

Abstract
Obtaining vesicular fractions from cell lines or animal tissue is both time and technically intensive. The presence of plasma membrane and nuclear contaminants within a preparation is often dependent upon the method of homogenization and is usually mitigated through the use of density gradients. We have developed a method that utilizes Balch homogenization and differential centrifugation to obtain two distinct vesicular fractions along with purified nuclear, cytoplasmic and ghost fractions within a three hour period of time without the use of density gradients. Importantly, these fractions maintain their biologic activity following isolation and may be used for both localization and biochemical analyses.
Keywords: Balch Homogenizer, Vesicle Isolation, Centrifugation, Endosome, STAT, IL-6
Introductory Statement
As signal transduction is increasingly associated with endosome trafficking, obtaining endosome-enriched vesicular fractions is of the utmost importance for biochemical characterization and localization. Numerous techniques have been developed for isolating vesicular fractions through the use of density gradients and centrifugation, detergent-based extraction or free-flow electrophoresis [1-6]. Several recurrent drawbacks, however, plague these methods. Excessive homogenization results in the formation of plasma membrane microsomes that contaminate vesicular fractions, and nuclear disruption releases nucleic acids and nuclear proteins into the cellular extract, altering sedimentation and also contaminating subcellular fractions [4]. Density gradient separations frequently employ Percoll, Ficoll or OptiPrep to mitigate contamination, but such methods add significant reagent cost and time to the acquisition of vesicle-enriched fractions [4; 6]. Finally, obtaining vesicular fractions often requires discarding or destroying other subcellular fractions of interest in signal transduction, such as the nucleus and plasma membrane [4; 7].
To address the issues of contamination, time and cellular waste, we have refined a method for isolating endosome-enriched vesicular fractions through the use of Balch homogenization, differential centrifugation, and a series of buffers to maintain endosome fidelity and structure. From this preparation five subcellular fractions are isolated rapidly, cleanly, and reproducibly within three hours. These fractions include a completely intact nuclear fraction, small vesicle fraction, large vesicle fraction, cytoplasmic fraction and a ghost fraction, which may then be used for various biochemical analyses and activity-based assays including western blotting, immunoprecipitation, multi-dimensional gel electrophoresis, electron microscopy and in vitro kinase assays.
Materials and Methods
Cell Lines and Reagents
3T3 mouse embryonic fibroblast (MEF) WT cells acquired from ATCC (CRL-2752) were grown in DMEM with 10% FBS and 40 μg/ml penicillin-streptomycin at 37° C in a 5% CO2 environment. All cell culture media and reagents were purchased from Mediatech (Monassas, VA). Antibodies against signal transducer and activator of transcription 3 (STAT3) and phospho-STAT3 were purchased from Cell Signaling Technologies (Danvers, MA). Antibodies against rab5 and dynein were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Clathrin heavy chain antibody was purchased from Affinity Bioreagents (Rockford, IL). Antibody against actin was purchased from Chemicon (Billerica, MA). Antibody against GAPDH was a kind gift from Dr. Richard Bram (Mayo Clinic) and antibody against histone H3 was a kind gift from Dr. Zhiguo Zhang (Mayo Clinic). HRP-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were purchased from Jackson Immunoresearch (West Grove, PA). Hyblot CL autoradiography film used in western blotting was purchased from Denville Scientific (Metuchen, NJ). Bicinchoninic acid protein assay kit, neutravidin beads and supersignal west pico solutions were purchased from Pierce (Rockford, IL). Araldite resin for electron microscopy was purchased from EMS (Fort Washington, PA). Ultracentrifuge tubes were purchased from Seton Scientific (Los Gatos, CA). Ultracentrifuges and rotors were from Beckman Coulter (Fullerton, CA). All chemicals were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise noted. The Balch homogenizer was custom ordered from and machined by EMBL precision engineering (Heidelberg, Germany) and tungsten-carbide ball bearings were purchased from Industrial Tectonics (Dexter, MI).
Buffers
All buffers were stored at 4° C. MES buffer contains 150 mM NaCl, and 25 nM 2-(N-Morpholino) ethanesulfonic acid sodium salt (Sigma M5057) pH to 6.5 in ddH2O with 1 mM PMSF, 10 μg/mL aprotinin, 1 μg/mL leupeptin, 1 mM NaVO3 and 1 mM NaF added fresh prior to use.
NDG lysis buffer contains 1% Nonidet P-40 (v/v), 0.5% deoxycholic acid sodium salt (w/v) and 10% glycerol (v/v) in 1X tris-buffered saline (20 mM Tris, 137 mM NaCl, pH 8.0 in ddH2O). On the day of use 1 mM PMSF, 10 μg/mL aprotinin, 1 μg/mL leupeptin, 1 mM NaVO3 and 1 mM NaF were added to the NDG Lysis Buffer.
