

Abstract
Persistent inflammatory response has adverse effects on left ventricular (LV) function and remodeling following acute myocardial infarction (AMI). We hypothesized that suppression of inflammation with IL-10 treatment attenuates LV dysfunction and remodeling after AMI. After the induction of AMI, mice were treated with either saline or recombinant IL-10, and inflammatory response and LV functional and structural remodeling changes were evaluated. IL-10 significantly suppressed infiltration of inflammatory cells and expression of inflammatory cytokines in the myocardium. These changes were associated with IL-10-mediated inhibition of p38 MAP kinase activation and repression of cytokine mRNA stabilizing protein, HuR. IL-10 treatment significantly improved LV functions, reduced infarct size and attenuated infarct wall thinning. MI-induced increase in MMP9 expression and activity was associated with increased fibrosis since IL-10 treatment reduced both MMP9 activity and fibrosis. siRNA knockdown of HuR mimicked IL-10 mediated reduction in MMP-9 expression and activity in NIH3T3 cells. Moreover, IL-10 treatment significantly increased capillary density in the infarcted myocardium which was associated with enhanced STAT3 phosphorylation. Taken together, our studies demonstrate that IL-10 suppresses inflammatory response and contributes to improved LV function and remodeling by inhibiting fibrosis via suppression of HuR/MMP9 and by enhancing capillary density through activation of STAT3.
Keywords: IL-10, myocardial infarction, cytokines, inflammation, cardiac remodeling
Introduction
Myocardial infarction is one of the leading causes of death in western societies and often results in the development of heart failure. Myocardial infarct size (acute phase), followed by adverse left ventricular (LV) remodeling (dilation and fibrosis) and cardiac dysfunction are major determinants in the pathogenesis of cardiac diseases 1, 2. A number of cardiac pathophysiological conditions including myocardial infarction and ischemia reperfusion injury leading to heart failure are associated with activation of inflammatory mediators in the heart 3, 4.
Expression of pro-inflammatory cytokines, for example, tumor necrosis factor-α (TNF), interleukin-1 (IL-1) and interleukin-6 (IL-6) and anti-inflammatory cytokine IL-10, mediate homeostasis within the heart in response to injury. However, a sustained expression of pro-inflammatory mediators at sufficiently high concentrations could lead to adverse outcome in the failing heart 2, 4, 5. Increased matrix metalloproteinase (MMPs) production associated with sustained inflammatory response may lead to excessive extracellular matrix (ECM) degradation in the early phase of MI, impairing infarct healing and aggravating early remodeling which in turn causes cardiac rupture 5–7. Also, the increased cytokine gene expression during acute phase of inflammation evokes a secondary, self-sustaining autocrine and paracrine growth factor and cytokine expression.
IL-10, a potent anti-inflammatory cytokine is a strong deactivator of monocytes and suppressor of various pro-inflammatory mediators 8, 9. However, the therapeutic effect of systemic IL-10 on inflammation mediated LV dysfunction and remodeling and the molecular signaling that governs these effects remains to be studied.
We have earlier reported that IL-10, a potent anti-inflammatory cytokine inhibits a panel of pro-inflammatory cytokine/chemokine mRNA expression in both human monocytic cell line (U937) and primary mouse macrophages via inhibiting cytokine mRNA stabilizing protein, HuR and suppression of p38 MAP kinase 10. p38 has also been implicated in oxidant stress-induced cardiac dysfunction and remodeling 11–13. Ischemic-oxidative stress of cardiomyocytes stimulate signal transducer and activator of transcription 3 (STAT3) exerts cardioprotection in the ischemic heart 14, 15. However, the potential role of IL-10 mediated anti-inflammatory responses and modulations in either STAT3 or HuR signaling during post-MI remodeling has not been explored. This study was undertaken to elucidate the role of IL-10 in modulating inflammation, LV dysfunction and remodeling, and the signaling mechanisms that regulate IL-10 mediated effects on remodeling.
Methods
Vertebrate Animals
All experiments conform to the protocols approved by the Institutional Animal Care and Use Committee. Nine weeks old mice of C57BL/6J background were procured from Jackson Research Laboratory (Bar Harbor, ME). The mice were allowed to acclimatize for 10 days under standard animal care conditions.
Antibodies and reagents
Antibodies against p-p38, p-STAT3, β-actin and total p38 were purchased from cell signaling Inc (Boston, MA). Anti-HuR antibodies from Upstate Inc (Lake Placid, NY). Phosphor-serine from Anaspec Inc (San Jose, CA). Total-STAT3 from Santa Cruz Biotechnology Inc (Santa Cruz, CA) Recombinant murine and human IL-10 and TNF-α was obtained from R&D Systems (Minneapolis, MN). LPS was obtained from Sigma Aldrich Inc (St. Louis, MO). and Curcurbitacin I from Calbiochem (San Diego, CA). Specific mouse HuR small interfering RNA (HuR siRNA) and a scrambled control siRNA were purchased from Ambion Inc (Austin, TX).
Cell Culture and antibodies
NIH3T3 Swiss albino fibroblasts (American Type Culture Collection) were cultured in DMEM (Mediatech, VA) with 10% FCS, L-glutamine, and 0.1 mmol/L β-mercaptoethanol. Human umbilical vein endothelial cells (HUVECs) were cultured in EBM-2 media (Clonitech, Palo Alto, CA) with 10%FCS. Cells were stimulated with LPS, TNF-alpha and or IL-10 at the dose of 10 ng/ml and curcurbitacin at 1μM concentration unless otherwise indicated.
Myocardial Infarction and study design
Mice were subjected to myocardial infarction (MI) by ligation of left anterior descending coronary artery (LAD) as described previously16. These mice were injected subcutaneously with mouse recombinant IL-10 (50 μg/kg b.w, MI+IL-10 group) or saline (MI group) on 0,1,3,5 and 7 days post-MI. The mice in sham group underwent the same procedure except for the LAD ligation. These mice received saline, subcutaneously. Inflammatory response was assessed after 3 days; LV functional changes on 14 and 28 days and structural remodeling at 28 days post-MI.
Echocardiography
Transthoracic two-dimensional M-mode echocardiogram was obtained using Vevo 770 (VisualSonics, Toronto, Canada) equipped with 30 MHz transducer. Echocardiographic studies were performed before (baseline) and at 14 and 28 days post MI on mice anesthetized with a mixture of 1.5% isoflurane and oxygen (1 L/min). M-mode tracings were used to measure LV wall thickness, end-systolic diameter (LVESD) and end-diastolic diameter (LVEDD). The mean value of 9 measurements was determined for each sample. Percent fractional shortening (%FS) was calculated as described 17.
Morphometric studies
The hearts were perfusion fixed with 10% buffered formalin. Hearts cut into 3 slices (apex, mid-LV and base) and paraffin embedded. The morphometric analysis including infarct size and wall thickness and percent fibrosis was performed on Masson’s trichrome stained tissue sections using ImageJ software (NIH, version 1.30, http://rsb.info.nih.gov/ij/). Wall thickness was measured perpendicular to the infarcted wall at three separate regions and averaged. Fibrosis area and total LV area was measured and expressed as percent fibrosis.
Immunohistochemistry
Immunohistochemistry for tissue sections were performed as described previously 18. Deparaffinized tissue sections were permeabilized and stained with anti-CD68 (Serotec, Raleigh, NC) for inflammatory cell infiltration, anti-CD31 antibody (BD Biosciences, San Jose, CA) for capillary density and alpha smooth muscle actin (α-SMA, Sigma Aldrich, St Louis, MO) for arteriole smooth muscle staining followed by incubation with respective secondary antibodies. Staining without the primary antibodies was used as control for non-specific fluorescence. Nuclei were counter-stained with 4',6-diamidino-2-phenylindole (DAPI, 1:5000, Sigma Aldrich, St Louis, MO), and sections were examined with a fluorescent microscope (Nikon ECLIPSE TE200). Capillary density (CD-31 positive), inflammatory cell infiltration (CD-68 positive) were assessed at 10 randomly selected high visual fields in the border zone of infarcted myocardium and expressed as number per high-power visual field (HVF). Arterioles were defined as round-shaped, α-SMA positive vessels with diameter of >10μm (small arterioles) to >20μm (large arterioles).
