

Abstract
The reduction potentials (Eh) for the redox couples GSH/GSSG and cysteine/cystine (Cys/CySS) in plasma are useful indicators of systemic oxidative stress and other medically relevant physiological states. This paper describes a sensitive method for determining plasma levels of GSH, GSSG, Cys and CySS used to calculate the in vivo Eh values. The method uses iodoacetate to alkylate free thiols; derivatization with dansyl chloride to fluorescently tag amino groups; and HPLC and fluorescence to separate, detect and quantify the molecules. Benefits of the method, such as sensitivity and dynamic range, are described; as are caveats, such as the importance of preventing red blood cell hemolysis and limitations in quantification of GSSG. General principles of redox chemistry and previous studies showing that the compounds are more oxidized than predicted from their standard reduction potentials are reviewed. The calculated in vivo reduction potential, Eh, is a convenient and informative way of summarizing the redox environment of plasma and is also useful for studies of cerebrospinal fluid, lymph, bronchoalveolar lavage fluid, human biopsies and a broad range of in vitro cell culture conditions.
Keywords: Glutathione, cysteine, oxidative stress, redox signaling, human plasma
Introduction
Thiol/disulfide couples have diverse functions in cell signaling, protein regulation and macromolecular structure and trafficking. Many of these couples, including both low molecular weight thiol/disulfides such as GSH/GSSG and Cys/CySS, and central protein dithiol/disulfide couples such as thioredoxin-1 and -2, exist under non-equilibrium states where the kinetics of oxidation and reduction determine the steady-state balance of reduced and oxidized forms [1]. This displacement from equilibrium allows rapid and dynamic regulation, supports redox signaling and represents a central target of non-radical mechanisms of oxidative stress [2]. The latter character makes the central GSH/GSSG and Cys/CySS couples in blood plasma useful as a clinical measure of oxidative stress [3].
A number of methods are available for study of the redox states of specific proteins in cells and subcellular compartments [4], typically using redox western blots [5-9], BIAM blots [10, 11] and related immunoassays (e.g., GSylation [12, 13]). Mass spectrometry-based redox proteomics methods are also becoming available [14-17]. However, to date, little information is available using these approaches to evaluate the steady-state redox potential in vivo. Direct measurement of GSH in vivo in humans is possible with magnetic resonance spectroscopy [18], but the sensitivity is limited and the approach has not been widely used.
In contrast, considerable information is available for in vivo redox states obtained by measuring the GSH/GSSG and Cys/CySS couples. The present manuscript describes a method which we have applied to thousands of human plasma samples for analysis of these redox couples under a range of health conditions. Prior to development of this assay, this laboratory assayed GSH by several methods: enzymatic recycling with GSSG reductase [19], spectrophotometry using the Saville assay [20], spectrophotometry with Ellmans reagent [21], fluorometry with orthophthaldehyde [22], and HPLC with UV-detection [23], electrochemical detection [24], spectrophotometric detection of S-carboxymethyl, N-DNP-derivatives [23], and fluorescent detection of bimane derivatives [25, 26]. With appropriate calibration and recognition of specific limitations, each of these methods provides acceptable and comparable estimates of GSH concentration. There are some exceptions, e.g. the thiol reagents (Saville assay and Ellman's assay) do not distinguish GSH, Cys and precursors such as N-acetylcysteine. However, an important conclusion is that there are many valid and convenient methods for measurement of GSH in biologic systems, a point further supported by the >80,000 scientific publications on GSH.
Despite the availability of these methods, useful clinical measurement of GSH is problematic for a number of reasons. In blood, most GSH is present in red blood cells, and the hematocrit is variable, especially in disease conditions of greatest interest. This difficulty can be overcome by isolating RBC for analysis or by correcting for hematocrit. However, even with such precautions, RBC have a relatively long and variable half-life in circulation so that the population of cells present is heterogeneous. Redox states vary with the life cycle of cells [27, 28], so changes in populations of RBC could change the measured values. Furthermore, on a cellular basis, GSH synthesis can be limited by availability of precursors, activity of synthetic enzymes and transporters, activity of the GSSG reductase, NADPH supply and other factors. Oxidatively damaged RBC are rapidly removed from circulation so that an inherent mechanism exists to prevent accumulation of cells with decreased GSH. Measures of GSH in RBC and plasma as a function of age showed that differences in RBC, although significant, are relatively less than in plasma [29]. Thus, measurement of GSH in whole blood or RBC represents a tissue measurement this complicates use as a systemic oxidative stress indicator.
GSH in plasma provides an alternative indicator of systemic oxidative stress because of the continuous interaction of plasma with tissues and organ systems. A main limitation is that the GSH, per se, is variable due to its relationship to the nutritional status of sulfur amino acids and use of GSH for detoxification purposes. Because of this, we use an expression of the reduced to oxidized forms of GSH as an indicator of oxidative stress. The commonly used expression of GSH/GSSG provides a simple means to summarize changes, but because oxidation-reduction reactions involving GSH consume 2 GSH to produce GSSG, the reducing force available from a GSH:GSSG ratio of 100 is substantially different for 10 mM GSH and 100 μM GSSG than it is for 100 μM GSH and 1 μM GSSG [30]. The use of a calculated redox potential (EhGSSG) for the GSH/GSSG couple, eliminates this problem and allows direct comparison of the “reducing force” available from the GSH/GSSG couple to that available from any other electron donor/acceptor couple.
The steady-state plasma redox potentials vary as a function of at least 3 processes, transport of thiol and disulfides between cells and plasma, redox reactions at the cell surface, and oxidation of thiols in the extracellular compartment [31]. Relative contributions of these processes are variable in association with tissues and disease state. However, all cells have transporters for Cys, CySS, GSH and/or GSSG, and these are clearly important in the overall balances maintained in plasma. Because the activity of these transporters maintains communication of the tissue redox potentials with those of plasma, plasma values provide useful indicators of tissue redox potentials [31]. On the other hand, because they are not in equilibrium with cell and tissue redox systems, these are only indirect indicators. Moreover, because the plasma interacts with the whole body, the overall balance may not reflect local effects in specific tissues.
Concentrations of GSH in plasma are much lower than in cells, and analysis of plasma, instead of whole blood or RBC, has a disadvantage that the low concentration relative to RBC makes it subject to artifact from even a small extent of hemolysis. With appropriate collection technique as described in the current protocol, samples can be obtained without hemolysis [32]. A second difficulty with plasma measurements is that GSH undergoes thiol-disulfide exchange with high concentrations of CySS and albumin-SS-Cys [33]. This problem can be prevented by rapid chemical modification of the thiols upon collection. Human plasma also contains oxidants and oxidative enzymes, such as ceruloplasmin, and the degradative enzyme, γ-glutamyl transpeptidase (GGT), which hydrolyzes GSH. Although the latter activity is low in most individuals, the activity increases with age and with certain diseases. Thus, although there are advantages to plasma measurements, there are specific requirements for sample collection to avoid artifacts [32, 34]. The protocol below was designed to address these limitations.
A problem for determination of plasma EhGSSG is that GSSG is difficult to measure at submicromolar concentrations. Thus, a major impetus for development of the current protocol was to provide a method to reliably measure GSSG in human plasma. GSH/GSSG recycling assays with GSSG reductase [19] overestimate GSSG in plasma due to the presence of the disulfide of Cys and GSH, CySSG, which reacts with GSH under the assay conditions to generate GSSG. Assay with HPLC and spectrophotometric detection of S-carboxymethyl, N-DNP-derivatives [23] is not sensitive enough to detect GSSG in human plasma. Methods obtaining GSSG as the difference between GSH and the total amount of GSH after reduction with DTT result in error due to the presence of micromolar concentrations of CySSG. The current assay measures GSSG separately from CySSG with a sensitivity to measure nanomolar concentrations in 10 to 50 μl aliquots. The capability to do this under conditions where GSH, Cys and CySS are also measured provides the most compelling reason to use the method described. Although not addressed in the current protocol, measurement of GSSG at nanomolar concentrations is also useful for studies with small tissue biopsies, bronchoalveolar lavage fluid, embryos, and primary cultures of small numbers of cells. Such applications have provided significant advances in the understanding of the thiol-dependent redox signaling and control mechanisms [1].
