROS and the PI3K-Akt Pathway (for review, see ref. 21). Also, data suggestive of a link between Akt activity and Grx status, implicating regulation of Akt via glutathionylation, have been reported in another context (207). Changes in Akt activity are connected also to multiple other signaling proteins potentially regulated by glutathionylation or interaction with Grx (e.g., PTEN, PP2A, and ASK-1; (273). Hence, further investigation is necessary to document the key control points that are regulated by reversible S-glutathionylation.
5. IKK-SSG
IκB kinase (IKK) activation is at the crux of insulin resistance in diabetes, and in particular, IKKβ mediates anti-inflammatory and antidiabetic effects of aspirin and aspirin derivatives (12, 276). Aspirin was first reported in 1876 to have tremendous benefits for lowering blood glucose concentrations, but the antithrombotic effects of such high doses prevent its use as an antidiabetic treatment (276). IKKβ inhibitors (e.g., salicylate) enhance insulin sensitivity in animals and humans, and heterozygous IKKβ mice are less resistant to insulin (12, 276, 276). Specifically, mice with IKKβ knocked down in skeletal muscle cells showed insulin responsiveness similar to wild type (255), but when it was knocked down in hepatocytes and myeloid cells, mice had selective hepatic sensitivity and overall sensitivity to insulin, respectively (12). Conversely, transgenic mice with constitutively active hepatocyte IKKβ developed diabetes (34). IKK is well established to exert inflammatory effects via NF-κB signaling (see below), and can exacerbate insulin resistance by direct phosphorylation of the insulin receptor substrate-1 (IRS-1) (95) (Fig. 3). Glutathionylation of IKKβ is demonstrated to regulate pro-inflammatory gene products of NF-κB activation in lung epithelial cells, reversible by glutaredoxin (251). By analogy, we speculate that inhibition of Grx would give rise to a desirable increase in IKK-SSG and corresponding inhibition in diabetes.
6. NF-κB-SSG
IKK phosphorylation of IκB promotes subsequent ubiquitination and degradation of IκB, freeing NF-κB for nuclear translocation where it binds DNA and activates transcription. NF-κB signaling is activated in many models of diabetes such as the retina (256, 274, 334), kidney (267), and liver (34). NF-κB activation is involved in major pathways leading to diabetic complications such as AGEs and PKC modulation, and inhibition of high glucose-induced mitochondrial superoxide levels decrease NF-κB hyperactivity (30). In vitro glutathionylation of p50 (p50-SSG) was shown to inhibit DNA binding (237). We found hypoxia and N-acetylcysteine treatment of pancreatic cancer cells led to inactivation of p65, and glutaredoxin restored the p65 transcriptional activity, indicating glutathionylation of p50, p65, or a transcriptional cofactor (242); (see Cancer section, below). Furthermore, glutaredoxin was reported to enhance NF-κB activation through NIK in HEK293 cells (118). S-glutathionylation has been reported for up to 13 proteins within the NF-κB pathway. For example, the activity of the ubiquitin/proteasome is increased in diabetic patients (189), and glutathionylation inhibits the ubiquitin-activating (E1) and ubiquitin-carrier (E2) enzymes (130, 219), and the 20S proteosome (66) (reviewed in more detail in the neurodegenerative section of this review).
7. PKC–SSG
Protein kinase C (PKC) is activated by increased diacylglyceride (DAG), and is one of the major pathways that leds to the pathogenesis of diabetic complications, primarily in vascular complications (154). PKCs are classified as calcium-dependent (cPKC), novel calcium-independent (nPKC), or atypical (aPKC) (273). cPKCs (α, β1, and β2) and nPKCs (ɛ and δ) all have tissue-specific implications in the diabetic retina, glomerulus, heart, and aorta, but the β-isoforms in the vasculature have the most significance (159). Ruboxistaurin is a PKC-β inhibitor being tested in clinical trials for vascular protection in diabetic retinopathy (46). Glutathionylation of PKC-α and reversal by Grx has been reported previously for NIH3T3 cells (320), and of the isoforms addressed above, glutathionylation inactivated PKC-α, β1, β2, ɛ, and δ in vitro (43). We speculate that, if Grx-mediated deglutathionylation of PKC leads to activation in vivo, Grx would be an additional therapeutic target for diabetic vascular complications. However, this is a limited view, which does not consider other control points potentially regulated by glutathionylation, such as aldose reductase (AR-SSG, see above) that can signal to PKC and NF-κB (NFκB-SSG, see above) (285).
H. Summary and discussion: Grx as a therapeutic target in diabetic complications
Reviewed here are many diabetes-related targets implicated in redox regulation by S-glutathionylation and Grx; however, the homeostatic disturbances that are generated in metabolic diseases are countless. Reports of protein glutathionylation are continually increasing in the literature, but often they are not documented in a physiologically relevant context.
Table 2 displays a sampling of additional proteins that have been reported to be glutathionylated under various conditions. These proteins are implicated in various aspects of diabetes, including metabolism, homeostatic and redox regulation, protein folding, leukocyte activation, transport, and cell death; however, most have not been studied under physiologically relevant conditions, and with the exception of pro-caspase 3, none have been tested for reversibility by Grx. In other cases, protein glutathionylation was observed in an endogenous milieu, but functional change and physiological impact were not studied. For example, glutathionylation of hemoglobin (HbSSG) has been reported in diabetic patients, and elevated HbSSG content correlates with increases in microangiopathy, but whether this serves only as a biomarker of oxidative stress or it has impact on redox homeostasis is not clear (262).
Table 2.
Proteins Reported to Undergo S-Glutathionylation and Implicated in Diabetes
| Protein | Function | Oxidizing stimulus | Milieu | Reference for glutathionylation | Implication in diabetes | Reference |
|---|---|---|---|---|---|---|
| Alcohol dehydrogenase | Catalyzes alcohol metabolism | GSH + diamide, or GSNO | In vitro | 155 | Increased activity in male rats | 54a |
| *Cu, Zn SOD | Catalyzes superoxide dismutation | GSH + diamide or GSNO; Decomposed GSNO | In vitro; In vitro | 155, 292a | Protect against alloxan-induced b-cell death | 107a |
| Malate dehydrogenase | Catalyzes malate-aspartate shuttle | Rose Bengal + 1 min white light | Heart homogenates | 79a | Decreased activity in leukocytes of dogs | 11a |
| Creatine kinase | Catalyzes creatine-P formation | GSH + diamide, or GSNO | In vitro | 155 | Decreased activity in rat heart | 239a |
| Glycogen phosphorylase b | Catalyzes glycogenolysis | GSH + diamide, or GSNO | In vitro | 155 | Eminent therapeutic target | 305a, 219a |
| Calbindin | Binds/regulates calcium | Decomposed GSNO | In vitro | 292a | Protects pancreatic b cells from death | 243a |
| Cathepsin K | Catabolism of bone & cartilage | GSNO | In vitro | 233b | Increased activity in rat bone | 117a |
| Fatty acid binding protein | Intracellular transporters in lipid metabolism | T cell blasts | Diamide + CHX | 89a | Increased levels in patients | 111a |
| HSP60 | Chaperones in protein folding | T cell blasts | Diamide + CHX | 89a | Downregulated in muscle | 38a |
| Pro-caspase 3 | Signals apoptotic cell death | GSH + diamide, or GSNO; TNFa + CHX | In vitro, Endothelial cells | 155, 227 | Activation & proliferation of b cell-specific T cells; activated in diabetic retinae | 170a, 202a |
| GAPDH | Housekeeping and cell signaling protein | Decomposed GSNO | In vitro | 292a | Inhibition leads to PKC activation, hexosamine flux, and AGEs in endothelial cells in high glucose | 77 |
| Hemoglobin | Transports oxygen | Diabetes | Human Subjects | 262 | Elevated in diabetic patients | 262 |
Altered protein function via S-glutathionylation may have impact on many different pathological areas in diabetes, but changes in S-glutathionylation status corresponding to functional differences and reversibility by glutaredoxin need to be documented.
Glutathionylation studies used CuZnSOD, but the type of SOD was not specified in the diabetes study. CHX, cycloheximide; GSH, glutathione; GSNO, nitrosylated glutathione, TNFα, tumor necrosis factor-alpha.
Diabetes has been the context of only a few studies implicating changes in glutathionylation status of key proteins and/or changes in glutaredoxin activity. The majority of these studies, including those on AR, potassium channels, IKK, and PTP-1B support the notion that inhibition of Grx would alleviate diabetic complications; however, the studies on insulin exocytosis and SERCA suggest the opposite effect. Therefore, determining the catalytic efficiency of endogenous Grx for deglutathionylation of specific protein-SSG substrates is critical to evaluating the outcomes of inhibiting Grx as a therapeutic strategy. Moreover, tissue, cell type, subcellular compartment, and microdomain specificities must be taken into consideration.
IX. Cardiovascular Diseases and Alterations in Protein–S-Glutathionylation Status
Within the cardiovascular system, protein S-glutathionylation is emerging as a critical signaling mechanism and consequence of oxidative insult, such as ischemia/reperfusion injury [(79), see below]. Protein S-glutathionylation regulates numerous physiological processes that are important in cardiovascular homeostasis and/or perturbed in disease, including myocyte contraction, oxidative phosphorylation, protein synthesis, vasodilation, glycolytic metabolism, and response to insulin (summarized in Table 3). This section discusses increasing evidence that perturbations in protein glutathionylation status—as well as Grx activity—contribute to the etiology of cardiovascular diseases such as myocardial infarction, cardiac hypertrophy, and atherosclerosis.
Table 3-1.
Cardiovascular Proteins Regulated by S-Glutathionylation
| Protein | Citation | Function | Model system | Stimulus | Detection | Effect(s) | Reversed by GRx? |
|---|---|---|---|---|---|---|---|
| Actin | 38 | Myocyte contraction | Male Sprague-Dawley rats | In vivo IR | 1-D PAGE + anti-GSH Ab; 2-D PAGE + anti-actin Ab; Actin IP + anti-GSH Ab | ↓ Polymerization rate; ↓ cooperativity in binding tropomyosin | Not shown |
| GAPDH | 79 | Glycolysis; apoptosis | Male Wistar rats | IR (isolated heart) | Bio-GSH perfusion + affinity purification + 1-D PAGE + band excision/digestion + peptide sequencing | ↓ GAP dehydrogenase activity | Not shown |
| Complex I | 293 | Mitochondrial respiration | Rat liver mitochondria; bovine heart mitochondria | [35S]-GSSG‡ | Fluorography | ↓ Rotenone-sensitive activity; ↑ O2 production | Not shown |
| 23 | Bovine heart mitochondria; purified complex I | [35S]-GSSG; GSH:GSSG = 0.67-12; GRx2/GSSG‡ | Fluorography; 1D PAGE + anti-GSH Ab; 1D PAGE + MS | ↓ Complex I activity | In vitro by GRx2 | ||
| Complex II* | 39 | Mitochondrial respiration | Male Sprague-Dawley rats; purified SQR | IR (isolated heart); in vivo IR; GSH:GSSG = 0.3-7 | 1D PAGE + anti-GSH Ab; MS | ↑ Electron transfer efficiency, ↓ electron leakage | Not shown |
Table 3-2.
Cardiovascular Proteins Regulated by S-Glutathionylation
| Protein | Citation | Function | Model system | Stimulus | Detection | Effect(s) | Reversed by GRx? |
|---|---|---|---|---|---|---|---|
| SERCA | 2 | cytosolic Ca2+ reuptake | SERCA isolated from rabbit heart; HEK cells w/transfected SERCA; aortic homogenates; aortic rings | In vitro: ONOO− + GSH-sepharose; ex vivo; ACh, NO, bradykinin | Affinity chromatography; Bio-GEE + 1D PAGE + anti-streptavidin; Mass Spec | ↑ Ca2+ uptake activity | Not shown |
| RyR | 265, 264 | SR Ca2+ release | SR vesicles from canine heart | Tachycardic pacing; NADPH + [35S]-GSH Exercise or tachycardia +/− I/R |
Tachycardia + 1D PAGE + anti-GSH Ab; phosphorimaging Tachycardia/Exercise + 1D PAGE + anti-GSH Ab |
↑ Ca2+ release rates (transient) ↑ Ca2+ release rates (transient); ↓ Ca2+ leak; ↓ infarct size |
Not shown Not shown |
| α-KGDH | 217 | TCA cycle | Rat heart mitochondria; purified αKGDH | H2O2; GSH/diamide | Reversibility by GRx1 | ↓ AKG dehydrogenase activity | via GRx1 |
| 8 | Cardiac mitochondria from male Sprague-Dawley rats | H2O2 | Reversibility by GRx1; NEM + GRx1 + Biotin-NEM; NEM + GRx1 + Biotin-SPDP | ↓ AKG dehydrogenase activity | via GRx1 |
Table 3-3.
Cardiovascular Proteins Regulated by S-Glutathionylation
| Protein | Citation | Function | Model system | Stimulus | Detection | Effect(s) | Reversed by GRx? |
|---|---|---|---|---|---|---|---|
| Ras | 2 | Hypertrophic signaling | Rat vascular smooth muscle cells | Angiotensin II H2O2 | Bio-GEE + streptavidin IP + 1D PAGE + anti-Ras Ab Ras IP + MS | ↑ Protein synthesis; ↑p-p38, p-Akt | Via GRx1 overexpression |
| 236 | Neonatal rat ventricular myocytes | Mechanical strain | Bio-GEE + streptavidin IP + 1D PAGE + anti-Ras Ab; Ras IP + MS | ↑ Ras-Raf binding; ↑ Ras-GTP binding; ↑ ERK activation; ↑ protein synthesis | Via GRx1 overexpression | ||
| 47 | Activation of MEK/ERK/Akt | Bovine aortic endothelial cells | ONOO− (some effects also shown with oxLDL) | Bio-GEE + streptavidin IP + 1D PAGE + anti-Ras Ab | ↑ Ras membrane translocation; ↑ ERK-P; ↑ Akt-P, ↑ guanine nucleotide exchange (in vitro) | Not shown | |
| 48 | Insulin resistance | Bovine aortic endothelial cells | oxLDL (some effects also shown with ONOO-) | Ras IP + anti-GSH Ab; Ras IP + MS | ↑ ERK-P (sustained); ↑ Akt-P (transient); ↓ insulin-induced Akt-P; ↑IRS-P | Via GRx1 overexpression |
Unusual example of a protein that is glutathionylated at baseline but deglutathionylated during oxidative stress (e.g., IR)
Excellent demonstrations of glutathionylation as a regulatory mechanism
Glutathionylation achieved under supraphysiological oxidant concentrations
A. Myocardial infarction
Eaton and colleagues (79) analyzed the effects of cardiac ischemia–reperfusion on glutathionylation of the cardiac proteome. Isolated rat hearts were perfused with biotinylated GSH following stop-flow ischemia, and glutathionylated proteins were detected via blotting with streptavidin-HRP. According to this analysis, overall protein glutathionylation was increased ∼15-fold following ischemia–reperfusion (IR), with the majority of the glutathionylation events occurring early in the reperfusion period. Some caution is required in interpreting these results because trapping the modified proteins as protein–SSG-biotin may inhibit their deglutathionylation and overestimate the degree of glutathionylation that would occur otherwise (273).
In the same study, glyceraldehydes-3-phosphate dehydrogenase (GAPDH) was identified as a prominent cardiac protein glutathionylated during IR. GAPDH immunopurified from ischemic tissue exhibited DTT-reversible loss of function, suggesting that GAPDH glutathionylation is likely inhibitory in vivo. While the consequences of GAPDH inhibition on cardiac function were not explored in this study, logical possibilities include: (a) contribution to the blockade of glycolysis characteristic of ischemic injury, (b) interference with translocation to the nucleus, resulting in increased apoptosis (51, 128), or (c) little to no effect, with the modified cysteine of GAPDH, serving primarily as a “sink” for excess oxidants rather than a site of homeostatic regulation. Importantly, GAPDH activity was restored by the end of the reperfusion period, suggesting that for this protein at least, glutathionylation may serve as a temporary modification to protect catalytic cysteines from irreversible oxidation.
Evidence for actin glutathionylation was demonstrated in a rat model of in vivo IR (38). Homogenates of ischemic tissue subjected to Western blot analysis with an anti-GSH antibody exhibited a prominent band corresponding to the molecular weight of actin, and immunoprecipitated actin reacted with the same antibody in a DTT-reversible manner. Studies on isolated G-actin indicated that glutathionylation delayed its rate of polymerization [consistent with a previous study on the effect of actin glutathionylation in A431 cells (315)], and decreased the cooperativity of its binding to tropomyosin, suggesting that actin-SSG formation contributes to the decline in cardiac contractility observed during ischemia. This interpretation could be strengthened by determining the glutathionylation status of actin following reperfusion. Since contractility is generally recovered in this model by the end of the reperfusion period, it would be expected that actin-SSG levels would decline with a similar time course, providing the basis for the improved contractility following IR insult.
In contrast to actin and GAPDH, mitochondrial complex II exhibits the opposite glutathionylation pattern following IR (i.e., deglutathionylation). In vivo IR, as well as stop-flow ischemia of isolated rat heart, resulted in decreased immunoreactivity of the 70 kD subunit of complex II (i.e., SQR) with an anti-GSH antibody (39). Studies of isolated SQR indicated that glutathionylation increased electron transfer activity somewhat and decreased leakage of superoxide (O2·−), suggesting that IR-associated deglutathionylation contributes to the decrease in SQR function observed during IR.
What could explain the divergence between the glutathionylation pattern of the general cardiac proteome (including actin and GAPDH) and that of SQR during IR? Possible contributing factors include the accessibility and intrinsic reactivities of modified cysteines, their proximity to sites of ROS production (as well as to Grx), and structural features that may stabilize the glutathionylated—or thiolate—status of the modified cysteine. Overall, the divergent results of these studies reinforce the concept that a single oxidative stimulus (e.g., IR) can affect glutathionylation status of different protein cysteines in different directions. Moreover, understanding the basis for these differences will require a greater understanding of the regulation of the factors regulating protein glutathionylation (e.g., concentrations of ROS, Grx1, and Grx2) within specific intracellular compartments, as well as quantitative relationships among protein–SSG events, the magnitude of alteration in protein activity, and resulting impact on cellular function.
Understanding the role of protein glutathionylation in myocardial infarction can also be approached by manipulating Grx, the primary intracellular deglutathionylating enzyme (41). To this end, mouse models with embryonic knockout of Grx1, as well as overexpression of Grx1 and Grx2 transgenes, were developed and subjected to in vivo and ex vivo IR. In general, experiments with transgenic animals suggest a cardioprotective role for both Grx isoforms; however, additional studies are needed to link the effects of each transgene to the protein glutathionylation status.
The first group to investigate the role of Grx on IR injury tested the effect of Grx1 embryonic knockout on infarct size and area at risk in an in vivo model of IR (120). No difference in either parameter was observed in Grx1 knockout (KO) vs. WT animals, even though the deglutathionylase activity of all of the mouse tissues, including heart, was essentially absent. One possible explanation for this unexpected outcome is that compensatory changes in the mechanisms of cellular homeostasis occurred during development, offsetting the detrimental effect of Grx1 knock-out. Therefore, to circumvent this complication, we recommend that future studies on the effects of Grx1 in IR injury utilize a tissue-specific, inducible KO model.
Malik et al. (183) further explored the role of Grx1 in IR injury by comparing the effects of embryonic global glrx1 KO with muscle-specific overexpression, and by widening the scope of injury to include ischemic pre-conditioning (IPC) prior to IR. IPC is widely recognized to decrease subsequent IR injury (reviewed in refs. 75 and 115), and there is evidence that Grx1 contributes to regulation of several signaling pathways implicated in the mechanism of IPC (see below). Unlike Ho et al. (120), Malik and colleagues reported a small but significant increase in infarct size, as well as decreased contractile performance, in glrx1 KO mice compared to controls. In contrast to the effects of Grx1 KO, Grx1 overexpression appeared to decrease infarct size and protect coronary function.
The basis for the distinct effects of glrx1 KO on infarct size reported by these two groups is not obvious, but likely reflects differences in experimental protocols. For example, Grx1 might play an important role in cardioprotection during early reperfusion, thus affecting infarct size after 2 h of reperfusion [observed by Malik et al. (183)] but exhibiting less of an effect after 4 h [when Ho et al. (120) measured infarct size]. Alternatively (or additionally), neurohumoral factors might have blunted the effect of glrx1 KO in the in vivo IR model of Ho et al. (120)., while their absence revealed an important cardioprotective role for Grx1 in the isolated heart model of Malik et al.(183). This scenario predicts that glrx1 KO indeed confers a detrimental effect on cardiac function during IR, and compensation by other organ systems may represent an important component of disease outcome when Grx1 activity is perturbed.
More dramatic than the role of Grx1 in IR injury was its contribution to IPC. Grx1 overexpression potentiated the protective effect of IPC on infarct size and cardiomyocyte apoptosis, while IPC failed to confer any cardiac protection in Grx1 KO mice. To explore the mechanism of Grx1-associated protection vs. IR injury, ROS production (via malondialdehyde (MDA) content), Akt phosphorylation, and Bcl-2 content were assayed. While there was no apparent change in Bcl-2 content associated with change in Grx1 content, Grx1 expression inversely correlated with MDA content following IR, and Grx1 KO was associated with decreased IPC-induced Akt phosphorylation. The latter observation led the authors to suggest that Grx1 could contribute to IPC via activation of Akt, which is consistent with other reports linking Grx activity to Akt phosphorylation (207, 211, 316), although the mechanism for this effect in vivo is not yet established (see Preconditioning section, below). However, the additional conclusion that Grx1's cardioprotective effects can be attributed to its role as an “antioxidant” is problematic. First, it is not consistent with the primary (and well-documented) function of Grx as a deglutathionylating enzyme (41, 108). In fact, studies of protein glutathionylation were not reported here, so it cannot be concluded that Grx1's cardioprotective effects were not related to its role in protein deglutathionylation. Second, a Grx1-mediated antioxidant activity is not explicitly demonstrated. One could imagine that, if Grx1 were expressed at high enough levels, it could serve as an antioxidant by supplying reduced cysteines in a manner analogous to N-acetylcysteine or DTT, but it is unlikely that such concentrations could be reproduced in vivo, even within a disease context. Is there a more likely explanation for the decreased MDA content observed in Grx1 TG mice subjected to IR? One possibility is that Grx1 may indirectly decrease ROS by deglutathionylating a ROS-generating enzyme, such as complex I, which exhibits increased superoxide production upon glutathionylation (293). This interpretation is more consistent with documented activities of Grx1, and could be investigated by determining the glutathionylation status of complex I during IR in WT and Grx1 TG mice. A similar scenario has been described for the cardioprotective role of Trx1 function in ischemic preconditioning. Analogous to glrx1 KO, inhibition of Trx1 resulted in increased MDA formation in the IR heart, suggesting an “antioxidant” function for the enzyme (62). Instead of proposing that Trx itself serves as an antioxidant, the authors suggested that it might promote antioxidant activity via up-regulation of antioxidant enzymes such as MnSOD. In light of their distinct enzymatic activities, it is intriguing that Grx1 and Trx1 confer similar cardioprotective effects during IPC, and determining the mechanism(s) for each enzyme's protective effects represents a fascinating avenue for future study.
The role of Grx2 in IR injury was also investigated using a transgenic animal model (211). As with Grx1 overexpression, Grx2 overexpressing mice exhibited decreased infarct size and improved myocardial function following ex vivo IR compared to WT mice. Unlike Grx1, Grx2 appeared to influence apoptosis, with Grx2 TG mice showing fewer apoptotic cardiomyocytes following IR compared to control animals. Additional evidence supporting a cardioprotective effect of Grx2 included decreases in caspase activation, cardiolipin loss, MDA formation, and diminution of the GSH:GSSG ratio following IR. The roles of Grx2 in cytochrome c release and Akt phosphorylation were less straightforward. Grx2 TG animals exhibited decreased IR-induced cytochrome c release, but the amount of cytosolic cytochrome c was much higher at baseline in TG animals and appeared to decline with IR. Grx2 overexpression has been linked to decreased cytochrome c release in oxidant-challenged HeLa cells (85), but the basis for its loss from the cytosol in Grx2 TG hearts with IR is puzzling. Phosphorylated Akt was also higher at baseline in TG vs. WT animals, with levels remaining steady following IR. Considering the distinct subcellular localizations of Grx2 and Akt [Grx2 in the mitochondrial matrix (226) and Akt in the cytosol], this regulation is most likely indirect; however, a direct role of Grx2 in regulating Akt activity [as has been proposed for Grx1 (207)] is possible if the Grx2 transgene was also expressed in cytosol. Since cytosolic Grx activity was not reported in the study, this explanation cannot be ignored. In fact, Enoksson and co-workers (85) showed that Grx2 targeted to the cytoplasm protected HeLa cells from doxorubicin-induced apoptosis, so verification of appropriate subcellular localization is a critical prerequisite for interpreting the findings of any study in which Grx2 is overexpressed.
Overall, the work of Nagy et al. (211) suggests a role for Grx2 in cardioprotection, but as in the case of other studies documenting the cytoprotective effect of Grx2 (85, 173), mechanism(s) remain unknown. Importantly, candidate target pathways were proposed, including Akt and NF-κB. Future work should focus on identifying the direct targets of Grx2 responsible for its pleiotropic effects on oxidant-induced signaling. An attractive candidate is mitochondrial complex I, in which glutathionylation of the 51- and 75-kD subunits is correlated with electron transport inhibition and increased production of superoxide (293). The 51kD subunit contains one conserved cysteine that is not bound to an FeS cluster, and this cysteine faces the mitochondrial matrix (119), making it a potential site of regulation by Grx2. Glutathionylation of complex I, with associated increases in superoxide production, would be expected to increase cytochrome c release and caspase activation, induce survival signals, and contribute to infarct size and cardiac dysfunction. Thus, complex I deglutathionylation by Grx2 is a conceivable upstream event responsible for modulating these effects in Grx2 transgenic animals.