RIPA Lysis Buffer contains 1% Nonidet P-40 (v/v), 1% deoxycholic acid sodium salt (w/v), 0.1% sodium dodecyl sulfate (w/v) and 0.002 M EDTA in 1X tris-buffered saline (20 mM Tris, 137 mM NaCl, pH 8.0 in ddH2O). On the day of use 1 mM PMSF, 10 μg/mL aprotinin, 1 μg/mL leupeptin, 1 mM NaVO3 and 1 mM NaF were added to the RIPA Lysis Buffer.
Cellular Fractionation
Prior to treatment, 8 × 106 3T3 MEF WT cells were starved in serum-free DMEM for three hours at 37° C. IL-6 (10 ng/mL) was then added directly to the starvation media for 5 or 10 minutes at 37° C. Following treatment, cells were immediately placed on ice, media was aspirated and the cells were washed twice with ice-cold Dulbecco's phosphate-buffered saline (DPBS) containing Ca2+ and Mg2+. 1 mL MES buffer was then added to each plate and cells were scraped into 1.5 mL microfuge tubes and placed on ice. During the cell starvation period, a Balch homogenizer with an 8.020 mm gap containing an 8.008 mm diameter tungsten-carbide ball bearing was assembled in ddH2O. Air was evacuated from the immersed homogenizer by passing ddH2O through the chamber several times in each direction using a 1 cc syringe. Before each sample was homogenized, 1 mL MES buffer was passed through the homogenizer to equilibrate the chamber and prevent sample hydrolysis.
Figure 1 provides a visual overview of the following cellular fractionation protocol. Cell samples were triturated gently with a 1 mL pipette to ensure single-cell suspension and then loaded into 1 cc syringes through 20 gauge needles. Cells were then passed six times each direction (12 passes total) through the Balch homogenizer using constant manual pressure. Following homogenization, samples were returned to fresh 1.5 mL microfuge tubes and kept on ice. Samples were then centrifuged at 4° C for 10 minutes at 1,000 × g. Supernatants (S1) were removed to new 1.5 mL microfuge tubes and pellets (P1) were washed with 1 mL MES buffer. S1 and P1 samples were spun down again at 4° C for 10 minutes at 1,000 × g to remove contaminants. Cleared P1 was transferred to new 1.5 mL microfuge tubes while washed P1 was lysed in 200 μL NDG lysis buffer. Both P1 and S1 were spun at 4° C for 10 minutes at 16,000 × g. The spun P1 sample at this point contained two fractions, the nuclear pellet and ghost supernatant. Ghost supernatants were transferred to new 1.5 mL microfuge tubes and nuclear pellets were washed with 200 μL NDG lysis buffer. The spun S1 sample contained the 16,000 × g large vesicle pellet and supernatant (S2). S2 was transferred to new 1.5 ml microfuge tubes while the large vesicle pellet was washed with 200 μL MES buffer. The nuclear pellet, ghost, large vesicle and S2 samples were then spun at 4° C for 10 minutes at 16,000 × g to remove contaminants. Supernatants were aspirated from the nuclear and large vesicle pellets and the nuclear pellet was resuspended in 200 μL RIPA buffer. If functional assays were performed with protein from the large vesicle samples, then the pellets are resuspended in 100 μL MES buffer. If large vesicle samples were run on western blot, then they were resuspended in 100 μL RIPA buffer. Supernatant from the cleared ghost samples was transferred to new 1.5 mL microfuge tubes. Cleared S2 samples were transferred to 14.29 mm × 88.9 mm polyallomer ultracentrifuge tubes (5030, Seton Scientific, Los Gatos, CA), placed in a Beckman SW-41Ti rotor and spun in a Beckman Optima LE 80K ultracentrifuge at 115,000 × g for 60 minutes at 4° C. Supernatant from this spin was removed to 15 mL conical tubes and remaining cytoplasmic proteins were precipitated at 4° C with 10% (v/v) trichloroacetic acid for one hour to overnight. Alternatively, cleared S2 samples were also spun at 115,000 × g for 60 minutes at 4° C in 1.5 mL polyallomer ultracentrifuge tubes with a Beckman TLA110 rotor in a Beckman Optima Max-E ultracentrifuge. Using this method avoided the need to precipitate cytoplasmic protein from the supernatant. The 115,000 × g pellet is the small vesicle sample and was resuspended in either 100 μL MES buffer or 100 μL RIPA buffer depending upon subsequent use. Following isolation, protein concentration from each sample was determined by BCA assay per vendor's instruction (Pierce; Rockford, IL) and all samples were kept at 4° C until assays were performed or frozen at -20° C for later use.