Principles
Redox Biology
Background information on the principles of thiol/disulfide reactions in redox biology are needed for effective use of the method described here. Clark's book on biological oxidation-reduction potentials [35], Gilbert's review on thiol-disulfide systems [30], and Schafer and Buettner's review on thiols and oxidative stress [36] are especially useful. More detailed summaries of the application of the methods described below for in vivo and cell culture studies are also available [2, 3, 31].
Oxidative stress and redox signaling involve electron transfer reactions, and redox potential (Eh) values provide convenient parameters to describe relationships between different biochemicals undergoing oxidation-reduction reactions. Eh is a measure of the tendency of redox couples to accept or donate electrons [34]. Eh is dependent upon both the inherent properties of the chemical to accept or donate electrons, expressed in the standard potential (Eo), and also the concentrations of the acceptor (oxidized) and donor (reduced) species of the couple, as defined by the Nernst equation (Eh = Eo + RT/nF ln([oxidized]/[reduced]). For an individual couple, such as that consisting of GSSG and 2H+/2GSH, the value is the half-cell potential expressed relative to a standard 2H+/H2 electrode. The redox potentials for thiol/disulfide couples are dependent upon pH (59 mV/pH unit for 1-electron transfers and 29.5 mV/pH unit for 2-electron transfers). In Eh calculations, this pH dependence is included in the standard potential Eo value; for plasma, values are expressed for pH 7.4. For cytoplasm and organelles, appropriate Eo values must be used for the local pH.
The difference in Eh values (ΔEh) between two couples is proportional to the free energy for electron transfer (ΔG = -nFΔEh, where n = number of electrons transferred and F is Faraday's constant), so the Eh values also provide information on the energetics of electron transfer. It is important to note that in biologic systems, electron transfer between redox couples is often kinetically limited so that even though ΔEh and ΔG values for a reaction may be favorable, electron transfer rates between two couples can be so slow as to be biologically unimportant. Thus, by having specific catalysts for electron transfer from NADH to water, it is possible to convert energy available from redox reactions into synthesis of ATP without a large array of competing non-energy yielding electron transfer reactions. Similarly for thiol-disulfide systems, under usual physiologic conditions, electron transfer pathways are highly controlled.
Although the concepts of electron transfer pathways as developed for mitochondrial bioenergetics are applicable to thiol systems, the relatively slow and common oxidation reactions of thiols are often depicted as autooxidation and experimental artifact rather than functional pathways operating at relatively slow rates [37]. Application of the principles used for mitochondrial electron transfer to thiol/disulfide redox reactions is complicated by the large number of distinct thiols in biologic systems, the considerable range of reactivity of the thiols, the heterogeneity of distribution of thiols and the limited means to measure Eh of thiol/disulfide couples. For instance, there are 214,000 unique Cys residues encoded by the human genome [2]. These protein thiols may be present in only a few copies per cell or high concentrations such as 10 mM GSH in liver. The thiols in proteins can be effectively inert or highly reactive due to their precise location within protein structure. And unlike NADH, flavoproteins and cytochromes, where redox changes can be directly measured by spectrophotometry or fluorometry, few means are available to measure thiols and disulfides without extraction or other major perturbations which can alter redox balance.
These concepts are relevant to the current assay because the results obtained with this assay provided the first clear evidence that thiol-disulfide systems are maintained under stable, non-equilibrium conditions in vivo [38]. Specifically, the assay provided unequivocal evidence for a disequilibrium of the Cys/CySS and GSH/GSSG couples in human plasma [38, 39], where the couples are substantially displaced from equilibrium despite the lack of membrane barriers or non-covalent binding of individual components. The second-order rate constant for thiol/disulfide exchange reactions [20 M-1min-1; [30]] is relatively slow at the prevailing concentrations of GSH (2 μM) and CySS (40 μM) in plasma [38], so that the calculated rate of reaction of GSH with CySS (0.0016 μmol·L-1·min-1) is much lower than the physiologic rate of Cys turnover in plasma [40-42]. Application of this method to a range of cell culture and animal models has now established that maintenance of thiol/disulfide couples at stable, non-equilibrium conditions (“redox poise”) is a general property of biologic systems. The span of redox potentials for different thiol/disulfide couples differs in subcellular compartments (Fig 1), with the most oxidized values being found in plasma. Fig 1 contains a logarithmic scale where 30 mV is equivalent to a 10-fold change in dithiol/disulfide ratio. Because the span is nearly 300 mV, protein thiol/disulfide redox states can be modulated over several orders of magnitude. Consequently, the energetic differences between couples are such that considerable error can occur in measurement if new electron transfer pathways are created during extraction. This means that all measures of redox poise must be considered operational because they reflect measurements of non-equilibrium steady-state conditions and involve methods which perturb the system.
Fig 1. Redox poise in biologic systems.

Steady-state redox potentials (Eh) of individual thiol/disulfide couples are displaced from equilibrium both within aqueous compartments and between subcellular compartments. For instance, the redox potential (Eh) for glutathione is the half-cell, 2-electron reduction potential of the couple consisting of GSSG and 2H+/2GSH, expressed relative to a standard 2H+/H2 electrode. The present assay was developed to determine the steady-state GSH/GSSG redox potential in plasma. Results show that plasma Cys/CySS redox potential is oxidized relative to that of GSH/GSSG, and the Eh values for each of these couples is highly oxidized relative to cellular and tissue thiol/disulfide redox potentials. Note that the steady-state Eh values are dependent upon the pH of the compartment. The values given in this figure are appropriate for the pH of each respective compartment. Other extracellular fluids are also more oxidized than cell and tissue values, but these have not been studied extensively.
Assay of redox poise
The assay described below is based upon accurate measurement of individual thiols (GSH, Cys) and disulfides (GSSG, CySS), and calculation of the Eh for each couple using the Nernst equation. These are non-equilibrium Eh values and represent steady-state values; hence, the terms “redox poise” or “redox state” are often used to avoid the technically incorrect use of “redox potential”. The method is described for measurement of plasma values and utilizes collection procedures and preservation solution which have been extensively tested [32]. Typical ranges of concentrations for the individual thiols and disulfides and calculated Eh values are given in Table 1. These data are summarized from a study of Eh as a function of age [39] and as a function of time of day [43]. In the former (top row), plasma samples were collected without regard to food consumption or time of day, with approximately even distributions of age across the age span. These samples were collected by laboratory personnel specifically dedicated to redox measurements. In the latter (bottom 4 lines), subjects were given meals with the RDA for sulfur amino acids at controlled times in the Emory Hospital General Clinical Research Center and plasma samples were collected by trained nurses within the unit. The results, from each study, presented as mean ± 1 SD, show the extent of variability among individuals. The distributions of analytes are skewed [39] and must be normalized for statistical evaluation. The data show significant effects of age and food consumption. The data also illustrate a difference in mean values obtained with samples collected in different facilities. The latter is noteworthy in that the translation of the method from the hands of redox biologists into those of skilled phlebotomists resulted in 5-10 mV more oxidized mean EhGSSG values, with lesser effect on EhCySS. Systematic studies in the laboratory do not provide an explanation for this, but the results show that in experimental studies, control and experimental subjects must be sampled by the same personnel under the same experimental conditions.
Table 1. Representative values for Cys/CySS and GSH/GSSG redox couples in human plasmaa.
| Age | Conditions | Cys (μM) | CySS (μM) | EhCySSb (mV) | GSH (μM) | GSSG (μM) | EhGSSGb (mV) |
|---|---|---|---|---|---|---|---|
| 18-93 y | No time-of-day control, Not fasting | 8.6 ± 3.4 | 47 ± 19 | -72 ± 14 | 1.92 ± 0.52 | 0.05 ± 0.04 | -140 ± 12 |
| 18-83 y | 0830, Fasting | 9.6 ± 2.9 | 66.0 ± 16.7 | -73.6 ± 8.4 | 1.39 ± 0.56 | 0.09 ± 0.04 | -123 ± 11 |
| 18-83 y | 2230, Postprandial | 12.0 ± 4.5 | 62.5 ± 20.7 | -79 ± 11 | 1.18 ± 0.56 | 0.08 ± 0.05 | -129 ± 13 |
| 60-83 y | 0830, Fasting | 9.8 ± 2.8 | 85.1 ± 16.8 | -70.5 ± 8.9 | 1.06 ± 0.24 | 0.08 ± 0.05 | -114 ± 8 |
| 60-83 y | 2230, Postprandial | 13.0 ± 4.3 | 81.3 ± 27.6 | -78.7 ± 7.3 | 0.98 ± 0.15 | 0.04 ± 0.01 | -124 ± 4 |
Data for mean Eh are averages of Eh values for individuals, calculated from the respective disulfide and thiol concentrations in the individual; hence, mean Eh values are not precisely the same that calculated from the mean thiol and disulfide concentrations given in this table.