Finally, the roles of both Grx isoforms in IR injury were explored in rats by Mukherjee et al. (204). These investigators examined the effects of broccoli extract (administered by oral gavage) on IR injury in isolated working rat hearts. Rats fed normal diets exhibited decreased Grx expression during IR, while rats given broccoli extract showed preservation of Grx1 and Grx2 content, which correlated with decreases in infarct size, cardiomyocyte apoptosis, cytochrome c release, and caspase-3 activation, as well as improved post-MI cardiac and hemodynamic function. It is difficult, however, to determine the specific contribution of Grx isoforms to these cardioprotective effects, since broccoli gavage also induced other genes known to regulate cellular survival and redox homeostasis (e.g., thioredoxins 1 and 2, thioredoxin reductase), and the effect of broccoli gavage on protein glutathionylation status was not reported.
In summary, cardiac ischemia-reperfusion results primarily in increased protein glutathionylation; however, some proteins (e.g., complex II) are deglutathionylated during an IR episode. The effects of glutathionylation on function of these proteins (e.g., GAPDH, actin) may protect them from irreversible damage, or contribute to IR-induced injury. The latter possibility is supported by the cardioprotective phenotype of Grx TG animals, which exhibit decreased infarct size and improved cardiac function following IR. While data from KO animals are not as straightforward, studies on mice with inducible, tissue-specific KO might clarify the roles of Grx in acute IR injury. A critical frontier in this endeavor is to determine the mechanism(s) of Grx-related cardioprotection, which will require linking the protective effects of Grx on cardiac function with glutathionylation status of its molecular targets.
B. Preconditioning
Ischemic preconditioning (IPC) describes the phenomenon in which a series of brief ischemic episodes protects against subsequent IR injury (75, 115, 208). Cardiac preconditioning can also be achieved by tachycardic stimuli, such as pacing or exercise (74, 264). Potential targets of glutathionylation in preconditioning will be discussed below.
Sanchez et al. (264, 265) provide evidence that glutathionylation of the cardiac ryanodine receptor (i.e., RyR2) contributes to tachycardia- and exercise-induced preconditioning. Exercise and tachycardic pacing both result in RyR2 glutathionylation, which is correlated with increased Ca2+ release rates (265) and increased colocalization with NADPH oxidase (264). NADPH oxidase inhibitors prevent these effects, suggesting that ROS production by this enzyme is the likely trigger for RyR2 glutathionylation in vivo. Importantly, NADPH oxidase inhibitors also abolish the protective effect of tachycardia or exercise on infarct size. Whereas RyR2 glutathionylation is not explicitly correlated with these observed changes, they are consistent with the concept that RyR2 glutathionylation contributes to tachycardic or exercise-induced preconditioning. This concept could be tested further by transfection of a mutant RyR2 that cannot be glutathionylated and determining the effect on tachycardic PC, or determining the role of Grx1 on RyR2 glutathionylation status (assuming the site of glutathionylation is exposed to the cytosol).
Unlike tachycardic or exercise-induced PC, a specific role for protein glutathionylation has not been established for IPC. However, the apparent contribution of Grx1 to IPC [via TG overexpression or KO, see (183) and above] implicates glutathionylation as a potential modulating mechanism. Numerous signaling intermediates have been implicated in the regulation of IPC, of which many components have been shown in other systems (reviewed by Shelton et al. (273) to be regulated by S-glutathionylation; hence, modulation of these pathways by glutathionylation in the heart represents a potential layer of regulation for cardiac IPC.
1. Protein kinase C (PKC)
PKC is considered to be a central regulator of IPC signaling (75), integrating multiple upstream signals (e.g., catecholamines, angiotensin II, and endothelin) and activating diverse downstream pathways implicated in IPC [reviewed by (115)]. Importantly, most PKC isoforms appear to be sensitive to inhibition by S-glutathionylation [reviewed in (273)], including those isoforms implicated in IPC signaling (115). Therefore, a potential mechanism by which Grx1 contributes to IPC may be via deglutathionylation (and subsequent activation) of PKC. This pathway could be tested by determining the effect of Grx manipulation on PKC activity, and correlating these changes to its glutathionylation status during brief ischemic challenges.
2. Protein kinase A (PKA)
Sanada et al. (263) provided evidence that PKA may contribute to IPC independently from PKC, presumably through inhibitory effects on Rho kinase and cytoskeletal reorganization. PKA is inhibited by glutathionylation in vitro, as well as in mouse fibroblasts treated with diamide (124) reviewed in (273). As for PKC, Grx could contribute to IPC via maintaining PKA in its active, deglutathionylated form. However, the effects of IPC on PKA glutathionylation status (and the additional role of Grx) have not yet been explored.
3. Nuclear factor κB (NF-κB)
NFκB is a pleiotropic transcription factor commonly activated during cellular stress (see also diabetes, neurodegenerative disease, and cancer sections). While the effects of NFκB activation are often dependent upon cell type and stress stimulus, its activation has been shown primarily to increase survival in cardiomyocytes (138). Two studies by Maulik and colleagues (193, 194) utilizing an inhibitor of NF-κB nuclear translocation suggest that the pathway is required for IPC in the isolated, perfused rat heart, and that the basis for its protective effect may be through activation of Bcl-2 transcription. Various groups have provided evidence that the NF-κB pathway is regulated at various foci by glutathionylation [e.g., p50 (237), p65 (242), IKK (251), and the proteasome (219)], with glutathionylation having an inhibitory effect in each case. Therefore, deglutathionylation of p50, p65, IKK, or the proteasome by Grx could maintain overall pathway activity during IPC. As in the examples discussed above, establishing such a role for Grx will require analysis of glutathionylation status of NF-κB pathway components during IPC, and correlating changes in glutathionylation status with corresponding changes in activity.
4. Akt
Akt, also known as PKB, has been implicated in cell growth, survival, and migration signals in diverse cell types. Numerous studies have identified Akt activation as contributing to both early (i.e., PKC activation) and late (i.e., mitochondrial) events in IPC (115). Wheres Akt itself has not been shown to be glutathionylated, it appears to be regulated (either directly or indirectly) by Grx1. Murata et al. (207) reported that H9c2 cells (rat embryonic cardiomyoblasts) in which Grx1 was overexpressed exhibited decreased H2O2-induced apoptosis, and this effect was correlated with an increased duration of Akt phosphorylation and decreased association with PP2A, the phosphatase associated with Akt inactivation. Studies on isolated Akt in the presence of H2O2, Grx, and/or GSH were interpreted to mean that Grx directly reduces an Akt intramolecular disulfide in vivo; however, the likelihood of this representing a physiological regulatory mechanism is questionable, since in vitro conditions did not approximate the cellular environment (e.g., GSH:GSSG = 20, lower than the typical ratio), kinetic competence was not demonstrated (endpoint assay was 30 min), turnover conditions were not used ([Grx1] was 80-fold higher than Akt), and Akt redox status was shown not to affect its kinase activity. A more likely mechanism of Grx-mediated regulation of Akt is via its phosphorylation status, as has been suggested by other groups exploring the effect of Grx on Akt activity and post-translational modification. For example, Akt phosphorylation is increased in Grx2 overexpressing mice (211), and Grx1 overexpression is correlated to increased Akt phosphorylation in bovine aortic endothelial cells (316). The mechanism of regulation of Akt phosphorylation by Grx is still not resolved. However, the latter authors appropriately consider that the level of regulation of Akt could be direct (as proposed by Murata's group) or indirect, via deglutathionylation of upstream activators such as PKA or PKC.
In summary, although a role for reversible glutathionylation in IPC has not yet been established, it is suggested by the contributory role of Grx1 in a transgenic animal model (183). Here, we identified and discussed signaling candidates implicated in IPC that are also established targets of regulation by S-glutathionylation, including PKC, PKA, and NFκB pathway components, all of which are rendered inactive upon glutathionylation. Akt was also discussed as being regulated by Grx, although the specific nature of the regulation is not yet understood. Importantly, no study to date has reported global or specific protein glutathionylation during IPC. Future work should focus on potential links between glutathionylation status of these proteins, and the effects of glutathionylation on their functions as well as on IPC in general.
A final consideration in the discussion of ischemic vs. tachycardic PC is the potential roles of Grx. Although the mechanism remains unknown, Grx1 appears to contribute to the protective effects of IPC (183); however, one might predict that Grx would hinder tachycardic PC via deglutathionylation of RyR2. Importantly, the effect of Grx1 activity on RyR2 glutathionylation status has not been reported, contrasting with documented Grx-dependent deglutathionylation of RyR1 [see above, (11)]. Determining the effect of manipulated Grx levels on RyR2 glutathionylation status (and Ca2+ release rates) would help address this question.
C. Nonspecific oxidative injury
IR and IPC are both conditions of oxidative stress. In IR, ROS are produced primarily from complexes I and III of the mitochondrial electron transport chain (167, 168), and ROS generation from mitochondria and/or NADPH oxidase appears to contribute to PC signaling (75, 264). Above, we discussed evidence for increased glutathionylation of the intracellular proteome—as well as individual proteins—with IR, and the potential for regulation of survival proteins by glutathionylation during IPC. Importantly, additional proteins are reported to be regulated by glutathionylation during generalized oxidative challenges, such as H2O2 treatment or exposure to decreased GSH:GSSG ratios. Although these oxidative stimuli do not necessarily model the physiological state or a specific disease condition, they identify candidate proteins for regulation by glutathionylation during pathological oxidative stresses such as IR, as well as other cardiovascular diseases associated with oxidative stress, such as hypertension (113) and atherosclerosis (151).
For example, mitochondrial complex I is glutathionylated in vitro upon exposure to GSSG (293) or low GSH:GSSG ratios [i.e., 0.67–12, (23)], and glutathionylation is reversed upon incubation with Grx2 and GSH. Complex I glutathionylation results in increased superoxide production (293), suggesting that this modification would increase ROS generation, leading to activation of redox signaling pathways and/or induction of cell death, depending upon the magnitude of modification. Key considerations regarding the possibility of complex I regulation by glutathionylation in vivo include its mechanism of formation and its potential role in cardiac disease.
Taylor et al. (293) propose that oxidative stress within the mitochondria alters the GSH:GSSG ratio sufficiently to cause complex I–SSG formation by thiol disulfide exchange; however, this mechanism is unlikely unless the modified cysteines display unusually low redox potentials (92, 99, 198). Thus, alternative mechanisms of glutathionylation (e.g., via nitrosothiol intermediate, sulfenic acid intermediate, etc., as described above) are more probable (Fig. 1). An additional mechanism of glutathionylation was proposed by Beer et al. (23), namely, catalysis by Grx2. Although mammalian glutaredoxins are efficient protein deglutathionylating enzymes, Grx1 promotes protein glutathionylation in the presence of GS· radical (287) and—to a much lesser extent—GSNO or GSSG. To determine if Grx2 exhibited similar behavior, it was incubated with 5 mM GSSG and mitochondrial membranes from rat heart containing complex I. Addition of Grx2 accelerated glutathionylation of membrane thiols over a short time course; however, when GSH was added to glutathionylated membrane proteins, Grx2 incubation led to overall deglutathionylation of protein–SSG. Since the latter conditions more closely represent the intermitochondrial milieu, they better reflect the potential environment in which Grx2 may regulate complex I–SSG in vivo. Thus, catalysis of glutathionylation by Grx2 appears not to be a likely mechanism of complex I–SSG formation.
Whether complex I is indeed regulated by glutathionylation in the intact heart has not yet been explored. Studies focused on documenting complex I–SSG formation in cardiac cells or tissue, with an oxidative stimulus relevant to cardiac disease (e.g., IR, angiotensin II treatment), and attention to the effects of Grx1 and Grx2 on complex I glutathionylation status will provide additional insight into this potential contribution to cardiac injury.
Another mitochondrial enzyme potentially regulated by glutathionylation during cardiac oxidative stress is α-ketoglutarate dehydrogenase (KGDH). Nulton–Persson and colleagues (217) demonstrated that H2O2 treatment of rat heart mitochondria led to inhibition of KGDH activity, which was reversed by Grx1 and GSH within minutes, but unaffected by the Trx system. Although KGDH glutathionylation was not shown directly, it was inferred from the recovery of activity by Grx1 treatment, and hypothesized to protect catalytic cysteine residues from irreversible damage during oxidative stress conditions, such as IR.
A later study by the same group (8) focused on the glutathionylation site of KGDH, and proposed an intriguing model in which glutathionylation occurs on a covalently bound lipoic acid moiety, rather than the typical cysteine sulfhydryl moiety. This conclusion was based on observations that H2O2 treatment prevented recognition of KGDH by an antilipoate antibody, as well as HNE-mediated oxidation. A key question concerning the proposal of mixed disulfide formation between KGDH-lipoic acid and GSH is its mechanism of stabilization. It would be expected that the vicinal cysteine on lipoic acid would undergo thiol–disulfide exchange with its neighboring Cys-SSG, forming lipoic acid intramolecular disulfide and GSH. Structural analysis or modeling studies might identify potential residues that stabilize the second, reduced Cys on lipoic acid, making it unavailable to react with the neighboring Cys-SSG. While stable mixed disulfide formation between protein-bound lipoic acid and GSH is a novel concept, and catalysis of lipoic acid-SSG would represent a new activity for Grx1, there are alternative interpretations to the authors' observations. For example, it is possible that KGDH is glutathionylated on a cysteine residue in close proximity to the bound lipoic acid, and steric interference by this glutathionylated cysteine blocks accessibility of lipoic acid to antibodies and HNE. Alternatively, glutathionylation on a distant Cys could induce a conformational change with the same effect on access of the lipoic acid to detection reagents. This possibility could be addressed by analyzing the glutathionylated product by mass spectrometry, and/or by isolating the lipoyl moiety prior to analysis for S-glutathionylation.
Overall, glutathionylation of complex I and KGDH appear to be facile upon exposure of mitochondria to oxidants in vitro; however, their relevance to oxidative stress-associated cardiac disease is not yet established. Determination of their glutathionylation status with IR, or other pathophysiological oxidative challenge, will provide insight into the likelihood of their regulation by glutathionylation in vivo.
D. Cardiac hypertrophy
As for IPC, multiple signaling pathways contribute to the development of pathological cardiac hypertrophy (reviewed by (116). Among them is the Raf/MEK/ERK pathway, which can be stimulated either by G protein-coupled receptor (GPCR) ligands (e.g., angiotensin II, endothelin) or by mechanical stretch, resulting in induction of protein synthesis.
Recently, Pimentel et al. (236) showed that mechanical strain-stimulated Raf/MEK/ERK pathway activation in neonatal rat ventricular myocytes was dependent upon glutathionylation (and subsequent activation) of Ras, a small GTPase implicated in myocyte growth signaling (Fig. 4, right). Notably, this study is one of few that fulfill most of the criteria for establishing S-glutathionylation as a regulatory mechanism. Specifically, the authors demonstrated that Ras–SSG formed in response to a physiological stimulus (mechanical strain) under physiological conditions (intact cells assumed to have a normal GSH:GSSG ratio); that Ras glutathionylation conferred effects on protein function (increased Raf and GTP binding); that Ras–SSG formation was reversed by Grx (via overexpression); and that Ras glutathionylation impacted a downstream pathway important in disease (protein synthesis in cardiac hypertrophy). The proposed role of Ras–SSG in hypertrophic signaling is summarized in Fig. 4.
FIG. 4.
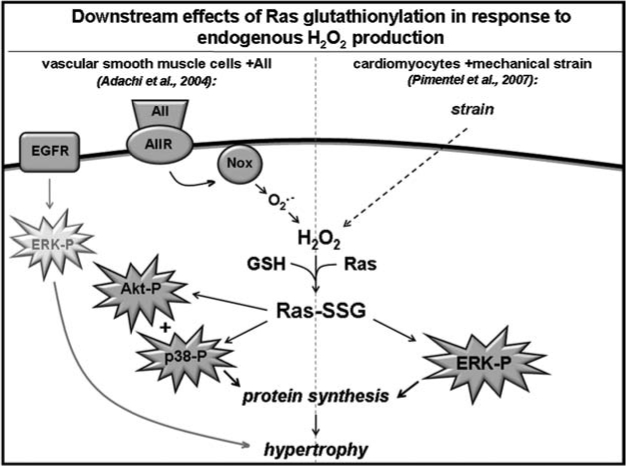
Downstream effects of Ras glutathionylation in response to endogenous H2O2 production. Two independent modes of Ras activation by glutathionylation are depicted here. On the left is shown how Ras-dependent and -independent pathways contribute to angiotensin II-induced hypertrophy in vascular smooth muscle cells (Adachi et al., 2004), namely (1) coupling of angiotensin II receptor activation to production of ROS by NADPH oxidase, followed by Ras glutathionylation and activation of Akt and p38, and (2) ROS-independent transactivation of EGFR and activation of the ERK signaling pathway. The right-hand scheme depicts a mechanism by which Ras-SSG mediates the hypertrophic response of cardiomyocytes to mechanical strain. Strain-stimulated cardiac myocytes exhibit ROS-dependent Ras glutathionylation, which activates the ERK pathway and results in increased protein synthesis. The basis for activation of Akt and p38 (left) vs. ERK (right) pathways by the same signaling intermediate (i.e., Ras-SSG) is not yet understood.
Determination of Ras glutathionylation status in animal models of cardiac hypertrophy would provide insight into the relevance of this pathway in progression of the disease in vivo. An animal model would also allow exploration of the effects of chronic strain—vs. acute strain—on Ras-SSG glutathionylation status, activity, and cardiac hypertrophy.
E. Atherosclerosis
Atherosclerosis is a complex disease process involving interactions between multiple cell types in the blood and vasculature. The precise role of glutathionylation in the development and progression of atherosclerosis is unknown; however, conditions within atherosclerotic plaques (e.g., hypoxia, oxidative stress, oxLDL, and inflammation) have been shown in other contexts to promote glutathionylation (79, 242, 290, 319), and Grx has been reported to associate with areas of oxidative stress within the vasculature (221). The following discussion explores further evidence for involvement of protein glutathionylation in atherogenesis.
Global protein glutathionylation increases in human monocyte-derived macrophages exposed to oxidized LDL (oxLDL) (319), a major component of atherosclerotic plaques also believed to contribute to their progression (151). Together with GSH depletion, increased protein–SSG content was implicated in oxLDL-induced macrophage death in vitro. Dying macrophages represent a major component of atherosclerotic plaques, and their presence in atherosclerotic lesions increases risk of rupture (164). The role(s) of specific glutathionylated proteins in macrophage cell death is not yet determined, nor is it known whether global protein glutathionylation increases in other cells types exposed to oxLDL; however, these questions form the basis for future studies.
Nonaka et al. (215) discovered that patients with arteriosclerosis of the extremities (i.e., arteriosclerosis obliterans, ASO), exhibit increased glutathionylation of serum proteins detected by SDS-PAGE followed by GST overlay. Remarkably, there was a positive correlation between disease progression and magnitude of protein glutathionylation measured, leading the authors to conclude that serum protein glutathionylation is both a sensitive and specific marker of ASO. However, many of the patients enrolled in the study had comorbid conditions also associated with increased serum protein–SSG, such as tobacco use (209), making the specificity of this marker for ASO unlikely. Importantly, these authors identified the serum protein apoB100 as a target for increased glutathionylation in ASO. ApoB100 is the major component of LDL, and it is tempting to speculate that its glutathionylation could affect its function, as has been shown for other post-translational modifications (292). Whether apoB100–SSG simply represents a disease marker, or contributes to the pathogenesis of ASO, remains an open question.
In the case of SERCA, the sarcoplasmic calcium ATPase, glutathionylation appears to be part of a normal regulatory mechanism that is disrupted during atherosclerosis. Adachi and colleagues (2) demonstrated that SERCA glutathionylation occurs in vascular cell lines and tissues in the presence of RNS and endogenous GSH. Glutathionylation could be stimulated by physiological ligands known to generate RNS (e.g., acetylcholine, bradykinin), led to increased SERCA ATPase activity, was correlated with vessel dilation, and was resistant to a cGMP inhibitor, leading the authors to propose that SERCA glutathionylation represents a physiological, cGMP-independent mechanism of vessel relaxation. Site-directed mutagenesis and mass spectroscopic analysis suggested that glutathionylation of Cys674, located in the cytosolic-facing hinge domain, was responsible for SERCA activation. Interestingly, analysis of cysteine modifications from atherosclerotic vs. normal rabbit aortas indicated increased sulfonate formation (including C674), which corresponded to decreased NO-induced relaxation, glutathionylation, and Ca2+ reuptake. Taken together, these observations suggest that irreversible oxidation (i.e., sulfonic acid formation) of SERCA's C674 during atherosclerosis prevents regulation of function by reversible glutathionylation and may contribute to the impaired vasodilation response to NO in atherosclerotic smooth muscle.
Adachi et al. (1) demonstrated that glutathionylation of Ras may contribute to vascular hypertrophy (implicated in atherosclerosis and hypertension) by activating protein synthesis in rat vascular smooth muscle cells (VSMCs). Treatment of VSMCs with angiotensin II (AII), an established stimulus for vascular hypertrophy, led to glutathionylation and activation of Ras, which resulted in increased phosphorylation of p38 and Akt, and increased protein synthesis (Fig. 4, left). These effects were dependent upon NADPH oxidase activation and ROS formation [shown separately to be activated by AII (162, 314)], and were blocked by overexpression of Grx1 or mutation of Ras at the site of glutathionylation (C118). Interestingly, AII-stimulated ERK activation, which contributes to AII-induced protein synthesis, was not redox-sensitive and proposed to occur independently of Ras glutathionylation. Like the work of Pimentel et al. (236), the work of Adachi and colleagues represents an excellent demonstration of protein regulation by S-glutathionylation: Ras–SSG formed in response to a physiological stimulus (AII treatment); glutathionylation resulted in a change in protein activity (increased Raf binding); was reversed by Grx1 (via overexpression); and was correlated to a physiological outcome (protein synthesis).
The work of Adachi and Pimentel point to several common events in hypertrophic signaling within the heart and vasculature: both require production of endogenous H2O2, result in Ras glutathionylation, and activate signaling pathways that ultimately result in increased protein synthesis. However, it is intriguing that the Ras–SSG-activated pathways implicated in hypertrophy differ in the two model systems. Why might H2O2-induced Ras glutathionylation activate the Raf/MEK/ERK pathway in cardiomyocytes vs. p38 and Akt–but not ERK–in vascular smooth muscle? (Fig. 4) The answer could reflect differences in signal transduction networking between cell types, different degrees of Ras glutathionylation resulting from each stimulus (assuming different thresholds of activation for downstream pathways), and/or distinct localization of ROS production (and subsequent Ras glutathionylation) depending upon the nature of the stimulus (i.e., NADPH oxidase vs. the source of strain-stimulated ROS).
In addition to modulating AII signaling in VSMCs, Ras-SSG may contribute to atherosclerosis by mediating the response to oxLDL in endothelial cells (Fig. 3). Clavreul et al. (47) demonstrated that treatment of bovine aortic endothelial cells (BAECs) with peroxynitrite led to Ras glutathionylation and activation of both ERK and Akt pathways (Fig. 5, left), and some of these observations were recapitulated with oxLDL treatment. Unlike mechanical strain- and AII-induced Ras glutathionylation, which required formation of H2O2, oxLDL-mediated glutathionylation was dependent upon peroxynitrite. The authors argue for a thiol–disulfide exchange mechanism with GSSG, based on proposed chemical reactions between peroxynitrite and GSH (producing GSSG), which exhibit a time course of GSSG formation compatible with that observed for Ras glutathionylation. This mechanism is feasible if the Kmix for Ras-Cys118-SSG formation is similar to the GSH:GSSG ratio achieved during peroxynitrite treatment (98, 99); but GSSG concentration was not measured in this study, and to the best of our knowledge, the Kmix for Ras-C118 has not been determined. Therefore, the mechanism of Ras glutathionylation associated with peroxynitrite treatment remains unclear.
FIG. 5.
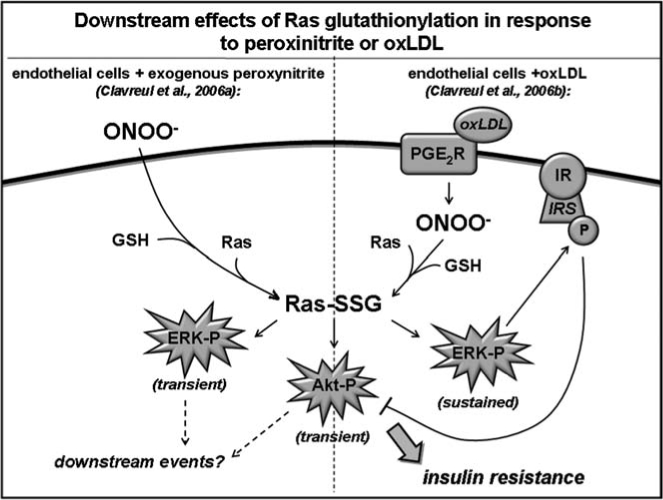
Downstream effects of Ras glutathionylation in response to exogenous peroxinitrite or oxLDL. This figure depicts distinct downstream events resulting from Ras glutathionylation in bovine aortic endothelial cells in response to peroxinitrite added exogenously (left) or generated endogenously in response to oxLDL exposure (right) as reported by Clavreul et al. (47, 48). In response to ONOO− treatment, Ras glutathionylation leads to transient phosphorylation of ERK and Akt; however, subsequent downstream signaling events remain unknown. oxLDL exposure (which leads to ONOO− production in situ) also caused Ras-SSG-dependent, transient Akt phosphorylation (center); however, it induced a sustained time course of ERK phosphorylation (right) as well as diminished Akt activation in response to a subsequent stimulus (insulin). The decreased insulin-induced Akt activation conferred by oxLDL pretreatmentcould be explained by ERK-induced phosphorylation (and inactivation) of IRS, which is upstream of Akt in the insulin signaling pathway.
Specific physiological effects of Ras glutathionylation (and subsequent activation of MEK/ERK and Akt) that might contribute to atherosclerosis were not reported here; however, it has been reported in other contexts that MEK/ERK activation within the endothelium may induce proliferation of endothelial cells, contributing to atherogenic vascular remodeling (238).