Terminal deoxynucleotidyl transferase mediated dUTP nick end-labeling (TUNEL) staining
TUNEL-staining was carried out on 4 μm thick paraffin-embedded sections as per manufacturer’s instructions (Cell death detection assay, Roche, Indianapolis, IN). Cardiac myocytes were identified using α-sarcomeric actinin antibodies (Sigma Chemicals, St Louis, MO). DAPI staining was used to count the total number of nuclei. The index of apoptosis was calculated as the percentage of apoptotic myocyte nuclei/total number of nuclei.
Western blot analysis
Tissue lysates were prepared from the LV infarct border zone using ice-cold radio immunoprecipitation assay buffer (RIPA; 158 mM NaCl, 10 mM Tris HCl, pH 7.2, 1 mM ethylene glycol tetra-acetic acid (EGTA), 1 mM sodium orthovanadate, 0.1% SDS, 1.0% Triton X-100, 1% Sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride). Proteins (50 μg) were electrophoresed and analyzed using anti-p-p38, anti-HuR and anti-p-ser (IP with STAT-3) antibodies. Equal protein loading in each lane was verified using antibodies against the corresponding total protein or β-actin.
Quantitative RT-PCR
Gene expression levels of TNF-α, IL-1β, IL-6, MCP-1, IP-10, TNF receptor 1 (TNFR1), TNF receptor 2 (TNFR2), Matrix metalloproteinases (MMP)-9, MMP-2 and Vascular endothelial growth factor (VEGF-A) were quantified in the border zone of infarct as described previously 19. RNA was collected from heart tissue, NIH3T3 and HUVEC cells with RNA STAT-60(TEL-TEST, Inc, Friendswood, Texas). Total RNA was reverse transcribed with iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA), and amplification was performed using Taqman 7300 (Applied Biosystems, Foster City, CA). Relative mRNA expression of target genes was normalized to the endogenous 18S control gene (Applied Biosystems).
In-gel zymography
MMP activity in LV tissue lysate or cell culture media was measured using gelatin in-gel zymography 6. After staining with Coomassie Blue and destaining, clear and digested regions representing MMPs activity was quantified by densitometry using a Kodak 2000 documentation system.
Statistical analyses
Data are presented as mean±SE. Data were analyzed using Student t tests or one-way analysis of variance (ANOVA) and a post hoc Tukey’s test. Probability (P) values of <0.05 were considered to be significant.
Results
IL-10 treatment suppresses inflammation in the myocardium at 3 days post-MI
Immunohistochemical staining of CD68-positive cells (CD68+ cells) on cardiac tissue sections was carried out to study the inflammatory cell infiltration on 3 days post-MI. Infiltration of CD68+ cells (Macrophage and monocyte) in the border zone of LV infarct increased at 3 days after MI (P<0.01 vs Sham, Fig 1). IL-10 treatment significantly inhibited CD68+ cells infiltration at the injury site (P<0.01 vs MI group, Fig 1).
Figure 1.
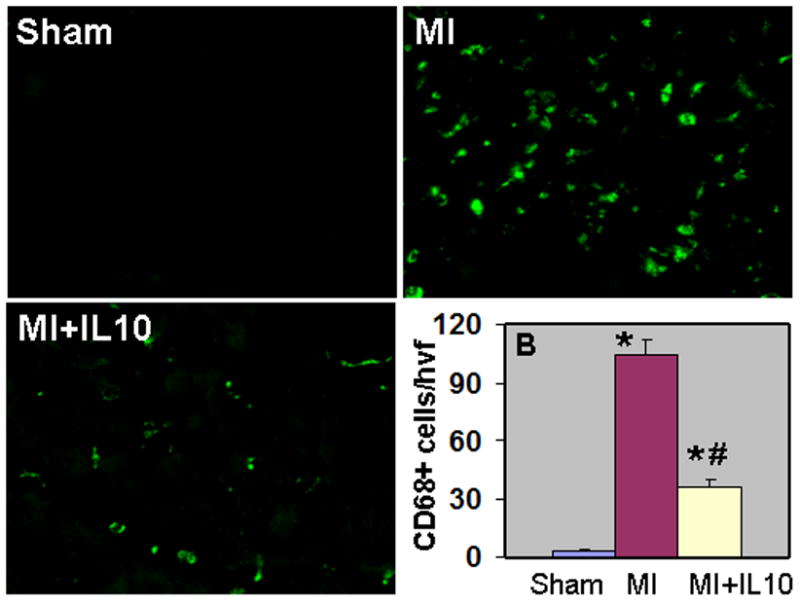
A. Immunofluorescent staining of inflammatory cells (CD68-positive, green fluorescence) in the heart at 3 days post MI. B. Quantitative analysis of infiltrating CD68-positive cells at 3 days post-MI. IL-10 inhibited CD68+ cells infiltration as compared to MI and Sham hearts. *P<0.01 vs Sham; #P<0.01 vs MI.
mRNA expression of various pro-inflammatory cytokines and chemokines (IL-1β, IL-6, TNF-α, MCP-1, IP-10) in the myocardium at 3 days post-MI was assessed by quantitative RT-PCR. Increased inflammatory cell infiltration was associated with increased mRNA expression of pro-inflammatory cytokines and chemokines in the myocardium (P<0.01 vs Sham, Fig 2). IL-10 treated group showed significant reduction in the mRNA expression levels (P<0.05 vs MI, Fig 2).
Figure 2.
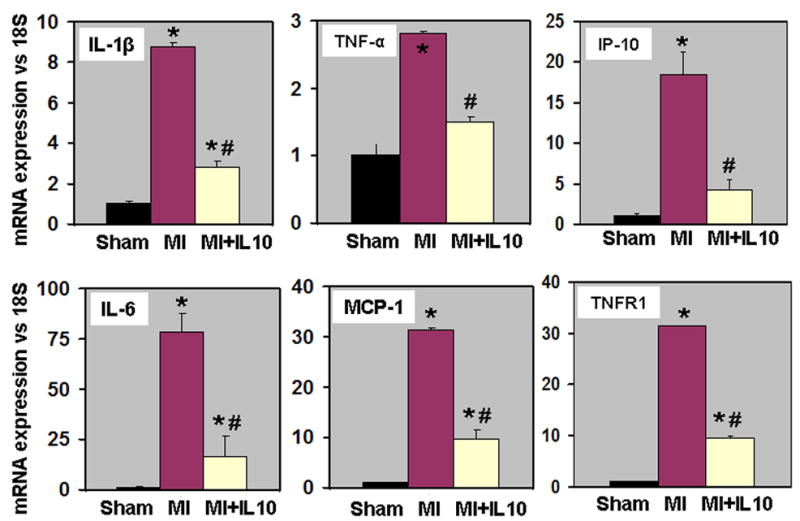
Stimulation of inflammatory responses of TNF-α is mediated through TNF receptors (TNFR1 and TNFR2) 20, 21. Ablation of TNFR1 blunts heart failure and improves survival, whereas ablation of the TNFR2 gene exacerbates heart failure and reduces survival 22. Quantitative RT-PCR analyses showed increased mRNA expression of TNF receptor-1 (TNFR1) in the myocardium at 3d post-MI (P<0.01 vs Sham, Fig 2). Interestingly, IL-10 treatment significantly reduced TNFR1 levels in the myocardium (P<0.01 vs MI, Fig 2). However, mRNA expression of TNFR2 increased after MI (fold change; MI, 2.53±0.12; MI+IL-10, 2.04±0.21; P<0.05 vs Sham,), but no significant difference was observed between the MI groups.