A summary of p values for significant associations of individual parameters with known health risk factors and disease obtained with this method is given in Table 2. Most of these results are for relatively small populations, and larger studies are needed for confirmation and extension of these findings. Of the individual chemicals, CySS concentration is most commonly associated with disease risk factors. A number of significant associations are also obtained for GSH concentration. As can also be seen from the table, oxidation of EhGSSG and/or EhCySS is commonly associated with disease risk. The differences between Cys and CySS in associations with antioxidants [45] and Zn2+ [46], and also in association with IL-1β and TNF-α [50], indicates that different redox mechanisms are involved. Similarly, the association of flow-mediated dilation with EhCySS and not with EhGSSG [51], while carotid intima media thickness was associated with EhGSSG and not with EhCySS [52], further suggests different redox mechanisms. These data underscore the need for detailed analysis of individual thiol/disulfide couples.
Table 2. Summary of p values for associations of amino thiol parameters with disease risk factors from publications using this method.
| Variable | GSH | GSSG | Cys | CySS | CySSG | EhGSH | EhCys | Ref |
|---|---|---|---|---|---|---|---|---|
| Age | <0.0001 | <.001 | 0.001 | <0.001 | a | <.001 | <.0001 | [39] |
| Time of Day | 0.05 | a | <.0001 | <.0001 | a | 0.02 | <.0001 | [43] |
| Med Diet | a | <0.05 | b | b | b | a | <0.05 | [44] |
| +Antioxidants | a | a | <0.01 | a | b | <0.05 | <0.05 | [45] |
| + Zn2+ | a | a | a | <0.05 | b | a | a | [46] |
| Smoking | <0.005 | a | <0.0001 | <0.01 | <0.0001 | 0.01 | <0.001 | [47] |
| Alcohol (BALF)c | <0.001 | <0.005 | b | b | b | <0.001 | b | [48] |
| Chemotherapy | <0.001 | b | a | <0.05 | b | <0.01 | Trend | [49] |
| IL-1β | b | b | <0.05 | a | b | b | <0.001 | [50] |
| TNF-α | b | b | a | <0.05 | b | b | <0.05 | [50] |
| Flow-mediated dilation | a | a | a | <0.01 | 0.01 | a | a | [51] |
| Carotid IMT | 0.01 | a | a | 0.02 | b | 0.02 | a | [52] |
| Persistent Atrial Fibrillation | b | b | b | b | b | <0.001 | <0.001 | [53] |
Refers to no significant association
Refers to no corresponding data were reported
Data for plasma did not show significant differences
Aspects of the rationale for development of the method and variations which were tested in development of this method are given below under “Caveats”. The key points of this protocol are 1) sample collection with a preservation solution and procedures to strictly avoid hemolysis are critical for accurate and useful data, 2) thiols are blocked by treatment with iodoacetic acid, which also introduces a negative charge useful for separation, 3) amines are modified with dansyl chloride to allow sensitive quantification by fluorescence, 4) derivatives are separated by HPLC on a weak anion exchange column with gradient elution, and 5) quantification is obtained by integration relative to the integral of an internal standard, with routine analysis of an external standard mix for validation and quality assurance.
Materials
Bathophenanthroline disulfonate sodium salt (BPDS)(Sigma B1375), iodoacetic acid (sodium salt)(Sigma I9148), dansyl chloride (Sigma D2625), L-serine (Sigma S4500), GSH (G4251), GSSG (G4501), Cys (C7352), CySS (C122009), γ-Glu-Cys (G0903), sodium acetate trihydrate (S8625) and heparin (sodium salt) were from Sigma Aldrich (St. Louis). The internal standard, γ-glutamylglutamate (γ-Glu-Glu or γEE) (157205) was from MP Biomedicals. The disulfide of Cys and GSH, CySH-GSH, was from Toronto Research Chemicals (Toronto, C995500). Boric acid, sodium tetraborate, potassium tetraborate, perchloric acid, acetone and chloroform are reagent grade and purchased locally. Methanol is HPLC grade (Fisher A452). Acetic acid is HPLC grade (Fisher A35). Distilled, deionized water is used throughout.
Collection of samples and initial processing involves use of 2 sets of microcentrifuge tubes which are termed “N-tubes” and “S-tubes”. These are prepared in advance and can be stored at -20° or -80° for several months prior to use. The “N” and “S” designations are arbitrary; they were originally used to distinguish a “new” formulation of the preservation solution and its respective “stop” solution from the original “A” and “B” tubes used for an earlier version of this method [32]. The original A and B solutions are still used for collection of mouse plasma and other applications [34].
Preservation Solution for N-tubes
-
Borate Buffer (0.5 M) Stock Solution with internal standard:
0.5 M Borate Buffer Stock Solution (this is stable in the refrigerator): Mix 12.4 g of boric acid and 19 g of Na2B4O7·10 H2O in 500 ml distilled, de-ionized H2O.
-
Addition of internal standard (final, 165 μM γ-glutamylglutamate; γ-Glu-Glu, or γEE)
Weigh precisely an amount of γ-glutamylglutamate (γEE) in the range of 2 to 5 mg.
Calculate the amount of Borate Buffer Stock Solution needed to make 165 μM γEE solution: Multiply the mg of γ-EE weighed times 500 ml and divide this number by 22.75 mg to obtain the volume needed.
As part of the quality assurance procedure, the γEE content of this solution must be verified to be correct relative to previous solutions used in the laboratory. This is done by performing analyses as described below of aliquots of new Stock Solution along with aliquots of previously used Stock Solution. Because the accuracy of this solution is critical for long-term comparisons and the integration of peaks represents a major source of variation, 4 aliquots of each solution should be analyzed for this comparison.
For enough N-solution for approx. 66 N-tubes: Mix 0.53 g L serine, 25 mg heparin (sodium salt), 50 mg BPDS (bathophenanthrolene disulfonate), 300 mg iodoacetic acid (sodium salt) into 10 ml Borate Buffer Stock with internal standard
Add 150 μl of N solution to each “N” tube. Prior to aliquoting N solution to each “N” tube, check accuracy of pipettor by pipetting 150 μl of water onto balance and fill out Internal Standard Solution Preparation Log form.
Store N-tubes in a freezer (-20 °C).
Preparation of “S” tubes
Preparation of 10% perchloric acid containing 0.2 M boric acid: Add 6.2 g boric acid into approximately 300 ml distilled, deionized H2O, add 71 ml perchloric acid (70%), add H2O to bring volume to 500 ml. This solution can be stored indefinitely in a refrigerator.
Add 200 μL per microcentrifuge tube. Store S-tubes in a freezer (-20 °C).
Materials for plasma collection
“N” tubes and “S” tubes as described above should be removed from the freezer; N tubes should be allowed to equilibrate to room temperature and S tubes should be maintained in a refrigerator or on ice. Because the timing of sampling, centrifugation and transfer of an aliquot of plasma to the S tube must be consistent, the following checklist should be used prior to initiation of sample collection:
23, 21 or 18 gauge butterfly needle
3 or 5 cc syringe
Ice bucket with ice
Microcentrifuge tube rack
2″ × 2″ gauze
Biohazard/sharps container
N and S tubes pre-labeled with freezer labels/cryo markers
Subject consent forms
Calibrated pipet
Microcentrifuge
200 μL pipetter tips
Gloves
Bandages
Alcohol wipes
Tourniquet
Data collection sheets
Solutions for sample derivatization
KOH/tetrahydroborate solution. A saturated tetrahydroborate solution containing KOH is used to adjust pH to 9.0 ± 0.2. This solution is prepared by adding 5.6 g KOH to a plastic bottle containing 50 g K2B4O7 ·4H2O and 100 ml water. This is mixed and allowed to stand at room temperature for several hours (typically overnight) before removing the solution from the remaining precipitate. This solution is stable indefinitely at room temperature.
