

Abstract
Purpose
To compare side-by-side the uptake of sorafenib and sunitinib in vitro by human uptake solute carriers of the SLC22A and SLCO families, transport by and inhibition of efflux ATP-binding cassette (ABC) transporters, and the role of ABCB1 in the plasma pharmacokinetics and brain penetration of these agents.
Experimental Design
Uptake of [3H]sorafenib or [3H]sunitinib was assessed in Xenopus laevis oocytes or mammalian cells transfected with cDNAs coding for human OATP1A2, OATP1B1, OATP1B3, OCT1, OAT2, OAT3, OCTN1 or OCTN2. Efflux and inhibition experiments were conducted in cells transfected with human ABCB1, ABCG2, ABCC2, or ABCC4. In vivo pharmacokinetic studies were performed in knockout mice lacking Abcb1-type transporters.
Results
Intracellular uptake was not appreciably affected by any of the studied solute carriers, and minute relative to the respective prototypical substrates. Sorafenib and sunitinib showed concentration-dependent (1 μM and 10 μM) low-to-moderate affinity for ABCB1, but were not affected by the other ABC transporters. Both agents inhibited all tested ABC transporters. The absence of Abcb1 had no affect on plasma pharmacokinetics, but brain penetration was moderately increased by 1.9- and 2.9-fold for sorafenib and sunitinib, respectively, in knockout animals versus controls.
Conclusions
Unlike other tyrosine kinase inhibitors, sorafenib and sunitinib do not appear to rely on active transport to enter the cell nor are they high affinity substrates for ABC efflux transporters. Based on these characteristics, these two drugs may be less susceptible to transporter-mediated alterations in systemic exposure and transporter-related resistance mechanisms.
Introduction
In recent years, eight orally administered small molecule tyrosine kinase inhibitors have been approved for the treatment of cancer in the United States. Among these, sorafenib and sunitinib are considered multikinase inhibitors since they inhibit multiple receptor and intracellular tyrosine kinases and exhibit antiangiogenic and antitumor activity (1-3). Sorafenib is an inhibitor of C-RAF, B-RAF, c-KIT, FLT-3, platelet-derived growth factor receptor-β (PDGFR-β), and vascular endothelial growth factor receptor (VEGFR) 1, 2, and 3, and is approved for the treatment of advanced renal cell carcinoma and hepatocellular carcinoma (2). Sunitinib, an inhibitor of c-Kit, FLT-3, PDGFR-α and β, and VEGFR 2, is approved for the treatment of advanced renal cell carcinoma and imatinib-resistant gastrointestinal stromal tumors (3). Sorafenib and sunitinib are being investigated for the treatment of other solid tumor malignancies (2, 3) and acute myelogenous leukemia (4, 5).
Studies have shown that tyrosine kinase inhibitors are substrates for and/or inhibit the function of various ATP-binding cassette (ABC) transporters, and these interactions may play an important role in modulating systemic pharmacokinetics of drugs, tissue and brain distribution, and cellular accumulation and resistance (6-16). Although our previous studies indicated that sorafenib and sunitinib had greater intracellular accumulation than imatinib in a panel of leukemia cell lines (17), no studies have aimed to identify mechanisms involved in cellular uptake and retention of these compounds.
The purpose of this study was to compare side-by-side 1) the uptake of sorafenib and sunitinib in vitro by human solute carriers of the SLC22A and SLCO families; 2) the transport of these compounds in vitro by human ABCB1, ABCG2, ABCC2, and ABCC4 and the ability of the tyrosine kinase inhibitors to inhibit these transporters; and 3) the plasma pharmacokinetics and brain penetration of sorafenib and sunitinib in Abcb1 knockout and wild-type mice.
Materials and Methods
Cell lines
The porcine kidney epithelial LLC-PK1 cell line containing empty vector (control) and stably expressed cells with human ABCB1 were kindly provided by Dr. John Schuetz (St. Jude Children’s Research Hospital, Memphis, TN). Human sarcoma Saos-2 cells containing pcDNA empty vector (control), ABCG2, or ABCC4 were also provided by Dr. John Schuetz. HEK293 cells stably transfected with OAT2 and OAT3 were provided by Dr. Yuichi Sugiyama (Tokyo, Japan) (18), and OCTN1 and OCTN2 cells were obtained from Dr. Akira Tsuji (Kanazawa, Japan) (19). Cells were cultured as previously described (12). Xenopus laevis oocytes injected with human OATP1A2, OATP1B1, OATP1B3, or OCT1 cRNA along with water-injected controls were obtained from BD Biosciences.
In vitro transport assays
Generally-labeled [3H]sorafenib, [3H]sunitinib, and [3H]docetaxel as well as [14C]adefovir dipivoxil (PMEA) were custom made by Moravek Biochemicals. In all in vitro experiments, radiolabeled drug was mixed with unlabeled drug (sorafenib, sunitinib: Toronto Research Chemicals; docetaxel: American RadioChemic; or PMEA: Moravek Biochemicals) to make the desired concentration.
Uptake experiments in oocytes expressing OATP1A2, OATP1B1, OATP1B3, or OCT1, or mammalian cells overexpressing OAT2, OAT3, OCTN1 or OCTN2 were performed as described previously (12, 20). Cells were incubated with sorafenib (concentration, 0.35-1.5 μM) or sunitinib (concentration, 0.15 - 0.45 μM). The selection of initial test concentration ranges was based on achievable unbound drug concentrations at steady-state in patients’ plasma (21), as well as feasibility based on the specific activity of the radiolabeled products. Prototypical substrates for each transporter were evaluated with each experiment as a positive control as follows: tetraethylammonium (10 μM) for OCT1, estradiol-17β-d-glucuronide (2 μM) for OATP1B3, estrone-3-sulfate (2 μM) for OATP1A2 and OATP1B1, p-aminohippuric acid (5 μM) for OAT2, methotrexate (1 μM) for OAT3, and L-carnitine (0.01 μM) for OCTN1 and OCTN2. Three or more independent experiments were performed in triplicate.