IL-10 attenuates post-MI left ventricular (LV) dysfunction, reduces infarct size and attenuates infarct wall thinning
LV function was assessed by echocardiography. M-mode tracings were analyzed at 14 and 28 days post-MI. Similar changes were observed at these two time points. MI increased LVESD and LVEDD (P<0.01 vs baseline; Fig 3A & 3B, Table 1) and decreased %FS and %EF at 28 days post-MI (P<0.05 vs baseline; Fig 3B), Table 1). IL-10 treatment attenuated cardiac dysfunction with significantly lowered LVESD and LVEDD, and increased %FS and %EF as compared to MI group (P<0.05 versus MI group, Fig 3B). Heart rates were not significantly different among the groups. Similar trends were observed at 14 days post-MI (Table 1). Infarct size was measured as percentage of the LV circumference from trichrome stained sections at 28 days post-MI. Infarct size of 43.13±0.65% was observed in MI group (Fig 4A). IL-10 treatment resulted in a significant reduction in the infarct size (30.86±1.24%, P<0.01 vs MI group, Fig 4A). Interestingly, IL-10 treatment showed an increase in the infarct wall thickness (0.33±0.01mm; P<0.01 vs MI, Fig 4B) as compared to a thin wall in MI group (0.19±0.02mm, Fig 4B).
Figure 3.
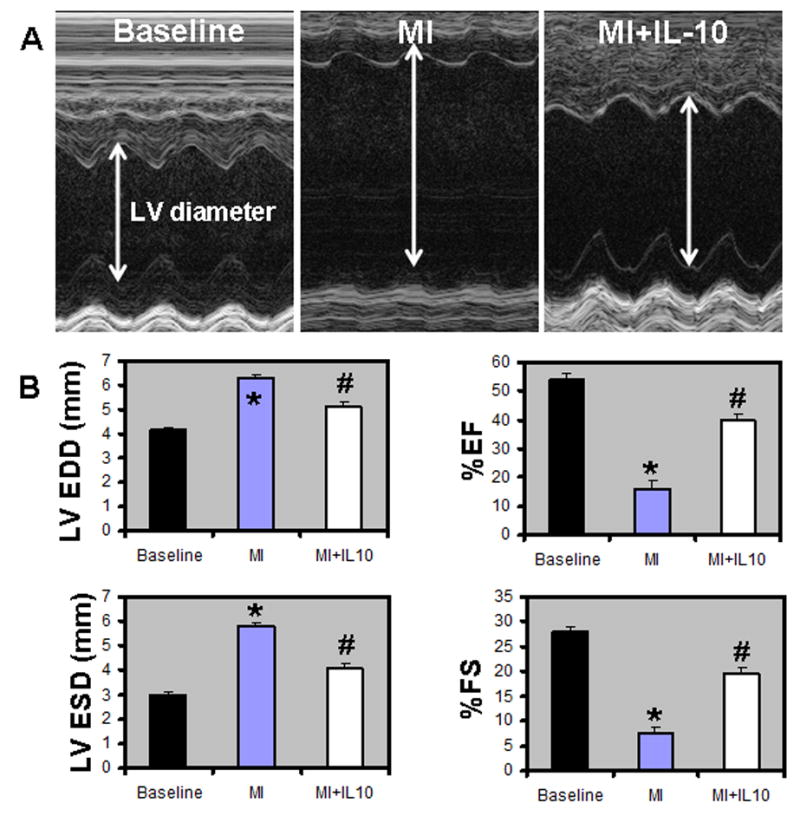
A. M-mode echocardiographic images obtained from baseline, MI and MI+IL-10 groups 28 days post-MI. Arrows indicate LV chamber diameter. B. Analysis of LV diameter in diastole and systole, and percent fractional shortening (%FS) and %EF calculations. LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; %EF, percent ejection fraction; %FS, percent fractional shortening; *P<0.05 vs baseline; #P<0.05 vs MI.
Table 1.
Echocardiographic measurements
| Parameters | Baseline (n=10) | 14 days Post-MI | 28 days Post-MI | P value | ||
|---|---|---|---|---|---|---|
| MI (n=8) | MI+IL-10 (n=8) | MI (n=8) | MI+IL-10 (n=8) | |||
| LVEDD (mm) | 4.15±0.11 | 5.72±0.31* | 4.92±0.19*† | 6.26±0.20 | 5.11±0.04# | *<0.01; †<0.05; #<0.01 |
| LVESD (mm) | 2.99±0.08 | 5.31±0.28* | 3.85±0.24*† | 5.79±0.17 | 4.08±0.20# | *<0.01; †<0.01; #<0.01 |
| EF (%) | 54.10±1.98 | 15.02±3.34* | 44.77±3.52*† | 16.07±2.74 | 39.89±2.17# | *<0.05; †<0.01; #<0.01 |
| FS (%) | 27.83±1.29 | 6.98±1.63* | 22.52±2.02*† | 7.48±1.33 | 19.61±1.20# | *<0.05; †<0.01; #<0.01 |
| Heart rate (bpm) | 424±7.93 | 456±16.72 | 447±12.25 | 480±21.67 | 480±10.69 | - |
Values are mean±SE; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; %EF, percent ejection fraction; %FS, percent fractional shortening; bpm, beats per minute;
comparison between baseline and MI groups;
comparison between MI groups at 14 days post-MI;
comparison between MI groups at 28 days post-MI.
Figure 4.
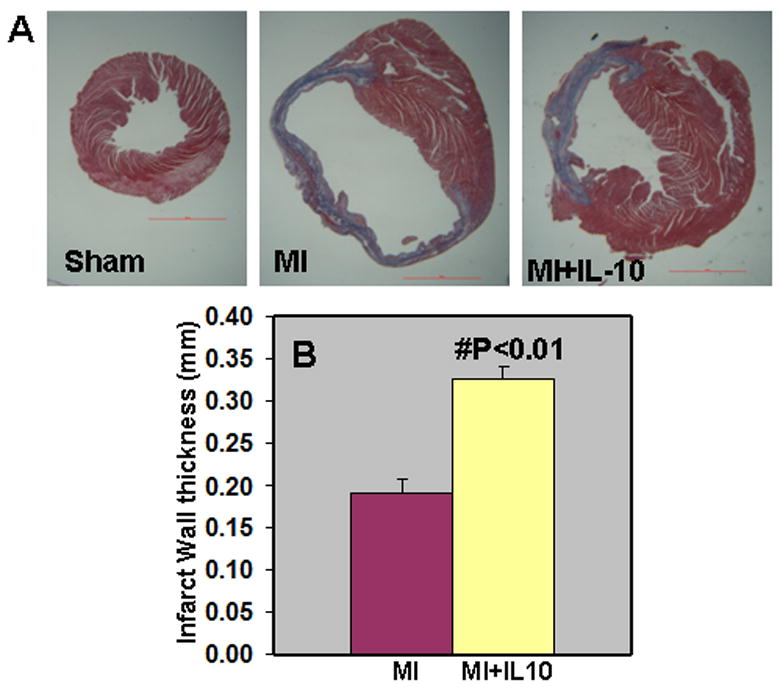
A. Trichrome stained heart sections (28 days post-MI). B. Quantitative analysis of infarct wall thickness at 28 days post-MI. IL-10 treatment attenuated infarct wall thinning when compared to MI group. #P<0.01 vs MI; n=8.