The dansyl chloride solution is prepared on the day of derivatization by dissolving 200 mg dansyl chloride in 10 ml of acetone. This is sufficient for 32 samples.
HPLC Solvents
Preparation of Acetate Stock: This is prepared by mixing 272 g sodium acetate trihydrate, 122 ml H2O and 378 ml glacial acetic acid and dissolving overnight on a stirplate. Note that there are many grades of acetic acid and acetate commercially available; only reagent grade or HPLC grade have been found to provide acceptable results.
-
Solvents A and B are filtered with a 0.45-mm pore size filter prior to use.
Solvent A. 80% (v/v) methanol/water.
Solvent B is an acetate-buffered (pH 4.6) methanol solution prepared by mixing 640 ml of methanol, 200 ml of acetate stock, 125 ml of glacial acetic acid and 50 ml of water.
External Standards
External standards are used to assure peak identification, and a standard mix is used routinely for both quality control and quality assurance. The following provides a general description for preparation of standards; the currently used protocol for the Clinical Biomarkers Laboratory is given in Appendix 1. Commercially available Cys and GSH typically have some fraction of respective disulfides so reduction is needed to obtain respective S-carboxymethyl, N-dansyl derivatives which are free from the disulfides. To eliminate these disulfide forms, Cys and GSH standards are prepared as 1 mM solutions with 2 mM dithiothreitol and diluted to10 μM into an appropriate dilution of the N solution to give 20 μM γEE. Note that if this is done at room temperature, appropriate aliquots of the other standards can be added after 10 min to give a mixed standard with 10 μM of each along with 20 μM γEE. This is convenient for identification because the γEE is 2-fold greater than the other standards, making peak identification easy and unambiguous. Because the disulfide of GSH and Cys is always present in plasma, we also include this in our standard mix. The standard mix is run as every 21st analysis so that any questions concerning either peak identification or quantification can be readily resolved. This can be important, for instance, when unknown chemicals are suspected of overlapping with the internal standard peak or raise questions concerning correct analyte peak identity. GSH and GSSG of biological origin also are contaminated with γ-Glu-Cys or its disulfide form with GSH, mono-des-Glu-GSSG. The latter are usually minor components which do not interfere with identification or quantification. It should be noted that the method as described can be used for quantification of with γ-Glu-Cys, Cys-Gly, CySS-mono-gly, mono-des-Glu-GSSG, cysteic acid, glutathione sulfonic acid, homocysteine, and homocysteine-SS-Cys. Standards are stable for about 1 year stored under the same conditions as samples. Standards prepared in larger lots, aliquoted and stored at -80° have not been found to noticeably degrade over periods of years. Degradation, when present, is most noticeable as doublet peaks, apparently due to sulfoxide generation. Crystallized standards can also be prepared and stored as dry powders, but this is not convenient because the amounts needed as standards are so small that this was found to be inefficient.
Instrumentation
The instrumentation currently used by the Clinical Biomarkers Laboratory, Emory University, is a gradient HPLC module 2695 (Waters) with Empower software. The Waters 2475 multi-fluorescence detector is used with excitation at 335 nm and emission detected at 518 nm. The laboratory has previously used instruments available from other companies which also performed satisfactorily for the HPLC separation. Sensitivity for fluorescence detection is a more critical matter because detection of GSSG can be limiting; when originally developed, comparisons showed that some detectors which used monochromators for variable wavelength capabilities were an order of magnitude less sensitive compared to the Gilson fluorescence detectors which used filters and were not satisfactory for routine use. Because the filter detectors were far less expensive, we relied upon the Gilson detectors (Gilson Medical Electronics, Middleton, WI) with 305-395 nm (excitation) and 510-650 nm (emission) bandpass filters until they were no longer available. In addition to the lower cost, these instruments also provided less of a baseline shift in the region of GSSG elution. To obtain the needed dynamic range, we used two detectors in tandem with different sensitivity settings to allow quantification of each of the desired analytes.
Separations are currently obtained on a 25 cm by 4.6 mm, 5 μm silica, LC-NH2 Supelco column from Sigma Aldrich (Cat # 58338). Over the past 15 years, the laboratory has successfully used weak anion exchange columns from multiple companies for this assay. Small differences in resolution and useful column life are observed, but these are not critical for the success of the assay. None of the manufacturers produce columns which are truly reproducible, apparently because the trivalent GSH and tetravalent GSSG have preferential binding to specific charged clusters on the column. The most important issue for successful separation is that the initial %B is adjusted (see next paragraph) according to the character of the column, which is largely determined by the age/use of the column. As columns age, the available positive charges decrease due to accumulated ion pairing and reaction of amines with chemicals in the plasma samples. The most important issue for successful quantification is that samples are analyzed in a routine sample set with an appropriate wash-in time for new columns. Specifically, the GSSG has 4 negative charges which allow very high affinity binding to a small fraction of sites on the column and lower affinity binding to most of the sites on the column. Thus, new columns show a non-linearity of recovery of elution of the GSSG derivative, which can mean that one can add the equivalent of a 10 nM GSSG sample and detect 0 nM while 50 nM would be detected as 40 nM. This problem can be avoided by running a number of wash-in samples and using a quality assurance sample to verify instrument response. A lesser problem of reproducibility occurs with the first sample run following ANY disruption of flow, i.e., stopped flow between sample sets. Consequently, we usually maintain continuous flow where possible and run the first analysis of a sample set as a blank run or a duplicate analysis of the external standard mix to assure proper equilibration.
Protocol
Collection of human plasma for glutathione analysis using N-tubes
The original validation for sample collection and analysis of GSH/GSSG was provided by [32] using a variation of the current protocol. This original method used “A” and “B” tubes and was based upon the same principles as the current method. However, a number of minor variations were adopted in the current method in which the A solution was replaced by the N solution and the B solution was replaced by the S solution, reported in [43]. Modifications included addition of the internal standard into the N solution, increased final IAA concentration, and increased concentrations of all components of the N solution to decrease plasma dilution. The latter change decreased error due to variation in hematocrit and also increased sensitivity to detect low concentrations of GSSG.
The procedure is described for a single sample prepared from a single collection with the syringe. We have evaluated the benefits for duplicate samples and concluded that there is not sufficient benefit gained in analytical accuracy to warrant routine analysis of duplicates. Collection of duplicates does provide protection against loss of sample during derivatization; however, this is rare and there can be artifactual changes due to delay in processing with duplicate collection.
Before starting blood draw, make sure the “N” tubes are at room temperature (to allow reaction of thiols with IAA) and the “S” tubes are on ice (0-4 °C).
Draw 3 ml blood with a butterfly needle and syringe, avoid a vacuum within the syringe.
Add blood from the syringe into a microcentrifuge tube labeled “N” to bring to the 1.5 ml line on the tube. Note that this step is one of the major sources of error for the method. Three points are critical. First, the volume of blood added to the preservation solution fixes the ratio of analytes to internal standard; thus, any error in volume added represent true error which is not corrected by calculation relative to the internal standard. This is designed to be 1350 μl of whole blood added to 150 μl of preservation solution. This can be achieved to within 50 μl accuracy by bringing the meniscus to the 1.5 ml line on clear microcentrifuge tubes, which is our usual approach. Greater accuracy can be obtained by delivering 1350 μl of blood using a better quality syringe or by using tared tubes and weighing to calculate volume added. Second, a delay in transfer to the N tube results in equilibration of the redox couples, meaning that the GSH/GSSG couple becomes more oxidized and the Cys/CySS couple becomes more reduced. Third, the N tube must be at room temperature; otherwise, the reaction of IAA with thiols will be incomplete and the results will be inaccurate. Timed studies show little variability if transfers are made within 1 min of the time blood was drawn, perhaps indicating that the presence of the cells helps preserve the plasma redox potential values.
Invert tube gently (to avoid hemolysis) 2× to mix.
-
Spin tube in microcentrifuge for 1 min to remove RBCs.