To assess temperature-dependent uptake of sorafenib and sunitinib, MV4-11 cells were washed with phosphate buffered saline (PBS) that was either chilled to 4 °C or warmed to 37 °C. The cells were centrifuged, the PBS removed, and the cell pellet was re-suspended in serum-free RPMI 1640 media containing 0.5 or 1.0 μM sorafenib or sunitinib that had been chilled to 4 °C or warmed to 37 °C. The final cell concentration was 1.5 million/mL. Two mL of the cell suspension were plated in each well of a 6-well plate. The plates were incubated at either 4 °C or 37 °C for 15 minutes. After drug incubations, plates were put on ice and the cells were collected and washed twice with cold PBS. The cell pellets were lysed with NaOH (1N) and the cellular accumulation was measured using a liquid scintillation counter and normalized to protein concentration, which was measured using a BCA protein estimation kit (Thermo Fisher Scientific, Rockford, IL). Two independent experiments were performed in triplicate.
Efflux experiments in cells overexpressing human ABCB1, ABCG2, ABCC2, and ABCC4 were performed as described previously (12, 22). Cells were incubated with sorafenib or sunitinib at an extracellular concentration of 1.0 μM. This initial concentration was selected to allow for a direct comparison with published results for other tyrosine kinase inhibitors using similar in vitro models (7, 10, 12, 13, 23), as well as ensuring concentrations were below those with the potential to inhibit ABC transporters. Prototypical substrates for each transporter were evaluated with each experiment as a positive control as follows: Hoechst 33342 (10 μM) for ABCG2, docetaxel (5 μM) for ABCC2 and PMEA (10 μM) for ABCC4. Two to three experiments were performed in triplicate.
Transport inhibition studies
Inhibition of ABCB1- and ABCG2-mediated transport by sorafenib and sunitinib was determined by flow cytometry using the fluorescent dye compounds calcein-AM and Hoechst 33342, respectively, as previously described (24). Briefly, 0.1 to 25 μM of sorafenib or sunitinib was added to LLC-PK1 cells expressing ABCB1, or 0.1 to 5 μM of sorafenib or sunitinib was added to Saos-2 cells expressing ABCB2 for 15 min followed by 45 min co-incubation with 1 μM calcein-AM or 10 μM Hoechst 33342 respectively at 37°C. Cells were washed and resuspended in buffer and cellular dye efflux was analyzed by flow cytometry. Two to three independent experiments were performed in duplicate.
Inhibition of ABCC2- and ABCC4-mediated transport was determined by assessing the effect of sorafenib, sunitinib, or MK571 (a general ABCC-transporter inhibitor) on the intracellular accumulation of the prototypical substrates docetaxel and PMEA, respectively. Saos-2 cells expressing ABCC2 or ABCC4 were incubated with 20 μM sorafenib or sunitinib or 50 μM MK571 for 15 min, followed by coincubation with 5 μM docetaxel or 10 μM PMEA for 4 h. The inhibitory effect of sorafenib, sunitinib, or MK571 was monitored as % change of cellular accumulation of prototypical substrates. Two to three independent experiments were performed in triplicate.
ATPase assay of ABCC2
PREDEASY™ ATPase Kit was obtained from XenoTech (Lenexa, Kansas), and was used to assess vanadate sensitive ATPase activity of ABCC2 in membrane vesicles from insect cells according to the manufacture’s protocol. Briefly, ABCC2-mediated efflux of substrates out of the cell uses ATP hydrolysis as an energy source, and the amount of inorganic phosphate released is quantified with a colorimetric reaction, which is proportional to the activity of the transporter (25). The assay is composed of two different tests which are performed on the same plate. In the activation test, transported substrates may stimulate baseline vanadate sensitive ATPase activity. In the inhibition test, which is carried out in the presence of a known activator of the transporter, inhibitors or slowly transported compounds may inhibit the maximal vanadate sensitive ATPase activity. Using the activation and inhibition tests, sorafenib was incubated with membrane vesicles at increasing drug concentrations (0.14, 0.41, 1.23, 3.70, 11.11, 33.33, 100, and 300 μM) for 10 minutes. Two to three experiments were performed in duplicate.
Animals
Abcb1a/1b-/- (Abcb1 knockout) mice and wild-type mice of identical genetic background (FVB) were obtained from Taconic (Hudson, NY). The protocol was approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital.
Drug formulation and administration
Sorafenib was dissolved in a 50% Cremophor EL (Sigma)-50% ethanol (Pharmaco Products) mixture to make a stock solution of sorafenib 24 mg/mL. The mixture was heated to 60 °C for 1 min and sonicated for 10 min to fully suspend the sorafenib. The sorafenib solution was diluted to 6 mg/mL using sterile water immediately before drug administration, as described previously (26). Sunitinib was dissolved in 80 mM citrate buffer at pH 3.5, for a final concentration 3 mg/mL. Mice received a single dose of 40 mg/kg sorafenib (6.67 mL/kg) or 20 mg/kg sunitinib (6.67 mL/kg) by oral gavage to produce clinically relevant concentrations (21). Three independent experiments were performed.