Figure 4. C. TUNEL staining for cardiac cell apoptosis (Red) and DAPI (blue) for nuclear staining in the border zone of LV infarct at 3 days post-MI. IL-10 treatment attenuated cardiac cell apoptosis after MI.
IL-10 attenuates MI-induced cardiac cell death by apoptosis
Heart section was stained with TUNEL method to detect cardiac apoptosis at 3 and 28 days post-MI. Similar trend was observed at both the time points after MI. MI increased the number of apoptotic cells in the border zone of infarction as compared to Sham group (percent apoptosis; MI, 3.07±0.24; Sham, 0.32±0.01; P<0.01 vs Sham, Fig 4C). However, IL-10 treatment significantly reduced the number of apoptotic cells in the border zone of LV infarct (percent apoptosis; MI+IL-10, 1.83±0.28; P<0.01 vs MI, Fig 4C). To determine the apoptotic cardiomyocytes, the sections were stained for alpha-sarcomeric actinin. The number of apoptotic myocytes increased after 28 days post-MI as compared to the Sham group (percent apoptosis; Sham, 0.34±0.02; MI, 1.67±0.11; P<0.01 vs sham). However, the myocyte apoptosis was not significantly different between the two MI groups (MI+IL-10, 1.56±0.07; P<0.01 vs sham). Whereas, it is interesting to note that there was increased number of TUNEL positive cells lining the capillaries after MI, which was significantly lower in mice treated with IL-10 (unpublished data).
IL-10 suppresses phosphorylation of p38 MAP kinase and mRNA stabilizing protein, HuR and activates STAT-3 in the myocardium, post-MI
To investigate intracellular signaling pathways that are affected by IL-10 treatment, we measured the protein levels of cytokine mRNA stabilizing protein, HuR and activation of p38 MAPKs at 3 days and STAT3 at 3 and 28 days in the LV after MI. Western blot analysis demonstrated that HuR protein expression was significantly up-regulated after 3 days of MI (P<0.01 vs Sham, Fig 5A). However, IL-10 significantly suppressed the protein expression of HuR (P<0.01, Fig 5A). Phosphorylation (activation) of p38 MAP kinase (p-p38) was increased in the myocardium at 3 days post-MI (P<0.01 vs Sham, Fig 5B). However, IL-10 treatment significantly reduced the phosphorylation of p38 MAP kinase (P<0.01 vs MI, Fig 5B). To study the effect of IL-10 on STAT3, tissue lysate was immuno-precipitated with STAT3 antibodies and Western blot performed using anti-p-serine antibodies. MI increased phosphorylation of STAT3 (p-ser) at 28 days (P<0.01 vs Sham, Fig 5C). Interestingly, STAT3 phosphorylation (p-ser) was further higher in myocardium of IL-10 treated mice after MI (P<0.05 vs MI, Fig 5C). Similar results were observed at 3 days post-MI.
Figure 5.
A. Western blot for HuR protein expression B. phosphorylation of p38 MAP kinase in LV at 3 days post-MI. Equal loading of proteins in each lane is shown by β-actin and total p38, respectively. C. Western blot for phosphor-serine (activation) against Total STAT3 in LV at 28 days post-MI. Total LV lysate was immunoprecipitated with anti-STAT3 antibodies and analyzed by Western blot using anti-phospho-serine (p-ser) antibodies.
IL-10 treatment enhances capillary density and attenuates arteriole smooth muscle hyperplasia in the LV at 28 days, post-MI
Immunohistochemical staining of CD31 positive cells (CD31+) was carried out to assess the neovascularization in border zone of infarct and remote non-infarct area at 28 days post-MI. Capillary density (CD31+ cells) in the border zone increased in the myocardium at 28 days post-MI (P<0.01 vs Sham, Fig 6A). Interestingly, IL-10 treatment further increased capillary density when compared to MI group (P<0.05 vs MI, Fig 6A). However, capillary density in the remote area was not different between the MI groups (data not shown). Previous report from our laboratory has shown that IL-10 significantly attenuated inflammation-mediated arterial smooth muscle hyperplasia in mice carotid artery injury model 10. To assess the effect of IL-10 on arterioles in the LV after MI, tissue sections were stained with anti-alpha-smooth muscle actin (SMA) and the vessels >20μm (large arteriole) were analyzed. It was interesting to note that the large arterioles were thicker with many layers of smooth muscle cells as compared to Sham group indicating arterial hyperplasia following MI (P<0.01 vs Sham, Fig 6B). However, IL-10 treatment attenuated SMC hyperplasia as compared to MI group (P<0.01 vs MI, Fig 6B). To determine the role of VEGF in IL-10 mediated enhancement of angiogenesis, quantitative RT-PCR analysis of the border zone of LV tissue at 28 days post-MI revealed increased VEGF-A expression, which was further higher in IL-10 treated group (P<0.05 vs MI, Fig 7A).
Figure 6.
A. Capillary density (CD31 staining, green fluorescence) in border zone of LV infarct at 28 days post-MI. Bar graph shows quantitative analysis of CD31+ capillaries per high visual field (hvf). MI increased capillaries, which was further higher in IL-10 treated mice. *P<0.01 vs Sham; #P<0.05 vs MI.
Figure 6. B. Arteriolar smooth muscle hyperplasia (smooth muscle actin stained, red fluorescence) in the LV at 28 days post-MI. Bar graph shows quantitative analysis of arteriolar wall thickness. MI increased inflammation induced SM hyperplasia, which was attenuated in IL-10 treated mice. *P<0.01 vs Sham; #P<0.01 vs MI.
Figure 7.
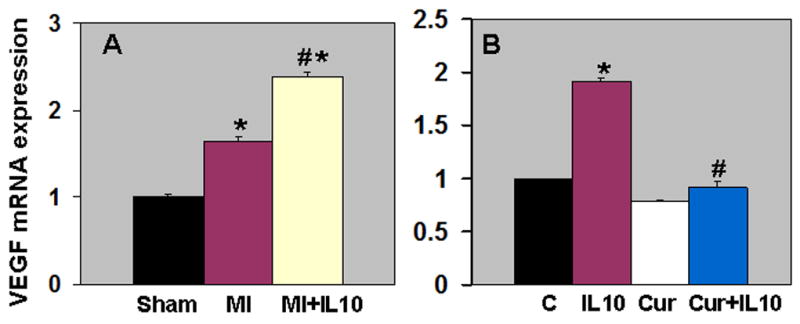
A. Quantitative analysis of mRNA expression of VEGF-A in the border zone of LV infarct at 28 days post-MI. Real time-PCR data shows that IL-10 treatment enhanced VEGF-A expression as compared to MI. *P<0.01 vs Sham; #P<0.05 vs MI B. Effect of STAT-3 inhibition on VEGF-A mRNA expression in HUVEC cells. Curcurbitacin I (Cur) treated cells inhibited IL-10 induced VEGF-A. mRNA expression normalized to 18S expression and depicted as fold change vs Sham. *P<0.01 vs control cells; #P<0.01 vs IL-10 treated cells.
IL-10-induced up-regulation of VEGF-A is mediated through STAT-3 in HUVEC cells
STAT3 is required for myocardial capillary growth and heart protection from ischemic injury 14. To determine the signaling mechanism involved in IL-10 mediated induction of VEGF, passage 4 HUVECs were grown until confluent and pretreated with curcurbitacin I (stat3 inhibitor, 1 μM) for 1 hr. The cells were further treated with IL-10 or saline for another 6hrs. RNA extracted from treated cells was subjected to quantitative RT-PCR analysis of VEGF-A gene expression. IL-10 treatment significantly increased VEGF-A expression (P<0.01 vs control cells, Fig 7B). However, inhibition of STAT-3 with curcurbitacin I significantly suppressed IL-10-induced VEGF-A gene expression (P<0.01 vs IL-10-treated cells, Fig 7B), therefore suggesting that IL-10-induced VEGF-A is mediated through STAT-3.