Although a mechanism for endothelial Ras–SSG-induced atherogenesis was not fully elucidated by Clavreul et al. (47), a role for oxLDL-induced Ras glutathionylation in insulin resistance was described in a subsequent study by the same group (48) (also see section on diabetes). Here, the effects of oxLDL-induced Ras glutathionylation were followed over a longer time course, and cross-talk with a second signaling pathway [insulin/insulin-receptor substrate (IRS)/Akt] was explored. Here, as in the previous study (47), oxLDL-induced Akt activation was transient, peaking at 15 min; however, ERK activation was sustained (>1 h). Moreover, subsequent activation of Akt by insulin/IRS was blunted by pretreatment with oxLDL, presumably via ERK-mediated phosphorylation (and inactivation) of IRS (Fig. 5, right). This work enhances understanding of downstream effects of Ras-SSG in endothelial cells, particularly within the context of insulin resistance. However, the distinctive effects of peroxynitrite and oxLDL on the time course of ERK activation raise questions about the use of exogenous peroxynitrite as a physiologically relevant stimulus.
While studied in different vascular cells using different oxidative stimuli, both Adachi and Clavreul report Ras—mediated phosphorylation of Akt, which is diminished by overexpression of Grx1. Interestingly, Wang et al. (316) report an opposite correlation between Grx1 activity and Akt activation in BAECs exposed to laminar flow. Grx1 activity approximately doubled within 5 min of exposure to physiological flow rates, and this activation correlated to increased phosphorylation of Akt and eNOS. Akt and eNOS phosphorylation were augmented with overexpression of Grx1, and diminished after treatment with Grx1 siRNA, suggesting that Grx1 activity precedes their activation, although a specific mechanism was not identified. These observations are consistent with those of Murata et al. (207), who reported increased Akt phosphorylation in H9c2 cardiomyoblasts overexpressing Grx1. Taken together, these studies highlight the complex relationship between Grx, protein glutathionylation, and Akt activity within the cardiovascular system. Importantly, Akt is emerging as a complicated signaling molecule within the heart and vasculature, implicated in various pathological signaling events, as well as in normal development and homeostasis (205). It is conceivable that Grx could participate in regulating the balance between physiological (i.e., laminar flow-induced) and pathophysiological (i.e., ATII-induced) Akt activation. Determining the status of Akt activation (as well as downstream effects such as eNOS activity and vessel hypertrophy) in Grx TG and KO animals would help address this complex situation.
An emerging contributor to atherogenesis is tumor necrosis factor-alpha (TNFα), which is thought to induce expression of adhesion molecules on endothelial cells and contribute to vascular smooth muscle cell apoptosis (73). Pan and Berk (227) treated endothelial cells with a combination of TNFα and cycloheximide (CHX), and observed Grx activation, pro-caspase-3 deglutathionylation, caspase-3 cleavage, and increased apoptosis. These effects were blocked by transfection with siRNA against Grx1, leading the authors to propose that Grx1-mediated deglutathionylation of pro-caspase-3 contributes to TNFα-induced apoptosis. Importantly, this study was the first to demonstrate glutathionylation of pro-caspase-3 and its effect on susceptibility to cleavage, and this report also identifies caspase-3 as another protein that exists in a glutathionylated state under resting conditions, becoming deglutathionylated by Grx1 in response to an ROS-generating stimulus.
Still, there are some difficulties in interpreting the results of this work. First, are the effects on caspase-3 glutathionylation due to TNFα or CHX? This is difficult to answer because the treatment given to control cells is not explicitly stated. Second, how closely does the TNFα concentration given to BAECs compare to circulating levels in diseased vessels? Here, a dose response curve of response to TNFα would be informative. Finally, this study raises an important question about the potential role of Grx in atheroprotection. In the case of Ras, Grx levels were correlated with decreased Ras–SSG and decreased hypertrophic signaling. In the case of pro-caspase-3, however, Grx overexpression (and deglutathionylation) led to increased apoptosis, a presumably atherogenic event. Taken together, these results highlight the fact that the role of Grx in cardiovascular disease may not be entirely straightforward, with its roles in disease protection or progression dependent upon cell type, extracellular stimuli, etc.
X. Implications of Protein S-Glutathionylation in Lung Disease
Oxidative stress is implicated in the etiology of various lung diseases, including chronic obstructive pulmonary disease (COPD), (245), asthma (253), cystic fibrosis (35), fibrotic diseases (313), and cancer (203). Although oxidative stress has been linked to increased protein glutathionylation in various experimental models [e.g., cardiac ischemia-reperfusion (79), H2O2 treatment of HeLa cells (290) or lymphocytes (41)], alterations in protein–SSG status and/or Grx activity in diseases of the lung remain largely unexplored. Below, we discuss preliminary evidence that protein glutathionylation and/or alterations in Grx activity may affect progression of various lung diseases; however, in most cases the specific roles and/or contributions of protein glutathionylation (or altered Grx activity) in disease etiology remains uncertain. These considerations are illustrated in Fig. 6.
FIG. 6.
Evidence for potential roles of protein glutathionylation and Grx activity in lung diseases. This figure depicts the major conclusions of studies focused on protein glutathionylation and/or Grx in specific lung diseases, or in cell lines derived from different regions of the lung.
A. Tobacco exposure
Muscat et al. (209) reported that the serum concentration of glutathionylated proteins is greater in smokers than in patients who never smoked, with protein–SSG concentrations correlated to cigarettes smoked per day and plasma concentrations of nicotine and cyanide metabolites. Whether a similar increase in protein–SSG content occurs within the lungs of smokers has not yet been reported. Nor is the significance of serum protein glutathionylation status well understood. For example, do glutathionylated serum proteins serve as markers of disease, or do they contribute to disease etiology, or both? Pertinent to this question is the identity of the serum glutathionylation targets. One could imagine that glutathionylation of hormone- or LDL-binding proteins (e.g., apoB100, see Cardiovascular section) may alter conformation and subsequent biological activity, having a greater impact on disease progression than glutathionylation of a protein such as albumin. Thus, identification of serum proteins that undergo increased glutathionylation in smokers, along with correlation of glutathionylation with a change in protein function, would provide insight into the contribution of glutathionylation events to tobacco-related lung disease.
A potential role for protein glutathionylation in the bronchial response to tobacco exposure is implied by Yoneda et al. (330), who observed a 10-fold induction of Grx1 mRNA during the first 10 h of exposure of a bronchial epithelial cell line to tobacco smoke. However, Grx1 protein levels and intracellular glutathionlyation status were not reported, so it is difficult to deduce the potential role of protein glutathionylation in this response. Interestingly, Hackett et al. (111) report no change in Grx levels (isoform not specified) via microarray analysis of airway epithelial cells collected from smokers vs. nonsmokers. The basis for Grx induction during acute vs. chronic tobacco exposure, as well as the role of both Grx isoforms in response to smoking, remain open questions for future study.
B. Hyperoxic injury
A role for Grx1 in hyperoxic lung injury was investigated by Ho et al. (120), who exposed WT and Grx1 KO mice to >99% O2 for 72 h. Lung hyperoxia has been shown to induce ROS production in mitochondria (90), and typically results in destruction of the alveolar epithelium, causing inflammation and edema (120). Grx1 KO did not alter the levels of hyperoxia-induced pulmonary edema or lung injury as measured by total protein and number of neutrophils in bronchoalveolar lavage fluid (BALF), leading the authors to conclude that changes in protein glutathionylation are probably not the basis for hyperoxia-induced lung injury. While this conclusion is a logical interpretation of the reported findings, it would be strengthened by determination of protein glutathionylation status in lungs from WT and Grx1 KO animals, particularly upon exposure to hyperoxia. In addition, utilization of a tissue-specific, inducible Grx1 KO animal model would obviate the possibility of compensatory mechanisms during development that could mask the effects of Grx and/or protein glutathionylation in acute response to pulmonary hyperoxia. Therefore, the potential roles of protein glutathionylation (and Grx) in response to hyperoxic lung injury remain uncertain.
C. Inflammation
While embryonic knockout of Grx1 did not contribute to hyperoxic lung injury (120), it appeared to confer an anti-inflammatory phenotype within the tracheal epithelium. Primary tracheal epithelial cells isolated from Grx1 KO mice exhibited decreased nuclear RelA, decreased LPS-induced NF-κB DNA binding, and decreased expression of NF-κB target genes macrophage inflammatory protein 2 and keratinocyte-derived chemokine (251). These effects were attributed to the inhibitory effect of glutathionylation on IκB kinase beta (IKKβ), demonstrated separately in a cell line derived from murine alveolar epithelium. To our knowledge, this work represents the only study performed in a pulmonary cell line in which regulation of a target protein by glutathionylation has been well documented. However, the role of IKK glutathionylation in inflammatory diseases of the lung is not yet established. This question could be addressed by determining NF-κB pathway activity, Grx1 activity, and the glutathionylation status of IKK, (as well as other NF-κB signaling pathway components potentially regulated by glutathionylation, see sections on diabetes and cancer) in models of pulmonary diseases associated with inflammation, such as pneumonia, asthma, and cystic fibrosis.
D. Fibrotic and granulomatous diseases
A role for protein glutathionylation in fibrotic and granulomatous lung disease was addressed indirectly by Peltoniemi et al. (232), who compared Grx1 expression levels in histological samples from healthy patients to those from patients with sarcoidosis, allergic alveolitis, and usual interstitial pneumonitis (UIP). In healthy lung, Grx1 was found primarily in alveolar macrophages and, to a lesser extent, in bronchial epithelium. Both granulomatous diseases (sarcoidosis and allergic alveolitis) were associated with decreased Grx1 immunoreactivity in macrophages and bronchial epithelium; however, the effect of UIP on Grx1 in these areas was not reported. In UIP, Grx1 staining was performed only in fibroblast foci, which exhibited negative to weak staining for Grx1. Importantly, this study was the first to explore the cell-type-specific expression of Grx isoforms in human lung, and documented decreased Grx1 protein expression in two granulomatous diseases. Important next steps include confirming that the decrease in Grx1 expression corresponds to a decrease in function, and identifying targets of glutathionylation that may be differentially regulated as a result of diminished Grx1 in granulomatous disease.
E. Chronic obstructive pulmonary disease (COPD)
A subsequent study by the same research group explored variations in Grx expression patterns associated with COPD (233). In lung homogenates and alveolar macrophages collected from COPD patients, Grx1 levels were slightly lower than in samples from healthy patients. In sputum samples, the opposite pattern was observed, with samples from COPD patients being somewhat enriched in Grx1. Interestingly, there was a positive correlation between the percentage of alveolar macrophages that stained positively for Grx1 and independent measures of lung function, such as forced expiratory volume in one second (FEV1). While the basis for this correlation is unknown, it provides a springboard for future studies.
What could explain the altered distribution of Grx1 between macrophages and the extracellular space in COPD? The authors speculate that in COPD, Grx1 is actively secreted from macrophages into the sputum and/or plasma, where it restores protein function via catalysis of deglutathionylation, simultaneously replenishing GSH in the oxidatively challenged airway. While this model is intriguing, evidence supporting each of the proposed events is limited, including (a) active secretion of Grx1 from macrophages, (b) maintenance of the deglutathionylation activity of Grx1 in the oxidative extracellular environment, and (c) the presence and activity of GSSG reductase and NADPH (required for reduction of the Grx reaction product GSSG to GSH) within the extracellular spaces of plasma and sputum. Besides confirming each of these events, the proposed role for Grx1 will require demonstration that the small increase in extracellular Grx1 observed in COPD patients is sufficient to contribute significantly to slowing disease progression.
An alternative interpretation to the observations of Peltoniemi et al. is that Grx1 content may increase in cell types other than alveolar macrophages. Indeed, the increase in Grx1 within lung homogenates of COPD patients appears greater than the increase observed in alveolar macrophages alone. Since Grx1 levels are low or undetectable in nonmacrophages from healthy lungs (232), a small increase in expression within the other cell types could confer significant functional effects. Therefore, it would be informative to compare Grx1 levels in other pulmonary cell types in patients with and without COPD.
F. Summary
Cigarette smoking is linked to an increase in global serum protein glutathionylation (209), although it is not yet known whether this change in protein glutathionylation status serves primarily as a disease marker, contributes to tobacco-induced lung disease, or both. Glutathionylation of IKK in tracheal epithelial cells inhibits TNFα- and LPS-induced NF-κB activation (251), suggesting that Grx may regulate inflammatory signaling in other lung diseases, such as infection, asthma, and cystic fibrosis. Grx1 is decreased in alveolar macrophages and bronchial epithelium in granulomatous diseases (232), although the specific role for protein glutathionylation in disease progression (or defense) remains unknown. Likewise, the basis for the correlation between Grx expression and lung function (233) remains uncertain; however, it does suggest potential roles for this enzyme (and reversible protein S-glutathionylation) in normal pulmonary function. Frontiers in the field of protein S-glutathionylation in pulmonary disease are similar to those identified for cardiovascular disease, including understanding connections between protein–SSG formation, Grx activity, and specific pathways that contribute to (or protect from) lung disease. Another emerging question is whether glutathionylated proteins in lung (or in the serum of those with lung disease) are inert clinical markers of disease or etiological participants in the disease process.
XI. Implications of Reversible Protein S-Glutathionylation in Cancer
Under normal physiological conditions, cells maintain a redox buffer consisting of high concentrations of antioxidants and antioxidant enzymes to minimize the oxidative action of ROS/RNS. Perturbations to this cellular redox state can lead to oxidative stress which has been acknowledged as a significant contributing factor to the pathogenesis of numerous human degenerative diseases, including cancer. Cancer cells differ from normal cells as they exhibit increased intrinsic oxidative stress, due to a number of factors including oncogenic stimulation, increased metabolic activity, and mitochondrial dysfunction (110).
Early characterization of tumor metabolism by Otto Warburg in 1924 showed that cancer cells exhibit two- to threefold increased glycolysis compared to normal cells, resulting in decrease in mitochondrial ATP formation (7). According to Warburg, cancer should be interpreted as a mitochondrial dysfunction, whereby cancerous cells exhibit an abnormally high ratio of glycolysis to respiration (Warburg effect). In fact some tumor cells can survive without functional mitochondria. Mitochondrial inactivation is an adaptive mechanism and is beneficial for carcinogenesis as it removes the mitochondrial apoptotic pathway and simultaneously decreases the toxicity of some chemotherapeutic drugs (taxol, gemcitabine) which rely on mitochondrial function (268). Recently, the biochemical mechanism for the Warburg effect was discovered showing that the embryonic M2 splice isoform of pyruvate kinase is exclusively present in cancer cells and is essential for the shift in cellular metabolism to aerobic glycolysis and this promotes tumorogenesis (42). Most solid tumors are characterized by regions of hypoxia. Thus, the Warburg effect is also interpreted as a consequence of the tumor's hypoxic environment making glycolysis the viable metabolic pathway for energy production. Interference with redox pathways by abnormal ROS/RNS levels is probably a key event in carcinogenesis. Whereas high concentrations of ROS/RNS can be toxic to cancer cells, substantial evidence suggests that lower concentrations are implicated in tumor progression (100). There is a growing body of evidence suggesting that ROS act as secondary messengers in intracellular signaling cascades that induce and maintain the oncogenic phenotype of cancer cells (84).
A. Thiol oxidation and cancer
ROS/RNS can react with and damage major intracellular macromolecules like proteins, nucleic acids, lipids, and carbohydrates. Inherent oxidative stress in cancer tissues may lead to reversible or irreversible oxidative modifications of cysteine residues on sensitive proteins causing alterations in the activity or function of the protein; (e.g., S-glutathionylation may occur within their catalytic centers or as part of protein–protein interaction interfaces). It has been reported recently that PTP1B which regulates tyrosine phosphorylation and is implicated in the development of cancer, undergoes reversible and irreversible thiol oxidation to sulfenic, sulfinic, and sulfonic acid derivatives that inactivates the protein in HepG2 and A431 cells (177). Dual-specific phosphatase Cdc25, which is overexpressed in different tumors, is reported to be highly susceptible to oxidation at the active site cysteine. The rate of thiolate conversion to sulfenic acid by H2O2 for Cdc25 was 15- and 400-fold faster than for PTP1B and GSH, respectively (280). Thiol oxidation has been shown to regulate the activity of various proteins involved in signal transduction, such as Ras, Jun N-terminal kinase (JNK)-2, c-Jun NF-κB, AP-1, PKC, caspase, and thioredoxin, all of which have important roles in the pathogenesis of cancer (84).
B. S-Glutathionylation and signal transduction in cancer
An early report of oxidation of cysteine thiols in tumor cells was published in 1968 (325). More recently, Cotgreave and Gerdes reviewed the relationship between protein–S-glutathionylation and cell proliferation (53). Over the past decade there have been numerous reports stating S-glutathionylation can modify several proteins and transcription factors some of which are involved in the cellular signaling of cancer (27). Mutations resulting in dysfunctional signaling pathways are frequently observed in cancerous cells. Figure 7 displays key signal transduction pathways in which regulation by reversible glutathionylation of signaling intermediates has been implicated.
FIG. 7.
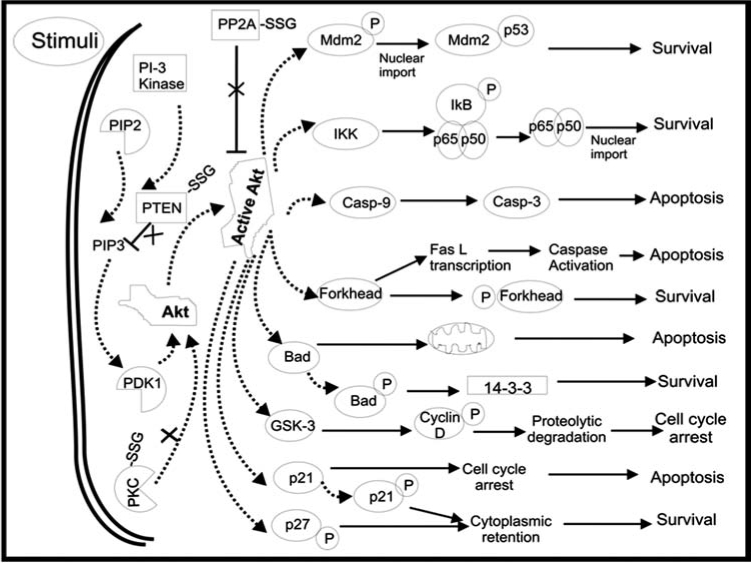
Apoptosis/survival signaling pathways that are regulated by reversible S-glutathionylation. This figure displays key signal transduction pathways in which regulation by reversible glutathionylation of signaling intermediates has been implicated. Target proteins that may play regulatory roles, and for which alteration in function due to glutathionylation has been reported in some context, are designated as protein-SSG [e.g., protein phosphatase 2A (PP2A)-SSG, phosphatase and tensin homologue deleted from chromosome 10 (PTEN)-SSG, protein kinase C (PKC)-SSG].
C. S-Glutathionylation and modulation of kinase/phosphatase signaling pathways
The state of unregulated cell growth, a typical characteristic of cancer, can occur by overactivation of proliferation and corresponding inhibition of apoptosis. A major signal transduction process that controls tumorogenesis involves kinase/phosphatase signaling pathways, described as follows.
1. Protein kinase C (PKC)
One of the prominent phosphorylation mechanisms in a cell is mediated by PKC, a family of ten Ser/Thr kinases that is involved in a variety of pathways that regulate cell growth, death, and stress responsiveness (271). PKCs control key events in tumor promotion and progression by modulating mitogenesis, cell adhesion, apoptosis, angiogenesis, invasion, and metastasis. Overall increased PKC levels are associated with malignant transformation in several cancer cell lines including breast, lung, and gastric carcinomas (218, 250, 266).
Phosphorylation, subcellular localization, and S-glutathionylation are the key modes of PKC regulation. Several unique structural aspects of PKC make it highly susceptible to oxidants, like the presence of cysteine-rich regions in the regulatory as well as catalytic domains, which are required for functional kinase activity. Human PKC isozymes have been found to contain 16–28 Cys residues that are distributed throughout the protein sequence (27). This includes one or two Cys-rich zinc finger regions (six cysteines per region) in the regulatory domain, and 5–8 Cys in the catalytic domain. Unlike the zinc–thiolates in the regulatory domain, the catalytic domain cysteines are uncoordinated and free to react with sulfhydryl modifying agents. Chemical modification in the catalytic domain inactivates PKC and in the regulatory domain activates it. In a study involving seven PKC isoenzymes, it was demonstrated that the thiol-specific oxidant diamide could induce S-glutathionylation in all the isoenzymes resulting in the formation of PKC-SSG (44). Humphries et al. reported that Cys499 of PKCα is S-glutathionylated which leads to its inactivation (124). Several other PKC isoenzymes (PKCβ1, β2, γ, δ, ɛ, and ζ) as well as protein kinase D were inactivated by S-glutathionylation, with PKC δ showing the most resistance (273). S-glutathionylation of one to three redox sensitive cysteines is sufficient to fully inactivate the kinase activity of most PKC isozymes (43). In view of the large cysteine contents in the catalytic domain of PKCs, it is expected that PKCs are highly susceptible for inactivation by S-glutathionylation. Activated PKCs can modulate several signal transduction cascades that are important in carcinogenesis and its inhibition by S-glutathionylation can modulate cancer cell survival or death. Due to its dual role in cell signaling, PKC can be a functional target for anticancer agents as well as tumor promoters.
2. PI3-kinase and Akt
Akt [also known as protein kinase B (PKB)] is a serine/threonine kinase that has an important role in cancer as it has a key position in the signaling cascade at the branchpoint at which the apoptotic and proliferative pathways diverge (Fig. 7). The initial stages of Akt pathway are the activation of membrane bound receptors which then recruit PI3-kinase (PI3K) that further recruits Akt to the membrane. At the membrane, Akt is activated by phosphorylation at Thr308 and Ser473 (58) by kinases including PDK1 (phosphoinosityl dependent kinase 1) and PKCs. This releases Akt from the membrane and the protein is translocated to the cytosol and the nucleus. Activated Akt phosphorylates a variety of substrates like IKK (involved in NF-κB pathway), murine double minute 2, also known as mdm2 (involved in p53 pathway), Bad (a proapoptotic member of the Bcl-2 family), caspase 9 (mitochondrial apoptotic pathway), forkhead transcription factors (death receptor pathway), GSK-3 (glycogen synthase kinase-3), p27 and p21 (cell cycle regulation) (100), all of which play significant roles in cancer. The activation of Akt increases both the rate of cellular proliferation and the resistance of cells to apoptosis.
The activity of Akt is directly or indirectly modulated by S-glutathionylation. The Akt pathway is negatively regulated by several phosphatases. The tumor suppressor PTEN (phosphatase and tensin homologue deleted from chromosome 10) directly opposes PI3K and is an important target for redox regulation. In a recent report it was suggested that in macrophages ATP-induced ROS inactivate PTEN by glutathionylation, which shifts the equilibrium towards the PI3K and activates the Akt pathway (56). Further downstream, Akt can be dephosphorylated by serine/threonine protein phosphatase PP2A which has been also reported to be regulated by S-glutathionylation. Evidence suggests that in human colon carcinoma Caco-2 cells, PP2A is inhibited by H2O2 and the process involves glutathionylation (247). Akt deactivation by phosphatses can trigger multiple cell death pathways. Since S-glutathionylation inactivates the phosphatases of the Akt pathway, it serves as a mechanism leading to the activation of the cell survival pathways in cancer. Therefore increasing glutathionylation of the phosphatases of the Akt pathway via inhibition of glutaredoxin would represent a strategy for chemoprevention.
3. Protein tyrosine phosphatase (PTP)
Protein tyrosine phosphatases regulate signal transduction pathways involving tyrosine phosphorylation, implicated in the development of cancer. Phosphorylation of tyrosine residues of various target proteins has been found to be mediated by generation of ROS (27). Lee et al. showed that EGF stimulation of human epidermoid carcinoma cells, A431 leads to ROS-mediated reversible inactivation of one of the PTP isoforms, PTP1B (165). Later Barrett et al. demonstrated that PTP1B was oxidized at its Cys215 to sulfenic acid which reacted with GSH to form a more stable mixed disulfide PTP1B-SSG (20). Furthermore they also documented by MS analysis that PTP1B isolated by immunoprecipitation from EGF-stimulated A431 cells was S-glutathionylated at its active site Cys215 which resulted in the inactivation of the phosphatase (19). Most recently, Lou et al. reported that in reponse to ROS produced constitutively by HepG2 and A431 human cancer cells resulted in oxidative inactivation of endogenous PTP1B. They also reported mass spectral analyses showing reversible and irreversible oxidation specifically on the Cys215 residue, whereas the other five cysteines of the PTP1B remained reduced (177). The authors reported the relative stochiometry of the reversible and irreversible thiol oxidation states of Cys215 that they analyzed. In HepG2 cells, Cys-SH (32%), Cys-SOH (25%), Cys-SO2H/Cys-SO3H (43%); and in A431 cells Cys-SH (13%), Cys-SOH (49%), Cys-SO2H/Cys-SO3H (38%). In the intracellular milieu, where GSH is abundant, the sulfenic acid (Cys-SOH) is expected to be converted facilely to the S-glutathionylated form, as shown previously by Barrett et al. in EGF-treated A 431 cells (20). Therefore, it is not clear why at least some amount of PTP1B-Cys215-SSG is not reported or discussed in the context of the study of the constitutive state of PTP1B in the same A431 cells (177). Nevertheless, reversible oxidation and S-glutathionylation of phosphotyrosine phosphatases resulting in their inactivation are likely regulatory mechanisms in tumor signal transduction.