This can be done for either 30 s or 1 min, depending upon acceleration/deceleration of the centrifuge, and should be established as a fixed parameter for the sample collection center. We have found 1 min to be satisfactory under most conditions. An important point is that the blood cells must be pelleted without hemolysis. If hemolysis occurs (pinkish color in plasma top layer), the sample should be discarded and another sample collected. We do not perform routine analysis to measure hemolysis because earlier studies with cyano-methemoglobin assay [32] showed no detectable hemolysis with the butterfly needle and syringe collection and care not to pull a vacuum with the syringe. In routine use, hemolysis was detected in only 2 out of more than 600 samples from individuals over 60 y, leading us to conclude that measurement of hemolysis is unnecessary with this collection procedure.
Transfer 200 μl of supernatant to a microcentrifuge tube labeled “S”. Routinely, this transfer should be done in 2 min after blood collection with consistent timing for each collection. In our experience, phlebotomists new to this procedure must perform 5-10 collections to achieve reproducibility.
Invert tubes 2× to mix.
Label tubes (using freezer safe labels, or permanent marker) with HIPAA-acceptable sample identifier, place tubes on ice and store at −80 °C in an appropriate storage container in the order in which they are listed on the master sample list. This should be done as soon as possible after collection. Samples at this step can be shipped on dry ice.
Derivatization
Samples in S tubes are thawed and protein is precipitated by spinning for 2 min in a microcentrifuge. Typically, samples are prepared in batches of 20-50, although larger numbers can be accommodated without difficulty. A 300 μl aliquot of each supernatant is transferred to a fresh microcentrifuge tube. The pH is adjusted to 9.0 ± 0.2 with the KOH/tetraborate solution (approximately 200 μl). About 3 min is required to allow complete precipitation of potassium perchlorate, after which time the pH should be verified to be in the correct range. After 20 min, 300 μl of the dansyl chloride solution is added, and the samples are mixed and placed in the dark at room temperature for 16 to 26 h. Chloroform (500 μl) is then added to each tube to extract the unreacted dansyl chloride, and samples are stored in the presence of both the perchlorate precipitate and the chloroform layer at 0–4° until assay by HPLC. Samples are analyzed within a few days of derivatization. Earlier stability tests using the A and B solutions [32] showed that samples could be stored under these conditions for up to 12 months; we have not repeated these stability tests using the N and S solutions, but anecdotal evidence suggests that some degradation occurs in 1-2 months. Consequently, analysis within 1 month is recommended.
HPLC separation
Samples are centrifuged for 2 min in a microcentrifuge prior to transfer of an aliquot of the upper (aqueous) layer to an autosampler. Typical injection volume is 50 μl, but this can be increased to 200 μl if necessary. In general, better consistency of analysis, particularly for Cys and CySS, is obtained with smaller injection volumes, presumably because of the accumulation of materials from the plasma on the column. Thus, if 50 μl injection volume does not allow quantification of GSSG, volumes are increased to 100 μl for repeat analysis. Typical detection limits for GSSG correspond to plasma values of 10 to 20 nM. A fraction of young, healthy individuals have values below this level; for these, we report a value equivalent to the detection limit. This avoids instrument-related exclusion of young, health individuals from studies and also avoids the unrealistic presentation of zero data. Samples are stable in an autosampler at room temperature for at least 3 days so that continuous weekend analysis is possible.
Flow rate for HPLC is maintained constant at 1.0 ml/min. A typical solvent gradient is as follows: Initial solvent conditions are 80% A, 20% B run at 1 ml/min for 10 min. A linear gradient to 20% A, 80% B is run over the period from 10 to 30 min. From 30 to 46 min, the conditions are maintained at 20% A, 80% B and returned to 80% A, 20% B from 46 to 48 min. Equilibration time for the next run is 12 min. For new columns, the same profile is used but the %B is 40% for initial conditions and the gradient is modified accordingly. As the column ages, the %B is similarly decreased to 10%; if resolution is retained, the %B can be further decreased to 5% before replacing the column. A sample chromatogram of human plasma, along with a corresponding external standard analysis, is shown in Fig 2.
Fig 2. HPLC separation and fluorescence detection of S-carboxymethyl, N-dansyl derivatives of GSH, GSSG, Cys and CySS.
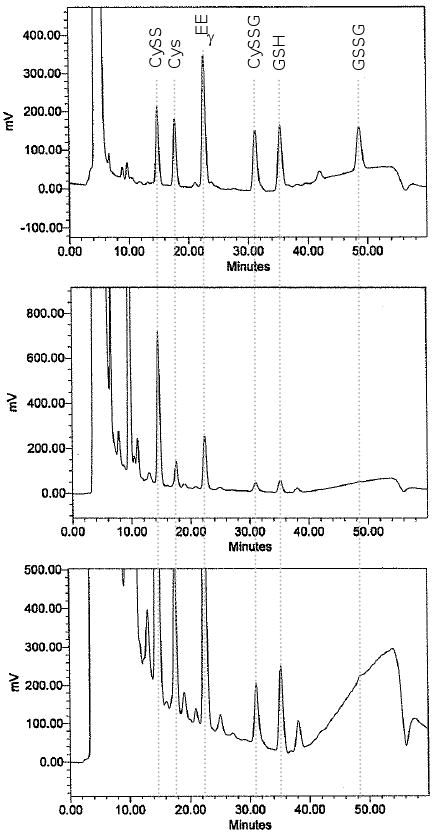
A. Standard mix of 10 μM of GSH, GSSG, Cys, CySS and the Cys-GSH disulfide along with 20 μM internal standard, γ-glutamylglutamate (γ-EE). B. Analysis of 50 μl of derivatized fasting plasma sample from young, healthy individual. C. Analysis of same sample as in B with sensitivity settings to quantify GSSG. The slope of the baseline in the vicinity of the GSSG peak as shown in this chromatogram is typical for this analysis. Within-day coefficient of variation for GSSG analysis under these conditions was found to be 9%, while the other analytes have coefficients of variation of 4-6%. Week-to-week coefficients of variation are comparable to within-day variation, consistent with peak integration being the most significant source of variability. Biological variation in young healthy individuals obtained by measurement of fasting samples at 9 am on Mondays for 4 consecutive weeks showed that average, within-individual variation was 2.3 mV (1 SD).
Calculations and Expected Results
For routine analyses, the integral values provided by the HPLC software for peaks corresponding to GSH, GSSG, Cys and CySS are used and concentrations are calculated relative to the integral of the internal standard, recognizing that the internal standard is only diluted into the plasma and not into the RBC volume present in whole blood. For GSH and Cys, this approach is considered completely valid because linearity of signals for GSH and Cys were established over 4 orders of magnitude and the fluorescence efficiency of the dansyl derivatives of GSH and Cys were found to be equivalent on a mole per mole basis with that for the internal standard, γEE. These conclusions were verified by direct comparisons of results obtained for unknowns calculated using the internal standard for reference and external standards for reference. The largest source of error in both cases was the accuracy of the software for integration of peaks. This error is typically about 5% and can be minimized by using an average value for the internal standard obtained from successive samples.
Quantification of disulfides is routinely obtained using the same approach [32, 34]. However, because the disulfides have two dansyl groups which are close enough to interact, the fluorescence efficiency is not identical to that of the internal standard. These values are typically are in the range of 1.2 for CySS and 1.4 for GSSG [34]. For more precise analysis, standard curves can be performed for each disulfide.
The individual concentrations, expressed in molar values are used with the Nernst equation to calculate redox potentials. For plasma at pH 7.4, an Eo value of -264 mV is used for the GSH/GSSG couple. An Eo value of -250 mV is used for the Cys/CySS couple. A histogram showing the distribution of plasma redox potential values is shown in Fig 3.
Fig 3. Histograms of plasma EhGSSG and plasma EhCySS without control for health risk, disease or time of day, compared to fasting, morning data from healthy, physically fit individuals with normal BMI.

Data are compiled to illustrate the range of values obtained with this assay. Samples compiled from several studies without regard to age, health status, time of day or food consumption are plotted with open squares for EhCySS and open diamonds for EhGSSG, each with a broken line. The relatively broad ranges of values are due to oxidation associated with BMI, type 2 diabetes, advanced age, alcohol abuse, cigarette smoking, proinflammatory cytokine levels and cardiovascular disease risk factors and also reduction as a consequence of eating foods containing Cys. The relatively broad ranges are contrasted with data for fasting, morning plasma EhCySS (filled circles) and EhGSSG (filled triangles) from young, healthy and physically fit individuals.