Pharmacokinetic studies
Following drug administration, 50-100 μL blood samples were collected with heparinized capillaries at 1, 2, and 4 h. For the 1 h and 4 h sample, mice were sampled twice with blood collected from the retro-orbital venus plexus at 1 h and via cardiac puncture at the terminal time point of 4 h. Blood was obtained via cardiac puncture at 2 h. Plasma was isolated by centrifugation at 3000 × g for 5 min and frozen at -80 °C until analysis. Brain samples were removed at 4 h and homogenized in 5 volumes (w/v) of human plasma and were then frozen at -80 °C until analysis. Sorafenib or sunitinib concentrations were measured by liquid chromatography-tandem mass spectrometry, as we described previously (27, 28). To account for drug in the brain vasculature contaminating brain tissue concentrations, the concentration of sorafenib or sunitinib in the brain vascular space (1.4% of the plasma concentration at 4hr) was subtracted from the brain concentration, as previously described (6). The area under the plasma concentration-time curve (AUC) was calculated from 0 to 4 h (AUC0-4h) using noncompartmental analysis and the linear-logarithmic trapezoidal method. Brain penetration of sorafenib or sunitinib was calculated as the ratio of the brain concentration at 4 h to the plasma AUC0-4h, as described previously for imatinib (6). AUC0-4h and brain penetration were compared between wild-type and knockout mice using a 2-tailed t-test using the statistical software program NCSS 2004.
Results
Uptake of sorafenib and sunitinib by solute carriers in vitro
To identify solute carriers involved in sorafenib and sunitinib transport, we evaluated drug accumulation in Xenopus laevis oocytes or HEK293 cells transfected with 7 different transporters, including OATP1A2, OATP1B1, OATP1B3, OCT1, OAT2, OCTN1 and OCTN2. Despite significant uptake of prototypical substrates by each transporter compared to control, none of the transporters tested facilitated sorafenib or sunitinib transport (Fig. 1). Sorafenib and sunitinib showed minimal differences (1% - 16%) in cellular uptake at 4 °C and 37 °C, indicating that active transport is not involved in this process (Supplemental Figure 1). Interestingly, sorafenib showed 3-4-fold higher uptake than sunitinib at both temperatures (Supplemental Figure 1).
Fig. 1.
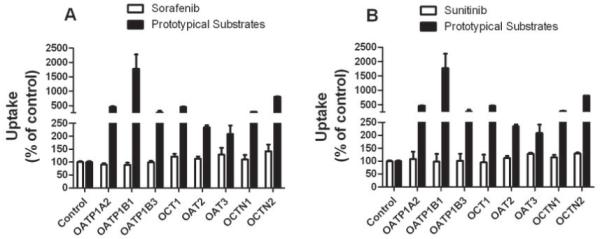
Uptake of sorafenib and sunitinib by solute carriers in vitro. Accumulation of sorafenib (A) and sunitinib (B) by Xenopus laevis oocytes expressing OATP1A2, OATP1B1, OATP1B3 and OCT1 or HEK293 cells expressing OAT1, OAT3, OCTN1 and OCTN2. Oocytes or HEK293 cells were incubated with sorafenib 0.35 -1.5 μM or sunitinib 0.15 - 0.45 μM for 1 h. Data represent the mean and standard deviations of 9-27 observations and are expressed as percent of water injected control; a single control bar is shown for all experiments combined. Prototypical substrates for each transporter were evaluated with each experiment as a positive control (black bar).
Transport of sorafenib and sunitinib by ABC transporters in vitro
In cells overexpressing ABCB1, sorafenib and sunitinib showed moderate affinity for this transporter at a concentration of 1 μM, with approximately 2-fold higher basal-to-apical transport compared to apical-to-basal transport (Fig. 2A). At a higher concentration of 10 μM, sorafenib was not transported by ABCB1, and sunitinib transport was substantially reduced compared with the lower concentration. This suggests that both drugs may exhibit a concentration-dependent autoinhibition of ABCB1 function. Subsequent investigation indicated that, in contrast to prototypical substrates, sorafenib was not transported by any of the other tested ABC transporters (ABCG2, ABCC2 and ABCC4) (Fig. 2B). Similarly, sunitinib was not transported by ABCG2 or ABCC2, although this agent was identified as a weak substrate for ABCC4.
Fig. 2.
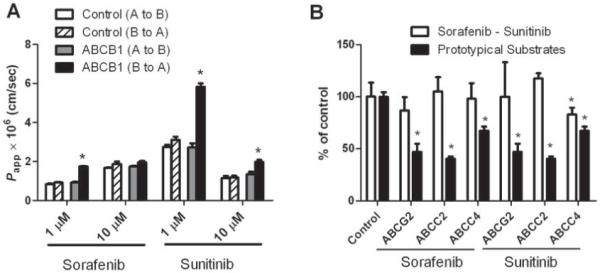
Transport of sorafenib and sunitinib by ABC transporters in vitro. (A) Transcellular transport of sorafenib and sunitinib in LLC-PK1 cells expressing ABCB1. Cells were incubated with 1μM drug for 1, 2, 3 and 4 h. Data represent 6 observations and are expressed as ABCB1-mediated Papp (B to A) / Papp (A to B) ratio. * p<0.05 versus ABCB1 (A to B) (B) Accumulation of sorafenib and sunitinib in Saos-2 cells overexpressing ABCG2 or ABCC4 or MDCKII cells overexpressing ABCC2. Cells were incubated with 1 μM drug for 4 h. Data represent the mean and standard deviations of 6-9 observations from 2-3 independent experiments and are expressed as percent control; a single control bar is shown for all experiments combined. Prototypical substrates for each transporter were evaluated with each experiment as a positive control (black bar). * p<0.05 versus control.
Inhibition of ABC transporter function by sorafenib and sunitinib in vitro
Since sorafenib and sunitinib showed concentration-dependent transport by ABCB1, with reduced transport at higher drug concentrations, we further evaluated if they could inhibit the function of ABC transporters. Both sorafenib and sunitinib decreased Hoechst 33342 efflux by cells overexpressing ABCG2, with concentrations inhibiting 50% of maximal efflux of 3.1 μM and 3.0 μM, respectively (Fig. 3A and 3B). Both drugs inhibited calcein-AM efflux by cells overexpressing ABCB1, although higher concentrations of sorafenib (16.6 μM) than sunitinib (6.7 μM) were required to inhibit half maximal efflux (Fig. 3C and 3D). We also evaluated the ability of sorafenib and sunitinib to inhibit ABCC2-mediated efflux of the prototypical substrate docetaxel in cells overexpressing this transporter. Sorafenib or sunitinib at a concentration of 20 μM inhibited docetaxel efflux by approximately 50% and 80%, respectively, with inhibition of up to approximately 90% observed by the potent inhibitor MK571 (Fig. 4A). In cells overexpressing ABCC4, sorafenib and sunitinib inhibited efflux of the substrate PMEA by approximately 70% and 80%, respectively, with near 100% inhibition by MK571 (Fig. 4B)
Fig. 3.