4. c-Jun N-terminal kinase (JNK)
JNK, a stress activated protein kinase (SAPK), has been implicated in pro-apoptotic signaling in cancer and may mediate the cytotoxicity of a variety of chemotherapeutic agents. JNK is a member of the MAPK family of protein kinases controlled by the serial activation of three upstream elements of a protein cascade. p38 MAPK and SAPK/JNK triggers the pro-apoptotic pathway, whereas ERK/MAPK signals the pro-survival pathway. In general, oxidative stress is correlated with the activation of the SAPK/JNK, p38 MAPK as well as ERK/MAPK kinases, but the relative control of one pathway over the other determines proliferation or apoptosis of the cancer cells and also their susceptibility or resistance to chemotherapeutics. The first characterized and well-known JNK kinase kinase is MEKK1 (MAPK/ERK kinase kinase 1), which is known to phosphorylate and activate the MKK4. ASK-1 (apoptosis signal-regulating kinase 1) is also reported to phosphorylate MKK4 at identical sites (SAPK/ERK kinase/mitogen-activated protein kinase kinase 4) which then further downstream activates JNK. ASK-1 has a clear role in signaling JNK towards apoptosis whereas activation of MEKK1 is known to transmit a survival signal. Using in vitro assay as well as direct analysis of MEKK1 purified from H2O2 and menandione-induced human prostate cancer cells LNCap, Cross and Templeton reported that the inhibition of MEKK1 involves S-glutathionylation at Cys1238, which is present in the ATP-binding pocket of the kinase, thus interfering with the catalytic activity of the enzyme (55). In another context ASK-1 was reported to be activated under oxidative conditions according to a mechanism involving dissociation of thioredoxin from a binding site at the N-terminus of ASK-1 which is inhibitory (260). In breast cancer and prostate cancer cells, Grx1 also has been reported to bind ASK-1, but at its C-terminus (281); and this association is also disrupted under oxidative conditions resulting in ASK-1 activation and stimulation of downstream pathways like JNK and p38 (282). These alternative mechanisms for regulation of MEKK and ASK-1 may explain the paradox whereby both MEKK1 and ASK-1 phosphorylate MKK4, the upstream kinase of JNK, ASK-1 promotes apoptosis while MEKK1 provides a cell survival signal. Thus, increasing S-glutathionylation and concomitant inactivation of MEKK1 by inhibiting glutaredoxin could represent a therapeutic approach against certain cancers (Fig. 8). Another mechanism for inactivation of JNK has been proposed, involving interaction with GSTpi. Various glutathione S-transferases (GST-alpha, -mu, -pi and -theta) have been reported to play a regulatory role in cell signaling by associating with critical kinases involved in cellular responses to stress, apoptosis, and proliferation (318). In particular, GST-pi was reported to bind and inhibit JNK. In unstressed cells, low JNK catalytic activity is maintained through its sequestration within a protein complex that includes GST-pi, JNK, and c-Jun. Under oxidative or nitosative stress (a common feature of cancer cells), all three of the above proteins are susceptible to S-glutathionylation, apparently disrupting the GSTpi–JNK complex so that free JNK is able to act on downstream gene targets like c-Jun; and GSTpi self-associates as oligomers (301). GSTpi undergoes S-glutathionylation at Cys47 and Cys101, both residues being crucial for the interaction with JNK.
FIG. 8.
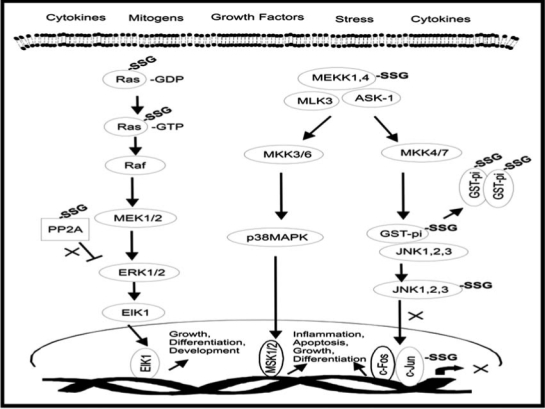
S-glutathionylation as a regulator of the JNK and MAPK signaling pathways in cancer. This figure depicts several sites in the JNK pathway and in other signaling pathways in cancer cells where reversible S-glutathionylation may serve as a potential regulatory mechanism (e.g., Ras-SSG, MAPK/ERK kinase kinase 1 (MEKK1)-SSG, c-Jun-SSG, glutathione S-transferase (GSTpi-SSG).
JNK activation has also been reported to be mediated by sulfenic acid modification in immortalized ovarian cancer cells (HeLa cells) following ROS generation by TNFα (141). In this report the authors reported that TNFα-induced ROS cause the oxidation and inhibition of JNK-inactivating phosphatases like PTPs by converting their catalytic cysteines (Cys293) to sulfenic acids. This resulted in sustained JNK activation that then could trigger the apoptotic cascade. In the intracellular environment of high GSH, it expected that the sulfenic acids would be readily converted to S-glutathionylated mixed disulfides, but only the sulfenic acid derivatives were reported in this study. Figure 8 depicts several sites in the JNK pathway and in other signaling pathways in cancer cells where reversible S-glutathionylation is a potential regulatory mechanism.
5. H-Ras
Ras is a member of the small GTPase super-family, its activity depending on the binding of GTP. Mutations of the Ras proto-oncogene (H-Ras, N-Ras, K-Ras) are found in ∼25% of all human tumors. Most of these mutations result in abrogation of normal GTPase activity of Ras. The Ras mutants can still bind to GTPase-activating protein (GAP), but cannot catalyze GTP hydrolysis. Malignant transformation may arise from the unregulated stimulation of Ras signaling pathways, which either stimulate growth or inhibit apoptosis. H-Ras has been implicated both as an initiator of oxidant-related signaling as well as a target of oxidative regulation. It has been suggested that the oxidative signals are relayed to the extracellular signal-related kinase (ERK) pathway through H-Ras. Out of the six cysteine residues of H-Ras, four (118, 181, 184, and 186) are exposed to the surface and can be potentially oxidized by oxidative and nitrosative stresses during normal and pathological cellular events (184). In vascular smooth muscle cells following angiotensin II treatment, there is S-glutathionylation of H-Ras at Cys118, which is an activating event (1). The finding that Cys118, serves as a redox-sensitive switch that mediates Ras activation by S-glutathionylation has broad implications for redox regulation of cell signaling and cancer. Though H-Ras is an important protein in the regulation of cancer, there are no studies of S-glutathionylation of Ras in cancer cells and hence definitely deserves attention.
D. S-glutathionylation and modulation of the proteasome pathway
Several signaling pathways in cancer, which involve critical cellular processes like cell cycle, cytokine-induced gene expression, differentiation, cell death, and stress response are known to be under regulation by the ubiquitin–proteasome pathway. Proteins once ubiquitinated are targeted to the pro-teasomes for further proteolytic cleavage in a cascade involving three enzymes E1, E2, and E3 (the proteolytic enzyme component of the proteasomes). C-Jun, NF-κB, and p53 are some of the well-known transcription factors that are involved in cell death and survival in cancer and are known to be regulated by the ubiquitin–proteasome pathway (49, 109). It has been demonstrated that the E1 and E2 components of the ubiquitin pathway are reversibly inhibited by S-glutathionylation during oxidative stress (219), which causes a shut down of the proteosomal degradation process. Glutathionylation of ubiquitin ligases can result in cancer promoting as well as cancer prevention effects. If ubiquitination and proteasomal degradation are a requirement for activation of a survival pathway [e.g., the NF-κB pathway (see below)], then glutathionylation/inactivation of ubiquitin ligases would have a tumor inhibitory effect. If instead ubiquitination and proteosomal degradation lead to inactivation of any of the proteins involved in a pro-apoptotic pathway like JNK/cJun, then glutathionylation and inhibition of ubiquitin ligases would have a tumor promoting effect.
There is some evidence that the ubiquitin–proteasome system is also involved in elimination or inactivation of carcinogenic species, suggesting a strategy for cancer chemo-prevention. The Kelch ECH associating protein 1-nuclear factor-E2-related factor 2-antioxidant response element (Keap 1-Nrf2-ARE) signaling pathway is an example (329). Under physiological conditions, Nrf2 is sequestered in the cytoplasm as an inactive complex with Keap1. Under oxidative stressed conditions, Keap 1 undergoes ubiquitination that allows Nrf2 to translocate to the nucleus, inducing the expression of antioxidant enzymes via its binding to ARE/EpRE (210). The Nrf2-mediated response diminishes susceptibility to carcinogenesis, acute chemical toxicity, oxidative stress, asthma, acute inflammation, septic shock, and neurodegenerative diseases. Hence inactivation of ubiquitin ligases by S-glutathionylation could have a negative impact on elimination and inactivation of carcinogenic species by inhibiting the Nrf pathway.
E. S-Glutathionylation modulation of transcription factors (c-Jun, NF-κB, p53, AP-1)
Many redox-responsive transcription factors, such as AP-1, p53, NF-kB, cyclic AMP-response element binding protein (CREB), and Sp-1 (specificity protein-1, involved in gene expression during early development), have conserved cysteines that are susceptible to thiol modification. A growing body of data indicates that modification of cysteine moieties in some redox-sensitive transcription factors interferes with their binding to DNA, thereby regulating target gene expression. Here we briefly review the effect of S-glutathionylation on transcriptional activity of NF-κB, c-Jun, AP-1, and p53.
1. NF-κB
NF-κB belongs to the Rel family and plays an important role in the transcription of anti-apoptotic genes in cancer. Among the five subunits that may comprise dimeric NF-κB, Rel B, and C-Rel activate transcription, and p50 and p52 possess DNA-binding properties. Rel A (p65) is known to participate in both DNA binding and transcription. The inactive form of NF-κB is localized in the cytoplasm through interaction with IκB repressor proteins (144). Upon oxidative stimulation, these IkB proteins are rapidly phosphorylated by IkB kinase α (IKKα) and β (IKKβ) and degraded via the ubiquitin–proteasome pathway. NF-κB then becomes free to translocate to the nucleus, where it binds to DNA and activates various anti-apoptotic genes, including Bcl-xL, Bcl-2, A1, tumor necrosis factor receptor-associated factors 1 and 2, cellular inhibitors of apoptosis (cIAP), and X chromosome-linked inhibitor of apoptosis (XIAP/hILP). NF-κB is activated during hypoxia and its inhibition by GSH supplementation induces apoptosis in mouse embryonic fibroblasts (MEFs), pancreatic adenocarcinoma, as well as lung cancer cells (242, 243). The NF-κB survival pathway has been shown to be regulated by S-glutathionylation at multiple steps. IKKs are upstream kinases that phosphorylate IκBs as well as p65-NF-κB itself. Raynaert et al. reported that, in alveolar epithelial cells, IKKβ is a prime target of oxidative inactivation by H2O2 through S-glutathionylation at Cys179, which likely occurs through a sulfenic acid precursor (251). Cys179 S-glutathionylation inactivates IKKβ and hence represses IκBα degradation as well as NF-κB transactivation. In contrast, in pancreatic cancer cells exposed to hypoxia and GSH supplementation (by N-acetylcysteine, NAC), the control point for inhibition of p65-NF-κB lies downstream of IκBα degradation. In these cells, nuclear translocation is not affected; instead, the data support a mechanism whereby p65-NFκB itself undergoes Grx-mediated S-glutathionylation, which inhibits p65-DNA binding and transcription of survival genes (242). Another group reported that p50–NF-κB is susceptible to S-glutathionylation on Cys62, thereby inhibiting DNA binding (237). Thus therapeutic strategies that would selectively enhance S-glutathionylation of components of the NF-κB pathway would be effective in treating cancers that depend on NF-κB as their major survival pathway.
2. AP-1, c-Jun
c-Jun is the major component of the AP-1 transcription factor that regulates a large number of genes involved in cell proliferation, differentiation, and apoptosis, thus playing a critical role in cancer. AP-1 is either homo- or heterodimers consisting of the members of Jun (c-Jun, Jun B, and Jun D) and Fos (c-Fos, FosB, Fra-1, and Fra-2) families, which interact each other through their basic leucine-zipper domain. Regulation of cellular AP-1 activity occurs via two mechanisms: one is an increase in the transcription of c-fos and c-jun, and the other is the phosphorylation of c-Fos and c-Jun proteins. The reduced state of critical cysteines present in the DNA-binding domain of both AP-1 and c-Jun is essential for effective DNA binding. Klatt et al. reported that binding of AP-1-c-Jun subunit to the DNA depended on the cellular GSH/GSSG ratio (156). A decrease in GSH/GSSG ratio provided a redox potential that induced S-glutathiolation of c-Jun at Cys269 as well as formation of an intermolecular disulfide bridge between conserved cysteines of the dimerization domain of the DNA binding site. Binding site analysis demonstrated that S-glutathionylation sterically blocked DNA binding. However, these experiments for S-glutathionylation were performed with isolated recombinant c-Jun. Hence it is not known whether this modification of c-Jun occurs intracellularly. Nonetheless, the observation has implications in cancer since a variety of carcinogenic insults, including phorbol ester and UV irradiation, stimulate AP-1 transcriptional activity (291). Therefore, suppression of abnormal activation of AP-1 through S-glutathionylation of c-Jun would be important from a therapeutic standpoint, but it requires more study.
3. p53
The tumor suppressor p53 is mutated in roughly 30%–50% of human cancers. p53 has been known to plays a vital role in maintaining genomic integrity by cell cycle control, DNA repair, differentiation, and apoptosis. Mice deficient in p53 expression develop normally but are highly susceptible to cancer, whereas mice that overexpress p53 exhibit a short lifespan and develop age-associated phenotype, including osteoporosis and organ atrophy (126, 289). p53 has 10 cysteine residues, all of which are present within the DNA-binding region and mutagenesis of these cysteines leads to complete loss of DNA binding (246). Several studies demonstrate that cysteine oxidation inhibits p53 DNA binding (271)
p53 mediates cell cycle regulation, DNA repair, and the cell's response to various stresses including oxidative stress, DNA damage, and chemotherapeutics. Recently, it was reported that p53 is S-glutathionylated in human colon cancer cells, and the sites of glutathionylation were identified by mass spectral analysis as the C124, C141, and C182, all present in the proximal DNA binding domain (310). Glutathionylation of p53 is also implicated in inhibition of oligomerization, thus preventing transcriptional activation (310). Marginal p53 glutathionylation was observed in the cancer cells under physiological thiol concentration, but it was increased during oxidant (H2O2) or anticancer drug (campothecin or cisplatin) treatment (310). Glutathionylated p53 was observed also in the nucleus, but p53-SSG is incapable of binding to transcription factors (310). Since p53 is a key player in the expression, of many proapoptotic genes (PUMA, Bax, etc.), it is tempting to speculate that inactivation of the tumor suppressor by glutathionylation may represent an acute adaptation strategy for suppressing the apoptotic signals generated in stressed cells; however, further studies are required to test this hypothesis.
4. Caspases
Caspases, a family of cysteine proteases, play an integral part in the execution phase of apoptosis and hence are important in cancer cell signaling. In this regard, there is one report that procaspase-3 cleavage is inhibited by S-glutathionylation, and its reactivation is regulated by glutaredoxin (227). Whether an analogous regulatory mechanism may be operable in cancer cells remains to be seen.
5. Ku
Ku is a multifunctional DNA repair protein that exists as a heterodimer, consisting of Ku 70 and Ku 80. Ku is also characterized as an anti-apoptotic factor, and as a participant in maintenance of telomere length. A recent minireview summarized the roles of Ku in cell proliferation and apoptosis in gastric cancer and pancreatic acinar cells (153). Under stressed conditions, decreases in nuclear Ku 70 and Ku 80 tip the balance toward apoptosis. There are various pathways that could lead to loss of Ku in the nucleus and inhibit its DNA binding and anti-apoptotic functions. Several lines of evidence suggest that glutathionylation could regulate Ku, at least indirectly. For example, under oxidative stress conditions in pancreatic acinar cells Ku 70 and Ku 80 were shown to be degraded by activated caspase-3 (153). Hence S-glutathionylation could regulate Ku indirectly under conditions where procaspase-3 activation is blocked by glutathionylation (described above). The ubiquitin–proteasome pathway also plays a role in decreasing Ku 70 levels in drug-induced apoptosis (94), implying that S-glutathionylation of ubiquitin ligases (described above) might also have an indirect effect on the anti-apoptotic function of Ku. Also Ku expression in gastric carcinoma cells is mediated by the NF-κB pathway which is known to be regulated by glutathionylation in other contexts (153, 237, 242, 251, 274). Ku also acts as a regulator of transcription by interacting with recombination signal binding protein Jκ and NFκB p50 homodimer to upregulate the p50 expression which may control proliferation of gastric cancer cells.
There is also one study that purports to provide evidence for direct glutathionylation on Ku. Here the authors described the role of glucose 6-phosphate dehydrogenase (G6PD) in control of NADPH production and global protein glutathionylation, and interpreted their results to relate glutathionylation to the loss of Ku 70 function (17). In G6PD-deficient cells where protein glutathionylation occurs to a greater extent in response to treatment with hydroxyethyl disulfide (HEDS), they observed inhibition of Ku binding to DNA, which may imply enhanced apoptosis. However, Ku binding to DNA was not affected by HEDS in the wild type or G6PD revertant cells, where there is less global protein glutathionylation. Thus, this article provides only indirect data for the interpretation that Ku 70-SSG formation occurs and is responsible for loss of Ku function, but several other interpretations are plausible (as described above).
F. Modulation of S-glutathionylation as a chemotherapeutic strategy for cancer
In humans, the time span between the first cellular modifications and diagnoses of cancer or death may involve several decades. Carcinogenic processes occur in three stages: initiation, promotion, and progression. Since cancer is usually not diagnosed until the progression phase, chemoprevention would have to cover almost the entire carcinogenic process and at later stages would not be specific of a causative carcinogen. Since reversible S-glutathionylation is likely to modulate an array of protein functions and signaling cascades that play an important role in tumor development, progression, and metastasis, it is an attractive target for drug design.
A primary reason for failure of cancer treatment and relapse in patients is the acquired or intrinsic resistance to anticancer therapies. Drug resistance in cancer has been attributed to various factors. Involvement of the PKC family in multidrug resistance was first reported in 1988 (88). Due to its widespread involvement in cancer and other cellular signaling processes, PKC, which is susceptible to regulation by S-glutathionylation, was considered as a viable target for cancer drug development. However, therapeutically targeting PKC has been unsuccessful at the Phase II and Phase III levels due to the complexity of the PKC family and their differing roles within different tumors (180).
Another factor responsible for drug resistance is the over-expression of detoxification enzymes like glutathione-S-transferases (GSTs). Over the last three decades it has been shown that chemoresistant tumors have an overexpression of GST isozymes which leads to accelerated detoxification of drug substrates causing the acquired resistance in cancer (196). GSTs are a family of Phase II detoxification enzymes that can regulate a variety of critical kinases like JNK, ASK1, MAPK, and it also has been interpreted that they may facilitate S-glutathionylation of target proteins under certain drug treatments (295); however this proposed mechanism is not well established (see ref. 92). Recent anticancer drug development programs using the platform of GSH, GSTs, and thiol homeostasis have demonstrated that, by promoting thiol oxidation locally, the tumor environment can be rendered toxic to malignant cells (100). Apart from designing GST inhibitors, another pharmacological approach is to design prodrugs which are inactive agents that are converted to active cytotoxics upon exposure to tumor tissues, minimizing toxicity towards surrounding normal tissues. Enhanced expression of GSTs in many tumors makes them a promising target for prodrug therapy. The GST-pi prodrug Telcyta (TLK286) has entered Phase III clinical trials (100). Two other agents in preclinical development utilize the prodrug approach and target S-glutathionylation of proteins. One is NOV-002, a pharmacologically stabilized form of GSSG, and the other is a nitric oxide (NO) releasing GST-pi activated prodrug PABA/NO, both of which can promote glutathionylation of a number of cellular proteins involved in cancer signaling (302, 303). Thus, therapeutic strategies that target S-glutathionylation may be particularly appropriate for incorporation into combination therapies and may offer new promise for cancer drug development. Figure 9 summarizes the potential glutathionylation targets that are implicated in cancer cell progression or apoptosis.
FIG. 9.

Potential targets of S-glutathionylation that are involved in cancer development and apoptosis. This block diagram lists various potential glutathionylation targets (central portion of the figure) and depicts how they are implicated in cancer cell progression or apoptosis (see text for further explanation).
G. Conclusions
Despite the substantial progress in identifying the biochemical events associated with the multistage process of carcinogenesis, the elucidation of molecular and cellular targets of chemopreventive agents still remains a major challenge. S-glutathionylation regulates a variety of cellular processes involved in cancer cell signaling. The list of S-glutathionylated proteins as well as the structural and functional significance of the modification is still incomplete. There is still a lot unknown in the field of S-glutathionylation and its significance in cancer. Future studies in understanding the mechanism and consequences of S-glutathionylation in cancer will not only improve our knowledge but also enable us to target this post-translational modification as a therapeutic strategy in cancer.
XII. Implications of Protein S-Glutathionylation in Neurodegenerative Diseases
Neurodegenerative diseases are characterized by a gradual but progressive loss of neurons. These diseases cause devastating effects on the patient and are currently only treated palliatively. The classical neurodegenerative diseases include AD, Parkinson's disease (PD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), and Friedriech's ataxia, each of which will be discussed further (see below). Multiple factors have been thought to contribute to neuronal loss through cell death including protein misfolding, excitotoxicity, activation of cell death pathways, mitochondrial dysfunction, increased iron deposition, and oxidative stress through the overexposure to RNS and ROS (29, 216). The major risk factor associated with neurodegeneration is aging (174). Aging is thought to allow for the accumulaton of mitochondrial DNA mutations coinciding with a diminished electron transport chain function and diminished production of ATP. Dysregulated cytosolic calcium and poor mitochondrial membrane integrity also occur with aging (216). These combined changes make an environment of elevated oxidative stress which must be counteracted by cellular antioxidants. As discussed above, S-glutathionylation of proteins is a posttranslational modification that appears to be vital for regulation of signal transduction in many cellular contexts. Oxidative stress, a hallmark of neurodegenerative diseases, promotes S-glutathionylation, resulting in disruption of normal signaling and potentially accelerating disease progression.
A. Oxidative stress and neurodegeneration
The brain is highly susceptible to oxidative stress as compared to other organs due to its requirement for excessive use of glucose for energy, as well as its inability to undergo cellular regeneration. These characteristics coupled with the major risk factor for neurodegeneration, aging, make it easy to conceive that oxidative challenges are involved in the disease process (6). Also, postmortem brain tissues from patients with neurodegenerative diseases show increased side effects of oxidative stress including oxidation of DNA, proteins, and lipids (127).
In neurodegeneration, oxidative challenges are met with an insufficient antioxidant defense mechanism. For example, in PD, glutathione, the cell's major thiol reducing agent, is diminished where the magnitude of depletion dictates the severity of the disease. Also in AD, antioixidant defenses are reduced including superoxide dismutase, catalase, and glutathione reductase (6). Paradoxically, in ALS, the superoxide dismutase gene (SOD1) is mutated causing overproduction of superoxide dismutase. Despite the elevation of SOD, transgenic mice expressing a familial variant of the SOD1 mutation known to produce ALS have increased lipid peroxidation, DNA oxidation, and protein oxidation within the ALS affected portion of the brain comprising the motor neurons (6).
B. Sources of reactive oxygen and nitrogen species in brain
The main source of reactive species within the brain is the mitochondrial electron transport chain. Because the brain requires ∼20% of the total body respiration despite its relatively small mass, the amount of ROS produced through leakage from the electron transport chain is meaningful (279). Through the electron transport chain the byproduct, superoxide, is produced mainly at complex I and complex III. Also, on the outer membrane of the mitochondria lies monoamine oxidase, which produces hydrogen peroxide as a byproduct of its oxidative deamination (174, 248). This may be of particular importance in PD with increased dopamine turnover increasing the amount of hydrogen peroxide produced through its oxidative deamination by monoamine oxidase B (222). Another source of ROS within the mitochondria occurs in the Krebs cycle, specifically through α-ketoglutarate dehyrogenase. Other sources of oxidants arise from NADPH oxidase, which is present in astrocytes, microglia, and neurons. Flavoproteins and other oxidases form hydrogen peroxide contributing to the overall oxidant content within the cell body. And lastly, inflammatory cells present within the brain are sources of RNS and ROS. Inhibition of nitric oxide synthase (NOS) has been shown to be neuroprotective, implicating NOS in the production of radicals which can produce peroxynitrite and hydrogen peroxide (127).
During neurodegenerative disease, aging is often a characteristic of the majority of patients. Aging affects mitochondria by increasing the number of mtDNA mutations and increased ROS production from the electron transport chain. The mutations may alter the accuracy of the electron transport chain resulting in increased production of ROS (249). Also, within aged neurons, calcium fluxes are less controlled, which creates a calcium overload leading to increased oxidative stress (192). Below is a discussion of major neurodegenerative diseases followed by the role of glutaredoxin and glutathionylation in the disease process.
1. Alzheimer's disease
Alzheimer's disease (AD) is the most common neurodegenerative disease where patients experience progressive cognitive deterioration, behavioral changes, and neuropsychiatric changes. Alzheimer's disease is characterized by amyloid β toxicity leading to senile plaques, mitochondrial dysfunction, and hyperphosphorylated Tau leading to neurofilbillary tangles, all of which contribute to oxidative stress (214, 216). It is noteworthy that mutations in presenilin, which plays a regulatory role with HIF-1α in pancreatic beta-cells, have been implicated in the development of AD and patients with type 2 diabetes are reported to develop amyloid plaques (76). Thus, there may be a connection between the oxidative stress conditions of AD and diabetes. Oxidative imbalances within AD lead to decreased abundance of free sulfhydryls on proteins from isolated hippocampal sections of AD postmortem brains as compared to controls. These postmortem samples from AD patients show an increased amount of glutathione reductase and glutathione peroxidase implicating an antioxidant defense response (4). Also, Grx levels are also upregulated in AD (5).
2. Parkinson's disease
Parkinson's disease (PD) is the second most common neurodegenerative disease characterized by loss of catecholaminergic neurons from the substantia nigra. PD patients experience bradykinesia, muscle rigidity, resting tremor, and postural instability (254). PD has been attributed to both genetic and environmental factors leading to overt oxidative stress, mitochondrial dysfunction, and protein aggregation (181). The pathogenesis of PD has been reported to include increased dopamine turnover, diminished glutathione content, and increase iron in the substantia nigra (222). Within PD, inhibition of complex I, the initial enzyme in the electron transport chain, impairs energy production, and increases oxidant production. Complex I inhibition has been shown as a hallmark characteristic of PD, and is currently the target of several chemical agents, including MPTP (N-methyl-4-phenyl-1,2,3,4-tetrahydropyridine), that are used to create animal models of PD (306).