Figure 1.

Visual overview of subcellular fractionation technique as described in the materials and methods.
Western Blotting
15 μg protein from each fraction in Laemmli sample buffer (1× contains 2% SDS (w/v), 10% glycerol, 60 mM Tris-Cl pH 6.8, 0.01% bromophenol blue (w/v)) containing 20% (v/v) DTT was loaded onto either 7.5% or 10% polyacrylamide Tris-HCl gels and run for 14 hours at 90V under constant voltage. Gels were then transferred to 0.4 μm nitrocellulose for 3 amp hours at 4° C, washed in ddH2O and blocked with 4% BSA in TBS (20 mM Tris, 137 mM NaCl, pH 7.5 in ddH2O) for one hour while rotating at room temperature. All antibodies were diluted 1:1000 in TBS and membranes were probed overnight at 4° C while rocking. The following day, primary antibody was removed and membranes were washed three times quickly with TBS-T (TBS with 0.002% Tween-20) and then once for 30 minutes at room temperature while shaking. HRP conjugated secondary antibody diluted 1:30,000 in TBS was then added to membranes for 1 hour at room temperature while shaking. Following secondary antibody application membranes were washed as above, developed with West Pico and then detected by film exposure.
Transmission Electron Microscopy
Subcellular fractions were prepared as described and incubated in Trump's fixative (4% paraformaldehyde (v/v) and 1% glutaraldehyde (v/v) in PBS) overnight at 4° C. The following day, agar was added to the samples and allowed to set. Agar pellets were then washed three times with phosphate buffer (PB; 0.038 M Na2HPO4-H2O and 0.162 M Na2HPO4 in ddH2O, pH 7.2) and stained with osmium (1% OsO4 in PB) while pulsing twice with vacuum for one minute each. Osmium was removed from the pellets that were then washed three times with ddH2O for 45 seconds. Pellets were sequentially dehydrated in 60%, 70%, 80%, 95% and 100% ethanol for 40 seconds at each step. Epon Araldite resin diluted in acetone was then used to embed the agar pellets. Pellets under vacuum were sequentially incubated for two one-minute pulses in 2:1 EtOH:resin, 1:1 EtOH:resin, 1:2 EtOH:resin and pure resin mixtures. Pellets were cured for one hour between EtOH:Resin mixtures. Once embedded with pure resin, samples were incubated overnight at room temperature under desiccation. Infiltrated pellets were then transferred to fresh molds with resin, baked at 45° C for 24 hours and then baked at 60° C for 24 hours. Araldite molds were then cut, placed on copper grids and imaged using a Jeol JEM-1400 transmission electron microscope.
Oligonucleotide Binding Assay
Two oligonucleotides containing a known STAT3 binding sequence [8] were synthesized as follows with the forward oligo biotinylated at the 3′ tail: Forward Oligo 5′ – TTTGGTATTCCCGGAAATGTTTTTGGTATTCCCGGAAATGTTTTTTTTTTTTTTT – 3′ Reverse Oligo 5′ – AAACCATAAGGGCCTTTACAAAAACCATAAGGGCCTTTACAAAAAAAAAAAAAAA – 3′ Prior to use, a 100 ng/μL oligo stock was made by mixing the two oligos in annealing buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 50 mM NaCl) and heating to 72° C for ten minutes in a water bath followed by slow cooling to 25° C. Stock was stored at -20° C until use.
3T3 MEF WT cells were starved in serum-free DMEM, treated with 10 ng/mL IL-6, and fractions were isolated in MES buffer as above. Protein concentration of each fraction was determined by BCA assay and 15 μg of sample from each fraction was mixed with 500 ng biotinylated oligo and rotated overnight at 4° C. The following day, samples were added to 50 μL neutravidin beads and rotated at room temperature for two hours. Samples were then spun at 1000 × g for 1 minute at room temperature and supernatant was removed from the beads as the unbound fraction and mixed with 50 μL sample buffer containing 20% (v/v) DTT. Beads were then washed, aspirated and spun down three times in MES buffer. Beads were then mixed with 75 μL sample buffer with 20% (v/v) DTT. Samples and unbound fractions were boiled at 100° C for 5 minutes, spun at 1000 × g for 1 minute and then loaded onto 7.5 % tris-HCl gels. Western blots for STAT3 were performed as described above.