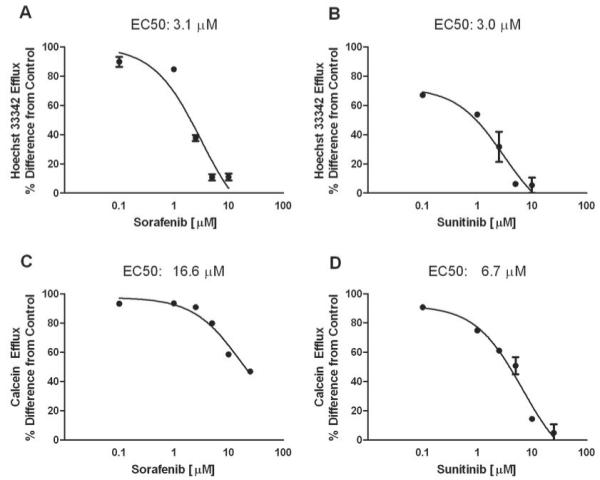
Inhibition of ABCB1 and ABCG2 function by sorafenib and sunitinib in vitro. Sorafenib (A) and sunitinib (B) decreased the efflux of hoechst 33342 in Saos-2 cells expressing human ABCG2; and sorafenib (C) and sunitinib (D) decreased the efflux of calcein in LLC-PK1 cells expressing ABCB1. Cells were incubated with increasing drug concentrations for 1 h. Flow cytometry was used to assess of hoechst 33342 and calcein cellular efflux. Data are the mean and standard deviation of two observations; representative figures of 2-3 independent experiments are shown. The lines represent the fit of a maximum effect model to the data.
Fig. 4.
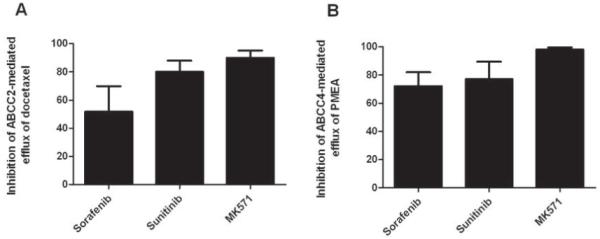
Inhibition of ABCC2 and ABCC4 function by sorafenib and sunitinib in vitro. Sorafenib inhibited the efflux of docetaxel in MDCKII cells overexpressing ABCC2 (A) and PMEA in Saos-2 cells overexpressing ABCC4 (B). Cells were incubated with 20 μM sorafenib or sunitinib or 50 μM MK571 for 15 min, followed by coincubation with 5 μM docetaxel or 1 μM PMEA for 4 h. Data represent the mean and standard deviation of 6-9 observations from 2-3 independent experiments and are expressed as % change of cellular accumulation of the prototypical substrates docetaxel or PMEA.
Effect of sorafenib on ATPase hydrolysis by ABCC2
To further examine the interaction with ABC transporters, we evaluated the effect of sorafenib on the ATPase activity of ABCC2. Sorafenib inhibited both the baseline ATPase activity and the maximal ATPase activity of ABCC2 in a concentration dependent manner (Fig 5). These data suggested that sorafenib is an inhibitor but not a substrate of ABCC2, consistent with our data in cells overexpressing ABCC2 (Fig. 2 and Fig. 4).
Fig. 5.
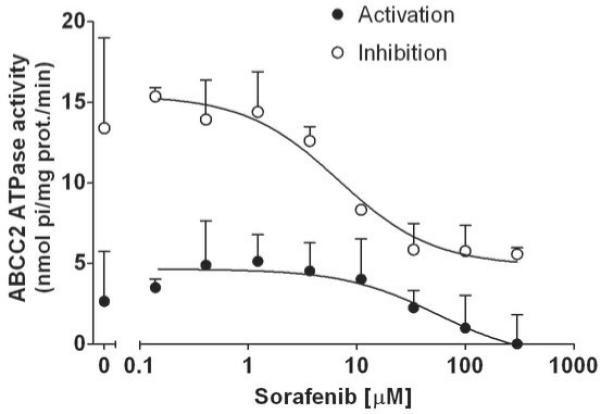
Effect of Sorafenib on ATPase hydrolysis by ABCC2. Sorafenib inhibits vanadate sensitive baseline and maximal ATPase activity of human ABCC2 expressed in membrane vesicles. Increasing concentrations of sorafenib were incubated with vesicles for 10 minutes. Data are a representative figure of 2-3 experiments performed in duplicate.
Role of Abcb1 in the pharmacokinetics and brain penetration of sorafenib and sunitinib in mice
The in vivo relevance of the interaction of sorafenib and sunitinib with ABCB1 was determined in mice lacking the mouse orthologue transporter Abcb1 (knockout) and wild-type mice. Minimal differences were observed in the plasma concentrations of sorafenib (Fig 6A) and sunitinib (Fig 6C) between wild type and Abcb1 knockout mice, and AUC0-4h values were not significantly different (P = 0.12 and P = 0.17, respectively). However, brain penetrations of sorafenib and sunitinib were moderately increased by 1.9-fold (P = 0.006) and 2.9-fold (P = 0.003), respectively, in the knockout animals versus controls (Fig 6B and 6D). Interestingly, in wild-type mice, sunitinib exhibited 10-fold greater brain penetration than sorafenib (31% versus 3.1%).
