

Abstract
During the pre-hibernation season, arctic ground squirrels (AGS) can tolerate 8 minutes of asphyxial cardiac arrest (CA) without detectable brain pathology. Better understanding of the mechanisms regulating innate ischemia tolerance in AGS has the potential to facilitate the development of novel, prophylactic agents to induce ischemic tolerance in patients at risk of stroke or cardiac arrest. We hypothesized that neuroprotection in AGS involves robust maintenance of ion homeostasis similar to anoxia-tolerant turtles. Ion homeostasis was assessed by monitoring ischemic depolarization (ID) in cerebral cortex during CA in vivo and during oxygen glucose deprivation in vitro in acutely prepared hippocampal slices. In both models, the onset of ID was significantly delayed in AGS compared to rats. The epsilon protein kinase C (εPKC) is a key mediator of neuroprotection and inhibits both Na+/K+-ATPase and voltage-gated sodium channels, primary mediators of the collapse of ion homeostasis during ischemia. The selective peptide inhibitor of εPKC (εV1–2) shortened the time to ID in brain slices from AGS but not in rats despite evidence that εV1–2 decreased activation of εPKC in brain slices from both rats and AGS. These results support the hypothesis that εPKC activation delays the collapse of ion homeostasis during ischemia in AGS.
Keywords: brain ischemia, heart arrest, tolerance, neuroprotection
Introduction
Out-of-hospital cardiac arrest (CA) affects more than 300,000 people per year in the USA, yet survival to discharge for these patients is 4.6% (Nichol et al. 2008). Survivors suffer a number of complications including neurological impairment related to neuron loss in the CA1 region of the hippocampus. From the approximately 70,000 patients/year that are resuscitated after CA, 60% die from extensive brain injury and only 3%–10% are able to resume their former life styles (Krause et al. 1986; Hypothermia-after-Cardiac-Arrest-Study-Group 2002). Arctic ground squirrels (AGS; Spermophilus parryii), a hibernating species, avoid brain damage caused by global cerebral ischemia during CA. We showed previously that during the pre-hibernation season female AGS tolerate 8 minutes of CA without brain pathology (Dave et al. 2006). This endogenous ischemic tolerance in AGS contrasts sharply with significant cell loss after global ischemia in rats and humans (Brierley and Cooper 1962; Siesjo 1981; Plum 1983; Dave et al. 2006). It remains unclear, however, if this remarkable ischemic tolerance depends on gender or adaptations associated with preparation for hibernation. Moreover, the mechanisms of ischemia tolerance in AGS were not explored.
During hibernation, ground squirrels experience blood-flow levels consistent with the clinical definition of lethal ischemia, but AGS arouse from prolonged torpor without deficits in memory or signs of neuropathology (Ma et al. 2005; Weltzin et al. 2006). This “ischemic tolerance” is due (at least in part) to preservation of energy balance by endogenous mechanisms that couple decreased energy demand with decreased energy supply (Lust et al. 1989; Frerichs et al. 1994; Frerichs et al. 1995). Ion channel arrest, i.e., functional down-regulation of ion conducting channels, is proposed to contribute to hypoxia and cold tolerance in hibernating animals (Hochachka 1986; Lutz and Milton 2004; Milton and Prentice 2007). Evidence for channel arrest arises from studies of anoxia-tolerant turtles where voltage-gated sodium channel (VGSC) density decreases and NMDA receptors are “silenced” during anoxia (Perez-Pinzon et al. 1992; Bickler et al. 2000). Similar studies of heterothermic mammals are generally lacking although one study shows that NMDA receptors are down regulated during torpor in brain tissue from hibernating AGS even when tissue is studied at 37 °C (Zhao et al. 2006). Downregulation of the Na+/K+-ATPase, when coupled to channel arrest, is also linked with the preservation of energy balance (Buck and Hochachka 1993). Na+/K+-ATPase activity is decreased during hibernation (MacDonald and Storey 1999) and this downregulation contributes to tolerance to oxygen glucose deprivation (OGD) in hippocampal slices in vitro (Ross et al. 2006). Due to the lack of studies of euthermic AGS, it remains unclear if ion channel arrest contributes to ischemic tolerance in AGS in the absence of hibernation.