Results and Discussion
Balch Homogenization
Freeze-thaw, dounce homogenization, blending, syringe trituration, nitrogen cavitation, and sonication are all techniques regularly utilized to disrupt the plasma membrane and produce the initial cellular homogenate from which subcellular fractions are isolated [1; 2; 4; 7; 9; 10]. Fraction contamination with lipids and proteins from the plasma membrane and nucleus is a common problem that arises from excessive disruption. Applying the appropriate amount of consistent pressure on cells to crack the plasma membrane without producing debris or disrupting internal structures is critical to maintain purity within subsequent subcellular fractions. We have employed Balch homogenization [11] to consistently disrupt the plasma membrane in a highly regulated fashion without micelle or microsome contamination of vesicular fractions, allowing us to bypass the use of gradient centrifugation. This method of homogenization has been used successfully, but not frequently [12; 13]. Additionally, this method does not disrupt the nucleus or other subcellular structures and may be easily calibrated to any cell type.
Balch homogenization functions by passing cells through a restricted passage while under constant pressure, creating small ruptures within the plasma membrane through the application of shear and compression forces. Vesicular structures readily pass through these gaps in the membrane during centrifugation while larger structures such as the nucleus, endoplasmic reticulum and plasma membrane sediment.
As schematically outlined in Figure 2, a Balch homogenizer, or “ball cracker”, consists of a tungsten-carbide ball bearing that sits within a stainless steel chamber and is held in place by two stainless steel end-caps. The end-caps are fitted with stainless steel square body luer hubs and o-rings to permit fluid injection into the chamber and held in place with thumb screws. In addition, rubber o-rings added into notches on the end-cap inserts play a critical role in sealing the chamber and must be well maintained to prevent leakage. Stainless steel is unlikely to warp under the rigor of regular use, functions as a heat sink to keep samples cool while placed in ice and may be easily machined, making it a good material choice for Balch homogenizer components. Fluid pressure entering the chamber is equally dispersed across the surface of the ball bearing as it is pushed against the opposite end-cap, ensuring the formation of a gap equal to one-half the difference in diameter between the chamber wall and ball bearing. As cells pass through this gap, fluid pressure on the plasma membrane corresponds directly to fluid viscosity and velocity. Both viscosity, controlled by cellular concentration within the MES buffer, and injection pressure should always be kept constant. Thus, shear force is most readily controlled by altering ball bearing size. We use a chamber with an 8.020 mm diameter and ball bearings that range in diameter from 8.004 mm to 8.014 mm, creating gaps 3 μm to 8 μm in size through which most cell types may be readily cracked.
Figure 2.

Schematic diagram of a Balch homogenizer. (A) Balch homogenizer components include a stainless steel central chamber, two stainless steel end-caps with 6.35 mm, long threaded square body female luer hubs inserted through rubber o-rings, four 25 mm M5 threaded thumb screws and a tungsten carbide ball. (B) The Balch homogenizer is constructed while submerged in water and air is evacuated prior to use. Fluid is then passed into the Balch homogenizer chamber using 1 cc syringes attached to the luer adapters. (C) Schematic diagram of the Balch homogenizer central chamber. All measurements are in millimeters. (D) Schematic diagram of the Balch homogenizer end- caps with all measurements in millimeters. Rubber O-rings must be placed into the grooves on the end-cap insert shafts to properly seal the chamber. Different luer adapters may be attached to the threaded end-caps allowing a wide variety of input sources. (E) 3T3 MEF WT cells prior to homogenization show complete exclusion of trypan blue. (F) Cells passed through an 8 μm gap show partial disruption. One cell completely excludes trypan blue while the others show varying inclusion. (G) Cells passed through a 7 μm gap show good disruption. Trypan blue readily enters the cell while the plasma membrane and cellular structure is largely intact. Further, no membrane or nuclear debris is found within the supernatant. (H) Cells passed through a 4 μm gap show signs of excessive homogenization. Nuclei are partially disrupted and separated from cell bodies while cellular debris may be seen within the supernatant.
Prior to endosome fractionation, the appropriate ball bearing for use in homogenization was determined by passing 8.0 × 106 3T3 MEF WT cells in 1 mL MES buffer six times each direction with constant pressure through the 8.020 mm channel with balls of various size. Two criteria were set in place to establish the appropriate ball size. First, trypan blue must be excluded from less than 1% of the cells following homogenization (Figure 2F & G). Second, sheared membrane debris or nuclei free from cellular ghosts must not be seen in the supernatant (Figure 2G & H). Following these criteria the 3T3 MEF WT cells, approximately 30 – 50 μm in diameter, were appropriately cracked using a ball size of 8.006 mm, yielding a gap size of 7 μm (Figure 2E – H).