Fig. 6.
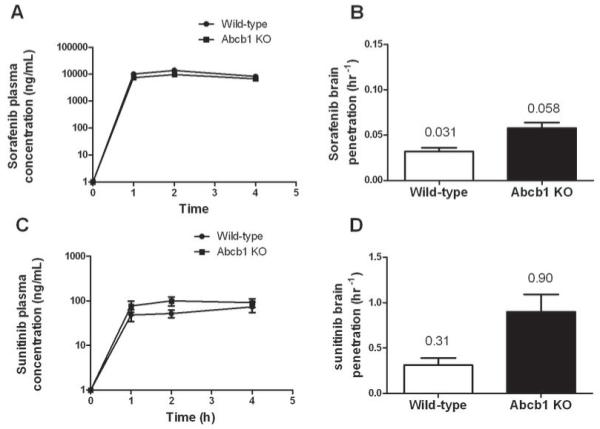
Role of ABCB1 in the plasma pharmacokinetics and brain penetration of sorafenib and sunitinib in mice. Plasma concentration-time curves for sorafenib (A) and sunitinib (C) in wild-type and Abcb1 knockout mice. Brain penetration of sorafenib (B) and sunitinib (C) in wild-type and Abcb1 knockout mice. Sorafenib 40mg/kg or sunitinib 20mg/kg was administered to mice, plasma samples were obtained at 1, 2 and 4 h after treatment, and whole brain tissue homogenate were collected at 4h. Brain penetration was determined as the brain concentration at 4 h divided by the plasma AUC0-4h. Data are the mean and standard deviations from 2-3 independent experiments (6-9 observations per time point).
Discussion
SLC and ABC transporters play an important role in drug absorption, distribution, elimination, drug interactions, and cellular accumulation and resistance. In this study, we explored the interaction of the multikinase inhibitors sorafenib and sunitinib with selected solute carriers and ABC transporters using in vitro and in vivo models. Overall, the current work indicates that neither sorafenib nor sunitinib are transported to an appreciable degree by the studied uptake carriers or efflux transporters, except for a low to moderate, concentration-dependent affinity for ABCB1. The in vivo relevance of the interaction of these drugs with ABCB1 was determined in mice lacking Abcb1-type transporters. Whereas the mouse experiments suggests that the systemic pharmacokinetics are not affected by a loss of Abcb1, the brain penetration of both drugs was moderately increased compared to wild-type counterparts. The results obtained from these studies highlight distinct features of sorafenib and sunitinib relative to other tyrosine kinase inhibitors that may potentially contribute to differential clinical activity in a variety of disease settings.
Unlike recent studies demonstrating active cellular uptake of imatinib by a number of solute carriers (12, 29), and an association of low expression of the SLC22A1 gene encoding OCT1 with resistance to imatinib in patients with chronic myeloid leukemia (30), no solute carrier was identified that was involved in the cellular uptake of sorafenib and sunitinib. In addition, neither agent showed temperature-dependent uptake. Combined, these results indicate that active transport processes are unlikely to play a significant role in the intracellular uptake of these agents. Sorafenib and sunitinib are primarily used for the treatment of solid tumors. Emerging data indicates that solute carriers are differentially expressed on solid tumor cells (31), and these differences have been associated with chemosensitivity and resistance (32, 33). Our data showing minimal active cellular uptake of sorafenib and sunitinib by a panel of SLC transporters suggests that these drugs may not be highly susceptible to solute carrier-mediated drug resistance mechanisms on solid tumor cells. Sorafenib and sunitinib also target tumor vasculature by inhibiting vascular endothelial growth factor receptors on normal endothelium. However, little is known regarding SLC transporter expression on tumor endothelial cells and how this may affect cellular uptake of sorafenib and sunitinib. Interestingly, OCTN2, a sodium-dependent transport protein for carnitine, is expressed in endothelial cells in heart tissue and was shown to contribute to the cardiac uptake of cardiovascular drugs (34). We showed increased transport of sorafenib and sunitinib over control by 141% and 130%, respectively, which although minimal, was similar to our previous studies of imatinib transport (12). It is possible that OCTN2-mediated drug uptake in heart tissue contributes to the cardiac toxicity observed with sorafenib, sunitinib, and imatinib (35, 36).
Most tyrosine kinase inhibitors in current clinical use, including imatinib, dasatinib, gefitinib and erlotinib have been associated with high substrate affinity for ABCB1 and ABCG2. In contrast, sorafenib and sunitinib showed only moderate affinity for ABCB1, with negligible transport observed in cells overexpressing ABCG2. Furthermore, these two tyrosine kinase inhibitors were not transported by ABCC2 or ABCC4. Therefore, sorafenib and sunitinib may be less susceptible to ABC transporter-mediated drug resistance in solid tumor cells (37). This possibility may also be relevant to the treatment of leukemia since ABC transporters have been shown to be expressed on hematopoietic and leukemic stem cells. For example, chronic myeloid leukemia cells transduced with ABCG2 exhibited lower intracellular accumulation of imatinib and nilotinib and were protected from drug-induced cytotoxicity, and hence, suggests a role of ABC transporters in stem cell resistance to tyrosine kinase inhibitors (7). These findings may be relevant to sorafenib, which is currently being evaluated for the treatment of acute myelogenous leukemia (AML) (5). Zhang et al. at MD Anderson Cancer Center recently published the results of an ongoing phase I trial of single-agent sorafenib in 16 adult patients with relapsed/refractory AML (4). Greater than 50% reduction in circulating blasts was observed in 6/6 (100%) of patients harboring a FLT3-ITD mutation and treated with sorafenib 400 or 600 mg twice daily. A modest clinical response in circulating blasts was observed in 3/7 (43%) patients with wild-type FLT3, despite the majority of them being treated at a dose level of 200 mg twice daily. Promising activity has been observed in 38 patients less than 65 years of age with newly diagnosed AML (13 had a FLT3 mutation and 25 were wild-type) given sorafenib concurrently with cytarabine/idarubicin. The overall response rate was 83% with 70% of patients achieving a complete response. At 9 months follow-up, the probability of survival was 82%, and remission duration was 72%, with high and durable response rate achieved in patients with both wild-type and mutated FLT3 AML.