IL-10 mediated attenuation of LV fibrosis is associated with suppressed mRNA expression and activity of MMP-9 in the LV post-MI
Quantitative analysis of trichrome-stained sections indicated increased fibrosis in the LV after 28 days post-MI as compared to Sham (P<0.01 vs Sham; Fig 8A). Interestingly, we observed significantly reduced fibrosis in the LV following IL-10 treatment (P<0.05 vs MI; Fig 8A). MMPs play an important role in ECM remodeling following MI. MMP (MMP-2 and-9) expression and activity increases rapidly in the heart post-MI 5. Here we examined mRNA expression by quantitative RT-PCR (Fig.8B) and MMP activity by in-gel zymography (Fig 8C) in the border zone at 28 days post-MI. Similar trend was observed at 3 and 28 days post-MI. Quantitative RT-PCR analysis indicated that the mRNA expression of MMP-9 increased after MI (P<0.01, MI vs Sham, Fig 8B). However, IL-10 treatment significantly reduced MMP-9 mRNA expression levels in the LV (P<0.01 vs MI, Fig 8B). However, mRNA expression and activity of MMP-2 was not significantly different between the MI groups at 3 and 28 days post-MI (unpublished data). Analysis of MMP activity using gelatin in-gel zymography (Fig 8C) indicated increased MMP-9 activity after MI (P<0.01 vs Sham, Fig 8C). IL-10 treatment showed reduced MMP-9 activity in the LV (P<0.01; Fig 8C).
Figure 8.
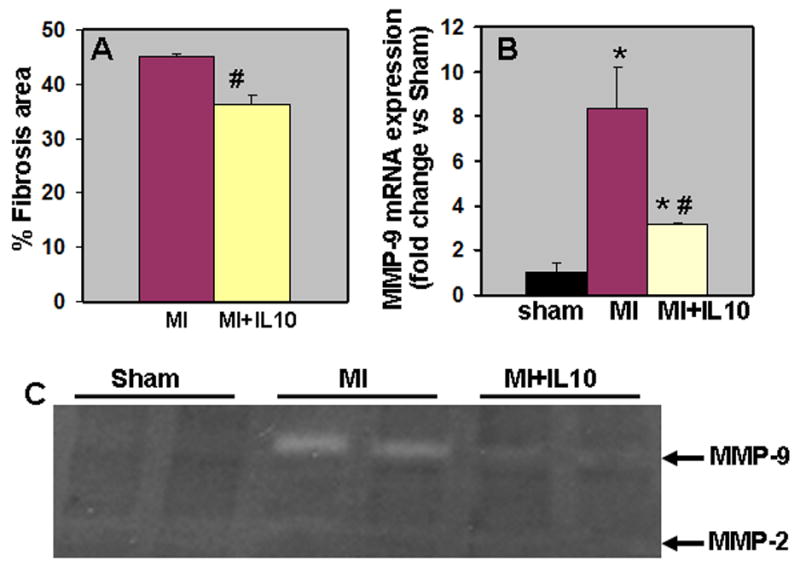
A. Quantitative analysis of fibrosis area at 28 days post-MI. IL-10 group showed lower fibrosis area when compared to MI group. #P<0.01 vs MI. B. Quantitative analysis of mRNA expression of MMP-9 (RT-PCR) in the myocardium at 28 days post-MI. mRNA expression normalized to 18S expression and depicted as fold change vs Sham. #P<0.01 vs MI. C. Gelatin in-gel zymography of LV infarct (border zone) tissue 28 days post-MI. Densitometry analysis showed that MMP-9 was higher in MI groups and reduced with IL-10 treatment. #P<0.01 vs MI.
siRNA-mediated HuR silencing mimics IL-10 mediated suppression of mRNA expression and activity of MMP-9 in NIH3T3 cells
Interleukin (IL)-1 beta induced MMP-9 expression in mesangial cells was mediated by mRNA-stabilizing factor, HuR 23. NIH3T3 cells were serum starved for 1 hr and transfected in duplicate dishes with either HuR siRNA or scrambled siRNA (0.1 μM) by using the Transfast Transfection Reagent (Promega) as per manufacturer's instructions. Twenty-four hours later, the DMEM medium was changed with fresh growth medium containing IL-10, TNF-α and their combination for another 24 hrs. RNA extracted from transfected and treated cells were analyzed for the knock-down of HuR and MMP-9 gene expression. As shown in Fig. 9A, IL-10 treatment significantly inhibited TNF-α mediated increases in HuR gene expression (P<0.01 vs TNF-α treated cells). It is interesting to note that silencing of HuR gene using siRNA mimicked the effects of IL-10 treatment. siRNA knock-down of HuR significantly abrogated TNF-α-induced HuR expression as compared to cells treated with scrambled siRNA (P<0.01, HuR-siRNA+TNF-α vs scrambled-siRNA+TNF-α, Fig 9A). The culture supernatants were collected and centrifuged at 12,500g with Microcon YM-3 centrifugal filter devices (cutoff molecules smaller than 3000 Da; Millipore, Bedford, Massachusetts) for 10 min to obtain a 10-fold concentrate culture supernatant. The supernatant was subjected to in-gel zymography analysis for MMP-9 activity. IL-10 significantly inhibited TNF-α-induced MMP-9 gene expression (P<0.01 vs TNF-α treated cells, Fig 9B) and activity (P<0.05 vs TNF-α treated cells, Fig 9C). Interestingly, knock-down of HuR lead to inhibited TNF-α-induced MMP-9 gene expression (P<0.01, HuR-siRNA+TNF-α vs scrambled-siRNA+TNF-α, Fig 9B) and activity (P<0.05, HuR-siRNA+TNF-α vs scrambled-siRNA+TNF-α, Fig 9C), therefore mimicking IL-10 effects on MMP-9.
Figure 9.
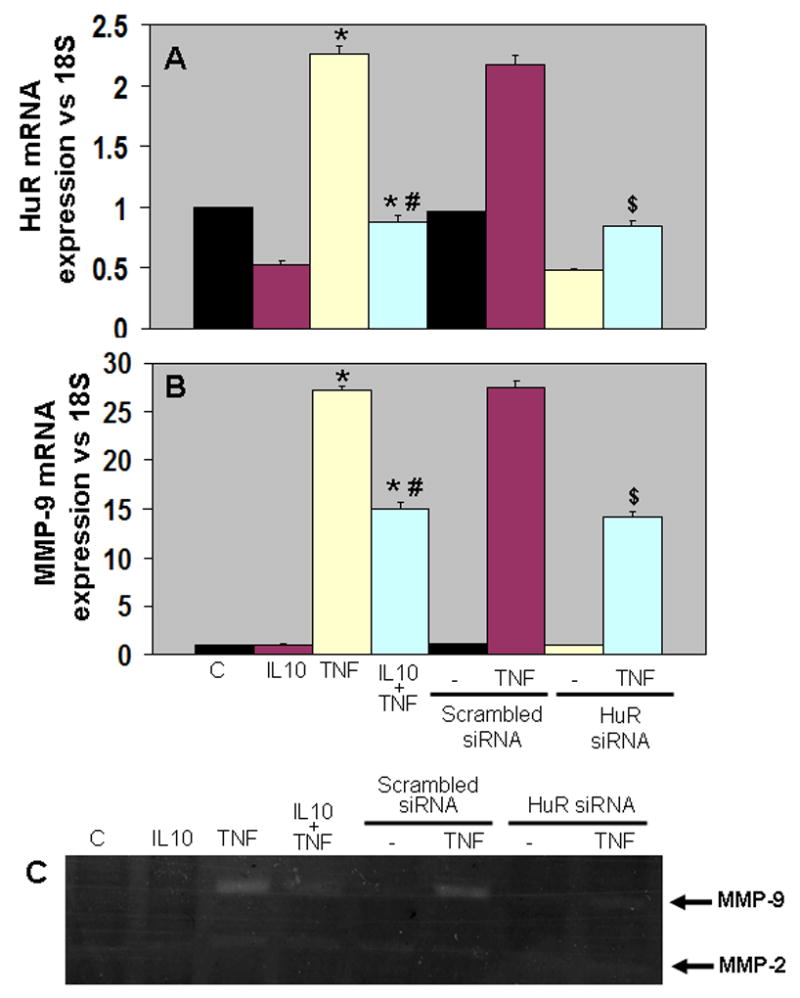
Quantitative analysis of mRNA expression of HuR (A) and MMP-9 (B) by RT-PCR in NIH3T3 cells. mRNA expression normalized to 18S expression and depicted as fold change vs Sham. IL-10 suppressed TNF-α induced HuR and MMP-9 expression (*P<0.01 vs control; #P<0.01 vs TNF-α treated cells). HuR siRNA mimicked IL-10 effects by suppressing TNF-α induced HuR and MMP-9 expression ($P<0.01, HuR siRNA+TNF-α vs scrambled siRNA+TNF-α treated cells). C. Gelatin in-gel zymography for the cell culture media. MMP-9 activity was higher in TNF-α treated cells and IL-10 and HuR siRNA inhibited TNF-α mediated increases in MMP-9 activity.