Caveats
As described above, a critical issue for sample collection is that hemolysis must be avoided. This is necessary because RBC's contain about 1 mM GSH and plasma contains about 2 μM GSH. Collecting without hemolysis can be achieved on a routine basis, even with sample collection from elderly and diseased individuals, by using a butterfly needle and syringe for collection. For instance, in over 600 samples collected with the described method from individuals with an average age of 71, data for only 2 subjects were discarded because of suspected hemolysis. Evacuated collection tubes are not used for this procedure because they result in higher GSH contents due to limited hemolysis [32]. In application of this method to routine use in seriously ill individuals where artifactual hemolysis is not likely to occur, i.e., blood collection by an experienced phlebotomist with a syringe from pre-existing and flushed cannulas, we have found aberrantly high plasma GSH to be relatively common. These high values probably reflect ongoing cell death and release of GSH, a pattern previously observed in studies of acute oxidative stress induced by CCl4 in rodents [54].
Rapid transfer of an appropriate blood volume into the preservation solution as described prevents oxidation and degradation. The preservation (“N” solution) solution was designed to contain an anticoagulant (heparin), a metal ion chelator (BPDS) to prevent metal ion-catalyzed oxidation, an inhibitor of GGT (serine·borate complex), an internal standard (γ-glutamylglutamate), a thiol reagent (iodoacetate) to block free thiols, and a borate buffer to control pH for chemical modification [32]. With this solution at room temperature, the thiols are derivatized during the time needed for centrifugation and transfer of an aliquot to an ice-cold acid solution (“S” solution). Serine·borate was used as an inhibitor of GGT because it is a competitive inhibitor with zero-time inhibition, unlike acivicin which requires a reaction time course for inhibition. Systematic studies in methods development showed: high concentrations of EDTA can interfere with GSSG detection; BPDS relative to no metal ion chelator had only a small benefit in a small fraction of samples, indicating that metal-ion catalyzed oxidative loss of thiols is not a major problem under the conditions of the assay; omission of serine (i.e., elimination of the inhibitory complex for GGT) altered detection of GSH with added GGT but had no effect on measured GSH in samples from healthy individuals; delay in addition or low concentrations of iodoacetate resulted in thiol-disulfide exchange of GSH with CySS; delay in removal of plasma after addition of the original “A” preservation solution had no effect on recovery of GSH or GSSG for periods up to 30 min [32], but this has not been re-evaluated with the “N” solution because of the adoption of the described 2-min time period for routine use in sample collection.
The “S” solution for protein removal was designed in conjunction with the planned derivatization and HPLC procedure. Although perchloric acid is an oxidizing acid, it has an important property in that perchlorate is soluble in the presence of Na+ but insoluble in the presence of K+. Thus, by using a K+-containing solution for neutralization after removal of protein, K+·perchlorate can be removed by centrifugation prior to HPLC. This improves chromatographic resolution and also prolongs the useful life of HPLC columns. Direct comparisons of GSH concentrations obtained with this method and with results obtained using sulfosalicylic acid or trichloroacetic acid showed no significant differences. Stability tests showed that samples could be stored in the presence of the S solution without neutralization and removal of the perchlorate for at least the following: overnight in the refrigerator, 2 weeks at -20° and 2 months at -80°. Because the pH adjustment for subsequent derivatization is optimized to pH 9, a condition where thiols are subject to oxidation, we did not evaluate stability after addition of KOH/K·borate but rather include pH adjustment just prior to dansylation. The relatively narrow pH optimum for dansylation prompted us to include borate in the “S” solution to maximize the buffering capacity at pH 9. Because of a relatively low solubility, the most convenient means to make this solution was to use a saturated solution.
The strategies for chemical modification of the thiols and amines and subsequent HPLC separations were based upon the assay of Reed et al [23]. In this assay, thiols are reacted with iodoacetate to introduce a negative charge at each thiol. The resulting S-carboxymethyl-cysteine has a net charge of -1 while GSH has a net charge of -2. To enhance sensitivity for detection, we replaced 1-fluoro-2,4-dinitrobenzene modification of amines with dansylchloride. Modification of the amines results in additional net negative charge so that N,N′-bis-dansyl-CySS (-2), S-carboxymethyl,N-dansyl-Cys (-2), S-carboxymethyl,N-dansyl-GSH (-3) and N,N′-bis-dansyl-GSSG (-4) are retained on weak anion exchange columns. The conditions for derivatization were extensively studied to assure complete modification of the thiols and because initial studies showed remarkable variability in detection of GSSG products.
In assay development, a number of variations were tested. One variation was to use N-ethylmaleimide for modification of thiols; this was not satisfactory because the modified thiols eluted too rapidly in the chromatography. A variation using bis-maleimido-propionic acid (BMPA) was found to be useful [55] and provides an alternative to the method described. An advantage of BMPA is that it reacts with thiols about 20-fold faster than IAA; however, the products are less stable and direct comparisons to the method described showed no significant differences in recovery of products [55]. Kinetic analyses of the carboxymethylation reaction in plasma samples showed that the reaction was complete in 2 min at room temperature. Hence, the procedure was adopted to collect blood, transfer an aliquot to the “N” tube, centrifuge, and transfer an aliquot to an S tube, as a single sequence of operations. This eliminated the separate carboxymethylation step in the original protocol using A and B tubes.
In development of the final protocol, the dansylation step was most problematic because the derivatization of CySS and GSSG had different kinetics for introduction of the second dansyl group. Modification of one amine on GSSG occurs with a rate constant identical to that of the amine in GSH. The product, mono-dansyl GSSG, elutes in the HPLC analysis approximately with the retention time of S-carboxymethyl-N-dansyl-γ-Glu-Cys, i.e., about halfway between the GSH derivative and bis-dansyl-GSSG. A similar problem occurs with the method of Reed et al [23] if the incubation time is shortened, but this can go unnoticed because the mono-DNP-GSSG coelutes with S-carboxymethyl-N-DNP-GSH. In the present protocol, conditions were optimized to accommodate the slower rate of reaction for the second amine within the limits of the stability of dansylchloride at high pH and the solubility of dansylchloride under the conditions of assay. For this, the borate buffer was important to easily achieve a pH of 9.0 ± 0.2. Time course studies showed that derivatization at room temperature overnight (16-24 h) was more flexible than using a higher temperature and shorter times because it allowed for derivatization at any time during the work day followed by termination of the reaction the following morning. Direct comparisons of samples incubated for 16 h and 24 h showed no significant differences. Chloroform was used to remove the dansyl chloride to terminate the reaction for kinetic analyses and was retained in the final protocol because it decreased the presence of other unidentified fluorescent products which interfered with measurement of peaks of interest. Additionally, this step improved the HPLC by removing phospholipids which accumulate on the weak anion exchange column and can cause variable retention time for N,N′-bis-dansyl-GSSG.
The HPLC conditions are basically those of Reed et al. [23] using a weak anion exchange column and a sodium acetate gradient in the presence of methanol. In the development of the current protocol, several variations were tested in attempts to decrease chromatography time without loss of ability to quantify Cys, CySS, GSH or GSSG. These studies showed that quantification of GSH and GSSG could be reliably obtained in 25 min chromatographic runs by increasing the initial percent B and shortening the gradient time. Alternatively, combination of acetate, succinate and citrate, but not acetate plus citrate or citrate alone, resulted in effective separation of GSH and GSSG with shorter run times. However, no combination was found, either by using direct mixtures or ternary gradients, which provided ability to reliably obtain Cys and CySS with shortened run times. This failure generally resulted because the Cys and CySS derivatives are similar in properties to related compounds in plasma (Cys-Gly, CySS-mono-Gly, CySS-bis-Gly, homoCys, homoCySS, homoCys-SS-Cys disulfide, homoCys-SS-Cys-Gly disulfide) [38] and are not readily resolved with variations in flow rate, pH, salt concentration or methanol content.