Similar to other tyrosine kinase inhibitors evaluated to date (9, 11, 14, 15, 38), sorafenib and sunitinib were shown to inhibit the function of ABC transporters, including ABCB1, ABCG2, ABCC2 and ABCC4, and thus this function appears to be a “class effect”. The mechanism of inhibition appears to be through direct contact at transport-substrate sites (7, 9, 15, 38). Another cellular effect common to tyrosine kinase inhibitors is that they can reverse multidrug resistance to a variety of chemotherapeutic agents. Recently, sunitinib was shown to partially reverse ABCB1-mediated resistance to romidepsin, a cylic depsipeptide, and completely reverse topotecan resistance mediated by ABCG2 (38).
Since sorafenib and sunitinib inhibit ABC transporters, the potential for drug-drug interactions exist. This is of particular concern when combining these tyrosine kinase inhibitors with cytotoxic anticancer that are substrates for ABC transporters such as doxorubicin, irinotecan, paclitaxel and docetaxel. In a phase I trial, plasma exposure to doxorubicin was increased by 20-30% in combination with sorafenib, but no increase in clinical toxicity was observed (39). In line with the clinical data, a significant pharmacokinetic drug interaction with doxorubicin mediated through ABCB1 or ABCC2 would not be expected since doxorubicin plasma exposure was unaltered in mice lacking one or both transporters compared to wild-type mice (40). When co-administered with sorafenib, minimal to no increases in plasma exposure were reported for irinotecan and its active metabolite SN-38 and paclitaxel (39). Pharmacokinetic data for docetaxel when given with sorafenib in vivo have not been published. Docetaxel is a weak substrate for human versus mouse ABCC2 in vitro (41), and thus in vivo interactions through this transporter are not expected. In addition, docetaxel plasma exposure was unchanged in mice lacking ABCB1 compared to wild-type mice (42), but significanty increased docetaxel exposure was observed in CYP3a-/- knockout mice compared to their wild-type counterparts (43). These data support the notion that metabolism is the predominant elimination mechanism for docetaxel, and indicate that a transporter-mediated pharmacokinetic drug interaction between docetaxel and sorafenib is not anticipated. However, drug interactions between sorafenib and docetaxel and other anticancer agents may occur in normal tissue expressing ABC transporters, which remains to be evaluated more thoroughly.
Since sorafenib and sunitinib exhibited low to moderate affinity for ABCB1, we evaluated the role of this transporter on the plasma pharmacokinetics of both agents in vivo. This was of particular interest because the absence of ABCB1 and ABCG2 significantly increased the plasma exposure of two other tyrosine kinsae inhibitors, imatinib and erlotinib (6, 23). In contrast, our data demonstrate that the absence of ABCB1 affected minimally the absorption and systemic disposition of sorafenib and sunitinib. However, this transporter may play a role in the CNS penetration of these agents. Indeed, brain penetration of sorafenib and sunitinib was increased by 1.9-fold and 2.9-fold, respectively, in knockout mice lacking Abcb1 compared to their wild-type counterparts. However, this effect appears moderate compared to that observed for imatinib whereby brain penetration was increase 3.6-fold in Abcb1 knockout mice and 12.6-fold in mice lacking both Abcb1 and Abcg2 compared to wild-type mice (6, 44). The presence of ABCG2 is expected to have a negligible effect on the plasma pharmacokinetics and brain penetration of sorafenib and sunitinib since neither agent was shown to be a substrate for this transporter. Thus, in contrast to associations noted between ABC transporter variants and adverse effects observed in cancer patients treated with tyrosine kinase inhibitors (13), pharmacogenetic studies of ABC transporters in relation to sorafenib and sunitinib pharmacokinetics are not expected to yield significant relationships. However, there is the possibility that associations exist between ABCB1 variants and organ-specific side effects.
Brain penetration of tyrosine kinase inhibitors has been reported to be low in wild-type murine models ranging from 2-10% for imatinib (6, 44) and 3-8% for dasatinib (45). CSF penetration of imatinib has been reported to be even lower at approximately 1% (46-48). The brain penetration of sorafenib was shown to be 3%, at the lower range reported for imatinib and dasatinib. In contrast, sunitinib exhibited a dramatically greater brain penetration of 31%. However, despite the relatively low brain penetration of sorafenib, concentrations that reach the brain (~300 ng/mL; 645 nM) may be sufficiently high to inhibit multiple tyrosine kinases and have clinical activity (2, 3). This possibility is consistent with a number of recent reports indicating that both sorafenib and sunitinib were active against cerebral metastases in patients with renal cell carcinoma (49, 50).
Conclusion
In conclusion, sorafenib and sunitinib appear to have several unique pharmacologic features relative to other tyrosine kinase inhibitors. In particular, these agent do not appear to rely significantly on active transport to enter the cell nor are they high affinity substrates for ABC efflux transporters. Based on these characteristics, sorafenib and sunitinib may be less susceptible to transporter-mediated alterations in plasma pharmacokinetics, tissue distribution, cellular accumulation and drug resistance.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kelly Filipski, Chaoxin Hu, and Torben Mikkelsen (St. Jude Children’s Research Hospital, Memphis, TN) for assistance with generating the in vitro data on solute carriers.