Buffer System
Each of the three buffers used in this protocol – MES buffer, NDG lysis buffer and RIPA lysis buffer – plays a unique role in harvesting subcellular fractions. MES buffer, originally formulated by Pearse [14], maintains protein coat stability on vesicular structures during isolation. Coat stability is pH dependent and best preserved in a neutral or mildly acidic environment less than pH 7.5 [15]. The pH 6.5 of MES buffer chosen for this protocol is low enough to ensure optimal protein coat maintenance yet high enough to sustain biological activity within an endosome, such as ligand-receptor binding. Divalent cations, included within the original MES buffer recipe, are unnecessary to maintain proteinaceous endosome structures and may obscure signals by enhancing kinase and phosphatase activity within the fractions [1; 16; 17]. Instead, sodium chloride has been added to ensure the isotonic state of our MES buffer. Finally, we add the protease and phosphatase inhibitors sodium orthovanadate, leupeptin, aprotinin and PMSF to prevent signal degradation during the isolation.
The difference in lytic strength between NDG lysis buffer and RIPA lysis buffer allows the isolation of a nuclear fraction. Consequently, NDG lysis buffer has been designed to strip the plasma membrane and other remaining ghost organelles from the nucleus in a detergent-dependent fashion without permeabilizing the nuclear membrane. Deoxycholic acid and NP-40, the primary detergents within this buffer, are at high enough concentration to induce micelle formation of the plasma membrane while leaving the nuclear and ER membranes intact [18]. The addition of SDS and increased deoxycholic acid concentration are necessary for nuclear membrane lysis and, as such, have been added to our RIPA lysis buffer, which is used to solubilize the nuclear pellet. The protease and phosphatase inhibitors added to the MES buffer are also included within the NDG and RIPA lysis buffers along with the addition of sodium fluoride to prevent enzymatic degradation of signals.
Subcellular Fractionation
Following treatment, 3T3 MEF WT cells were placed on ice, washed, scraped into microfuge tubes and Balch homogenized as described within the materials and methods section. Prior to homogenization, cells display an intact plasma membrane and undisturbed subcellular architecture (Figure 3A). Cells collected immediately following homogenization (Figure 3B) show signs of mild but notable plasma membrane disruption (black arrow) through which vesicles will separate from the nuclear and ghost fractions during centrifugation. Importantly, the nuclear envelope shows no signs of disruption following homogenization (Figure 3B, white arrrow). Differential centrifugation of samples as described above and visually in Figure 1 yields five fractions – nuclear (Figure 3C), ghost (Figure 3F), cytosol (Figure 3E), large vesicle (Figure 3D) and small vesicle (Figure 3G) – within three hours, not including time of starvation or treatment. The nuclear fraction contains intact, non-disrupted nuclei along with smooth and rough endoplasmic reticulum (ER) (Figure 3C). The preservation of intact nuclear membranes, as confirmed by EM (Figure 3C) and suggested by ER co-isolation, is an enormous benefit gained through the use of this method. Following isolation, the nuclear pellet may be resuspended in MES buffer or NDG lysis buffer, but only RIPA lysis buffer will permeabilize the nuclear membrane. Once lysed in RIPA, bath sonication is necessary to clear congealed nucleic acids before the fraction may be used for western blotting or any other biochemical analysis. Western blot analysis of histone H3 (H3) (Figure 4A), a known nuclear protein, demonstrates that nuclei are highly enriched within the nuclear fraction and do not contaminate other fractions. This is as also confirmed by EM.
Figure 3.
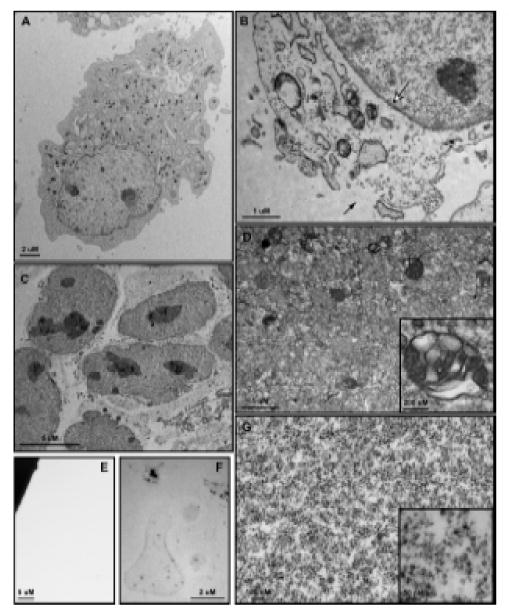
Transmission electron microscopy of 3T3 MEF WT cells and subcellular fractions. (A) Whole cells are approximately 30 – 50 μm in diameter and show a clearly defined plasma membrane with normal subcellular architecture. (B) Following Balch homogenization, small tears are formed within the plasma membrane (black arrow) through which vesicles pass during centrifugation. The nuclear envelope is entirely intact (white arrow) and subcellular structures show few signs of disturbance. (C) The nuclear fraction consists of nuclei with unbroken envelopes and attached endosplasmic reticulum, both smooth and rough. (D) The large vesicle fraction contains a heterogeneous population of membrane-bound organelles 50 nm – 0.5 μm in size. Large, multivesicular structures are well-represented within this fraction (Inset). The small vesicle fraction contains membrane-bound structures 15 nm – 25 nm in size. Electron density within this population varies widely (Inset). (F) After NDG lysis the ghost fraction contains plasma membrane micelles as well as some electron dense protein clusters. (G) No membrane or protein structures are visualized within the cytosolic fraction.