A potential mediator of ion channel arrest is the epsilon PKC isozyme (εPKC). εPKC modulates VGSCs that drive action potentials in neurons. VGSCs contribute to loss of ion homeostasis, neuronal depolarization and the release of neurotransmitters including excitotoxic glutamate during cerebral ischemia (Hodgkin and Huxley 1952; Stuart 1999; Chen et al. 2005). Earlier we and others have shown that εPKC is a key neuroprotective pathway activated in different models of ischemic tolerance (Di-Capua et al. 2003; Raval et al. 2003; Lange-Asschenfeldt et al. 2004; Bright and Mochly-Rosen 2005; Chou and Messing 2005; Li et al. 2005; Perez-Pinzon et al. 2005; Long et al. 2006). In view of these observations, the goal of this study was to assess the role of εPKC in brain ion homeostasis in euthermic AGS during global cerebral ischemia in vivo and during OGD in vitro. Loss of ion homeostasis was detected by onset of the DC shift of the extracellular potential (ischemic depolarization: I D). Our experimental design compared the ischemic tolerant AGS with ischemic intolerant Sprague-Dawley rat.
Experimental procedures
Animals
All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and approved by the Animal Care Committee of the University of Miami (UM) and the University of Alaska Fairbanks (UAF). Arctic ground squirrels (AGS, Spermophilus parryii) were wild-trapped as juveniles in July at approximately (68°38' N, 149°38' W; elevation 809 m), transported to UAF, quarantined for 2 weeks and housed at 17°C under natural lighting conditions until the fall equinox after which lighting was maintained at 12:12 L:D. In April-May, AGS were flown to UM where they were housed under similar conditions (17°C, and 12L:12D). Food was available ad libitum at all times. Experiments were conducted during late April and May. Male AGS and male Sprague-Dawley rats weighing 622 – 1246 g and 300–392 g, respectively, were used for the study with 5–6 animals per experimental group.
Induction of CA
Procedures for induction of cardiac arrest and resuscitation were described previously (Dave et al. 2004; Dave et al. 2006). For non-survival (in vivo DC shift, ischemic depolarization: ID) experiments, animals were monitored for 15 min after the onset of CA. For survival experiments resuscitation was initiated after 10 min of asphyxia. Head and body temperatures were maintained at 37 °C using heating lamps for 1 hr. Because no histopathology was noted after CA in AGS in our previous study (Dave et al. 2006), histopathology in AGS after CA in the present study was compared to naïve AGS not subjected to surgical procedures with the intention of including sham surgical controls if histopathology was noted in the CA group.
Determination of onset of ischemic depolarization in vivo
All surgical procedures were similar to those described above except that a 1.8 mm diameter hole was drilled in the skull to accommodate an extracellular recording electrode. Electrodes were positioned in cortex overlying dorsal hippocampus defined by stereotaxic coordinates for rats with nosebar set to −2.5 mm and replicated in AGS with nosebar set to −2.0 mm. AP coordinates were relative to ear bar zero in AGS and to bregma in rats. Lateral coordinates were relative to midline suture in AGS and to bregma in rats. The ventral coordinate was relative to the dura surface in both rats and AGS. Coordinates for rats were (APb −4.2; Lb +3.0) and for AGS were (APEBZ +4.5mm; Lmls +3.0). The dura was broken with a 26-ga needle and a NaCl-filled (150 mM) glass micropipette was inserted through the dura to a cortical depth of 1.0 mm. A ground electrode (Ag/AgCl) was placed subcutaneously on the top of the head. Field potentials were amplified using BMA-931 bioamplifier with Super-Z headstage (CWE Ardmore, PA), low-pass filtered (DC, 10 kHz), converted to digital form using Digidata 1200 ADC board at 5 KHz (Axon Instruments, Sunnyvale, CA) and stored on a computer using pClamp suite of acquisition and analysis software (Molecular Devices).