3. Huntington's disease
Huntington's disease (HD) is an autosomal dominant disease that causes degeneration of medium spiny GABAergic neurons in the striatum. Its characteristic neurodegerentation causes a progressive atrophy of the caudate nucleus, putamen, and global pallidus. HD is a genetic disease caused by a CAG repeat expansion of exon 1 in the gene for the Huntington protein. Oxidative stress is present in HD as represented by increased DNA oxidative products and elevated DNA strand breaks. Oxidative challenges present with HD are thought to arise from poor mitochondrial membrane integrity. Complex II and complex III of the electron transport chain show impaired function (185). HD patients also show decreased ATP levels probably due to the inhibition of complexes II and III. An increase in cytosolic Ca2+ is also seen (216).
4. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a devastating disease resulting in progressive skeletal muscle weakness, muscle atrophy, paralysis, and ultimately death within 2–5 years of onset. ALS results from a progressive degeneration of motor neurons within the ventral horn of the spinal cord, brainstem, and motor cortex. Degeneration also occurs within the corticospinal tract. Approximately 20% of patients have a mutation in the superoxide dismutase 1 (SOD1) gene, however, the majority of patients have normal SOD1 activity. Other cellular characteristics of ALS include a loss of mitochondrial membrane potential and increased cytosolic calcium (216).
5. Freidreich's ataxia
Freidreich's ataxia is the most common hereditary ataxia leading to degeneration of large sensory neurons and spinocerebellar tracts. Other characteristics of this disease include cardiomyopathy, and increased incidence of diabetes. Patients most often are homozygous for a hyperexpansion of the first intron in the frataxin gene. This leads to increased accumulation of iron in the mitochondria leading to increased oxidative stress (230). In Freidreich's ataxia, blood samples from patients showed decreased abundance of free glutathione with very little changes in total glutathione content. Also, an increase in glutathione bound to hemogoblin was discovered (235).
C. Glutaredoxin and neurodegeneration
Glutaredoxin has been shown to be expressed throughout the brain giving its high activity in the brain similar to that found in liver (81). Grx2 was also recently shown to be active in both mouse and human brain (147). Despite Grx activity in brain, it has not been extensively studied in many neurodegenerative disease models. PD is modeled in mice with the treatment of a specific complex I inhibitor, 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP). Upon treatment with MPTP, mice show an increase in Grx activity followed by a restoration of complex I activity implicating the necessity of Grx to complex I regulation. Using antisense to Grx1 recovery of complex I activity was inhibited (150). Immunohistochemical studies performed on AD brains showed increased expression of Grx in healthy neurons, however in neurons showing signs of neurodegeneration, Grx immunoreactivity was decreased compared to neurons from control brains (5). As discussed previously, Grx is the specific and efficient deglutathionylase allowing redox regulation of multiple proteins through S-glutathionylation. Alternatively, Grx has been shown to regulate activation of apoptosis signaling kinase 1 (ASK1) through its ability to bind to the C-terminus of ASK1. ASK1 has been shown to be associated with dopaminergic cell death in PD models. Under low glucose conditions, ASK1 activates a signaling cascade that includes activation of SEK1 and JNK (281). This pathway is also activated in the human neuroblastoma cell line SHSY5Y upon treatment with 6-hydroxydopamine leading to cell death (224). ASK1 has also been shown to be activated in mouse models of PD upon treatment with MPTP. This activation is inhibited with addition of α-lipoic acid, a thiol antioxidant (146). ASK1 has been shown to be regulated by Grx in various cell lines where, upon excess oxidative stimuli, including H2O2 or glucose deprivation, leads to dissociation of Grx from ASK1 and allows for its translocation to the nucleus to activate apoptosis. This dissociation was inhibited with excess catalase or N-acetyl cysteine addition, implicating ASK1 activation as a response to oxidative challenges. Depletion of cellular glutathione with the addition of butathionine sulfoximine (BSO) also leads to inhibition of Grx dissociation. The authors state that glutathione is thus necessary for the dissociation of Grx creating an intramolecular disulfide bond amongst the vicinal thiols in Grx. Excess glutathione that exists in the cell should be able to reduce this disulfide bond rapidly, making it readily available for continuous binding to ASK1 implicating that Grx dissociation must occur through some other currently unknown mechanism (282). Trx binds the N terminus of ASK1 and is also shown to regulate ASK1 activation (260). Trx binding is increased with overexpression of Grx (282). The focus of this section, however, will be dedicated to redox regulation by Grx of proteins implicated in neurodegenerative processes.
D. Proteins associated with neurodegeneration that are redox regulated through S-glutathionylation
Each of the proteins listed in Table 4 and illustrated in Fig. 10 has been implicated in the development of neurodegenerative diseases, as delineated in the following paragraphs.
Table 4.
Glutathionylation Targets in Neurodegenerative Diseases
| Protein | Challenge to induce glutathionylation | Effect on activity | Evidence in disease | Reference |
|---|---|---|---|---|
| Actin | Increased iron found in fibroblasts of patients | Inhibitory | Freidriech's ataxia | 230 |
| GAPDH | Isolated fibroblasts from HD and AD patients | Inhibitory | Alzheimer's disease, Huntington's disease | 195 |
| Complex I | Increased GSSG | Inhibitory | Parkinson's disease | 23 |
| IDPm | MPTP in mice | Inhibitory | Parkinson's disease | 152 |
| E1 Ubiquitin Enzyme | BSO in PC12 Cells | Inhibitory | Parkinson's disease | 133 |
| α-Ketoglutarate dehydrogenase | H2O2 in HEK293 cells | Inhibitory | Alzheimer's disease | 275 |
The table provides a listing of various proteins that have been implicated as targets of glutathionylation and corresponding inhibition of protein function within the context of oxidative stress associated with neurodegenerative diseases.
FIG. 10.
FIG. 10. Potential glutathionylation targets implicated in neurodegenerative diseases. This figure illustrates the variety of cellular compartments influenced by protein glutathionylation. Evidence for changes in function associated with glutathionylation is discussed in the text for each of the target proteins depicted here.
1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
GAPDH is pertinent to glycolysis forming 1,3-bispho-phoglycerate from glyceraldehyde-3-phosphate along with the production of NADH. GAPDH also has been implicated in functioning in roles in membrane fusion, microtubule binding, nuclear RNA transport, calcium homeostasis, and transcription. If excess GAPDH accumulates in the nucleus, it may also lead to apoptosis.(112) GAPDH has been shown to accumulate in many insoluble plaques in neurodegenerative diseases including neurofibrillary tangles, Lewy bodies, and detergent insoluble extracts from AD (213). GAPDH consists of four identical subunits containing four cysteine residues each. Two of these cysteine residues (Cys149 and Cys153) are found in the catalytic site of GAPDH. Mohr et al. discovered GAPDH can become S-glutathionylated upon treatment with GSNO, potentially through the formation of GS radicals. This glutathionylation leads to a decrease in GAPDH activity (202). Furthermore, isolated fibroblasts from skin of patients with AD or HD compared to their age-matched controls showed no change in content of GAPDH; however, a decrease in GAPDH activity was reported for nuclear fractions in both AD and HD patients as well as in post-nuclear fractions in AD (195). In postmortem brain sections from the inferior parietal lobule of AD patients compared to age matched controls, increased glutathionylated proteins were observed through 2D gel electrophoresis with the major proteins identified being deoxyhemoglobin, α cyrstallin B, α enolase, and GAPDH (214). Others have reported an increase in GAPDH accumulation in neurons in a HD mouse model with elevated levels occuring in the nucleus of neurons in the affected areas. The role of GAPDH in apoptosis coincides with a functional alteration in protein activity (270). Glutathionylation results in a diminution of GAPDH activity, which may be important to neurodegeneration; however, further investigation must be performed for a complete mechanism of GAPDH and its role in neurodegeneration.
2. Actin
Actin is an abundant protein involved in maintaining the infrastructure of the cytoplasmic matrix. Its reorganization is involved in cell growth, cell movement, and cell division. Wang et al. reported actin deglutathionylation occurs as an activating reduction allowing for cytosketal changes to be made. Actin deglutathionylation is mediated by Grx. Glutathionylation occurs on Cys374 of actin, which remains in the oxidized, glutathionylated state under normal cellular conditions; however, upon stimulation with growth factors such as epidermal growth factor, actin is reduced to the nonglutathionylated state, allowing for polymerization of actin filaments and cytoskeletal reorganization (315, 317). Actin glutathionylation has been reported in Freidreich's ataxia. Analysis of fibroblasts from Freidreich's ataxia patients compared to controls showed increased actin glutathionylation along with enlarged cells and cytoplasmic area. Staining for actin showed a disarrangement of actin filaments as a consequence of glutathionylation. Also noted was a decreased GSH:GSSG ratio (230). Actin glutathionylation has been shown to slow polymerization and result in disorganization, which may play a role in the progression of disease.
3. Mitochondrial NADP+-dependent isocitrate dehydrogenase (IDPm)
IDPm is responsible for the catalysis of the oxidative decarboxylation of isocitrate to form α ketoglutarate producing NADPH. IDPm has been reported to protect against oxidative stress. It contains a critical cys residue within the active site. Glutathionylation at this cysteine (Cys269) inhibits function and is reversed by either DTT or Grx2 using isolated protein. An alteration of in vitro GSH:GSSG ratio from 100 to 10 or 1 results in 15–25% inhibition of IDPm. Using HEK293 cells, H2O2 or diamide induced glutathionylation of IDPm in a dose-dependent fashion; however, these treatments are far from physiological. In the PD model using MPTP to induce dopaminergic cell death and parkinsonism in mice, an increase in glutathionylation of IDPm was observed as determined by reactivity to the anti-GSH antibody. This increase in glutathionylation occurred in a dose-dependent fashion (152). The consequence of glutathionylation of IDPm in neurodegeneration has yet to be determined.
4. Tau
Tau functions to aid in the assembly of micro-tubules as a microtubule-associated protein. Existing mainly in neurons, Tau promotes neurite extension and axonal growth. Tau is also responsible for the maintenance of microtubules within the cytoskeleton of mature axons. Tau is the major component found in AD neurofibrillary tangles. Tau requires a 3-repeat minimal sequence required for filament assembly. In the third repeat, Tau has a cys responsible for microtubule binding (67–69). This cysteine (Cys322) is oxidizable where oxidation promotes dimerization into regular filaments. In vitro studies performed using only the microtubule binding fragment of Tau showed that Tau can interact with GSH and to a lesser extent DTT. Upon reduction with either GSH or DTT, Tau homodimers were reduced to monomers. Upon its association with GSH, Tau is no longer able to form dimers, but can form regular filaments, which detracts from a protective role of GSH (67). Glutathionylation of Tau and the ability of Tau-SSG to form neurofibrillary tangles are still undetermined; however, this modification may alter the normal ability of Tau to oligomerize.
5. Complex I
Complex I functions as a NADH-ubiquinone oxidoreductase, the first enzyme participating in the electron transport chain responsible for the majority of the ATP production within the cell (125). Inhibition of complex I is the hallmark of PD where inhibition by only 25% causes impaired oxidative phosphorylation. Animal models of PD utilize complex I inhibitors such as MPTP and rotenone to induce parkinsonism in mice. Upon treatment of mice with MPTP or rotenone, decreased ATP and increased ROS was discovered adding to the oxidative stress existing on the system and potentially leading to cell death (306). Also, MPTP treatment of mice resulted in an increase in Grx1 working as potential antioxidant protective mechanism. Knocking down either Grx1 or Grx2 also resulted in decreased complex I activity, implicating the importance of Grx in the regulation of complex I activity (147, 150). In vitro studies have shown complex I is inhibited by glutathionylation occuring through the addition of excess GSSG resulting in a rapid production of superoxide. Addition of excess GSH and Grx2 only partially reversed the inhibition of complex I (23, 293). The glutathionylation status of complex I in patients or animal models of PD has not been determined. The ability of complex I to become inhibited by glutathionylation under a physiological stress has also yet to be determined.
6. Tyrosine hydroxylase
Tyrosine hydroxylase (TH) catalyzes the rate limiting step in dopamine anabolism converting tyrosine to L-dopamine. Depletion of dopamine is the cause of the devastating motor symptoms that occur in PD (234). Tyrosine hydroxylase function decreases to a greater extent than the loss of dopaminergic neurons, suggesting a contribute of TH function to the disease state (9). Tyrosine hydroxylase contains 7 cysteines, however it is unknown which ones are essential to the catalytic function of tyrosine hydroxylase. In PC12 cells treated with diamide, tyrosine hydroxylase becomes glutathionylated, leading to its inactivation (28). The glutathionylation status of tyrosine hydroxylase in animal models of PD or in human samples is unknown. Inhibition of tyrosine hydroxylase through glutathionylation can easily exacerbate the disease through a diminution of endogenous production of dopamine.
7. p53
p53 mediates the cell's response to DNA damage through initiation of DNA repair, halting the cell cycle, or activating an apoptotic signaling cascade. Levels of p53 have been reported to be elevated in AD and mild cognitive impairment; and brain samples from AD patients showed increased oxidation of p53 and elevated nitrotyrosine levels compared to controls (36). In addition, p53 levels have been reported to be elevated in other neurodegenerative contexts, especially in PD in the caudate nucleus as measured through immunoreactivity in postmortem brain samples compared to control. p53 can be activated upon oxidative stress and initiates apoptosis through binding to mitochondrial Bcl-2, displacing Bax, which induces apoptosis and cytochrome C release from the mitochondria. Models of PD, specifically involving 6-hydroxydopamine or MPTP, show alterations in p53 content (96, 201). The p53 protein has been reported to be S-glutathionylated in human cancer cell lines, with concomitant inhibition of its oligimerization and transcriptional activation (see Cancer section, above). Since neurons are postmitotic cells, p53 may activate a mitochondrial apoptotic cascade; however, it is currently unknown whether glutathionylation affects p53 binding to Bcl-2 or Bcl-XL (310).
8. Cytosolic calcium regulators
Ryanodine receptor type 3 is located in brain and allows for the release of calcium from the endoplasmic reticulum into the cytosol. Calcium homeostasis is altered in neurodegeneration, probably as a function of aging, where control of calcium fluxes is impaired and is thought to contribute to the overall oxidative stress in the disease process (192). Also, MPTP has been shown to induce intracellular calcium concentrations leading to cell death (166). Inhibition of the IP3 receptor or the ryanodine receptor also inhibits neuronal cell death from excitotoxic injury (192). Ryanodine receptor type 1 has been shown to be glutathionylated in vitro, allowing it to open and release calcium from the endoplasmic reticulum. This glutathionylation occurred under a number of oxidants including superoxide, hydrogen peroxide, nitric oxide, and oxidized glutathione (11). It is currently unknown if this same type of glutathionylation occurs on the ryanodine receptors present in the brain (ryanodine receptor type 3), but one could conceive how glutathionylation of the ryanodine receptors, leading to a release of calcium from ER stores, can lead to increased intracellular calcium levels and contribute to neuronal death. Calcium regulation by glutathionylation is further discussed in the cardiac and diabetes sections.
9. Ras
Ras has been shown to be glutathionylated within the cardiovascular system in response to angiotensin II (as discussed previously, see cardiovascular section) through its interaction with the angiotensin I receptor (1). Inhibition of this receptor with losartan in an MPTP PD model prevents dopaminergic cell loss (106). Also, application of captopril, an angiontensin converting enzyme inhibitor, in a MPTP model of PD inhibits dopamingeric cell death and oxidative stress (206). A mechanism is not currently known for the dopaminergic neuronal protection but it may occur through Ras inhibiting its glutathionylation and activation by angiotensin II. However, angiotensin II has been reported to be protective. In an in vitro model of PD using overexpression of α-synuclein in the neuroglioma H4 cell line, angiotensin II protected cells from α-synuclein toxicity through its interaction with the angiotensin 4 receptor (107). This field has much to be learned with only preliminary implications for Ras and its activity, which is regulated by glutathionylation.
10. Proteasome degradation pathway
The proteasome degradation pathway is central to the maintenance of homeostasis during stress by signaling damaged or misfolded proteins for degradation. Each protein marked for degradation is tagged with ubiquitin, a highly conserved 8.5 kDa protein, through a series of reactions. The E1 activating enzyme uses its conserved thiol group to create an ester bond between it and the carboxy terminal glycine of ubiquitin. The ubiquitin is then transferred to the E2 conjugating enzyme. This enzyme either directly tags the protein substrate with ubiquitin or transfers the ubiquitin protein to a substrate via the E3 ligating enzyme. Once a substrate is initially tagged with ubiquitin, a polyubiquitin chain is formed linking ubiquitin to a lysine residue in the protein. This signals the substrate for degradation in the 26S proteosome complex (133) Protein glutathionylation has been implicated in the regulation of multiple steps of this pathway (219) (Fig. 11).
FIG. 11.
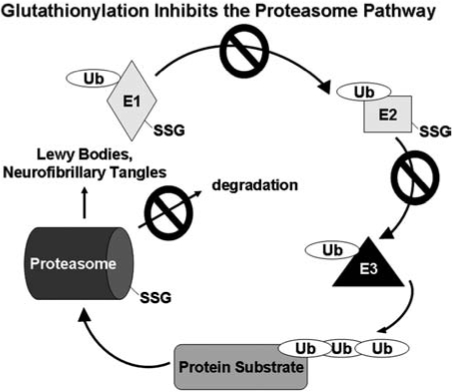
Glutathionylation targets within the proteasome pathway. Glutathionylation occurs at multiple levels of the proteasome pathway resulting in inhibition of function. This oxidant-induced post translational modification leading to deactivation of the protein degradation process could contribute to protein aggregation that is prevalent in neurodegenerative diseases, including formation of Lewy bodies (PD) and neurofibrillary tangles (AD).
The E1 activating enzyme and the E2 conjugating enzyme contain cysteines in their respective active sites which must be maintained in the reduced state in order to remain active. Using retinal pigment epithelial cells, treatment with excess hydrogen peroxide (millimolar levels) led to a decrease in ubiquitin-E1 conjugation along with an increase in the GSSG:GSH ratio. A similar effect was observed using whole retina. Also, an increase in the uptake of [35S] was observed after hydrogen peroxide treatment and was reversed to control levels upon DTT treatment. Increased exposure of hydrogen peroxide caused a decrease in E1 and E2 activity, implicating incorporation of GSH to be inhibitory. The authors do not directly show glutathionylation but do correlate an increase in oxidants, albeit in a rather nonphysiological concentration, to a decrease in activity through interaction with the active site cysteines, since they observe a similar affect with NEM treatment. Retinal supernatants incubated with either NEM, a sulfhydryl blocking agent, or GSSG, glutathione disulfide, also resulted in a decrease in ubiquitin conjugation. This implies that modifications of the active site cysteine result in a diminution of function. Since GSH is the abundant thiol in the cell, this modification is likely S-glutathionylation, however specific data showing S-glutathionylation has yet to be seen (130). In a PD cell culture model, PC12 cells, inhibition of GSH synthesis resulted in a decrease in ubiquitin conjugation to the E1 enzyme (132). This implicates a possible inhibition in PD, where diminished levels of GSH are observed (277).
The proteasome has also been implicating in regulation by S-glutathionylation. In yeast, the 20S proteasome was shown to be inhibited upon treatment with hydrogen peroxide and glutathione in excess. This was attenuated with the substitution of GSH for GSSG. The authors propose the mechanism of glutathionylation to occur through the formation of the sulfenic acid (P-SOH), which then quickly interacts with glutathione in the cell to form the S-glutathionylated protein. The physiological relevance of this observation is somewhat questionable due to the requirement of excess hydrogen peroxide (millimolar levels), which will not be seen in a pathological state (66).
T lymphocytes isolated from elderly patients show decreased proteasome activity compared to the young. Inhibition of glutathione synthesis by BSO caused a diminution of proteasome activity in T lymphocytes from the young donors comparable to the activity seen in the elderly; however, T lymphocytes from the elderly did not show a further decrease in activity upon BSO treatment (63). Decreased proteasome function has been observed as a consequence of aging and may contribute to diseases, especially neurodegeneration, where build up of proteins are seen. Furthermore, in PD, Lewy bodies comprised of proteinacious products in the brain contain ubiquitin, implicating a possible inhibition at the proteasome level impairing their degradation (132).
11. α-Ketoglutarate dehydrogenase
Alzheimer's patients show inhibition in the tricarboxylic acid (TCA) cycle specifically with the decline in α-ketoglutarate dehydrogenase activity; however, the mechanism of this decline is unknown. Oxidants, including hydrogen peroxide, inhibit the function of α-ketoglutarate dehydrogenase in other systems, as discussed in the cardiovascular section (275). Its glutathionylation may occur in neurodegeneration, however no studies have been performed in neurodegenerative models. The relative importance of its inhibition in the disease state is not known and a potential area for further investigation.
XIII. Summary and Conclusions
Glutathionylation of proteins within neurodegeneration may play a pivotal role in initiation of disease state as well as simply be a consequence of the degenerative progression. Protein glutathionylation may occur in the early stages of the disease as a protective mechanism combating oxidative challenges within the brain and may be unable to be reversed. Further study of signaling cascades will have to be done in a neurodegenerative disease model under physiological or pathological stresses to further understand its contribution to disease progression.
XIV. Frontier Areas of Investigation
There remain several shortcomings in our understanding of reversible S-glutathionylation as a regulatory mechanism. From the point of view of regulatory targets, prediction of sensitive cysteine residues on specific proteins is not possible because consensus sequences for glutathionylation sites have not been identified, contrasting with other types of post-translational modification like phosphorylation. Also the mechanisms of formation of protein–SSG are not well understood, and it is not known whether they are enzyme-mediated. Furthermore, the possibility of redox scaffolds that would position specific proteins in juxtaposition to sites of ROS generation has not been characterized as yet. Thus, many new discoveries lie ahead in this intense area of investigation.
As noted above, there are an increasing number of paradoxical examples of specific proteins existing in the highly reducing environment of the cytoplasm in the form of protein–SSG mixed disulfides; and a number of these proteins respond to oxidant-generating stimuli, like growth factors and cytokines, by being deglutathionylated by glutaredoxin. These proteins include actin, aldose reductase, pro-caspase 3, mitochondrial complex II (39, 227, 285, 317), and most recently a report on IRF-3 which has implicated IRF-3-SSG in the regulation of interferon signaling in virus-infected host cells (241). These observations raise additional questions about reversible S-glutathionylation, including: how are the disulfide moieties of specific protein–SSG sequestered from the action of Grx? What are the mechanisms of activation or induction of Grx that cause its activity to be increased under various conditions? Because the action of glutaredoxin is implicated in many disease processes reviewed above, it seems to become an attractive therapeutic target; however selective inhibition of glutaredoxin in a tissue selective manner poses a daunting challenge owing to its broad role in sulfhydryl homeostasis.
Footnotes
Reviewing Editors: Allan Butterfield, Isabella Dalle-Donne, Santiago Lamas, and Danyelle M. Townsend.
Abbreviations
AII, angiotensin II; Ab, antibody; AD, Alzheimer's disease; AGE, advanced glycation end products; ALS, amyotrophic lateral sclerosis; AP-1, activator protein 1; apoB100, apolipoprotein B100; ASO, arteriosclerosis obliterans; AR, aldose reductase; ARE, antioxidant response element; ASK-1, apoptosis signaling kinase-1; BAEC, bovine aortic endothelial cell; BALF, bronchoalveolar lavage fluid; Bcl2, B-cell CLL/lymphoma 2; BCl-xL, B-cell lymphoma x long; BSO, buthionine sulfoximine; cAMP, cyclic adenosine monophosphate; Cdc25, cell division cycle 25; CHX, cycloheximide; cIAP, cellular inhibitors of apoptosis; COPD, chronic obstructive pulmonary disease; 1-D, 1-dimensional; 2-D, 2-dimensional; DTT, dithiolthreitol; EGF, epidermal growth factor; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal-regulated kinase; ETC, electron transport chain; FEV1, forced expiratory volume in 1 second; GAP, GT-Pase-activating protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; G6PD, glucose 6-phosphate dehydrogenase; Grx, glutaredoxin; GSH, glutathione; GSK-3, glycogen synthase kinase 3; GSNO, nitrosylated glutathione; GSSG, glutathione disulfide; GST, glutathione-S-transferase; HD, Huntington's disease; HEDS, hydroxyethyl disulfide; HNE, 4-hydroxy-2-nonenal; HRP, horseradish peroxidase; IDPm, isocitrate dehydrogenase-mitochondrial NADP+-dependent; IKK, I kappa B kinase; IP, immunoprecipitation; IP3R, inosital-1,4,5 triphosphate receptor; IPC, ischemic preconditioning; I-R, ischemia-reperfusion; IRF-3, interferon regulatory factor-3; IRS, insulin receptor substreat; JNK-2, Jun N-terminal kinase-2; Keap1, Kelch ECH associating protein 1; α KGDH, alpha ketoglutarate dehydrogenase; KO, knockout; LPS, lipopolysaccharide; MAO, monoamine oxidase; MAPK, mitogen-activated protein kinase; MDA, malonaldehyde; Mdm2, murine double minute 2; MEF, mouse embryonic fibroblast; MEKK1, MAPK/ERK kinase kinase 1; MKK4, Mitogen-activated protein kinase kinase 4; MPTP, N-methyl-4-phenyl-1,2,3,4-tetrahydropyridine; NEM, N-ethyl malamide; NF-κB, nuclear factor kappa? B; Nrf2, nuclear factor-E2-related factor 2; NOS, nitric oxide synthase; O2·−, superoxide; 6-OHDA, 6-hydroxydopamine; ONOO−, peroxynitrite; oxLDL, oxidized low density lipoprotein; PAGE, polyacrylamide gel electrophoresis; PARP, poly (ADP-ribose) polymerase, PD, Parkinson's disease; PDK1, phosphoinosityl dependent kinase 1; PI3K, phosphoinositide 3 kinase; PKA, protein kinase A; PKB, protein kinase B; PKC, protein kinase C; PP2A, protein phosphatase 2A; P-SSG (protein–SSG), protein glutathione mixed disulfide (glutathionylated protein); PSNO, S-nitrosylated protein; PTEN, phosphatase and tensin homologue deleted from chromosome 10; PTP, protein tyrosine phosphatase; PTP1B, protein tyrosine phosphatase 1B; RNS, reactive nitrogen species; ROS, reactive oxygen species; RyR, ryanodine receptor; SAPK, stress-activated protein kinase; SEK1, SAPK/Erk kinase; SERCA, sarco/endoplasmic reticulum calcium AT-Pase; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SOD, superoxide dismutase (sod1, superoxide dismutase 1 gene); SQR, succinate ubiquinone reductase; TDOR, thiol disulfide oxidoreductase; TG, transgenic; TH, tyrosine hydroxylase; TNFα, tumor necrosis factor alpha; Trx, thioredoxin; UIP, usual interstitial pneumonitis; VSMC, vascular smooth muscle cell; WT, wild-type; XIAP/hILP, X chromosome-linked inhibitor of apoptosis.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) research grants 1 R01 AG024413 and 1 PO1 AG 15885 (JJM), and a Merit Review research grant from the Department of Veteran's Affairs (JJM); and by NIH training grants 5 T32 EY07157 (MDS); 1 F30 AG029687, T32 GM008803, and T32 GM07250 (MMG); and 2 T32 GM008803 (EAS) for Ph.D. training support. We are grateful to David W. Starke for critical review of the manuscript.