Figure 4.

Biochemical characterization and functional assay utilizing subcellular fractions. (A) Western blot analysis of subcellular fractions isolated from 3T3 MEF WT cells treated with 10 ng/ml IL-6 for the indicated period as described within the materials and methods section. STAT3, found in all subcellular fractions, is enriched within the nuclear fraction following IL-6 treatment. Activated STAT3 may also be found within the ghost, small vesicle, and large vesicle fractions. Rab5, dynein and clathrin are all found within the small vesicle fraction, indicating this fraction is enriched with early endosomes and clathrin-coated vesicles. The complete localization of e-cadherin and histone H3 to the ghost and nuclear fractions, respectively, indicates these fractions do not contaminate the vesicular fractions. The cytosolic fraction is characterized by the presence of actin, rab5, dynein, and GAPDH, suggesting the accumulation of monomeric cytoskeletal proteins and a pool of latent proteins that interact with vesicular structures. The large vesicle fraction does not contain structures associated with early endosomes and is more thoroughly described within the text and Figure 3. (B) Western blot analysis of STAT3 DNA binding capacity following 10 ng/ml IL-6 treatment of 3T3 MEF WT cells and fraction isolation. Fractions were incubated with a biotinylated oligo containing a known STAT3 binding region and then pulled down with neutravidin agarose beads as described in the materials and methods. STAT3 isolated through oligo binding is shown in the top panel labeled ‘Oligo Pulldown’ whereas the remaining STAT3 that was unable to bind the oligo is shown in the bottom panel ‘No Pulldown’. STAT3 within the small vesicle fraction has the greatest DNA binding capacity that increases upon IL-6 treatment.
The ghost fraction consists of those subcellular components that do not pass through the sheared plasma membrane during the 1000 × g sedimentation and are readily stripped from the nuclear components by NDG lysis. Of the five fractions, only the ghost requires detergent based lysis to obtain. EM evaluation of the ghost fraction following detergent lysis shows lightly stained membrane micelles lacking any observable substructure and a scattering of electron dense clusters (Figure 3F). Western blot analysis reveals the fraction rich in the integral plasma membrane protein e-cadherin and the cytoskeletal protein actin (Figure 4A).
The large vesicle fraction consists of membrane-bound vesicular structures 50 nm to 0.5 μm in size as confirmed by EM (Figure 3D). Multivesicular bodies are the most prominent features found within this fraction (Figure 3D, Inset), though round, homogenously stained vesicles are the most numerous. Western blot analysis of this fraction confirms the absence of rab5 endosomal structures, suggesting late endosomes as a primary component (Figure 4A). Notably, no mitochondria were seen within the large vesicle pellet, suggesting that they remain within the ghost fraction upon centrifugation. The large vesicle pellet is readily lysed in both NDG and RIPA lysis buffers though may be easily resuspended in MES buffer to maintain vesicular structures.
The small endosome fraction consists of membrane-bound vesicles approximately 15 nm to 25 nm in size (Figure 3G). A heterogeneously stained population of vesicles are isolated within this fraction (Figure 3G and Inset) and western blot analysis show enrichment of clathrin and rab5, indicating the presence of clathrin coated vesicles and early endosomes, respectively (Figure 4A). The dependence of endosome maturation, from the early to late stage, upon retrograde transport makes the presence of dynein and rab5 within this fraction expected and demonstrates physiologic relevance recapitulated within our enrichment [19]. Interestingly, glyceraldehdye-3-phosphate dehydrogenase (GAPDH) was found within the small and large vesicular fractions as well as the cytosolic fraction, which corresponds with reports that this protein may function as a novel transferrin receptor (Figure 4A) [20]. As with the large vesicle fraction, the small vesicle fraction may be resuspended with NDG or RIPA lysis buffers as well as MES buffer.
Finally, our cytosolic fraction contains proteins not associated with any vesicular structure or membrane-defined organelle. Within this fraction we observe actin, rab5, dynein and GAPDH by western blot (Figure 4A) but we do not observe structure by EM (Figure 3E), suggesting the presence of monomeric cytoskeletal components and a pool of proteins that may interact with vesicles while performing their primary known function. Depending upon the method of ultracentrifugation, the total protein content of this fraction may require TCA precipitation followed by RIPA lysis buffer resuspension for an appreciable concentration to be reached.