In vitro electrophysiology experiments
AGS and rat brain slices were prepared as described earlier (Perez-Pinzon et al. 1998; Perez-Pinzon et al. 1999). In brief, AGS and rats were deeply anesthetized with sodium pentobarbital (60 mg/kg) i.p. and placed on ice to induce mild hypothermia (~ 33 – 35°C) for approximately 10 min. The animals were perfused with sucrose substituted saline in mM (250 sucrose, 3.5 KCl, 26 NaHC03, 10 glucose, 1.25 NaHPO4, 1.2 MgSO4 and 2.5 MgCl2). The perfusate was delivered into the root of the ascending aorta using a 60 ml syringe. The animals were decapitated and the brain rapidly removed. The brain was hemissected and slices of 400 µm thickness sectioned with a Leica VT1000S vibrating microtome, in artificial cerebrospinal fluid (ACSF oxygenated with 95% O2/5% CO2). The ACSF contained in mM: 126 NaCl, 3.5 KCl, 2.0 CaCl2, 2.0 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose. Hippocampi were dissected from slices and stored at room temperature (~ 22°C) for at least 1 h before they were transferred to the recording chamber. Slices were pre-treated with the TAT carrier (vehicle) or a selective TAT-conjugated εPKC inhibitor peptide (εV1–2) (200 nM each) (KAI Pharmaceuticals Inc., San Francisco, CA) for 30 min (Gray et al. 1997). Individual slices were then transferred to an interface-type recording chamber where they were superfused with warmed (35–36 °C) ACSF at a rate of 1 ml/min, and oxygenated with humidified 95% O2–5% CO2. General population measurements of excitatory post-synaptic field potentials (fEPSP) were recorded with a NaCl-filled (150 mM) glass micropipette inserted into the cellbody layer of the CA1 hippocampus subfield. Orthodromic field potentials were elicited by stimulating the Schaffer collaterals with bipolar tungsten electrodes insulated with teflon except at the tip. Stimulation consisted of constant-current, square-wave pulses (0.2 ms in duration) delivered at 30 s intervals. After the baseline evoked response was stable, OGD was induced by switching to glucose-free ACSF (substituted with equimolar sucrose) and by switching the gas mixture in the atmosphere above the slice from 95% O2–5% CO2 to 95% N2–5% CO2. As soon as an ID was noted, the OGD conditions were switched back to normal glucose and oxygen to model reperfusion. Field potentials were amplified using BMA-931 bioamplifier with Super-Z headstage (CWE Ardmore, PA), low-pass filtered (DC, 10 kHz), converted to digital form using Digidata 1200 ADC board at 5 KHz (Axon Instruments, Sunnyvale, CA) and stored on a computer using pClamp suite of acquisition and analysis software (Molecular Devices).
Histology
Seven days after restoration of spontaneous circulation (ROSC), AGS were perfused with FAM (a mixture of 40 % formaldehyde, glacial acetic acid, and methanol, 1:1:8 by volume) for 19 min following a 1 min initial perfusion with physiologic saline. The perfusate was delivered into the root of the ascending aorta at a constant pressure of 110–120 mm Hg as previously described (Perez-Pinzon et al. 1997). The heads were removed and immersed in FAM at 4 °C for 1 day. The brains were then removed from the skull, and coronal brain blocks were embedded in paraffin; coronal sections of 10 µm thickness were cut and stained with hematoxylin and eosin. In AGS, an area equivalent to 3.8 mm posterior to bregma in rats was examined. Ischemic neurons were those exhibiting ischemic cell change (ICC) including (1) eosinophilic cytoplasm, (2) dark-staining triangular shaped nuclei, and (3) eosinophilic-staining nucleolus. Normal neuronal counts were made within the CA1 region of hippocampus by an investigator blinded to the experimental conditions and the counts expressed as number of normal neurons present per mm of CA1 region.