References
- 1.Adachi T. Pimentel DR. Heibeck T. Hou X. Lee YJ. Jiang B. Ido Y. Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 2.Adachi T. Weisbrod RM. Pimentel DR. Ying J. Sharov VS. Schoneich C. Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 3.Aguirre V. Uchida T. Yenush L. Davis R. White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 4.Aksenov MY. Markesbery WR. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer's disease. Neurosci Lett. 2001;302:141–145. doi: 10.1016/s0304-3940(01)01636-6. [DOI] [PubMed] [Google Scholar]
- 5.Akterin S. Cowburn RF. Miranda–Vizuete A. Jimenez A. Bogdanovic N. Winblad B. Cedazo–Minguez A. Involvement of glutaredoxin-1 and thioredoxin-1 in betaamyloid toxicity and Alzheimer's disease. Cell Death Differ. 2006;13:1454–1465. doi: 10.1038/sj.cdd.4401818. [DOI] [PubMed] [Google Scholar]
- 6.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 7.Andjelkovic M. Jakubowicz T. Cron P. Ming XF. Han JW. Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci USA. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Applegate MA. Humphries KM. Szweda LI. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 2008;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- 9.Ara J. Przedborski S. Naini AB. Jackson–Lewis V. Trifiletti RR. Horwitz J. Ischiropoulos H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Proc Natl Acad Sci USA. 1998;95:7659–7663. doi: 10.1073/pnas.95.13.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aracena P. Sanchez G. Donoso P. Hamilton SL. Hidalgo C. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278:42927–42935. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- 11.Aracena–Parks P. Goonasekera SA. Gilman CP. Dirksen RT. Hidalgo C. Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionyxlation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- 11a.Arai T. Nakamura M. Magori E. Fukuda M. Sako T. Decrease in malate dehydrogenase activities in peripheral leucocytes of type 1 diabetic dogs. Res Vet Sci. 2003;74:183–185. doi: 10.1016/s0034-5288(02)00177-7. [DOI] [PubMed] [Google Scholar]
- 12.Arkan MC. Hevener AL. Greten FR. Maeda S. Li ZW. Long JM. Wynshaw–Boris A. Poli G. Olefsky J. Karin M. IKKbeta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 13.Arnelle DR. Stamler JS. NO+, NO, and NO- donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 14.Arredouani A. Guiot Y. Jonas JC. Liu LH. Nenquin M. Pertusa JA. Rahier J. Rolland JF. Shull GE. Stevens M. Wuytack F. Henquin JC. Gilon P. SERCA3 ablation does not impair insulin secretion but suggests distinct roles of different sarcoendoplasmic reticulum Ca(2+) pumps for Ca(2+) homeostasis in pancreatic beta-cells. Diabetes. 2002;51:3245–3253. doi: 10.2337/diabetes.51.11.3245. [DOI] [PubMed] [Google Scholar]
- 15.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115:2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Askelof P. Axelsson K. Eriksson S. Mannervik B. Mechanism of action of enzymes catalyzing thiol-disulfide interchange. Thioltransferases rather than transhydrogenases. FEBS Lett. 1974;38:263–267. doi: 10.1016/0014-5793(74)80068-2. [DOI] [PubMed] [Google Scholar]
- 17.Ayene IS. Biaglow JE. Kachur AV. Stamato TD. Koch CJ. Mutation in G6PD gene leads to loss of cellular control of protein glutathionylation: mechanism and implication. J Cell Biochem. 2008;103:123–135. doi: 10.1002/jcb.21394. [DOI] [PubMed] [Google Scholar]
- 18.Bandyopadhyay S. Starke DW. Mieyal JJ. Gronostajski RM. Thioltransferase (glutaredoxin) reactivates the DNA-binding activity of oxidation-inactivated nuclear factor I. J Biol Chem. 1998;273:392–397. doi: 10.1074/jbc.273.1.392. [DOI] [PubMed] [Google Scholar]
- 19.Barrett WC. DeGnore JP. Keng YF. Zhang ZY. Yim MB. Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 20.Barrett WC. DeGnore JP. Konig S. Fales HM. Keng YF. Zhang ZY. Yim MB. Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 21.Barthel A. Klotz LO. Phosphoinositide 3-kinase signaling in the cellular response to oxidative stress. Biol Chem. 2005;386:207–216. doi: 10.1515/BC.2005.026. [DOI] [PubMed] [Google Scholar]
- 22.Beauvois MC. Merezak C. Jonas JC. Ravier MA. Henquin JC. Gilon P. Glucose-induced mixed [Ca2+]c oscillations in mouse beta;-cells are controlled by the membrane potential and the SERCA3 Ca2+ -ATPase of the endoplasmic reticulum. Am J Physiol Cell Physiol. 2006;290:C1503–C1511. doi: 10.1152/ajpcell.00400.2005. [DOI] [PubMed] [Google Scholar]
- 23.Beer SM. Taylor ER. Brown SE. Dahm CC. Costa NJ. Runswick MJ. Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 24.Berndt C. Hudemann C. Hanschmann EM. Axelsson R. Holmgren A. Lillig CH. How does iron-sulfur cluster coordination regulate the activity of human glutaredoxin 2? Antioxid Redox Signal. 2007;9:151–157. doi: 10.1089/ars.2007.9.151. [DOI] [PubMed] [Google Scholar]
- 25.Biaglow JE. Donahue J. Tuttle S. Held K. Chrestensen C. Mieyal J. A method for measuring disulfide reduction by cultured mammalian cells: relative contributions of glutathione-dependent and glutathione-independent mechanisms. Anal Biochem. 2000;281:77–86. doi: 10.1006/abio.2000.4533. [DOI] [PubMed] [Google Scholar]
- 26.Bidasee KR. Dincer UD. Besch HR., Jr. Ryanodine receptor dysfunction in hearts of streptozotocin-induced diabetic rats. Mol Pharmacol. 2001;60:1356–1364. doi: 10.1124/mol.60.6.1356. [DOI] [PubMed] [Google Scholar]
- 27.Biswas S. Chida AS. Rahman I. Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol. 2006;71:551–564. doi: 10.1016/j.bcp.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 28.Borges CR. Geddes T. Watson JT. Kuhn DM. Dopamine biosynthesis is regulated by S-glutathionylation. Potential mechanism of tyrosine hydroxylast inhibition during oxidative stress. J Biol Chem. 2002;277:48295–48302. doi: 10.1074/jbc.M209042200. [DOI] [PubMed] [Google Scholar]
- 29.Bredesen DE. Rao RV. Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 31.Buetler TM. Gallagher EP. Wang C. Stahl DL. Hayes JD. Eaton DL. Induction of phase I and phase II drug-metabolizing enzyme mRNA, protein, and activity by BHA, ethoxyquin, and oltipraz. Toxicol Appl Pharmacol. 1995;135:45–57. doi: 10.1006/taap.1995.1207. [DOI] [PubMed] [Google Scholar]
- 32.Burwell LS. Nadtochiy SM. Tompkins AJ. Young S. Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cabiscol E. Levine RL. The phosphatase activity of carbonic anhydrase III is reversibly regulated by glutathiolation. Proc Natl Acad Sci USA. 1996;93:4170–4174. doi: 10.1073/pnas.93.9.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai D. Yuan M. Frantz DF. Melendez PA. Hansen L. Lee J. Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantin AM. White TB. Cross CE. Forman HJ. Sokol RJ. Borowitz D. Antioxidants in cystic fibrosis. Conclusions from the CF antioxidant workshop, Bethesda, Maryland, November 11-12, 2003. Free Radic Biol Med. 2007;42:15–31. doi: 10.1016/j.freeradbiomed.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cenini G. Sultana R. Memo M. Butterfield DA. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008;45:81–85. doi: 10.1016/j.freeradbiomed.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandra A. Srivastava S. Petrash JM. Bhatnagar A. Srivastava SK. Modification of aldose reductase by S-nitrosoglutathione. Biochemistry. 1997;36:15801–15809. doi: 10.1021/bi9714722. [DOI] [PubMed] [Google Scholar]
- 38.Chen FC. Ogut O. Decline of contractility during ischemia-reperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am J Physiol Cell Physiol. 2006;290:C719–C727. doi: 10.1152/ajpcell.00419.2005. [DOI] [PubMed] [Google Scholar]
- 38a.Cheu HS. Shan YX. Yang TL. Lin HD. Chen JW. Lin SJ. Wang PH. Insulin deficiency downregulated heat shock protein 60 and IGF-1 receptor signaling in diabetic myocardium. Diabetes. 2005;54:175–181. doi: 10.2337/diabetes.54.1.175. [DOI] [PubMed] [Google Scholar]
- 39.Chen YR. Chen CL. Pfeiffer DR. Zweier JL. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 40.Cheng HP. Wei S. Wei LP. Verkhratsky A. Calcium signaling in physiology and pathophysiology. Acta Pharmacol Sin. 2006;27:767–772. doi: 10.1111/j.1745-7254.2006.00399.x. [DOI] [PubMed] [Google Scholar]
- 41.Chrestensen CA. Starke DW. Mieyal JJ. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J Biol Chem. 2000;275:26556–26565. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- 42.Christofk HR. Vander Heiden MG. Harris MH. Ramanathan A. Gerszten RE. Wei R. Fleming MD. Schreiber SL. Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 43.Chu F. Ward NE. O'Brian CA. Potent inactivation of representative members of each PKC isozyme subfamily and PKD via S-thiolation by the tumor-promotion/progression antagonist glutathione but not by its precursor cysteine. Carcinogenesis. 2001;22:1221–1229. doi: 10.1093/carcin/22.8.1221. [DOI] [PubMed] [Google Scholar]
- 44.Chu F. Ward NE. O'Brian CA. PKC isozyme S-cysteinylation by cystine stimulates the pro-apoptotic isozyme PKC delta and inactivates the oncogenic isozyme PKC epsilon. Carcinogenesis. 2003;24:317–325. doi: 10.1093/carcin/24.2.317. [DOI] [PubMed] [Google Scholar]
- 45.Claiborne A. Miller H. Parsonage D. Ross RP. Proteinsulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993;7:1483–1490. doi: 10.1096/fasebj.7.15.8262333. [DOI] [PubMed] [Google Scholar]
- 46.Clarke M. Dodson PM. PKC inhibition and diabetic microvascular complications. Best Pract Res Clin Endocrinol Metab. 2007;21:573–586. doi: 10.1016/j.beem.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 47.Clavreul N. Adachi T. Pimental DR. Ido Y. Schoneich C. Cohen RA. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006;20:518–520. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- 48.Clavreul N. Bachschmid MM. Hou X. Shi C. Idrizovic A. Ido Y. Pimentel D. Cohen RA. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2006;26:2454–2461. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]
- 49.Clegg HV. Itahana K. Zhang Y. Unlocking the Mdm2-p53 loop: Ubiquitin is the key. Cell Cycle. 2008;7:287–292. doi: 10.4161/cc.7.3.5358. [DOI] [PubMed] [Google Scholar]
- 50.Coetzee WA. Nakamura TY. Faivre JF. Effects of thiolmodifying agents on KATP channels in guinea pig ventricular cells. Am J Physiol. 1995;269:H1625–H1633. doi: 10.1152/ajpheart.1995.269.5.H1625. [DOI] [PubMed] [Google Scholar]
- 51.Colell A. Ricci JE. Tait S. Milasta S. Maurer U. Bouchier–Hayes L. Fitzgerald P. Guio–Carrion A. Waterhouse NJ. Li CW. Mari B. Barbry P. Newmeyer DD. Beere HM. Green DR. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–997. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 52.Coppock DL. Thorpe C. Multidomain flavin-dependent sulfhydryl oxidases. Antioxid Redox Signal. 2006;8:300–311. doi: 10.1089/ars.2006.8.300. [DOI] [PubMed] [Google Scholar]
- 53.Cotgreave IA. Gerdes RG. Recent trends in glutathione biochemistry–glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem Biophys Res Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- 54.Coughlan MT. Cooper ME. Forbes JM. Renal microvascular injury in diabetes: RAGE and redox signaling. Antioxid Redox Signal. 2007;9:331–342. doi: 10.1089/ars.2006.1469. [DOI] [PubMed] [Google Scholar]
- 54a.Crabb DW. The effect of experimental diabetes on liver alcohol dehydrogenase activity in rats. Alcohol Clin Exp Res. 1986;10:77–80. doi: 10.1111/j.1530-0277.1986.tb05619.x. [DOI] [PubMed] [Google Scholar]
- 55.Cross JV. Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cruz CM. Rinna A. Forman HJ. Ventura AL. Persechini PM. Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dafre AL. Sies H. Akerboom T. Protein S-thiolation and regulation of microsomal glutathione transferase activity by the glutathione redox couple. Arch Biochem Biophys. 1996;332:288–294. doi: 10.1006/abbi.1996.0344. [DOI] [PubMed] [Google Scholar]
- 58.Dai Y. Wei Z. Sephton CF. Zhang D. Anderson DH. Mousseau DD. Haloperidol induces the nuclear translocation of phosphatidylinositol 3′-kinase to disrupt Akt phosphorylation in PC12 cells. J Psychiatry Neurosci. 2007;32:323–330. [PMC free article] [PubMed] [Google Scholar]
- 59.Dalle–Donne I. Giustarini D. Rossi R. Colombo R. Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34:23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- 60.Dalle–Donne I. Milzani A. Gagliano N. Colombo R. Giustarini D. Rossi R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid Redox Signal. 2008;10:445–473. doi: 10.1089/ars.2007.1716. [DOI] [PubMed] [Google Scholar]
- 61.Dalle–Donne I. Rossi R. Giustarini D. Colombo R. Milzani A. Actin S-glutathionylation: evidence against a thiol-disulphide exchange mechanism. Free Radic Biol Med. 2003;35:1185–1193. doi: 10.1016/s0891-5849(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 62.Das DK. Thioredoxin regulation of ischemic preconditioning. Antioxid Redox Signal. 2004;6:405–412. doi: 10.1089/152308604322899477. [DOI] [PubMed] [Google Scholar]
- 63.Das R. Ponnappan S. Ponnappan U. Redox regulation of the proteasome in T lymphocytes during aging. Free Radic Biol Med. 2007;42:541–551. doi: 10.1016/j.freeradbiomed.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davis DA. Dorsey K. Wingfield PT. Stahl SJ. Kaufman J. Fales HM. Levine RL. Regulation of HIV-1 protease activity through cysteine modification. Biochemistry. 1996;35:2482–2488. doi: 10.1021/bi951525k. [DOI] [PubMed] [Google Scholar]
- 65.Davis DA. Newcomb FM. Starke DW. Ott DE. Mieyal JJ. Yarchoan R. Thioltransferase (glutaredoxin) is detected within HIV-1 and can regulate the activity of glutathionylated HIV-1 protease in vitro. J Biol Chem. 1997;272:25935–25940. doi: 10.1074/jbc.272.41.25935. [DOI] [PubMed] [Google Scholar]
- 66.Demasi M. Silva GM. Netto LE. 20 S proteasome from Saccharomyces cerevisiae is responsive to redox modifications and is S-glutathionylated. J Biol Chem. 2003;278:679–685. doi: 10.1074/jbc.M209282200. [DOI] [PubMed] [Google Scholar]
- 67.Di Noto L. University of Florida: Gainsville, FL; 2002. Biochemical Investigation of Tau and MAP2: polymerization implications for neuropathic filament assembly in Alzheimer's disease (Dissertation) [Google Scholar]
- 68.Di Noto L. Deture MA. Purich DL. Disulfide-crosslinked tau and MAP2 homodimers readily promote microtubule assembly. Mol Cell Biol Res Commun. 1999;2:71–76. doi: 10.1006/mcbr.1999.0153. [DOI] [PubMed] [Google Scholar]
- 69.Di Noto L. Deture MA. Purich DL. Structural insights into Alzheimer filament assembly pathways based on site-directed mutagenesis and S-glutathionylation of three-repeat neuronal Tau protein. Microsc Res Tech. 2005;67:156–163. doi: 10.1002/jemt.20195. [DOI] [PubMed] [Google Scholar]
- 70.Di Simplicio P. de Giorgio LA. Cardaioli E. Lecis R. Miceli M. Rossi R. Anichini R. Mian M. Seghieri G. Franconi F. Glutathione, glutathione utilizing enzymes and thioltransferase in platelets of insulin-dependent diabetic patients: relation with platelet aggregation and with microangiopatic complications. Eur J Clin Invest. 1995;25:665–669. doi: 10.1111/j.1365-2362.1995.tb01983.x. [DOI] [PubMed] [Google Scholar]
- 71.Di Simplicio P. Jensson H. Mannervik B. Effects of inducers of drug metabolism on basic hepatic forms of mouse glutathione transferase. Biochem J. 1989;263:679–685. doi: 10.1042/bj2630679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diwakar L. Kenchappa RS. Annepu J. Ravindranath V. Downregulation of glutaredoxin but not glutathione loss leads to mitochondrial dysfunction in female mice CNS: implications in excitotoxicity. Neurochem Int. 2007;51:37–46. doi: 10.1016/j.neuint.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 73.Dixon WG. Symmons DP. What effects might anti-TNFalpha treatment be expected to have on cardiovascular morbidity and mortality in rheumatoid arthritis? A review of the role of TNFalpha in cardiovascular pathophysiology. Ann Rheum Dis. 2007;66:1132–1136. doi: 10.1136/ard.2006.063867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Domenech RJ. Macho P. Velez D. Sanchez G. Liu X. Dhalla N. Tachycardia preconditions infarct size in dogs: role of adenosine and protein kinase C. Circulation. 1998;97:786–794. doi: 10.1161/01.cir.97.8.786. [DOI] [PubMed] [Google Scholar]
- 75.Downey JM. Davis AM. Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12:181–188. doi: 10.1007/s10741-007-9025-2. [DOI] [PubMed] [Google Scholar]
- 76.Dror V. Kalynyak TB. Bychkivska Y. Frey MH. Tee M. Jeffrey KD. Nguyen V. Luciani DS. Johnson JD. Glucose and ER-calcium channels regulate HIF-1beta via presenilin in pancreatic beta cells. J Biol Chem. 2008;283:9909–9916. doi: 10.1074/jbc.M710601200. [DOI] [PubMed] [Google Scholar]
- 77.Du X. Matsumura T. Edelstein D. Rossetti L. Zsengeller Z. Szabo C. Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Du ZX. Wang HQ. Zhang HY. Gao DX. Involvement of glyceraldehyde-3-phosphate dehydrogenase in tumor necrosis factor-related apoptosis-inducing ligand-mediated death of thyroid cancer cells. Endocrinology. 2007;148:4352–4361. doi: 10.1210/en.2006-1511. [DOI] [PubMed] [Google Scholar]
- 79.Eaton P. Wright N. Hearse DJ. Shattock MJ. Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J Mol Cell Cardiol. 2002;34:1549–1560. doi: 10.1006/jmcc.2002.2108. [DOI] [PubMed] [Google Scholar]
- 79a.Eaton P. Shattock MJ. Purification of proteins susceptible to oxidation at cysteine residues: identification of malate dehydrogenase as a target for S-glutathiolation. Ann NY Acad Sci. 2002;973:529–532. doi: 10.1111/j.1749-6632.2002.tb04694.x. [DOI] [PubMed] [Google Scholar]
- 80.Efrat S. Fleischer N. Hanahan D. Diabetes induced in male transgenic mice by expression of human H-ras oncoprotein in pancreatic beta cells. Mol Cell Biol. 1990;10:1779–1783. doi: 10.1128/mcb.10.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ehrhart J. Gluck M. Mieyal J. Zeevalk GD. Functional glutaredoxin (thioltransferase) activity in rat brain and liver mitochondria. Parkinsonism Relat Disord. 2002;8:395–400. doi: 10.1016/s1353-8020(02)00020-2. [DOI] [PubMed] [Google Scholar]
- 82.Eiserich JP. Patel RP. O'Donnell VB. Pathophysiology of nitric oxide and related species: free radical reactions and modification of biomolecules. Mol Aspects Med. 1998;19:221–357. doi: 10.1016/s0098-2997(99)00002-3. [DOI] [PubMed] [Google Scholar]
- 83.Ejima K. Nanri H. Araki M. Uchida K. Kashimura M. Ikeda M. 17beta-estradiol induces protein thiol/disulfide oxidoreductases and protects cultured bovine aortic endothelial cells from oxidative stress. Eur J Endocrinol. 1999;140:608–613. doi: 10.1530/eje.0.1400608. [DOI] [PubMed] [Google Scholar]
- 84.England K. Cotter TG. Direct oxidative modifications of signalling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005;10:237–245. doi: 10.1179/135100005X70224. [DOI] [PubMed] [Google Scholar]
- 85.Enoksson M. Fernandes AP. Prast S. Lillig CH. Holmgren A. Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem Biophys Res Commun. 2005;327:774–779. doi: 10.1016/j.bbrc.2004.12.067. [DOI] [PubMed] [Google Scholar]
- 86.Erkasap N. SERCA in genesis of arrhythmias: what we already know and what is new? Anadolu Kardiyol Derg. 2007;7(Suppl 1):43–46. [PubMed] [Google Scholar]
- 87.Fernando MR. Lechner JM. Lofgren S. Gladyshev VN. Lou MF. Mitochondrial thioltransferase (glutaredoxin 2) has GSH-dependent and thioredoxin reductase-dependent peroxidase activities in vitro and in lens epithelial cells. FASEB J. 2006;20:2645–2647. doi: 10.1096/fj.06-5919fje. [DOI] [PubMed] [Google Scholar]
- 87a.Findlay VJ. Townsend DM. Morris TE. Fraser JP. He L. Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fine RL. Patel J. Chabner BA. Phorbol esters induce multidrug resistance in human breast cancer cells. Proc Natl Acad Sci USA. 1988;85:582–586. doi: 10.1073/pnas.85.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Flohe L. The glutathione peroxidase reaction: molecular basis of the antioxidant function of selenium in mammals. Curr Top Cell Regul. 1985;27:473–478. doi: 10.1016/b978-0-12-152827-0.50047-5. [DOI] [PubMed] [Google Scholar]
- 89a.Fratelli M. Demol H. Puype M. Casagrande S. Eberini I. Salmona M. Bonetto V. Mengozzi M. Duffieux F. Miclet E. Bachi A. Vandekerckhove J. Gianazza E. Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Freeman BA. Crapo JD. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J Biol Chem. 1981;256:10986–10992. [PubMed] [Google Scholar]
- 91.Friling RS. Bergelson S. Daniel V. Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc Natl Acad Sci USA. 1992;89:668–672. doi: 10.1073/pnas.89.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gallogly MM. Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 93.Gallogly MM. Starke DW. Leonberg AK. Ospina SE. Mieyal JJ. Kinetic and mechanistic characterization, and versatile catalytic properties of mammalian glutaredoxin 2 (Grx 2): implications for intracellular roles. Biochemistry in press. 2008 doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gama V. Yoshida T. Gomez JA. Basile DP. Mayo LD. Haas AL. Matsuyama S. Involvement of the ubiquitin pathway in decreasing Ku70 levels in response to drug-induced apoptosis. Exp Cell Res. 2006;312:488–499. doi: 10.1016/j.yexcr.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 95.Gao Z. Hwang D. Bataille F. Lefevre M. York D. Quon MJ. Ye J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277:48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- 96.Garden GA. Morrison RS. The multiple roles of p53 in the pathogenesis of HIV associated dementia. Biochem Biophys Res Commun. 2005;331:799–809. doi: 10.1016/j.bbrc.2005.03.185. [DOI] [PubMed] [Google Scholar]
- 97.Gaston B. Nitric oxide and thiol groups. Biochim Biophys Acta. 1999;1411:323–333. doi: 10.1016/s0005-2728(99)00023-7. [DOI] [PubMed] [Google Scholar]
- 98.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 99.Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 100.Giles GI. The redox regulation of thiol dependent signaling pathways in cancer. Curr Pharm Des. 2006;12:4427–4443. doi: 10.2174/138161206779010549. [DOI] [PubMed] [Google Scholar]
- 101.Giustarini D. Milzani A. Aldini G. Carini M. Rossi R. Dalle–Donne I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid Redox Signal. 2005;7:930–939. doi: 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- 102.Gladyshev VN. Liu A. Novoselov SV. Krysan K. Sun QA. Kryukov VM. Kryukov GV. Lou MF. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J Biol Chem. 2001;276:30374–30380. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 103.Gnudi L. Frevert EU. Houseknecht KL. Erhardt P. Kahn BB. Adenovirus-mediated gene transfer of dominant negative ras(asn17) in 3T3L1 adipocytes does not alter insulin-stimulated P13-kinase activity or glucose transport. Mol Endocrinol. 1997;11:67–76. doi: 10.1210/mend.11.1.9866. [DOI] [PubMed] [Google Scholar]
- 104.Goldstein BJ. Protein-tyrosine phosphatase 1B (PTP1B): a novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr Drug Targets Immune Endocr Metabol Disord. 2001;1:265–275. doi: 10.2174/1568008013341163. [DOI] [PubMed] [Google Scholar]
- 105.Goldstein BJ. Mahadev K. Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grammatopoulos TN. Jones SM. Ahmadi FA. Hoover BR. Snell LD. Skoch J. Jhaveri VV. Poczobutt AM. Weyhenmeyer JA. Zawada WM. Angiotensin type 1 receptor antagonist losartan, reduces MPTP-induced degeneration of dopaminergic neurons in substantia nigra. Mol Neurodegener. 2007;2:1. doi: 10.1186/1750-1326-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grammatopoulos TN. Outeiro TF. Hyman BT. Standaert DG. Angiotensin II protects against alpha-synuclein toxicity and reduces protein aggregation in vitro. Biochem Biophys Res Commun. 2007;363:846–851. doi: 10.1016/j.bbrc.2007.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107a.Grankvist K. Marklund S. Sehlin J. Taljedal IB. Superoxide dismutase, catalase and scavengers of hydroxyl radical protect against the toxic action of alloxan on pancreatic islet cells in vitro. Biochem J. 1979;182:17–25. doi: 10.1042/bj1820017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gravina SA. Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32:3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 109.Gu Q. Bowden GT. Normolle D. Sun Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol. 2007;178:1009–1023. doi: 10.1083/jcb.200612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gupte A. Mumper RJ. Copper chelation by D-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic Biol Med. 2007;43:1271–1278. doi: 10.1016/j.freeradbiomed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 111.Hackett NR. Heguy A. Harvey BG. O'Connor TP. Luettich K. Flieder DB. Kaplan R. Crystal RG. Variability of antioxidant-related gene expression in the airway epithelium of cigarette smokers. Am J Respir Cell Mol Biol. 2003;29:331–343. doi: 10.1165/rcmb.2002-0321OC. [DOI] [PubMed] [Google Scholar]