Biochemical Activity of Subcellular Fractions
Recently, signal transducer and activator of transcription 3 (STAT3) has been shown to interact with endosomal structures following interleukin-6 (IL-6) induced activation [21-23]. To demonstrate the capacity of our enrichment protocol in tracking intracellular signals through subcellular compartments, 3T3 MEF WT cells were treated with 10 ng/ml IL-6 across a short time course and STAT3 activity, as measured by tyrosine 705 phosphorylation, was assessed by western blotting. Within five minutes, STAT3 was activated within the ghost, small vesicle and large vesicle fractions, with some accumulation within the nucleus (Figure 4A). Relative to other subcellular fractions, little STAT3 was activated within the cytoplasmic fraction at any time throughout the treatment course. Ten minutes following activation, phosphorylated STAT3 maintained a presence within the vesicle fractions and accumulated within the nuclear fraction (Figure 4A). STAT3 is unique in that it serves as both a mediator of signal activation and a transcription factor. Our capacity to track STAT3 through peripheral subcellular fractions and demonstrate nuclear accumulation following IL-6 induced activation is in accordance with known biologic function. This suggests our isolation protocol may be of significant use in localizing second-messenger proteins to subcellular compartments during signal transduction.
To demonstrate that second-messenger proteins isolated from the vesicular fractions are biologically active, the capacity of isolated STAT3 protein to bind DNA was tested by performing an oligo pulldown assay from the large and small vesicular fractions. Vesicular fractions isolated from 3T3 MEF WT cells before and after IL-6 treatment were resuspended in MES buffer and incubated with a biotinylated double- stranded DNA oligo containing a known STAT3 binding sequence [8]. As compared to the untreated control, STAT3 within the IL-6 treated small vesicle fraction increasingly bound the biotinylated oligo and pulled down with streptavidin beads whereas STAT3 from the large vesicle and nuclear fractions did not (Figure 4B). A small amount of STAT3 within the untreated ghost fraction bound the oligo and this capacity is reduced with treatment. This suggests that activated STAT3 within the vesicular fractions was biologically active upon isolation and capable of binding DNA, a fundamental function of this transcription factor. Further, vesicular STAT3 DNA binding capacity suggests subsequent transcriptional activity and biologic relevance arising from this structure. Together, the capacity to track STAT3 activation through subcellular fractions and apply the isolated fractions to activity-dependent assays demonstrates the utility of this fractionation technique in evaluating vesicle-dependent biology.
Conclusions
Our objective in this work was to develop a protocol that: (1) limits plasma membrane contamination of subcellular fractions, (2) avoids the expense, time, and variability involved with sucrose, Percoll, Ficoll or OptiPrep gradients, and (3) allows us to separate as many subcellular fractions as possible for rapid protein localization and functional analysis of the vesicle fractions. To this end we have employed Balch homogenization, centrifugation without gradients, and a calibrated buffering system to maximize yield of the vesicular fractions and minimize contamination, while additionally obtaining nuclear, ghost and cytoplasmic fractions.
From the products of this subcellular fractionation any number of further protocols may be developed or employed to evaluate the character and function of vesicular fractions and the proteins within. Immunoprecipitation of whole vesicles may be a powerful means of showing protein association on a membrane-bound structure without direct interaction. As demonstrated, DNA binding assays may provide insight into the vesicular dependence of activated transcription factors. Finally, in vitro kinase assays may be performed with vesicle associated protein kinases to demonstrate signal activation from a vesicular source.
Acknowledgments
We thank Dr. Richard Bram and Dr. Zhiguo Zhang for providing antibodies, Dr. Larry Pease and Dr. Dave Katzmann for the use of their ultracentrifuges and rotors, Trace Christensen for his assistance with electron microscopy, and Kimberly Cook and Mary Pendergast for their critique of this manuscript. This work was supported by Donald and Frances Herdrich (CLH) and an early career development award from Mayo Clinic (CLH). CLG was supported by the Mayo Graduate School and the Kern Predoctoral Neuroscience Fellowship.