Discussion
Cardiac pathophysiological conditions like myocardial infarction and reperfusion injury have been associated with the activation of proinflammatory cytokines such as interleukin 1β, tumor necrosis factor (TNF)-α and interleukin-6 24. Various reports have shown that strategies targeting inflammatory response in the heart had promising results with attenuation of cardiac remodeling 13, 25–28. Here, we inhibited inflammatory response in the heart with mouse recombinant IL-10, a potent anti-inflammatory cytokine and studied LV function and remodeling. The important findings of this study are that IL-10 administration attenuated myocardial inflammation and MMP-9 (expression and activity) at 3 days after MI followed by attenuation of LV dysfunction and remodeling with effects on fibrosis and capillary density at 28 days post-MI. The above effects were suggested to be partly due to IL-10 mediated suppression of cytokine mRNA stabilizing protein, HuR and STAT-3 activation.
Inflammatory mediators have been implicated in LV remodeling, including cardiomyocyte hypertrophy 29 with alterations in fetal gene expression and contractile abnormalities 7, 26, 27. Our study has shown that the infiltration of CD68-positive monocyte/macrophages in the border zone of the myocardium was increased at 3 days post-MI. Inflammatory cell infiltration was associated with an increase in mRNA expression of various pro-inflammatory cytokines and chemokines (IL-1β, IL-6, TNF-α, IP-10, MCP-1) after MI. These “stress activated” cytokines are produced by various cell types in the myocardium, including cardiomyocytes or by the infiltrating inflammatory cells.
IL-10, a potent anti-inflammatory cytokine has been shown to limit the infiltration of inflammatory cells in vascular injury model 10. In agreement with the above study, IL-10 treatment inhibited inflammatory cells infiltration and pro-inflammatory cytokines and chemokines (P<0.05 vs MI) in the myocardium. The failure of anti-TNF-α therapy in humans using antibody was attributed partly due to unaffected IL-6, IL-1β and MCP-1 levels within the myocardium 30. Moreover, IL-1β has also been shown to have similar effects as that of TNF-α 27. Important finding of the present study is that IL-10 not only inhibits TNF-α, but also other pro-inflammatory cytokines, which are shown to have adverse cardiac remodeling effects. In addition, IL-10 may attenuate remodeling through direct effects on protease activity without involvement of other cytokines.
Myocardial expression of cytokines contributes to depression of contractile performance and adverse LV remodeling 7, 24. In the present study, echocardiography showed increase in LV end-diastolic (LVEDD) and end-systolic diameters (LVESD) and decrease in %FS and EF% after MI and IL-10 attenuated these effects at 28 days post-MI (P<0.05 vs MI). Cytokine-induced depression of contractile performance was suggested to be a result of interference with myocardial calcium handling 31, 32. But whether IL-10 affects calcium mediated cardiac contractility is not known from the present study. The significant upregulation of proinflammatory cytokines (at 3 days) could trigger a second phase of elevated cytokines levels in the non-infarcted myocardium that promotes interstitial fibrosis and collagen deposition leading to ventricular dysfunction 24. Our findings corroborated well with the above changes. Most importantly a recent study 33 has shown that transplantation of bone marrow mononuclear cells (BM-MNCs) in infarcted mouse hearts led to a significant improvement in cardiac function. These BM-MNCs secreted significant amounts of IL-10 and the cardiac protection was associated with decreased T lymphocyte accumulation, reactive hypertrophy, and myocardial collagen deposition. Also, various studies have reported that activation of MMPs and p38, and reduced angiogenesis and STAT-3 have affected remodeling and cardiac dysfunction 6, 12–14.
Homeostasis of ECM (degradation and accumulation) play an important role in the pathogenesis of LV remodeling, which is controlled by matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) 34, 35. Increased matrix metalloproteinase (MMPs) production associated with sustained inflammatory response may lead to excessive extracellular matrix (ECM) degradation leading to cardiac rupture (in the acute phase) and adverse remodeling changes later 5–7.
Our study shows that MI increased fibrosis (P<0.01 vs Sham) associated with increased mRNA expression and gelatinolytic activity of MMP-9 in the LV (P<0.01 vs Sham). These findings corroborate well with the previous finding, where both IL-1β and TNF-α have been associated with increased MMPs and fibrosis in the heart 29. Interesting finding of the present study is that IL-10 treatment significantly reversed these effects. However, MMP-2 expression and activity was not affected between the groups. IL-10 mediated MMP-9 suppression in the LV was also associated with inhibition of mRNA stabilizing protein, HuR. Since MMP-9 expression in mesangial cells is mediated by mRNA-stabilizing factor, HuR 23, we examined the role of HuR in TNF-α mediated MMP-9 induction in NIH3T3 (mouse fibroblast) cell line. Interestingly, siRNA mediated HuR knock-down resulted in the suppression of TNF-α-mediated increases in MMP-9 expression and activity. This suggests that IL-10 mediated HuR suppression could have lead to MMP-9 inhibition. TNF-α augments IL-1β stimulated release of MMP-9, but not MMP-2 36. Earlier reports have shown that targeted deletion of MMP attenuates early infarct rupture and cardiac contractility and remodeling in mice post MI 6. In vitro experiments demonstrated that isolated canine mononuclear cells with neutralizing antibody to IL-10 inhibited TIMP-1 mRNA expression, which suggested that IL-10 may have a significant role in healing by inducing TIMP-1 expression 8. These findings along with previous reports suggest that IL-10 attenuation of LV remodeling might partly be due to reduced fibrosis and inhibition of MMP-9.
Neovascularization is an integral part of the infarct healing process. MI increased capillary density in the border zone as compared to sham group. Interestingly, IL-10 treatment further increased CD31-positive capillaries at 28 days post-MI. Reports suggest that suppression of IP-10 synthesis during the healing phase may allow formation of the wound neovessels, a critical process for infarct healing 37. In the present study, though IL-10 suppressed IP-10 mRNA expression, whether enhanced neovascularization in IL-10 treated infarcts was mediated through suppression of IP-10 is not clear. TNF-α has been shown to have different effects on proliferation and differentiation (tube formation) of human umbilical vein endothelial cells 38. TNF-α showed both proangiogenic (at low TNF-α concentration for short period) and antiangiogenic (at high TNF-α concentration for long period) effects. Since high level of TNF-α was expressed in the myocardium, anti-angiogenic effects as mentioned above might be a possibility in the present study. MMP plays an important role in angiogenesis. IL-10 inhibited MMP-9 levels and increased neovascularization in the myocardium at 28 days post-MI. This process is not clear, but reports have suggested that matrix metalloproteinase inhibition reduces left ventricular remodeling but did not inhibit angiogenesis after myocardial infarction 39, 40. Although MMPs can directly stimulate angiogenesis, MMPs can also generate inhibitors of angiogenesis such as angiostatin 41. But no such attempt to look at angiostatin was made in the present study.