- 111a.Haluzik M. Anderlova K. Dolezalova R. Adamikova A. Haluzikova D. Housova J. Svacina S. Haluzik M. Serum adipocyte fatty acid binding protein levels in patients with type 2 diabetes mellitut and obesity the influence of fenofibrate treatment. Physiol Res. 2008 doi: 10.33549/physiolres.931371. [DOI] [PubMed] [Google Scholar]
- 112.Hara MR. Agrawal N. Kim SF. Cascio MB. Fujimuro M. Ozeki Y. Takahashi M. Cheah JH. Tankou SK. Hester LD. Ferris CD. Hayward SD. Snyder SH. Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 113.Harrison D. Gongora MC. Guzik TJ. Widder J. Oxidative Stress and Hypertension. J Am Soc Hypert. 2007;1:30–44. doi: 10.1016/j.jash.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 114.Hashemy SI. Johansson C. Berndt C. Lillig CH. Holmgren A. Oxidation and S-nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: effects on structure and activity. J Biol Chem. 2007;282:14428–14436. doi: 10.1074/jbc.M700927200. [DOI] [PubMed] [Google Scholar]
- 115.Hausenloy DJ. Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 116.Heineke J. Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 117.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 117a.Hie M. Shimono M. Fujii K. Tsukamoto I. Increased cathepsin K and tartrate-resistant acid phosphatase expression in bone of streptozotocin-induced diabetic rats. Bone. 2007;41:1045–1050. doi: 10.1016/j.bone.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 118.Hirota K. Matsui M. Murata M. Takashima Y. Cheng FS. Itoh T. Fukuda K. Yodoi J. Nucleoredoxin, glutaredoxin, and thioredoxin differentially regulate NF-kappaB, AP-1, and CREB activation in HEK293 cells. Biochem Biophys Res Commun. 2000;274:177–182. doi: 10.1006/bbrc.2000.3106. [DOI] [PubMed] [Google Scholar]
- 119.Hirst J. Carroll J. Fearnley IM. Shannon RJ. Walker JE. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim Biophys Acta. 2003;1604:135–150. doi: 10.1016/s0005-2728(03)00059-8. [DOI] [PubMed] [Google Scholar]
- 120.Ho YS. Xiong Y. Ho DS. Gao J. Chua BH. Pai H. Mieyal JJ. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic Biol Med. 2007;43:1299–1312. doi: 10.1016/j.freeradbiomed.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hogg N. The biochemistry and physiology of S-nitrosothiols. Annu Rev Pharmacol Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 122.Holmgren A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc Natl Acad Sci USA. 1976;73:2275–2279. doi: 10.1073/pnas.73.7.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huang KP. Huang FL. Glutathionylation of proteins by glutathione disulfide S-oxide. Biochem Pharmacol. 2002;64:1049–1056. doi: 10.1016/s0006-2952(02)01175-9. [DOI] [PubMed] [Google Scholar]
- 124.Humphries KM. Juliano C. Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277:43505–43511. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 125.Hurd TR. Costa NJ. Dahm CC. Beer SM. Brown SE. Filipovska A. Murphy MP. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 126.Inoue T. Wu L. Stuart J. Maki CG. Control of p53 nuclear accumulation in stressed cells. FEBS Lett. 2005;579:4978–4984. doi: 10.1016/j.febslet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 127.Ischiropoulos H. Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ishitani R. Tajima H. Takata H. Tsuchiya K. Kuwae T. Yamada M. Takahashi H. Tatton NA. Katsube N. Proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase: a possible site of action of antiapoptotic drugs. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:291–301. doi: 10.1016/S0278-5846(03)00024-1. [DOI] [PubMed] [Google Scholar]
- 129.Ivarsson R. Quintens R. Dejonghe S. Tsukamoto K. in't Veld P. Renstrom E. Schuit FC. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes. 2005;54:2132–2142. doi: 10.2337/diabetes.54.7.2132. [DOI] [PubMed] [Google Scholar]
- 130.Jahngen–Hodge J. Obin MS. Gong X. Shang F. Nowell TR., Jr. Gong J. Abasi H. Blumberg J. Taylor A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem. 1997;272:28218–28226. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- 131.Jao SC. English Ospina SM. Berdis AJ. Starke DW. Post CB. Mieyal JJ. Computational and mutational analysis of human glutaredoxin (thioltransferase): probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry. 2006;45:4785–4796. doi: 10.1021/bi0516327. [DOI] [PubMed] [Google Scholar]
- 132.Jha N. Jurma O. Lalli G. Liu Y. Pettus EH. Greenamyre JT. Liu RM. Forman HJ. Andersen JK. Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson's disease. J Biol Chem. 2000;275:26096–26101. doi: 10.1074/jbc.M000120200. [DOI] [PubMed] [Google Scholar]
- 133.Jha N. Kumar MJ. Boonplueang R. Andersen JK. Glutathione decreases in dopaminergic PC12 cells interfere with the ubiquitin protein degradation pathway: relevance for Parkinson's disease? J Neurochem. 2002;80:555–561. doi: 10.1046/j.0022-3042.2001.00009.x. [DOI] [PubMed] [Google Scholar]
- 134.Ji Y. Akerboom TP. Sies H. Thomas JA. S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Arch Biochem Biophys. 1999;362:67–78. doi: 10.1006/abbi.1998.1013. [DOI] [PubMed] [Google Scholar]
- 135.Johansson C. Kavanagh KL. Gileadi O. Oppermann U. Reversible sequestration of active site cysteines in a 2Fe-2Sbridged dimer provides a mechanism for glutaredoxin 2 regulation in human mitochondria. J Biol Chem. 2007;282:3077–3082. doi: 10.1074/jbc.M608179200. [DOI] [PubMed] [Google Scholar]
- 136.Johansson C. Lillig CH. Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J Biol Chem. 2004;279:7537–7543. doi: 10.1074/jbc.M312719200. [DOI] [PubMed] [Google Scholar]
- 137.Johnson JD. Kuang S. Misler S. Polonsky KS. Ryanodine receptors in human pancreatic beta cells: localization and effects on insulin secretion. FASEB J. 2004;18:878–880. doi: 10.1096/fj.03-1280fje. [DOI] [PubMed] [Google Scholar]
- 138.Jones WK. Brown M. Ren X. He S. McGuinness M. NFkappaB as an integrator of diverse signaling pathways: the heart of myocardial signaling? Cardiovasc Toxicol. 2003;3:229–254. doi: 10.1385/ct:3:3:229. [DOI] [PubMed] [Google Scholar]
- 139.Jourd'heuil D. Jourd'heuil FL. Feelisch M. Oxidation and nitrosation of thiols at low micromolar exposure to nitric oxide. Evidence for a free radical mechanism. J Biol Chem. 2003;278:15720–15726. doi: 10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- 140.Jung CH. Thomas JA. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch Biochem Biophys. 1996;335:61–72. doi: 10.1006/abbi.1996.0482. [DOI] [PubMed] [Google Scholar]
- 141.Kamata H. Honda S. Maeda S. Chang L. Hirata H. Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 142.Kanda M. Ihara Y. Murata H. Urata Y. Kono T. Yodoi J. Seto S. Yano K. Kondo T. Glutaredoxin modulates platelet-derived growth factor-dependent cell signaling by regulating the redox status of low molecular weight protein-tyrosine phosphatase. J Biol Chem. 2006;281:28518–28528. doi: 10.1074/jbc.M604359200. [DOI] [PubMed] [Google Scholar]
- 143.Kaneto H. Katakami N. Kawamori D. Miyatsuka T. Sakamoto K. Matsuoka TA. Matsuhisa M. Yamasaki Y. Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid Redox Signal. 2007;9:355–366. doi: 10.1089/ars.2006.1465. [DOI] [PubMed] [Google Scholar]
- 144.Karin M. Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 145.Karoui H. Hogg N. Frejaville C. Tordo P. Kalyanaraman B. Characterization of sulfur-centered radical intermediates formed during the oxidation of thiols and sulfite by peroxynitrite. ESR-spin trapping and oxygen uptake studies. J Biol Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 146.Karunakaran S. Diwakar L. Saeed U. Agarwal V. Ramakrishnan S. Iyengar S. Ravindranath V. Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson's disease: protection by alpha-lipoic acid. FASEB J. 2007;21:2226–2236. doi: 10.1096/fj.06-7580com. [DOI] [PubMed] [Google Scholar]
- 147.Karunakaran S. Saeed U. Ramakrishnan S. Koumar RC. Ravindranath V. Constitutive expression and functional characterization of mitochondrial glutaredoxin (Grx2) in mouse and human brain. Brain Res. 2007;1185:8–17. doi: 10.1016/j.brainres.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 148.Kelliher M. Fastbom J. Cowburn RF. Bonkale W. Ohm TG. Ravid R. Sorrentino V. O'Neill C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer's disease neurofibrillary and beta-amyloid pathologies. Neuroscience. 1999;92:499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- 149.Kenchappa RS. Diwakar L. Boyd MR. Ravindranath V. Thioltransferase (glutaredoxin) mediates recovery of motor neurons from excitotoxic mitochondrial injury. J Neurosci. 2002;22:8402–8410. doi: 10.1523/JNEUROSCI.22-19-08402.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kenchappa RS. Ravindranath V. Glutaredoxin is essential for maintenance of brain mitochondrial complex I: studies with MPTP. FASEB J. 2003;17:717–719. doi: 10.1096/fj.02-0771fje. [DOI] [PubMed] [Google Scholar]
- 151.Kher N. Marsh JD. Pathobiology of atherosclerosis–a brief review. Semin Thromb Hemost. 2004;30:665–672. doi: 10.1055/s-2004-861509. [DOI] [PubMed] [Google Scholar]
- 152.Kil IS. Park JW. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem. 2005;280:10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- 153.Kim H. DNA repair Ku proteins in gastric cancer cells and pancreatic acinar cells. Amino Acids. 2008;34:195–202. doi: 10.1007/s00726-006-0411-1. [DOI] [PubMed] [Google Scholar]
- 154.King GL. Shiba T. Oliver J. Inoguchi T. Bursell SE. Cellular and molecular abnormalities in the vascular endothelium of diabetes mellitus. Annu Rev Med. 1994;45:179–188. doi: 10.1146/annurev.med.45.1.179. [DOI] [PubMed] [Google Scholar]
- 155.Klatt P. Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 156.Klatt P. Molina EP. De Lacoba MG. Padilla CA. Martinez–Galesteo E. Barcena JA. Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 157.Klatt P. Molina EP. Lamas S. Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J Biol Chem. 1999;274:15857–15864. doi: 10.1074/jbc.274.22.15857. [DOI] [PubMed] [Google Scholar]
- 158.Kowluru V. Kowluru RA. Increased oxidative stress in diabetes regulates activation of a small molecular weight G-protein, H-Ras, in the retina. Mol Vis. 2007;13:602–610. [PMC free article] [PubMed] [Google Scholar]
- 159.Koya D. King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 160.Kumar S. Holmgren A. Induction of thioredoxin, thioredoxin reductase and glutaredoxin activity in mouse skin by TPA, a calcium ionophore and other tumor promoters. Carcinogenesis. 1999;20:1761–1767. doi: 10.1093/carcin/20.9.1761. [DOI] [PubMed] [Google Scholar]
- 161.Kwak HS. Yim HS. Chock PB. Yim MB. Endogenous intracellular glutathionyl radicals are generated in neuroblastoma cells under hydrogen peroxide oxidative stress. Proc Natl Acad Sci USA. 1995;92:4582–4586. doi: 10.1073/pnas.92.10.4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Landmesser U. Cai H. Dikalov S. McCann L. Hwang J. Jo H. Holland SM. Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lee K. Ozanne SE. Hales CN. Ashford ML. Effects of chemical modification of amino and sulfhydryl groups on KATP channel function and sulfonylurea binding in CRIG1 insulin-secreting cells. J Membr Biol. 1994;139:167–181. doi: 10.1007/BF00232621. [DOI] [PubMed] [Google Scholar]
- 164.Lee RT. Libby P. The unstable atheroma. Arterioscler Thromb Vasc Biol. 1997;17:1859–1867. doi: 10.1161/01.atv.17.10.1859. [DOI] [PubMed] [Google Scholar]
- 165.Lee SR. Kwon KS. Kim SR. Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 166.Leist M. Volbracht C. Fava E. Nicotera P. 1-Methyl-4-phenylpyridinium induces autocrine excitotoxicity, protease activation, and neuronal apoptosis. Mol Pharmacol. 1998;54:789–801. doi: 10.1124/mol.54.5.789. [DOI] [PubMed] [Google Scholar]
- 167.Lesnefsky EJ. Chen Q. Moghaddas S. Hassan MO. Tandler B. Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 168.Lesnefsky EJ. Hoppel CL. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch Biochem Biophys. 2003;420:287–297. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 169.Li J. Huang FL. Huang KP. Glutathiolation of proteins by glutathione disulfide S-oxide derived from S-nitrosoglutathione. Modifications of rat brain neurogranin/RC3 and neuromodulin/GAP-43. J Biol Chem. 2001;276:3098–3105. doi: 10.1074/jbc.M008260200. [DOI] [PubMed] [Google Scholar]
- 170.Li X. Xu Z. Li S. Rozanski GJ. Redox regulation of Ito remodeling in diabetic rat heart. Am J Physiol Heart Circ Physiol. 2005;288:H1417–H1424. doi: 10.1152/ajpheart.00559.2004. [DOI] [PubMed] [Google Scholar]
- 170a.Liadis N. Murakami K. Eweida M. Elford AR. Sheu L. Gaisano HY. Hakem R. Ohashi PS. Woo M. Caspase-3-dependent beta-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol. 2005;25:3620–3629. doi: 10.1128/MCB.25.9.3620-3629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lillig CH. Berndt C. Vergnolle O. Lonn ME. Hudemann C. Bill E. Holmgren A. Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc Natl Acad Sci U S A. 2005;102:8168–8173. doi: 10.1073/pnas.0500735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lillig CH. Holmgren A. Thioredoxin and related molecules–from biology to health and disease. Antioxid Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 173.Lillig CH. Lonn ME. Enoksson M. Fernandes AP. Holmgren A. Short interfering RNA-mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc Natl Acad Sci USA. 2004;101:13227–13232. doi: 10.1073/pnas.0401896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lin MT. Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 175.Lind C. Gerdes R. Schuppe–Koistinen I. Cotgreave IA. Studies on the mechanism of oxidative modification of human glyceraldehyde-3-phosphate dehydrogenase by glutathione: catalysis by glutaredoxin. Biochem Biophys Res Commun. 1998;247:481–486. doi: 10.1006/bbrc.1998.8695. [DOI] [PubMed] [Google Scholar]
- 176.Lonn ME. Hudemann C. Berndt C. Cherkasov V. Capani F. Holmgren A. Lillig CH. Expression pattern of human glutaredoxin 2 isoforms: identification and characterization of two testis/cancer cell-specific isoforms. Antioxid Redox Signal. 2008;10:547–557. doi: 10.1089/ars.2007.1821. [DOI] [PubMed] [Google Scholar]
- 177.Lou YW. Chen YY. Hsu SF. Chen RK. Lee CL. Khoo KH. Tonks NK. Meng TC. Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. FEBS J. 2008;275:69–88. doi: 10.1111/j.1742-4658.2007.06173.x. [DOI] [PubMed] [Google Scholar]
- 178.Lundberg M. Johansson C. Chandra J. Enoksson M. Jacobsson G. Ljung J. Johansson M. Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J Biol Chem. 2001;276:26269–26275. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 179.Lysell J. Stjernholm VY. Ciarlo N. Holmgren A. Sahlin L. Immunohistochemical determination of thioredoxin and glutaredoxin distribution in the human cervix, and possible relation to cervical ripening. Gynecol Endocrinol. 2003;17:303–310. doi: 10.1080/gye.17.4.303.310. [DOI] [PubMed] [Google Scholar]
- 180.Mackay HJ. Twelves CJ. Targeting the protein kinase C family: are we there yet? Nat Rev Cancer. 2007;7:554–562. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 181.Maguire–Zeiss KA. Short DW. Federoff HJ. Synuclein, dopamine and oxidative stress: co-conspirators in Parkinson's disease? Brain Res Mol Brain Res. 2005;134:18–23. doi: 10.1016/j.molbrainres.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 182.Major CD. Wolf BA. Interleukin-1beta stimulation of c-Jun NH(2)-terminal kinase activity in insulin-secreting cells: evidence for cytoplasmic restriction. Diabetes. 2001;50:2721–2728. doi: 10.2337/diabetes.50.12.2721. [DOI] [PubMed] [Google Scholar]
- 183.Malik G. Nagy N. Ho YS. Maulik N. Das DK. Role of glutaredoxin-1 in cardioprotection: an insight with Glrx1 transgenic and knockout animals. J Mol Cell Cardiol. 2008;44:261–269. doi: 10.1016/j.yjmcc.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 184.Mallis RJ. Buss JE. Thomas JA. Oxidative modification of H-ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem J. 2001;355:145–153. doi: 10.1042/0264-6021:3550145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mancuso M. Coppede F. Migliore L. Siciliano G. Murri L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J Alzheimers Dis. 2006;10:59–73. doi: 10.3233/jad-2006-10110. [DOI] [PubMed] [Google Scholar]
- 186.Manevich Y. Feinstein SI. Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci USA. 2004;101:3780–3785. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mannick JB. Schonhoff CM. Nitrosylation: the next phosphorylation? Arch Biochem Biophys. 2002;408:1–6. doi: 10.1016/s0003-9861(02)00490-3. [DOI] [PubMed] [Google Scholar]
- 188.Maples KR. Kennedy CH. Jordan SJ. Mason RP. In vivo thiyl free radical formation from hemoglobin following administration of hydroperoxides. Arch Biochem Biophys. 1990;277:402–409. doi: 10.1016/0003-9861(90)90596-q. [DOI] [PubMed] [Google Scholar]
- 189.Marfella R. Cacciapuoti F. Grassia A. Manfredi E. De Maio G. Caruso G. Pepe M. Nittolo G. Cacciapuoti F. Role of the ubiquitin-proteasome system in carotid plaque instability in diabetic patients. Acta Cardiol. 2006;61:630–636. doi: 10.2143/AC.61.6.2017962. [DOI] [PubMed] [Google Scholar]
- 190.Matsui M. Oshima M. Oshima H. Takaku K. Maruyama T. Yodoi J. Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178:179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 191.Matsuo M. Tucker SJ. Ashcroft FM. Amachi T. Ueda K. NEM modification prevents high-affinity ATP binding to the first nucleotide binding fold of the sulphonylurea receptor, SUR1. FEBS Lett. 1999;458:292–294. doi: 10.1016/s0014-5793(99)01170-9. [DOI] [PubMed] [Google Scholar]
- 192.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 193.Maulik N. Engelman RM. Rousou JA. Flack JE., III Deaton D. Das DK. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation. 1999;100:II369–II375. doi: 10.1161/01.cir.100.suppl_2.ii-369. [DOI] [PubMed] [Google Scholar]
- 194.Maulik N. Sato M. Price BD. Das DK. An essential role of NFkappaB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1998;429:365–369. doi: 10.1016/s0014-5793(98)00632-2. [DOI] [PubMed] [Google Scholar]
- 195.Mazzola JL. Sirover MA. Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer's disease and in Huntington's disease fibroblasts. J Neurochem. 2001;76:442–449. doi: 10.1046/j.1471-4159.2001.00033.x. [DOI] [PubMed] [Google Scholar]
- 196.McIlwain CC. Townsend DM. Tew KD. Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene. 2006;25:1639–1648. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Meyer DJ. Kramer H. Ozer N. Coles B. Ketterer B. Kinetics and equilibria of S-nitrosothiol-thiol exchange between glutathione, cysteine, penicillamines and serum albumin. FEBS Lett. 1994;345:177–180. doi: 10.1016/0014-5793(94)00429-3. [DOI] [PubMed] [Google Scholar]
- 198.Mieyal JJ. Srinivasan U. Starke DW. Gravina SA. Mieyal P.A. Glutathionyl Specificity of the Thioltransferases: Mechanistic and Physiological Implications. In: Packer L, editor; Cadenas E, editor. Biothiols in Health and Disease. Marcel Dekker, Inc.; 1995. pp. 305–372. [Google Scholar]
- 199.Mieyal JJ. Starke DW. Gravina SA. Dothey C. Chung JS. Thioltransferase in human red blood cells: purification and properties. Biochemistry. 1991;30:6088–6097. doi: 10.1021/bi00239a002. [DOI] [PubMed] [Google Scholar]
- 200.Mieyal JJ. Starke DW. Gravina SA. Hocevar BA. Thioltransferase in human red blood cells: kinetics and equilibrium. Biochemistry. 1991;30:8883–8891. doi: 10.1021/bi00100a023. [DOI] [PubMed] [Google Scholar]
- 201.Mogi M. Kondo T. Mizuno Y. Nagatsu T. p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci Lett. 2007;414:94–97. doi: 10.1016/j.neulet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 202.Mohr S. Hallak H. de Boitte A. Lapetina EG. Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 202a.Mohr S. Xi X. Tang J. Kern TS. Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes. 2002;51:1172–1179. doi: 10.2337/diabetes.51.4.1172. [DOI] [PubMed] [Google Scholar]
- 203.Mossman BT. Lounsbury KM. Reddy SP. Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am J Respir Cell Mol Biol. 2006;34:666–669. doi: 10.1165/rcmb.2006-0047SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mukherjee S. Gangopadhyay H. Das DK. Broccoli: a unique vegetable that protects mammalian hearts through the redox cycling of the thioredoxin superfamily. J Agric Food Chem. 2008;56:609–617. doi: 10.1021/jf0728146. [DOI] [PubMed] [Google Scholar]
- 205.Mullonkal CJ. Toledo–Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg. 2007;20:195–203. doi: 10.1080/08941930701366471. [DOI] [PubMed] [Google Scholar]
- 206.Munoz A. Rey P. Guerra MJ. Mendez–Alvarez E. Soto–Otero R. Labandeira–Garcia JL. Reduction of dopaminergic degeneration and oxidative stress by inhibition of angiotensin converting enzyme in a MPTP model of parkinsonism. Neuropharmacology. 2006;51:112–120. doi: 10.1016/j.neuropharm.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 207.Murata H. Ihara Y. Nakamura H. Yodoi J. Sumikawa K. Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 208.Murry CE. Jennings RB. Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 209.Muscat JE. Kleinman W. Colosimo S. Muir A. Lazarus P. Park J. Richie JP., Jr. Enhanced protein glutathiolation and oxidative stress in cigarette smokers. Free Radic Biol Med. 2004;36:464–470. doi: 10.1016/j.freeradbiomed.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 210.Na HK. Surh YJ. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol Carcinog. 2006;45:368–380. doi: 10.1002/mc.20225. [DOI] [PubMed] [Google Scholar]
- 211.Nagy N. Malik G. Tosaki A. Ho YS. Maulik N. Das DK. Overexpression of glutaredoxin-2 reduces myocardial cell death by preventing both apoptosis and necrosis. J Mol Cell Cardiol. 2008;44:252–260. doi: 10.1016/j.yjmcc.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 212.Nakai J. Ogura T. Protasi F. Franzini–Armstrong C. Allen PD. Beam KG. Functional nonequality of the cardiac and skeletal ryanodine receptors. Proc Natl Acad Sci USA. 1997;94:1019–1022. doi: 10.1073/pnas.94.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Nakajima H. Amano W. Fujita A. Fukuhara A. Azuma YT. Hata F. Inui T. Takeuchi T. The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death. J Biol Chem. 2007;282:26562–26574. doi: 10.1074/jbc.M704199200. [DOI] [PubMed] [Google Scholar]
- 214.Newman SF. Sultana R. Perluigi M. Coccia R. Cai J. Pierce WM. Klein JB. Turner DM. Butterfield DA. An increase in S-glutathionylated proteins in the Alzheimer's disease inferior parietal lobule, a proteomics approach. J Neurosci Res. 2007;85:1506–1514. doi: 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]
- 215.Nonaka K. Kume N. Urata Y. Seto S. Kohno T. Honda S. Ikeda S. Muroya T. Ikeda Y. Ihara Y. Kita T. Kondo T. Serum levels of S-glutathionylated proteins as a riskmarker for arteriosclerosis obliterans. Circ J. 2007;71:100–105. doi: 10.1253/circj.71.100. [DOI] [PubMed] [Google Scholar]
- 216.Norenberg MD. Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50:983–997. doi: 10.1016/j.neuint.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nulton–Persson AC. Starke DW. Mieyal JJ. Szweda LI. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry. 2003;42:4235–4242. doi: 10.1021/bi027370f. [DOI] [PubMed] [Google Scholar]
- 218.O'Brian CA. Liskamp RM. Solomon DH. Weinstein IB. Inhibition of protein kinase C by tamoxifen. Cancer Res. 1985;45:2462–2465. [PubMed] [Google Scholar]
- 219.Obin M. Shang F. Gong X. Handelman G. Blumberg J. Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–569. doi: 10.1096/fasebj.12.7.561. [DOI] [PubMed] [Google Scholar]
- 219a.Oikonomakos NG. Glycogen phosphorylase as a molecular target for type 2 diabetes therapy. Curr Protein Pept Sci. 2002;3:561–586. doi: 10.2174/1389203023380422. [DOI] [PubMed] [Google Scholar]