Financial Support: This work was supported by the United States Public Health Service Cancer Center Support Grant 3P30CA021765 and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Statement of Translation Relevance:
Unlike other tyrosine kinase inhibitors registered for the treatment of cancer, such as those that inhibit the BCR-Abl oncogene or epidermal growth factor receptor, the multikinase inhibitors sorafenib and sunitinib do not appear to rely on active transport to enter the cell nor are they high affinity substrates for ABC efflux transporters. Although both drugs showed moderate affinity for ABCB1, the lack of ABCB1 in knockout mice did not affect the absorption and systemic disposition of sorafenib and sunitinib and resulted in moderate increases in brain penetration relative to other tyrosine kinase inhibitors. Based on these characteristics, sorafenib and sunitinib may be less susceptible to transporter-mediated alterations in plasma pharmacokinetics, tissue distribution, cellular accumulation and drug resistance. These distinct pharmacological features may contribute to differential clinical activity in a variety of cancers including solid tumors, central nervous system tumors, and hematological malignancies. The findings also have relevance to clinical pharmacogenetic studies.
References
- 1.Fabian MA, Biggs WH, 3rd, Treiber DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 2.Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–44. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 3.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–96. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Konopleva M, Shi YX, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100:184–98. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 5.Mori S, Cortes J, Kantarjian H, Zhang W, Andreef M, Ravandi F. Potential role of sorafenib in the treatment of acute myeloid leukemia. Leukemia & lymphoma. 2008;49:2246–55. doi: 10.1080/10428190802510349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breedveld P, Pluim D, Cipriani G, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer research. 2005;65:2577–82. doi: 10.1158/0008-5472.CAN-04-2416. [DOI] [PubMed] [Google Scholar]
- 7.Brendel C, Scharenberg C, Dohse M, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21:1267–75. doi: 10.1038/sj.leu.2404638. [DOI] [PubMed] [Google Scholar]
- 8.Burger H, van Tol H, Boersma AW, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- 9.Dai CL, Tiwari AK, Wu CP, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer research. 2008;68:7905–14. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiwase DK, Saunders V, Hewett D, et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin Cancer Res. 2008;14:3881–8. doi: 10.1158/1078-0432.CCR-07-5095. [DOI] [PubMed] [Google Scholar]
- 11.Houghton PJ, Germain GS, Harwood FC, et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer research. 2004;64:2333–7. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- 12.Hu S, Franke RM, Filipski KK, et al. Interaction of imatinib with human organic ion carriers. Clin Cancer Res. 2008;14:3141–8. doi: 10.1158/1078-0432.CCR-07-4913. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Cusatis G, Brahmer J, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6:432–8. doi: 10.4161/cbt.6.3.3763. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura Y, Oka M, Soda H, et al. Gefitinib (“Iressa”, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2-mediated drug resistance. Cancer research. 2005;65:1541–6. doi: 10.1158/0008-5472.CAN-03-2417. [DOI] [PubMed] [Google Scholar]
- 15.Shi Z, Peng XX, Kim IW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer research. 2007;67:11012–20. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- 16.Yang CH, Huang CJ, Yang CS, et al. Gefitinib reverses chemotherapy resistance in gefitinib-insensitive multidrug resistant cancer cells expressing ATP-binding cassette family protein. Cancer research. 2005;65:6943–9. doi: 10.1158/0008-5472.CAN-05-0641. [DOI] [PubMed] [Google Scholar]
- 17.Hu S, Niu H, Minkin P, et al. Comparison of antitumor effects of multitargeted tyrosine kinase inhibitors in acute myelogenous leukemia. Mol Cancer Ther. 2008;7:1110–20. doi: 10.1158/1535-7163.MCT-07-2218. [DOI] [PubMed] [Google Scholar]
- 18.Tahara H, Kusuhara H, Maeda K, Koepsell H, Fuse E, Sugiyama Y. Inhibition of oat3-mediated renal uptake as a mechanism for drug-drug interaction between fexofenadine and probenecid. Drug metabolism and disposition: the biological fate of chemicals. 2006;34:743–7. doi: 10.1124/dmd.105.008375. [DOI] [PubMed] [Google Scholar]
- 19.Nezu J, Tamai I, Oku A, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21:91–4. doi: 10.1038/5030. [DOI] [PubMed] [Google Scholar]
- 20.Franke RM, Baker SD, Mathijssen RH, Schuetz EG, Sparreboom A. Influence of solute carriers on the pharmacokinetics of CYP3A4 probes. Clin Pharmacol Ther. 2008;84:704–9. doi: 10.1038/clpt.2008.94. [DOI] [PubMed] [Google Scholar]
- 21.Baker S, Hu S. Pharmacokinetic considerations for new targeted therapies. Clin Pharmacol Ther. 2009 doi: 10.1038/clpt.2008.242. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker SD, Verweij J, Cusatis G, et al. Pharmacogenetic Pathway Analysis of Docetaxel Elimination. Clin Pharmacol Ther. 2008 doi: 10.1038/clpt.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchetti S, de Vries NA, Buckle T, et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther. 2008;7:2280–7. doi: 10.1158/1535-7163.MCT-07-2250. [DOI] [PubMed] [Google Scholar]
- 24.Wierdl M, Wall A, Morton CL, et al. Carboxylesterase-mediated sensitization of human tumor cells to CPT-11 cannot override ABCG2-mediated drug resistance. Mol Pharmacol. 2003;64:279–88. doi: 10.1124/mol.64.2.279. [DOI] [PubMed] [Google Scholar]
- 25.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–14. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 26.Hakime A, Hines-Peralta A, Peddi H, et al. Combination of radiofrequency ablation with antiangiogenic therapy for tumor ablation efficacy: study in mice. Radiology. 2007;244:464–70. doi: 10.1148/radiol.2442061005. [DOI] [PubMed] [Google Scholar]