Several studies have shown involvement of inflammatory mediators in progressive myocytes loss due to necrosis and/or apoptosis suggesting that these cytokines are involved in the progression of cardiac remodeling 28, 42. In the present study, the number of apoptotic myocytes increased after 28 days post-MI as compared to the Sham group but there was no significant difference between the two MI groups. However, it is interesting to note that there was increased number of TUNEL positive cells lining the capillaries after MI, which was significantly lower in mice treated with IL-10 (data not shown). The role of IL-10 in apoptosis cannot be overlooked for the important reason that MI group showed increased p38 MAP kinase activation and reduced STAT3 phosphorylation, the two important pathways that play an anti-apoptosis role 13, 14.
p38 mitogen activated protein kinase mediates both death signaling and functional depression in the heart 13. The p38 MAPK pathway indeed mediates myocardial apoptosis and functional depression by caspase-1, caspase-3 and caspase-11 activation, and tumor necrosis factor, interleukin-1β, interleukin-6 production after myocardial ischemia 13. In a rat model of myocardial injury, inhibition of p38 MAPK lead to cardio-protective effects with improvement of cardiac function, reduction of inflammatory cell infiltration, and cardiomyocyte apoptosis 11, 12. Therefore, in the present study, IL-10 mediated attenuation of LV dysfunction and remodeling might be in part due to suppression of p38 phophorylation.
Signal transducer and activator of transcription 3 (STAT3) is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury 14. Cardiomyocyte-restricted knockout of STAT3 resulted in higher sensitivity to inflammation, cardiac fibrosis, and heart failure. STAT3-deficient mice treated with lipopolysaccharide demonstrated significantly more cardiac TNF production, apoptosis and fibrosis 15. CMC-restricted STAT3 transgenic mice showed reduced myocardial capillary density and increased interstitial fibrosis followed by dilated cardiomyopathy with impaired cardiac function and premature death. STAT3-deficient mice showed enhanced susceptibility to myocardial infarction with increased cardiac apoptosis, increased infarct sizes, and reduced cardiac function and survival 14. In the present study, STAT3 activation in the IL-10 treated group was associated with increased expression of VEGF-A and capillary density. Furthermore, inhibition of STAT3 with curcurbitacin I in HUVEC cells resulted in reduced IL-10-mediated VEGF-A expression. Taken together, our study suggests that IL-10 mediated increased angiogenesis in LV after MI might be through STAT3 activation.
IL-10 in clinical therapy: Although not specifically used for cardiovascular disease, the powerful immunomodulatory properties of IL-10 and the promising results from IL-10 therapy on the course of several inflammatory diseases in experimental models induced the interest on clinical application of IL-10. To our knowledge, human recombinant-IL-10 has been tested in healthy volunteers, patients with Crohn’s disease, rheumatoid arthritis, psoriasis, hepatitis C infection, HIV infection and for the inhibition of therapy associated cytokine release in organ transplantation and Jarisch-Herxheimer reaction 43. These and other studies established the safety of IL-10 treatment, however, the results from clinical trials met with varied success. The varied response may represent the limiting effect of mode of therapeutic IL-10 delivery (systemic vs. local) and the large size of protein (recombinant IL-10). A better understanding of IL-10 mode of action and identification of downstream targets (such as HuR and or p38 MAPK in our studies) that may mediate the effects of this cytokine may in future be suitable for small molecule based pharmacological interventions. Also, understanding the role of IL-10 in transplanted progenitor cells in the heart could enhance cell based therapeutic approaches in cardiac interventions.
In conclusion, the data presented here suggest that IL-10 reduces severity of pro-inflammatory responses and contributes to improved LV function and remodeling with effects on MMP-9 activation, fibrosis and angiogenesis after myocardial infarction, possibly via the activation of Stat-3 and suppression of p38 MAP kinases.
Acknowledgments
Sources of Funding: Work described in this manuscript was in part supported by American Heart Association grant 0530350N and National Institute of Health grant AA014575 to R.K.
Footnotes
Disclosures: None
Publisher's Disclaimer: This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the "Fair Use of Copyrighted Materials" (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
References
- 1.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem. 2006;13:1877–1893. doi: 10.2174/092986706777585086. [DOI] [PubMed] [Google Scholar]
- 3.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol. 2008;180:2625–2633. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 4.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, Doevendans PA, van Echteld CJ, Joles JA, Quax PH, Piek JJ, Pasterkamp G, de Kleijn DP. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257–264. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 5.Tao ZY, Cavasin MA, Yang F, Liu YH, Yang XP. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004;74:1561–1572. doi: 10.1016/j.lfs.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. 2005;115:599–609. doi: 10.1172/JCI22304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun M, Dawood F, Wen WH, Chen M, Dixon I, Kirshenbaum LA, Liu PP. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110:3221–3228. doi: 10.1161/01.CIR.0000147233.10318.23. [DOI] [PubMed] [Google Scholar]
- 8.Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–2808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 9.Yao L, Huang K, Huang D, Wang J, Guo H, Liao Y. Acute myocardial infarction induced increases in plasma tumor necrosis factor-alpha and interleukin-10 are associated with the activation of poly(ADP-ribose) polymerase of circulating mononuclear cell. Int J Cardiol. 2008;123:366–368. doi: 10.1016/j.ijcard.2007.06.069. [DOI] [PubMed] [Google Scholar]
- 10.Rajasingh J, Bord E, Luedemann C, Asai J, Hamada H, Thorne T, Qin G, Goukassian D, Zhu Y, Losordo DW, Kishore R. IL-10-induced TNF-alpha mRNA destabilization is mediated via IL-10 suppression of p38 MAP kinase activation and inhibition of HuR expression. Faseb J. 2006;20:2112–2114. doi: 10.1096/fj.06-6084fje. [DOI] [PubMed] [Google Scholar]
- 11.Bellahcene M, Jacquet S, Cao XB, Tanno M, Haworth RS, Layland J, Kabir AM, Gaestel M, Davis RJ, Flavell RA, Shah AM, Avkiran M, Marber MS. Activation of p38 mitogen-activated protein kinase contributes to the early cardiodepressant action of tumor necrosis factor. J Am Coll Cardiol. 2006;48:545–555. doi: 10.1016/j.jacc.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Ma JY, Kerr I, Chakravarty S, Dugar S, Schreiner G, Protter AA. Selective inhibition of p38alpha MAPK improves cardiac function and reduces myocardial apoptosis in rat model of myocardial injury. Am J Physiol Heart Circ Physiol. 2006;291:H1972–1977. doi: 10.1152/ajpheart.00043.2006. [DOI] [PubMed] [Google Scholar]
- 13.Wang M, Tsai BM, Turrentine MW, Mahomed Y, Brown JW, Meldrum DR. p38 mitogen activated protein kinase mediates both death signaling and functional depression in the heart. Ann Thorac Surg. 2005;80:2235–2241. doi: 10.1016/j.athoracsur.2005.05.070. [DOI] [PubMed] [Google Scholar]