- 220.Okamoto T. Akaike T. Sawa T. Miyamoto Y. van der Vliet A. Maeda H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J Biol Chem. 2001;276:29596–29602. doi: 10.1074/jbc.M102417200. [DOI] [PubMed] [Google Scholar]
- 221.Okuda M. Inoue N. Azumi H. Seno T. Sumi Y. Hirata K. Kawashima S. Hayashi Y. Itoh H. Yodoi J. Yokoyama M. Expression of glutaredoxin in human coronary arteries: its potential role in antioxidant protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:1483–1487. doi: 10.1161/hq0901.095550. [DOI] [PubMed] [Google Scholar]
- 222.Olanow CW. Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 223.Ostrowski MC. Kistler WS. Properties of a flavoprotein sulfhydryl oxidase from rat seminal vesicle secretion. Biochemistry. 1980;19:2639–2645. doi: 10.1021/bi00553a016. [DOI] [PubMed] [Google Scholar]
- 224.Ouyang M. Shen X. Critical role of ASK1 in the 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J Neurochem. 2006;97:234–244. doi: 10.1111/j.1471-4159.2006.03730.x. [DOI] [PubMed] [Google Scholar]
- 225.Packer L. Kraemer K. Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. 2001;17:888–895. doi: 10.1016/s0899-9007(01)00658-x. [DOI] [PubMed] [Google Scholar]
- 226.Pai HV. Starke DW. Lesnefsky EJ. Hoppel CL. Mieyal JJ. What is the functional significance of the unique location of glutaredoxin 1 (Grx1) in the intermembrane space of mitochondria? Antioxid Redox Signal. 2007;9:2027–2033. doi: 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
- 227.Pan S. Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- 228.Park JB. Levine M. The human glutaredoxin gene: determination of its organization, transcription start point, and promoter analysis. Gene. 1997;197:189–193. doi: 10.1016/s0378-1119(97)00262-x. [DOI] [PubMed] [Google Scholar]
- 229.Park JW. Reaction of S-nitrosoglutathione with sulfhydryl groups in protein. Biochem Biophys Res Commun. 1988;152:916–920. doi: 10.1016/s0006-291x(88)80127-x. [DOI] [PubMed] [Google Scholar]
- 230.Pastore A. Tozzi G. Gaeta LM. Bertini E. Serafini V. Di Cesare S. Bonetto V. Casoni F. Carrozzo R. Federici G. Piemonte F. Actin glutathionylation increases in fibroblasts of patients with Friedreich's ataxia: a potential role in the pathogenesis of the disease. J Biol Chem. 2003;278:42588–42595. doi: 10.1074/jbc.M301872200. [DOI] [PubMed] [Google Scholar]
- 231.Pei Z. Liu G. Lubben TH. Szczepankiewicz BG. Inhibition of protein tyrosine phosphatase 1B as a potential treatment of diabetes and obesity. Curr Pharm Des. 2004;10:3481–3504. doi: 10.2174/1381612043382954. [DOI] [PubMed] [Google Scholar]
- 232.Peltoniemi M. Kaarteenaho–Wiik R. Saily M. Sormunen R. Paakko P. Holmgren A. Soini Y. Kinnula VL. Expression of glutaredoxin is highly cell specific in human lung and is decreased by transforming growth factor-beta in vitro and in interstitial lung diseases in vivo. Hum Pathol. 2004;35:1000–1007. doi: 10.1016/j.humpath.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 233.Peltoniemi MJ. Rytila PH. Harju TH. Soini YM. Salmenkivi KM. Ruddock LW. Kinnula VL. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir Res. 2006;7:133. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233a.Peltoniemi MJ. Karala AR. Jurvansuu JK. Kinnula VL. Ruddock LW. Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the gamma-linkage present in glutathione. J Biol Chem. 2006;281:33107–33114. doi: 10.1074/jbc.M605602200. [DOI] [PubMed] [Google Scholar]
- 233b.Percival MD. Ouellet M. Campagnolo C. Claveau D. Li C. Inhibition of cathepsin K by nitric oxide donors: evidence for the formation of mixed disulfides and a sulfenic acid. Biochemistry. 1999;38:13574–13583. doi: 10.1021/bi991028u. [DOI] [PubMed] [Google Scholar]
- 234.Pickel VM. Joh TH. Field PM. Becker CG. Reis DJ. Cellular localization of tyrosine hydroxylase by immunohistochemistry. J Histochem Cytochem. 1975;23:1–12. doi: 10.1177/23.1.234988. [DOI] [PubMed] [Google Scholar]
- 235.Piemonte F. Pastore A. Tozzi G. Tagliacozzi D. Santorelli FM. Carrozzo R. Casali C. Damiano M. Federici G. Bertini E. Glutathione in blood of patients with Friedreich's ataxia. Eur J Clin Invest. 2001;31:1007–1011. doi: 10.1046/j.1365-2362.2001.00922.x. [DOI] [PubMed] [Google Scholar]
- 236.Pimentel DR. Adachi T. Ido Y. Heibeck T. Jiang B. Lee Y. Melendez JA. Cohen RA. Colucci WS. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J Mol Cell Cardiol. 2006;41:613–622. doi: 10.1016/j.yjmcc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 237.Pineda–Molina E. Klatt P. Vazquez J. Marina A. Garcia dL. Perez–Sala D. Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 238.Pintus G. Tadolini B. Posadino AM. Sanna B. Debidda M. Carru C. Deiana L. Ventura C. PKC/Raf/MEK/ERK signaling pathway modulates native-LDL-induced E2F-1 gene expression and endothelial cell proliferation. Cardiovasc Res. 2003;59:934–944. doi: 10.1016/s0008-6363(03)00526-1. [DOI] [PubMed] [Google Scholar]
- 239.Poole LB. Karplus PA. Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 239a.Popovich BK. Sayen MR. Dillmann WH. Insulin responsiveness of CK-M and CK-B mRNA in the diabetic rat heart. Am J Physiol. 1991;261:E377–E381. doi: 10.1152/ajpendo.1991.261.3.E377. [DOI] [PubMed] [Google Scholar]
- 240.Prestera T. Zhang Y. Spencer SR. Wilczak CA. Talalay P. The electrophile counterattack response: protection against neoplasia and toxicity. Adv Enzyme Regul. 1993;33:281–296. doi: 10.1016/0065-2571(93)90024-8. [DOI] [PubMed] [Google Scholar]
- 241.Prinarakis E. Chantzoura E. Thanos D. Spyrou G. S-glutathionylation of IRF3 regulates IRF3-CBP interaction and activation of the IFNbeta pathway. EMBO J. 2008;60:254–257. doi: 10.1038/emboj.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Qanungo S. Starke DW. Pai HV. Mieyal JJ. Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 243.Qanungo S. Wang M. Nieminen AL. N-Acetyl-L-cysteine enhances apoptosis through inhibition of nuclear factor-kappaB in hypoxic murine embryonic fibroblasts. J Biol Chem. 2004;279:50455–50464. doi: 10.1074/jbc.M406749200. [DOI] [PubMed] [Google Scholar]
- 243a.Rabinovitch A. Suarez-Pinzon WL. Sooy K. Strynadka K. Christakos S. Expression of calbindin-D(28k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology. 2001;142:3649–3655. doi: 10.1210/endo.142.8.8334. [DOI] [PubMed] [Google Scholar]
- 244.RACKER E. Glutathione-homocystine transhydrogenase. J Biol Chem. 1955;217:867–874. [PubMed] [Google Scholar]
- 245.Rahman I. Antioxidant therapies in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:15–29. doi: 10.2147/copd.2006.1.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Rainwater R. Parks D. Anderson ME. Tegtmeyer P. Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rao RK. Clayton LW. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem Biophys Res Commun. 2002;293:610–616. doi: 10.1016/S0006-291X(02)00268-1. [DOI] [PubMed] [Google Scholar]
- 248.Reddy PH. Mitochondrial dysfunction in aging and Alzheimer's disease: strategies to protect neurons. Antioxid Redox Signal. 2007;9:1647–1658. doi: 10.1089/ars.2007.1754. [DOI] [PubMed] [Google Scholar]
- 249.Reddy S. Jones AD. Cross CE. Wong PS. van der Vliet A. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. Biochem J. 2000;347:821–827. [PMC free article] [PubMed] [Google Scholar]
- 250.Reed JA. Albino AP. Update of diagnostic and prognostic markers in cutaneous malignant melanoma. Dermatol Clin. 1999;17:631–643. doi: 10.1016/s0733-8635(05)70112-1. [DOI] [PubMed] [Google Scholar]
- 251.Reynaert NL. van der Vliet A. Guala AS. McGovern T. Hristova M. Pantano C. Heintz NH. Heim J. Ho YS. Matthews DE. Wouters EF. Janssen–Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci USA. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Rhee SG. Chae HZ. Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 253.Riedl MA. Nel AE. Importance of oxidative stress in the pathogenesis and treatment of asthma. Curr Opin Allergy Clin Immunol. 2008;8:49–56. doi: 10.1097/ACI.0b013e3282f3d913. [DOI] [PubMed] [Google Scholar]
- 254.Riedlerer PF. Views on neurodegeneration as a basis for neuroprotective strategies. Med Sci Monit. 2004;10:RA287–RA290. [PubMed] [Google Scholar]
- 255.Rohl M. Pasparakis M. Baudler S. Baumgartl J. Gautam D. Huth M. De LR. Krone W. Rajewsky K. Bruning JC. Conditional disruption of IkappaB kinase 2 fails to prevent obesity-induced insulin resistance. J Clin Invest. 2004;113:474–481. doi: 10.1172/JCI18712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Romeo G. Liu WH. Asnaghi V. Kern TS. Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–2248. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- 257.Rosen CF. Poon R. Drucker DJ. UVB radiation-activated genes induced by transcriptional and posttranscriptional mechanisms in rat keratinocytes. Am J Physiol. 1995;268:C846–C855. doi: 10.1152/ajpcell.1995.268.4.C846. [DOI] [PubMed] [Google Scholar]
- 258.Rozell B. Barcena JA. Martinez–Galisteo E. Padilla CA. Holmgren A. Immunochemical characterization and tissue distribution of glutaredoxin (thioltransferase) from calf. Eur J Cell Biol. 1993;62:314–323. [PubMed] [Google Scholar]
- 259.Sahlin L. Wang H. Stjernholm Y. Lundberg M. Ekman G. Holmgren A. Eriksson H. The expression of glutaredoxin is increased in the human cervix in term pregnancy and immediately post-partum, particularly after prostaglandin-induced delivery. Mol Hum Reprod. 2000;6:1147–1153. doi: 10.1093/molehr/6.12.1147. [DOI] [PubMed] [Google Scholar]
- 260.Saitoh M. Nishitoh H. Fujii M. Takeda K. Tobiume K. Sawada Y. Kawabata M. Miyazono K. Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Salmeen A. Andersen JN. Myers MP. Meng TC. Hinks JA. Tonks NK. Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 262.Sampathkumar R. Balasubramanyam M. Sudarslal S. Rema M. Mohan V. Balaram P. Increased glutathionylated hemoglobin (HbSSG) in type 2 diabetes subjects with microangiopathy. Clin Biochem. 2005;38:892–899. doi: 10.1016/j.clinbiochem.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 263.Sanada S. Asanuma H. Tsukamoto O. Minamino T. Node K. Takashima S. Fukushima T. Ogai A. Shinozaki Y. Fujita M. Hirata A. Okuda H. Shimokawa H. Tomoike H. Hori M. Kitakaze M. Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation. 2004;110:51–57. doi: 10.1161/01.CIR.0000133390.12306.C7. [DOI] [PubMed] [Google Scholar]
- 264.Sanchez G. Escobar M. Pedrozo Z. Macho P. Domenech R. Hartel S. Hidalgo C. Donoso P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res. 2008;77:380–386. doi: 10.1093/cvr/cvm011. [DOI] [PubMed] [Google Scholar]
- 265.Sanchez G. Pedrozo Z. Domenech RJ. Hidalgo C. Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39:982–991. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 266.Sandler AB. Molecular targeted agents in non-small-cell lung cancer. Clin Lung Cancer. 2003;5(Suppl 1):S22–S28. [PubMed] [Google Scholar]
- 267.Schmid H. Boucherot A. Yasuda Y. Henger A. Brunner B. Eichinger F. Nitsche A. Kiss E. Bleich M. Grone HJ. Nelson PJ. Schlondorff D. Cohen CD. Kretzler M. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55:2993–3003. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- 268.Schniewind B. Christgen M. Kurdow R. Haye S. Kremer B. Kalthoff H. Ungefroren H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int J Cancer. 2004;109:182–188. doi: 10.1002/ijc.11679. [DOI] [PubMed] [Google Scholar]
- 269.Schoneich C. Kinetics of thiol reactions. Methods Enzymol. 1995;251:45–55. doi: 10.1016/0076-6879(95)51109-1. [DOI] [PubMed] [Google Scholar]
- 270.Senatorov VV. Charles V. Reddy PH. Tagle DA. Chuang DM. Overexpression and nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase in a transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2003;22:285–297. doi: 10.1016/s1044-7431(02)00013-1. [DOI] [PubMed] [Google Scholar]
- 271.Shackelford RE. Heinloth AN. Heard SC. Paules RS. Cellular and molecular targets of protein S-glutathiolation. Antioxid Redox Signal. 2005;7:940–950. doi: 10.1089/ars.2005.7.940. [DOI] [PubMed] [Google Scholar]
- 272.Sheehan D. Detection of redox-based modification in two-dimensional electrophoresis proteomic separations. Biochem Biophys Res Commun. 2006;349:455–462. doi: 10.1016/j.bbrc.2006.08.124. [DOI] [PubMed] [Google Scholar]
- 273.Shelton MD. Chock PB. Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 274.Shelton MD. Kern TS. Mieyal JJ. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Muller cells: model of diabetic retinopathy. J Biol Chem. 2007;282:12467–12474. doi: 10.1074/jbc.M610863200. [DOI] [PubMed] [Google Scholar]
- 275.Shi Q. Xu H. Kleinman WA. Gibson GE. Novel functions of the alpha-ketoglutarate dehydrogenase complex may mediate diverse oxidant-induced changes in mitochondrial enzymes associated with Alzheimer's disease. Biochim Biophys Acta. 2008;1782:229–238. doi: 10.1016/j.bbadis.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Shoelson SE. Lee J. Yuan M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S49–S52. doi: 10.1038/sj.ijo.0802501. [DOI] [PubMed] [Google Scholar]
- 277.Sian J. Dexter DT. Lees AJ. Daniel S. Agid Y. Javoy–Agid F. Jenner P. Marsden CD. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 278.Singh SP. Wishnok JS. Keshive M. Deen WM. Tannenbaum SR. The chemistry of the S-nitrosoglutathione/glutathione system. Proc Natl Acad Sci USA. 1996;93:14428–14433. doi: 10.1073/pnas.93.25.14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Smith MA. Nunomura A. Zhu X. Takeda A. Perry G. Metabolic, metallic, and mitotic sources of oxidative stress in Alzheimer disease. Antioxid Redox Signal. 2000;2:413–420. doi: 10.1089/15230860050192198. [DOI] [PubMed] [Google Scholar]
- 280.Sohn J. Rudolph J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 281.Song JJ. Lee YJ. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem J. 2003;373:845–853. doi: 10.1042/BJ20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Song JJ. Rhee JG. Suntharalingam M. Walsh SA. Spitz DR. Lee YJ. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 283.Song P. Wu Y. Xu J. Xie Z. Dong Y. Zhang M. Zou MH. Reactive nitrogen species induced by hyperglycemia suppresses Akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation. 2007;116:1585–1595. doi: 10.1161/CIRCULATIONAHA.107.716498. [DOI] [PubMed] [Google Scholar]
- 284.Srinivasan U. Mieyal PA. Mieyal JJ. pH profiles indicative of rate-limiting nucleophilic displacement in thioltransferase catalysis. Biochemistry. 1997;36:3199–3206. doi: 10.1021/bi962017t. [DOI] [PubMed] [Google Scholar]
- 285.Srivastava SK. Ramana KV. Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- 286.Stamler JS. Singel DJ. Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 287.Starke DW. Chock PB. Mieyal JJ. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem. 2003;278:14607–14613. doi: 10.1074/jbc.M210434200. [DOI] [PubMed] [Google Scholar]
- 288.Stavreus-Evers A. Masironi B. Landgren BM. Holmgren A. Eriksson H. Sahlin L. Immunohistochemical localization of glutaredoxin and thioredoxin in human endometrium: a possible association with pinopodes. Mol Hum Reprod. 2002;8:546–551. doi: 10.1093/molehr/8.6.546. [DOI] [PubMed] [Google Scholar]
- 289.Strosznajder RP. Jesko H. Banasik M. Tanaka S. Effects of p53 inhibitor on survival and death of cells subjected to oxidative stress. J Physiol Pharmacol. 2005;56:215–221. [PubMed] [Google Scholar]
- 290.Sullivan DM. Wehr NB. Fergusson MM. Levine RL. Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 291.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 292.Swift LL. Role of the Golgi apparatus in the phosphorylation of apolipoprotein B. J Biol Chem. 1996;271:31491–31495. [PubMed] [Google Scholar]
- 292a.Tao L. English AM. Protein S-glutathiolation triggered by decomposed S-nitrosoglutathione. Biochemistry. 2004;43:4028–4038. doi: 10.1021/bi035924o. [DOI] [PubMed] [Google Scholar]
- 293.Taylor ER. Hurrell F. Shannon RJ. Lin TK. Hirst J. Murphy MP. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J Biol Chem. 2003;278:19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 294.Terada LS. Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol. 2006;174:615–623. doi: 10.1083/jcb.200605036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Tew KD. Redox in redux: Emergent roles for glutathione S-transferase P (GSTP) in regulation of cell signaling and S-glutathionylation. Biochem Pharmacol. 2007;73:1257–1269. doi: 10.1016/j.bcp.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 296.Thomas JA. Poland B. Honzatko R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch Biochem Biophys. 1995;319:1–9. doi: 10.1006/abbi.1995.1261. [DOI] [PubMed] [Google Scholar]
- 297.Thorpe C. Hoober KL. Raje S. Glynn NM. Burnside J. Turi GK. Coppock DL. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch Biochem Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 298.Tomlinson DR. Gardiner NJ. Glucose neurotoxicity. Nat Rev Neurosci. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- 299.Tong X. Ying J. Pimentel DR. Trucillo M. Adachi T. Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol. 2008;44:361–369. doi: 10.1016/j.yjmcc.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Tonks NK. PTP1B: from the sidelines to the front lines! FEBS Lett. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- 301.Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv. 2007;7:313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Townsend DM. Findlay VJ. Fazilev F. Ogle M. Fraser J. Saavedra JE. Ji X. Keefer LK. Tew KD. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006;69:501–508. doi: 10.1124/mol.105.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Townsend DM. Findlay VL. Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307. doi: 10.1016/S0076-6879(05)01019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Trapp S. Proks P. Tucker SJ. Ashcroft FM. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J Gen Physiol. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Trapp S. Tucker SJ. Ashcroft FM. Mechanism of ATP-sensitive K channel inhibition by sulfhydryl modification. J Gen Physiol. 1998;112:325–332. doi: 10.1085/jgp.112.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305a.Treadway JL. Mendys P. Hoover DJ. Glycogen phosphorylase inhibitors for treatment of type 2 diabetes mellitus. Expert Opin Investig Drugs. 2001;10:439–454. doi: 10.1517/13543784.10.3.439. [DOI] [PubMed] [Google Scholar]
- 306.Tretter L. Sipos I. Adam–Vizi V. Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson's disease. Neurochem Res. 2004;29:569–577. doi: 10.1023/b:nere.0000014827.94562.4b. [DOI] [PubMed] [Google Scholar]
- 307.Urata Y. Ihara Y. Murata H. Goto S. Koji T. Yodoi J. Inoue S. Kondo T. 17Beta-estradiol protects against oxidative stress-induced cell death through the glutathione/glutaredoxin-dependent redox regulation of Akt in myocardiac H9c2 cells. J Biol Chem. 2006;281:13092–13102. doi: 10.1074/jbc.M601984200. [DOI] [PubMed] [Google Scholar]
- 308.van den Berghe N. Ouwens DM. Maassen JA. van Mackelenbergh MG. Sips HC. Krans HM. Activation of the Ras/mitogen-activated protein kinase signaling pathway alone is not sufficient to induce glucose uptake in 3T3-L1 adipocytes. Mol Cell Biol. 1994;14:2372–2377. doi: 10.1128/mcb.14.4.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.van Montfort RL. Congreve M. Tisi D. Carr R. Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 310.Velu CS. Niture SK. Doneanu CE. Pattabiraman N. Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Viner RI. Williams TD. Schoneich C. Peroxynitrite modification of protein thiols: oxidation, nitrosylation, and S-glutathiolation of functionally important cysteine residue(s) in the sarcoplasmic reticulum Ca-ATPase. Biochemistry. 1999;38:12408–12415. doi: 10.1021/bi9909445. [DOI] [PubMed] [Google Scholar]
- 312.Vojtek AB. Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–19928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 313.Walters DM. Cho HY. Kleeberger SR. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxid Redox Signal. 2008;10:321–332. doi: 10.1089/ars.2007.1901. [DOI] [PubMed] [Google Scholar]
- 314.Wang HD. Xu S. Johns DG. Du Y. Quinn MT. Cayatte AJ. Cohen RA. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res. 2001;88:947–953. doi: 10.1161/hh0901.089987. [DOI] [PubMed] [Google Scholar]
- 315.Wang J. Boja ES. Tan W. Tekle E. Fales HM. English S. Mieyal JJ. Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 316.Wang J. Pan S. Berk BC. Glutaredoxin mediates Akt and eNOS activation by flow in a glutathione reductase-dependent manner. Arterioscler Thromb Vasc Biol. 2007;27:1283–1288. doi: 10.1161/ATVBAHA.107.144659. [DOI] [PubMed] [Google Scholar]
- 317.Wang J. Tekle E. Oubrahim H. Mieyal JJ. Stadtman ER. Chock PB. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc Natl Acad Sci USA. 2003;100:5103–5106. doi: 10.1073/pnas.0931345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Wang T. Arifoglu P. Ronai Z. Tew KD. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 319.Wang Y. Qiao M. Mieyal JJ. Asmis LM. Asmis R. Molecular mechanism of glutathione-mediated protection from oxidized low-density lipoprotein-induced cell injury in human macrophages: role of glutathione reductase and glutaredoxin. Free Radic Biol Med. 2006;41:775–785. doi: 10.1016/j.freeradbiomed.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 320.Ward NE. Stewart JR. Ioannides CG. O'Brian CA. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39:10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- 321.Wardman P. von Sonntag C. Kinetic factors that control the fate of thiyl radicals in cells. Methods Enzymol. 1995;251:31–45. doi: 10.1016/0076-6879(95)51108-3. [DOI] [PubMed] [Google Scholar]
- 322.Weik R. Neumcke B. ATP-sensitive potassium channels in adult mouse skeletal muscle: characterization of the ATP-binding site. J Membr Biol. 1989;110:217–226. doi: 10.1007/BF01869152. [DOI] [PubMed] [Google Scholar]
- 323.Wells WW. Rocque PA. Xu DP. Meyer EB. Charamella LJ. Dimitrov NV. Ascorbic acid and cell survival of adriamycin resistant and sensitive MCF-7 breast tumor cells. Free Radic Biol Med. 1995;18:699–708. doi: 10.1016/0891-5849(94)00188-p. [DOI] [PubMed] [Google Scholar]
- 324.West MB. Hill BG. Xuan YT. Bhatnagar A. Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. FASEB J. 2006;20:1715–1717. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- 325.Wheldrake JF. Pasternak CA. The oxidation of cyst(e)ine by mast-cell tumour P815 in culture. Biochem J. 1968;106:437–444. doi: 10.1042/bj1060437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Winterbourn CC. Metodiewa D. Reaction of superoxide with glutathione and other thiols. Methods Enzymol. 1995;251:81–86. doi: 10.1016/0076-6879(95)51112-1. [DOI] [PubMed] [Google Scholar]
- 327.Yang Y. Jao S. Nanduri S. Starke DW. Mieyal JJ. Qin J. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry. 1998;37:17145–17156. doi: 10.1021/bi9806504. [DOI] [PubMed] [Google Scholar]
- 328.Yaras N. Ugur M. Ozdemir S. Gurdal H. Purali N. Lacampagne A. Vassort G. Turan B. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes. 2005;54:3082–3088. doi: 10.2337/diabetes.54.11.3082. [DOI] [PubMed] [Google Scholar]
- 329.Yates MS. Kensler TW. Chemopreventive promise of targeting the Nrf2 pathway. Drug News Perspect. 2007;20:109–117. doi: 10.1358/dnp.2007.20.2.108343. [DOI] [PubMed] [Google Scholar]
- 330.Yoneda K. Chang MM. Chmiel K. Chen Y. Wu R. Application of high-density DNA microarray to study smoke- and hydrogen peroxide-induced injury and repair in human bronchial epithelial cells. J Am Soc Nephrol. 2003;14:S284–S289. doi: 10.1097/01.asn.0000078023.30954.05. [DOI] [PubMed] [Google Scholar]
- 331.Yoshitake S. Nanri H. Fernando MR. Minakami S. Possible differences in the regenerative roles played by thioltransferase and thioredoxin for oxidatively damaged proteins. J Biochem (Tokyo) 1994;116:42–46. doi: 10.1093/oxfordjournals.jbchem.a124500. [DOI] [PubMed] [Google Scholar]
- 332.Zdychova J. Vesela J. Kazdova L. Komers R. Renal activity of Akt kinase in experimental Type 1 diabetes. Physiol Res. 2007 doi: 10.33549/physiolres.931337. [epub Oct. 11, 2007]. [DOI] [PubMed] [Google Scholar]
- 333.Zhang ZY. Lee SY. PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. Expert Opin Investig Drugs. 2003;12:223–233. doi: 10.1517/13543784.12.2.223. [DOI] [PubMed] [Google Scholar]
- 334.Zheng L. Szabo C. Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]