The current method was independently validated by simultaneous measurement of samples with a stable isotope dilution mass spectrometry [56]. The latter was developed with the expectation that a mass spectrometry method could shorten the analysis time and provide more rigorous data. However, the procedure developed did not have sufficient dynamic range to consistently provide good quantification of both GSSG and CySS in plasma samples. Because multiple analysis were required to reliably obtain GSSG values, the method ultimately saved little analysis time over the HPLC method and cost more. Hence, although this method is available, it is not as useful as the currently described HPLC method for routine analysis.
Summary of most critical points for accurate measurements
The initial blood collection and transfer to the preservation solution fixes the ratio of analytes to the internal standard and also fixes the oxidized to reduced ratios. As indicated in the preparation of the N tube, the internal standard concentration must be made to very precise standards, i.e., < 1% error and the volume of preservation added to the N tube must be very accurate, i.e., < 1% error. If this is done, then the most critical step in the procedure is transfer of the correct volume of blood into the N tube. For human studies, this step is often performed by phlebotomists who are collecting samples for a number of different purposes, often with competing demands in terms of collection order and timing. Hence, there can be variability which is not easily controlled. However, the critical nature of this step can be emphasized. When possible, we have adopted protocols for collection of redox potential samples as the first in series of sample collections.
A simple diagnostic procedure to verify accuracy of sample collection involves comparison of the integral for the internal standard measurements within a sample set. If accuracy of the integration has been validated and the internal standard concentration in the N tube has been verified according to the standard operating procedures, variability in integrals for the internal standard peaks most likely means that the volume of blood added to N tube was inconsistent. The collection center must be notified of this problem. For irreplaceable samples, calculations are performed relative to the external standard with correction for the extent of dilution with the preservation solution based upon the internal standard recovery.
After addition to the N tubes, the ratios of analytes to internal standard are fixed so that pipetting errors do not affect results. However, a second major source of error is in the integration of peaks; error in integration of the unknowns is amplified by the error in the integration of the internal standard. Thus, routine evaluation of integration parameters and validation relative to the external standards are needed to minimize error. Direct comparisons of duplicate samples and repeat measures showed that coefficients of variation are less if the average value for the internal standard for a 20-sample set is used instead of the internal standard for each individual sample. However, we have found this to be cumbersome and have adopted a standard operating procedure in which we verify that internal standard recoveries are consistent and perform calculations based upon the internal standard peak of each respective sample.
Assay of GSSG is the most problematic of the analytes measured. The major source of variation is in the accurate pH adjustment, which is necessary because of the narrow pH optimum for GSSG reaction with dansyl chloride. For quality control, chromatograms are examined for a mono-dansyl-GSSG peak in the vicinity of the γ-Glu-Cys peak. If derivatization is incomplete, this peak will be evident. Another indicator of the likelihood of incomplete derivatization is the color of the chloroform layer. The chloroform layer will be yellow if the pH adjustment is accurate but clear if the pH is too alkaline. An additional problem can occur with new columns, where high affinity sites must be saturated before quantitative recovery of the GSSG derivative is obtained.
Perspectives
The present method has been used to measure GSH/GSSG and Cys/CySS redox potentials in thousands of human plasma samples under different disease, nutritional and toxicologic conditions. The same method has been used in human studies to measure redox potentials in the intestinal lumen, bronchoalveolar lavage fluid, lymph, cerebrospinal fluid and tissue biopsies from heart, liver, lung and small intestines. The method has also been used to study thiol/disulfide redox systems in cell culture and corresponding in vivo preparations in laboratory animals. Results show that the redox poise of GSH/GSSG and Cys/CySS are fundamental parameters of biologic systems which are perturbed in disease and toxicity. The present method to measure the redox poise of GSH/GSSG and Cys/CySS couples provides a reliable means to quantify perturbations linked to disease, diet and environmental exposures. The method has a number of limitations, and improved methods are needed both for convenience and shorter analysis time. However, the advantage of the current method is that the dynamic range is sufficient to allow quantification of redox potentials of GSH/GSSG and Cys/CySS couples in a single analysis. More general application of these methods can be expected to clarify redox mechanisms of oxidative stress and provide improved strategies for interventions to protect against related disease processes.
Supplementary Material
Acknowledgments
Support for research in the authors' laboratory was provided by NIEHS grants ES009047, ES011195 and ES016731. The authors have no conflicts of interest concerning the protocol or instruments used. The authors gratefully acknowledge the helpful suggestions of the anonymous reviewers of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kemp M, Go YM, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 4.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bersani NA, Merwin JR, Lopez NI, Pearson GD, Merrill GF. Protein electrophoretic mobility shift assay to monitor redox state of thioredoxin in cells. Methods Enzymol. 2002;347:317–326. doi: 10.1016/s0076-6879(02)47031-0. [DOI] [PubMed] [Google Scholar]
- 6.Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmgren A, Fagerstedt M. The in vivo distribution of oxidized and reduced thioredoxin in Escherichia coli. J Biol Chem. 1982;257:6926–6930. [PubMed] [Google Scholar]
- 8.Nkabyo YS, Ziegler TR, Gu LH, Watson WH, Jones DP. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1352–1359. doi: 10.1152/ajpgi.00183.2002. [DOI] [PubMed] [Google Scholar]
- 9.Watson WH, Jones DP. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003;543:144–147. doi: 10.1016/s0014-5793(03)00430-7. [DOI] [PubMed] [Google Scholar]
- 10.Go YM, Ziegler TR, Johnson JM, Gu L, Hansen JM, Jones DP. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic Biol Med. 2007;42:363–370. doi: 10.1016/j.freeradbiomed.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JR, Lee SM, Cho SH, Kim JH, Kim BH, Kwon J, Choi CY, Kim YD, Lee SR. Oxidation of thioredoxin reductase in HeLa cells stimulated with tumor necrosis factor-alpha. FEBS Lett. 2004;567:189–196. doi: 10.1016/j.febslet.2004.04.055. [DOI] [PubMed] [Google Scholar]
- 12.Cotgreave IA, Gerdes R, Schuppe-Koistinen I, Lind C. S-glutathionylation of glyceraldehyde-3-phosphate dehydrogenase: role of thiol oxidation and catalysis by glutaredoxin. Methods Enzymol. 2002;348:175–182. doi: 10.1016/s0076-6879(02)48636-3. [DOI] [PubMed] [Google Scholar]
- 13.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 14.Sultana R, Reed T, Butterfield DA. Detection of 4-hydroxy-2-nonenal- and 3-nitrotyrosine-modified proteins using a proteomics approach. Methods Mol Biol. 2009;519:351–361. doi: 10.1007/978-1-59745-281-6_22. [DOI] [PubMed] [Google Scholar]
- 15.Go YM, Pohl J, Jones DP. Quantification of redox conditions in the nucleus. In: Hancock R, editor. Methods in Molecular Biology: The Nucleus. Totawa, USA: Human Press; 2008. [DOI] [PubMed] [Google Scholar]
- 16.Go YM, Pohl J, Jones DP. Quantification of redox conditions in the nucleus. Methods Mol Biol. 2009;464:303–317. doi: 10.1007/978-1-60327-461-6_17. [DOI] [PubMed] [Google Scholar]
- 17.Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao T, Heberlein K, Jonas C, Jones DP, Hu X. New double quantum coherence filter for localized detection of glutathione in vivo. Magn Reson Med. 2006;55:676–680. doi: 10.1002/mrm.20788. [DOI] [PubMed] [Google Scholar]