- 14.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 15.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishnamurthy P, Subramanian V, Singh M, Singh K. Deficiency of beta1 integrins results in increased myocardial dysfunction after myocardial infarction. Heart. 2006;92:1309–1315. doi: 10.1136/hrt.2005.071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finsen AV, Christensen G, Sjaastad I. Echocardiographic parameters discriminating myocardial infarction with pulmonary congestion from myocardial infarction without congestion in the mouse. J Appl Physiol. 2005;98:680–689. doi: 10.1152/japplphysiol.00924.2004. [DOI] [PubMed] [Google Scholar]
- 18.Ii M, Nishimura H, Iwakura A, Wecker A, Eaton E, Asahara T, Losordo DW. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via "imported" nitric oxide synthase activity. Circulation. 2005;111:1114–1120. doi: 10.1161/01.CIR.0000157144.24888.7E. [DOI] [PubMed] [Google Scholar]
- 19.Qin G, Kishore R, Dolan CM, Silver M, Wecker A, Luedemann CN, Thorne T, Hanley A, Curry C, Heyd L, Dinesh D, Kearney M, Martelli F, Murayama T, Goukassian DA, Zhu Y, Losordo DW. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc Natl Acad Sci U S A. 2006;103:11015–11020. doi: 10.1073/pnas.0509533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goukassian DA, Qin G, Dolan C, Murayama T, Silver M, Curry C, Eaton E, Luedemann C, Ma H, Asahara T, Zak V, Mehta S, Burg A, Thorne T, Kishore R, Losordo DW. Tumor necrosis factor-alpha receptor p75 is required in ischemia-induced neovascularization. Circulation. 2007;115:752–762. doi: 10.1161/CIRCULATIONAHA.106.647255. [DOI] [PubMed] [Google Scholar]
- 21.Luo D, Luo Y, He Y, Zhang H, Zhang R, Li X, Dobrucki WL, Sinusas AJ, Sessa WC, Min W. Differential functions of tumor necrosis factor receptor 1 and 2 signaling in ischemia-mediated arteriogenesis and angiogenesis. Am J Pathol. 2006;169:1886–1898. doi: 10.2353/ajpath.2006.060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation. 2004;109:1892–1897. doi: 10.1161/01.CIR.0000124227.00670.AB. [DOI] [PubMed] [Google Scholar]
- 23.Huwiler A, Akool el S, Aschrafi A, Hamada FM, Pfeilschifter J, Eberhardt W. ATP potentiates interleukin-1 beta-induced MMP-9 expression in mesangial cells via recruitment of the ELAV protein HuR. J Biol Chem. 2003;278:51758–51769. doi: 10.1074/jbc.M305722200. [DOI] [PubMed] [Google Scholar]
- 24.Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts: possible implication in left ventricular remodeling. Circulation. 1998;98:149–156. doi: 10.1161/01.cir.98.2.149. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Takemura G, Okada H, Miyata S, Maruyama R, Li L, Higuchi M, Minatoguchi S, Fujiwara T, Fujiwara H. Reduction of inflammatory cytokine expression and oxidative damage by erythropoietin in chronic heart failure. Cardiovasc Res. 2006;71:684–694. doi: 10.1016/j.cardiores.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Mayer B, Holmer SR, Hengstenberg C, Lieb W, Pfeifer M, Schunkert H. Functional improvement in heart failure patients treated with beta-blockers is associated with a decline of cytokine levels. Int J Cardiol. 2005;103:182–186. doi: 10.1016/j.ijcard.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki K, Murtuza B, Smolenski RT, Sammut IA, Suzuki N, Kaneda Y, Yacoub MH. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation. 2001;104:I308–I303. doi: 10.1161/hc37t1.094871. [DOI] [PubMed] [Google Scholar]
- 28.Yeh CH, Chen TP, Wu YC, Lin YM, Jing Lin P. Inhibition of NFkappaB activation with curcumin attenuates plasma inflammatory cytokines surge and cardiomyocytic apoptosis following cardiac ischemia/reperfusion. J Surg Res. 2005;125:109–116. doi: 10.1016/j.jss.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 29.Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398–1407. doi: 10.1161/CIRCULATIONAHA.106.643585. [DOI] [PubMed] [Google Scholar]
- 30.Aukrust P, Yndestad A, Damas JK, Gullestad L. Inflammation and chronic heart failure-potential therapeutic role of intravenous immunoglobulin. Autoimmun Rev. 2004;3:221–227. doi: 10.1016/S1568-9972(03)00103-4. [DOI] [PubMed] [Google Scholar]
- 31.Duncan DJ, Hopkins PM, Harrison SM. Negative inotropic effects of tumour necrosis factor-alpha and interleukin-1beta are ameliorated by alfentanil in rat ventricular myocytes. Br J Pharmacol. 2007;150:720–726. doi: 10.1038/sj.bjp.0707147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugishita K, Kinugawa K, Shimizu T, Harada K, Matsui H, Takahashi T, Serizawa T, Kohmoto O. Cellular basis for the acute inhibitory effects of IL-6 and TNF-alpha on excitation-contraction coupling. J Mol Cell Cardiol. 1999;31:1457–1467. doi: 10.1006/jmcc.1999.0989. [DOI] [PubMed] [Google Scholar]
- 33.Burchfield JS, Iwasaki M, Koyanagi M, Urbich C, Rosenthal N, Zeiher AM, Dimmeler S. Interleukin-10 from transplanted bone marrow mononuclear cells contributes to cardiac protection after myocardial infarction. Circ Res. 2008;103:203–211. doi: 10.1161/CIRCRESAHA.108.178475. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006;113:2089–2096. doi: 10.1161/CIRCULATIONAHA.105.573865. [DOI] [PubMed] [Google Scholar]
- 35.Mori S, Gibson G, McTiernan CF. Differential expression of MMPs and TIMPs in moderate and severe heart failure in a transgenic model. J Card Fail. 2006;12:314–325. doi: 10.1016/j.cardfail.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 36.Brown RD, Jones GM, Laird RE, Hudson P, Long CS. Cytokines regulate matrix metalloproteinases and migration in cardiac fibroblasts. Biochem Biophys Res Commun. 2007;362:200–205. doi: 10.1016/j.bbrc.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frangogiannis NG. The role of the chemokines in myocardial ischemia and reperfusion. Curr Vasc Pharmacol. 2004;2:163–174. doi: 10.2174/1570161043476375. [DOI] [PubMed] [Google Scholar]
- 38.Heba G, Krzeminski T, Porc M, Grzyb J, Ratajska A, Dembinska-Kiec A. The time course of tumor necrosis factor-alpha, inducible nitric oxide synthase and vascular endothelial growth factor expression in an experimental model of chronic myocardial infarction in rats. J Vasc Res. 2001;38:288–300. doi: 10.1159/000051057. [DOI] [PubMed] [Google Scholar]
- 39.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM, Jr, Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, Spinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;290:H232–239. doi: 10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]
- 40.Lindsey ML, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti-Morrow L, Crawford J, Black S, Libby P, Mitchell PG, Lee RT. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002;105:753–758. doi: 10.1161/hc0602.103674. [DOI] [PubMed] [Google Scholar]
- 41.Cornelius LA, Nehring LC, Harding E, Bolanowski M, Welgus HG, Kobayashi DK, Pierce RA, Shapiro SD. Matrix metalloproteinases generate angiostatin: effects on neovascularization. J Immunol. 1998;161:6845–6852. [PubMed] [Google Scholar]
- 42.Ing DJ, Zang J, Dzau VJ, Webster KA, Bishopric NH. Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bak, and Bcl-x. Circ Res. 1999;84:21–33. doi: 10.1161/01.res.84.1.21. [DOI] [PubMed] [Google Scholar]
- 43.Asadullah K, Sterry W, Volk HD. Interleukin-10 therapy--review of a new approach. Pharmacol Rev. 2003;55:241–269. doi: 10.1124/pr.55.2.4. [DOI] [PubMed] [Google Scholar]