Support: This work was supported by Donald and Frances Herdrich (CLH) and an early career development award from Mayo Clinic (CLH). CLG was supported by the Mayo Graduate School and a Kern Pre-doctoral Neuroscience Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Daiss JL, Roth TF. Isolation of coated vesicles: comparative studies. Methods Enzymol. 1983;98:337–49. doi: 10.1016/0076-6879(83)98162-4. [DOI] [PubMed] [Google Scholar]
- 2.Pearse BM. Isolation of coated vesicles. Methods Enzymol. 1983;98:320–6. doi: 10.1016/0076-6879(83)98160-0. [DOI] [PubMed] [Google Scholar]
- 3.Marsh M, Schmid S, Kern H, Harms E, Male P, Mellman I, Helenius A. Rapid analytical and preparative isolation of functional endosomes by free flow electrophoresis. J Cell Biol. 1987;104:875–86. doi: 10.1083/jcb.104.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morand JN, Kent C. A one-step technique for the subcellular fractionation of total cell homogenates. Anal Biochem. 1986;159:157–62. doi: 10.1016/0003-2697(86)90321-0. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs R, Ellinger I. Free-flow electrophoretic analysis of endosome subpopulations of rat hepatocytes. Curr Protoc Cell Biol. 2002;Chapter 3(Unit 3):11. doi: 10.1002/0471143030.cb0311s14. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Donowitz M. Fractionation of subcellular membrane vesicles of epithelial and nonepithelial cells by OptiPrep density gradient ultracentrifugation. Methods Mol Biol. 2008;440:97–110. doi: 10.1007/978-1-59745-178-9_8. [DOI] [PubMed] [Google Scholar]
- 7.Beaumelle BD, Gibson A, Hopkins CR. Isolation and preliminary characterization of the major membrane boundaries of the endocytic pathway in lymphocytes. J Cell Biol. 1990;111:1811–23. doi: 10.1083/jcb.111.5.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horvath CM, Wen Z, Darnell JE., Jr A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995;9:984–94. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- 9.Beaumelle BD, Hopkins CR. High-yield isolation of functionally competent endosomes from mouse lymphocytes. Biochem J. 1989;264:137–49. doi: 10.1042/bj2640137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horstkorte R, Fan H, Reutter W. Rapid isolation of endosomes from BHK cells: identification of DPP IV (CD26) in endosomes. Exp Cell Res. 1996;226:398–401. doi: 10.1006/excr.1996.0241. [DOI] [PubMed] [Google Scholar]
- 11.Balch WE, Rothman JE. Characterization of protein transport between successive compartments of the Golgi apparatus: asymmetric properties of donor and acceptor activities in a cell-free system. Arch Biochem Biophys. 1985;240:413–25. doi: 10.1016/0003-9861(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 12.Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–64. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howe CL, Valletta JS, Rusnak AS, Mobley WC. NGF signaling from clathrin-coated vesicles: evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron. 2001;32:801–14. doi: 10.1016/s0896-6273(01)00526-8. [DOI] [PubMed] [Google Scholar]
- 14.Pearse BM. Coated vesicles from pig brain: purification and biochemical characterization. J Mol Biol. 1975;97:93–8. doi: 10.1016/s0022-2836(75)80024-6. [DOI] [PubMed] [Google Scholar]
- 15.Woodward MP, Roth TF. Coated vesicles: characterization, selective dissociation, and reassembly. Proc Natl Acad Sci U S A. 1978;75:4394–8. doi: 10.1073/pnas.75.9.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waas WF, Dalby KN. Physiological concentrations of divalent magnesium ion activate the serine/threonine specific protein kinase ERK2. Biochemistry. 2003;42:2960–70. doi: 10.1021/bi027171w. [DOI] [PubMed] [Google Scholar]
- 17.Han GS, Wu WI, Carman GM. The Saccharomyces cerevisiae Lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J Biol Chem. 2006;281:9210–8. doi: 10.1074/jbc.M600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neugebauer JM. Detergents: an overview. Methods Enzymol. 1990;182:239–53. doi: 10.1016/0076-6879(90)82020-3. [DOI] [PubMed] [Google Scholar]
- 19.Driskell OJ, Mironov A, Allan VJ, Woodman PG. Dynein is required for receptor sorting and the morphogenesis of early endosomes. Nat Cell Biol. 2007;9:113–20. doi: 10.1038/ncb1525. [DOI] [PubMed] [Google Scholar]
- 20.Raje CI, Kumar S, Harle A, Nanda JS, Raje M. The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J Biol Chem. 2007;282:3252–61. doi: 10.1074/jbc.M608328200. [DOI] [PubMed] [Google Scholar]
- 21.Devergne O, Ghiglione C, Noselli S. The endocytic control of JAK/STAT signalling in Drosophila. J Cell Sci. 2007;120:3457–64. doi: 10.1242/jcs.005926. [DOI] [PubMed] [Google Scholar]
- 22.Bild AH, Turkson J, Jove R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. Embo J. 2002;21:3255–63. doi: 10.1093/emboj/cdf351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah M, Patel K, Mukhopadhyay S, Xu F, Guo G, Sehgal PB. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem. 2006;281:7302–8. doi: 10.1074/jbc.M508527200. [DOI] [PubMed] [Google Scholar]