- 19.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Analytical Biochemistry. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 20.Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst. 1958;83:670–672. [Google Scholar]
- 21.Ellman G, L H. A precise method for the determination of whole blood and plasma sulfhydryl groups. Analytical Biochemistry. 1979;93:98–102. [PubMed] [Google Scholar]
- 22.Hissin PJ, H R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Analytical Biochemistry. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 23.Reed DJ, B JR, Beatty PW, Brodie AE, Ellis WW, Potter DW. High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Analytical Biochemistry. 1980;106:55–62. doi: 10.1016/0003-2697(80)90118-9. [DOI] [PubMed] [Google Scholar]
- 24.Lakritz J, Plopper CG, Buckpitt AR. Validated high-performance liquid chromatography-electrochemical method for determination of glutathione and glutathione disulfide in small tissue samples. Anal Biochem. 1997;247:63–68. doi: 10.1006/abio.1997.2032. [DOI] [PubMed] [Google Scholar]
- 25.Chinn PC, Pigiet V, Fahey RC. Determination of thiol proteins using monobromobimane labeling and high-performance liquid chromatographic analysis: application to Escherichia coli thioredoxin. Anal Biochem. 1986;159:143–149. doi: 10.1016/0003-2697(86)90319-2. [DOI] [PubMed] [Google Scholar]
- 26.Fenton SS, Fahey RC. Analysis of biological thiols: determination of thiol components of disulfides and thioesters. Anal Biochem. 1986;154:34–42. doi: 10.1016/0003-2697(86)90492-6. [DOI] [PubMed] [Google Scholar]
- 27.Kirlin WG, Cai J, DeLong MJ, Patten EJ, Jones DP. Dietary compounds that induce cancer preventive phase 2 enzymes activate apoptosis at comparable doses in HT29 colon carcinoma cells. J Nutr. 1999;129:1827–1835. doi: 10.1093/jn/129.10.1827. [DOI] [PubMed] [Google Scholar]
- 28.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med. 1999;27:1208–1218. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 29.Samiec PS, Drews-Botsch C, Flagg EW, Kurtz JC, Sternberg P, Jr, Reed RL, Jones DP. Glutathione in human plasma: decline in association with aging, age-related macular degeneration, and diabetes. Free Radic Biol Med. 1998;24:699–704. doi: 10.1016/s0891-5849(97)00286-4. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 31.Moriarty-Craige SE, Jones DP. Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr. 2004;24:481–509. doi: 10.1146/annurev.nutr.24.012003.132208. [DOI] [PubMed] [Google Scholar]
- 32.Jones DP, Carlson JL, Samiec PS, Sternberg P, Jr, Mody VC, Jr, Reed RL, Brown LA. Glutathione measurement in human plasma. Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clin Chim Acta. 1998;275:175–184. doi: 10.1016/s0009-8981(98)00089-8. [DOI] [PubMed] [Google Scholar]
- 33.Lash LH, Jones DP. Distribution of oxidized and reduced forms of glutathione and cysteine in rat plasma. Arch Biochem Biophys. 1985;240:583–592. doi: 10.1016/0003-9861(85)90065-7. [DOI] [PubMed] [Google Scholar]
- 34.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 35.Clark WM. Oxidation-Reduction Potentials of Organic Systems. Baltimore: Williams and Wilkins; 1960. [Google Scholar]
- 36.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 37.Jones DP. Disruption of mitochondrial redox circuitry in oxidative stress. Chem Biol Interact. 2006;163:38–53. doi: 10.1016/j.cbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free Radic Biol Med. 2000;28:625–635. doi: 10.1016/s0891-5849(99)00275-0. [DOI] [PubMed] [Google Scholar]
- 39.Jones DP, Mody VC, Jr, Carlson JL, Lynn MJ, Sternberg P., Jr Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic Biol Med. 2002;33:1290–1300. doi: 10.1016/s0891-5849(02)01040-7. [DOI] [PubMed] [Google Scholar]
- 40.Fukagawa NK, Ajami AM, Young VR. Plasma methionine and cysteine kinetics in response to an intravenous glutathione infusion in adult humans. Am J Physiol. 1996;270:E209–214. doi: 10.1152/ajpendo.1996.270.2.E209. [DOI] [PubMed] [Google Scholar]
- 41.Lyons J, Rauh-Pfeiffer A, Yu YM, Lu XM, Zurakowski D, Tompkins RG, Ajami AM, Young VR, Castillo L. Blood glutathione synthesis rates in healthy adults receiving a sulfur amino acid-free diet. Proc Natl Acad Sci U S A. 2000;97:5071–5076. doi: 10.1073/pnas.090083297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raguso CA, Regan MM, Young VR. Cysteine kinetics and oxidation at different intakes of methionine and cystine in young adults. Am J Clin Nutr. 2000;71:491–499. doi: 10.1093/ajcn/71.2.491. [DOI] [PubMed] [Google Scholar]
- 43.Blanco RA, Ziegler TR, Carlson BA, Cheng PY, Park Y, Cotsonis GA, Accardi CJ, Jones DP. Diurnal variation in glutathione and cysteine redox states in human plasma. Am J Clin Nutr. 2007;86:1016–1023. doi: 10.1093/ajcn/86.4.1016. [DOI] [PubMed] [Google Scholar]
- 44.Dai J, Jones DP, Goldberg J, Ziegler TR, Bostick RM, Wilson PW, Manatunga AK, Shallenberger L, Jones L, Vaccarino V. Association between adherence to the Mediterranean diet and oxidative stress. Am J Clin Nutr. 2008;88:1364–1370. doi: 10.3945/ajcn.2008.26528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moriarty-Craige SE, Adkison J, Lynn M, Gensler G, Bressler S, Jones DP, Sternberg P., Jr Antioxidant supplements prevent oxidation of cysteine/cystine redox in patients with age-related macular degeneration. Am J Ophthalmol. 2005;140:1020–1026. doi: 10.1016/j.ajo.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 46.Moriarty-Craige SE, Ha KN, Sternberg P, Jr, Lynn M, Bressler S, Gensler G, Jones DP. Effects of long-term zinc supplementation on plasma thiol metabolites and redox status in patients with age-related macular degeneration. Am J Ophthalmol. 2007;143:206–211. doi: 10.1016/j.ajo.2006.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moriarty SE, Shah JH, Lynn M, Jiang S, Openo K, Jones DP, Sternberg P. Oxidation of glutathione and cysteine in human plasma associated with smoking. Free Radic Biol Med. 2003;35:1582–1588. doi: 10.1016/j.freeradbiomed.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Yeh MY, Burnham EL, Moss M, Brown LA. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am J Respir Crit Care Med. 2007;176:270–276. doi: 10.1164/rccm.200611-1722OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jonas CR, Puckett AB, Jones DP, Griffith DP, Szeszycki EE, Bergman GF, Furr CE, Tyre C, Carlson JL, Galloway JR, Blumberg JB, Ziegler TR. Plasma antioxidant status after high-dose chemotherapy: a randomized trial of parenteral nutrition in bone marrow transplantation patients. Am J Clin Nutr. 2000;72:181–189. doi: 10.1093/ajcn/72.1.181. [DOI] [PubMed] [Google Scholar]
- 50.Iyer SS, Accardi CJ, Ziegler TR, Blanco RA, Ritzenthaler JD, Rojas M, Roman J, Jones DP. Cysteine redox potential determines pro-inflammatory IL-1beta levels. PLoS One. 2009;4:e5017. doi: 10.1371/journal.pone.0005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashfaq S, Abramson JL, Jones DP, Rhodes SD, Weintraub WS, Hooper WC, Vaccarino V, Alexander RW, Harrison DG, Quyyumi AA. Endothelial Function and Aminothiol Biomarkers of Oxidative Stress in Healthy Adults. Hypertension. 2008;52:80–85. doi: 10.1161/HYPERTENSIONAHA.107.097386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashfaq S, Abramson JL, Jones DP, Rhodes SD, Weintraub WS, Hooper WC, Vaccarino V, Harrison DG, Quyyumi AA. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J Am Coll Cardiol. 2006;47:1005–1011. doi: 10.1016/j.jacc.2005.09.063. [DOI] [PubMed] [Google Scholar]
- 53.Neuman RB, Bloom HL, Shukrullah I, Darrow LA, Kleinbaum D, Jones DP, Dudley SC., Jr Oxidative stress markers are associated with persistent atrial fibrillation. Clin Chem. 2007;53:1652–1657. doi: 10.1373/clinchem.2006.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kadiiska MB, Gladen BC, Baird DD, Dikalova AE, Sohal RS, Hatch GE, Jones DP, Mason RP, Barrett JC. Biomarkers of oxidative stress study: are plasma antioxidants markers of CCl(4) poisoning? Free Radic Biol Med. 2000;28:838–845. doi: 10.1016/s0891-5849(00)00198-2. [DOI] [PubMed] [Google Scholar]
- 55.Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM, Jr, Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. Faseb J. 2004;18:1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- 56.Johnson JM, Strobel FH, Reed M, Pohl J, Jones DP. A rapid LC-FTMS method for the analysis of cysteine, cystine and cysteine/cystine steady-state redox potential in human plasma. Clin Chim Acta. 2008;396:43–48. doi: 10.1016/j.cca.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.