- 27.Minkin P, Zhao M, Chen Z, Ouwerkerk J, Gelderblom H, Baker SD. Quantification of sunitinib in human plasma by high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;874:84–8. doi: 10.1016/j.jchromb.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao M, Rudek MA, He P, et al. A rapid and sensitive method for determination of sorafenib in human plasma using a liquid chromatography/tandem mass spectrometry assay. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;846:1–7. doi: 10.1016/j.jchromb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Thomas J, Wang L, Clark RE, Pirmohamed M. Active transport of imatinib into and out of cells: implications for drug resistance. Blood. 2004;104:3739–45. doi: 10.1182/blood-2003-12-4276. [DOI] [PubMed] [Google Scholar]
- 30.White DL, Saunders VA, Dang P, et al. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108:697–704. doi: 10.1182/blood-2005-11-4687. [DOI] [PubMed] [Google Scholar]
- 31.Okabe M, Szakacs G, Reimers MA, et al. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol Cancer Ther. 2008;7:3081–91. doi: 10.1158/1535-7163.MCT-08-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Anderle P, Bussey KJ, et al. Membrane transporters and channels: role of the transportome in cancer chemosensitivity and chemoresistance. Cancer research. 2004;64:4294–301. doi: 10.1158/0008-5472.CAN-03-3884. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Lovejoy KS, Shima JE, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer research. 2006;66:8847–57. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grube M, Meyer zu Schwabedissen HE, Prager D, et al. Uptake of cardiovascular drugs into the human heart: expression, regulation, and function of the carnitine transporter OCTN2 (SLC22A5) Circulation. 2006;113:1114–22. doi: 10.1161/CIRCULATIONAHA.105.586107. [DOI] [PubMed] [Google Scholar]
- 35.Lenihan DJ. Tyrosine kinase inhibitors: can promising new therapy associated with cardiac toxicity strengthen the concept of teamwork? J Clin Oncol. 2008;26:5154–5. doi: 10.1200/JCO.2008.18.5439. [DOI] [PubMed] [Google Scholar]
- 36.Mann DL. Targeted cancer therapeutics: the heartbreak of success. Nature medicine. 2006;12:881–2. doi: 10.1038/nm0806-881. [DOI] [PubMed] [Google Scholar]
- 37.Szakacs G, Annereau JP, Lababidi S, et al. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer cell. 2004;6:129–37. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 38.Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent(R), SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ABC transporters, P-glycoprotein (ABCB1) and ABCG2. Drug metabolism and disposition: the biological fate of chemicals. 2008 doi: 10.1124/dmd.108.024612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takimoto CH, Awada A. Safety and anti-tumor activity of sorafenib (Nexavar) in combination with other anti-cancer agents: a review of clinical trials. Cancer chemotherapy and pharmacology. 2008;61:535–48. doi: 10.1007/s00280-007-0639-9. [DOI] [PubMed] [Google Scholar]
- 40.Vlaming ML, Mohrmann K, Wagenaar E, et al. Carcinogen and anticancer drug transport by Mrp2 in vivo: studies using Mrp2 (Abcc2) knockout mice. The Journal of pharmacology and experimental therapeutics. 2006;318:319–27. doi: 10.1124/jpet.106.101774. [DOI] [PubMed] [Google Scholar]
- 41.Zimmermann C, van de Wetering K, van de Steeg E, Wagenaar E, Vens C, Schinkel AH. Species-dependent transport and modulation properties of human and mouse multidrug resistance protein 2 (MRP2/Mrp2, ABCC2/Abcc2) Drug metabolism and disposition: the biological fate of chemicals. 2008;36:631–40. doi: 10.1124/dmd.107.019620. [DOI] [PubMed] [Google Scholar]
- 42.Bardelmeijer HA, Ouwehand M, Buckle T, et al. Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer research. 2002;62:6158–64. [PubMed] [Google Scholar]
- 43.van Herwaarden AE, Wagenaar E, van der Kruijssen CM, et al. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. The Journal of clinical investigation. 2007;117:3583–92. doi: 10.1172/JCI33435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oostendorp RL, Buckle T, Beijnen JH, van Tellingen O, Schellens JH. The effect of P-gp (Mdr1a/1b), BCRP (Bcrp1) and P-gp/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Invest New Drugs. 2008 doi: 10.1007/s10637-008-9138-z. [DOI] [PubMed] [Google Scholar]
- 45.Porkka K, Koskenvesa P, Lundan T, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112:1005–12. doi: 10.1182/blood-2008-02-140665. [DOI] [PubMed] [Google Scholar]
- 46.Leis JF, Stepan DE, Curtin PT, et al. Central nervous system failure in patients with chronic myelogenous leukemia lymphoid blast crisis and Philadelphia chromosome positive acute lymphoblastic leukemia treated with imatinib (STI-571) Leukemia & lymphoma. 2004;45:695–8. doi: 10.1080/10428190310001625728. [DOI] [PubMed] [Google Scholar]
- 47.Petzer AL, Gunsilius E, Hayes M, et al. Low concentrations of STI571 in the cerebrospinal fluid: a case report. British journal of haematology. 2002;117:623–5. doi: 10.1046/j.1365-2141.2002.03523.x. [DOI] [PubMed] [Google Scholar]
- 48.Takayama N, Sato N, O’Brien SG, Ikeda Y, Okamoto S. Imatinib mesylate has limited activity against the central nervous system involvement of Philadelphia chromosome-positive acute lymphoblastic leukaemia due to poor penetration into cerebrospinal fluid. British journal of haematology. 2002;119:106–8. doi: 10.1046/j.1365-2141.2002.03881.x. [DOI] [PubMed] [Google Scholar]
- 49.Medioni J, Cojocarasu O, Belcaceres JL, Halimi P, Oudard S. Complete cerebral response with sunitinib for metastatic renal cell carcinoma. Ann Oncol. 2007;18:1282–3. doi: 10.1093/annonc/mdm275. [DOI] [PubMed] [Google Scholar]
- 50.Valcamonico F, Ferrari V, Amoroso V, et al. Long-lasting successful cerebral response with sorafenib in advanced renal cell carcinoma. J Neurooncol. 2009;91:47–50. doi: 10.1007/s11060-008-9676-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.