Cell fractionation and Western blot analysis
To measure the translocation of εPKC from the cytosol to the nucleus, cytoskeleton or membrane compartments, tissue was fractionated into soluble and particulate fractions. The latter contained nuclear, cytoskeletal and some membrane components. This method is adapted from one described previously (Raval et al. 2003). Hippocampal slices were frozen in liquid nitrogen at the end of electrophysiology experiments and stored at −80°C until the analysis. At the time of Western blot analysis, the hippocampal slices were resuspended in 400 µl of cell lysis buffer (4 mM ATP, 100 mM KCl, 10 mM imidazole, 2 mM EGTA, 1 mM MgCl2, 20% glycerol, 0.05% Triton X-100, 17 µg/ml PMSF, 20 µg/ml soybean trypsin inhibitor, 25 µg/ml leupeptin, and 25 µg/ml aprotinin). The suspended slices were homogenized using an all-glass homogenizer. The homogenate was then centrifuged at 4°C at 1000 × g for 10 min. The supernatant (soluble fraction) was carefully taken off and recentrifuged at 16,000 × g for 15 min to exclude any contaminating pellet material. The initial pellet was resuspended in 250 µl of cell lysis buffer containing 1% Triton X-100 and was extracted on ice for 60 min. Samples were centrifuged at 16,000 × g for 15 min. The supernatant is the particulate fraction. Both fractions were analyzed for protein content by the Bradford assay, and 40 µg of protein from each fraction was separated by 12% SDS-PAGE. Protein was transferred to Immobilon-P (Millipore) membrane and incubated with the primary antibody anti-epsilon PKC (Calbiochem, La Jolla, CA) (1:500). Immunoreactivity was detected using enhanced chemiluminescence (ECL Western blotting detection kit; Amersham Biosciences, Little Chalfont, UK). Chemiluminescence images were digitized at eight-bit precision by means of a CCD-based camera (8–12 bits) (Xillix Technologies, Vancouver, British Columbia, Canada) equipped with a 55 mm Micro-Nikkor lens (Nikon, Tokyo, Japan). The camera was interfaced to an advanced image-analysis system (MCID model M2; Imaging Research, St. Catherines, Ontario, Canada). The digitized immunoblots were subjected to densitometric analysis using MCID software.
Statistics
All data are expressed as mean ± SEM. Statistical evaluation of the data of heart rate, mean arterial blood pressure, systolic blood pressure and diastolic blood pressure was performed using repeated measures ANOVA followed by a Student-Newman-Keuls. Other statistical evaluation was performed using ANOVA followed by Bonferroni’s post hoc test using Sigmastat software (Systat Software Inc., San Jose, CA).
Results
AGS brain is resistant to injury from 10 min cardiac arrest
In a previous study, we observed that non-hibernating (euthermic) female AGS tolerate 8 min of global cerebral ischemia during the pre-hibernation season (in late August and early September) without evidence of neuropathology. These results contrasted with the substantial neuropathology in sex-matched Sprague Dawley rats following the same duration of CA (Dave et al. 2006). Here we asked whether or not female gender or the pre-hibernation season was necessary for this species’ specific tolerance to global cerebral ischemia. We tested this hypothesis by exposing male AGS to 10 min of CA during the early post-hibernation season. As observed earlier in female AGS (Dave et al. 2006), prior to CA the mean plasma glucose in AGS was higher by 56 – 63 % as compared to rats, despite comparable periods of overnight fasting (supplement Table 1). In rats the blood PO2 and PCO2 levels before induction of CA were 110 ± 5 and 39 ± 1 mgHg, respectively (Supplement Table 1). In contrast, blood PO2 was lower and PCO2 was significantly higher in AGS before induction of CA (63 ± 6 and 55 ± 4 mmHg, respectively), consistent with normal values reported for this species (Ma et al. 2005; Dave et al. 2006). In spite of the differences in plasma PO2 and PCO2 levels in both species, plasma pH was similar (AGS 7.43 ± 0.03 vs. rat 7.43 ± 0.01) (Supplement Table 1). After cessation of ventilation, both species showed an immediate bradycardia followed by hypotension to 50 mmHg. Within approximately four minutes, mean arterial pressure (MAP) decreased to below 15 mmHg and heart rate decreased to less than 40 beats per minute (bpm) (Figure 1). Systolic and diastolic pressures decreased in parallel in both species. Upon resuscitation, MAP returned to 60 mmHg within two minutes (Figure 1). The ECG pattern was restored to normal within five minutes of ROSC. Histological assessment of neuronal death in the brain was performed seven days after ROSC following 10 minutes of CA (Figure 2). No ischemic neurons were found in naïve control AGS or in AGS subjected to CA. The numbers of normal neurons were statistically similar in control and CA groups (control 103±0.1 vs. CA 90±6; n=5) (Figure 2A). These results demonstrate that AGS tolerate as much as 10 minutes of CA. In the CA group, we were able to resuscitate all animals and all of them survived 7 days of reperfusion. We did not study 10 minutes of CA in rats since we already observed near total CA1 pyramidal cell death in rats after 8 minutes of CA (Raval et al. 2005; Dave et al. 2006)
Figure 1. Heart rate and blood pressure during cardiac arrest in AGS and rat.
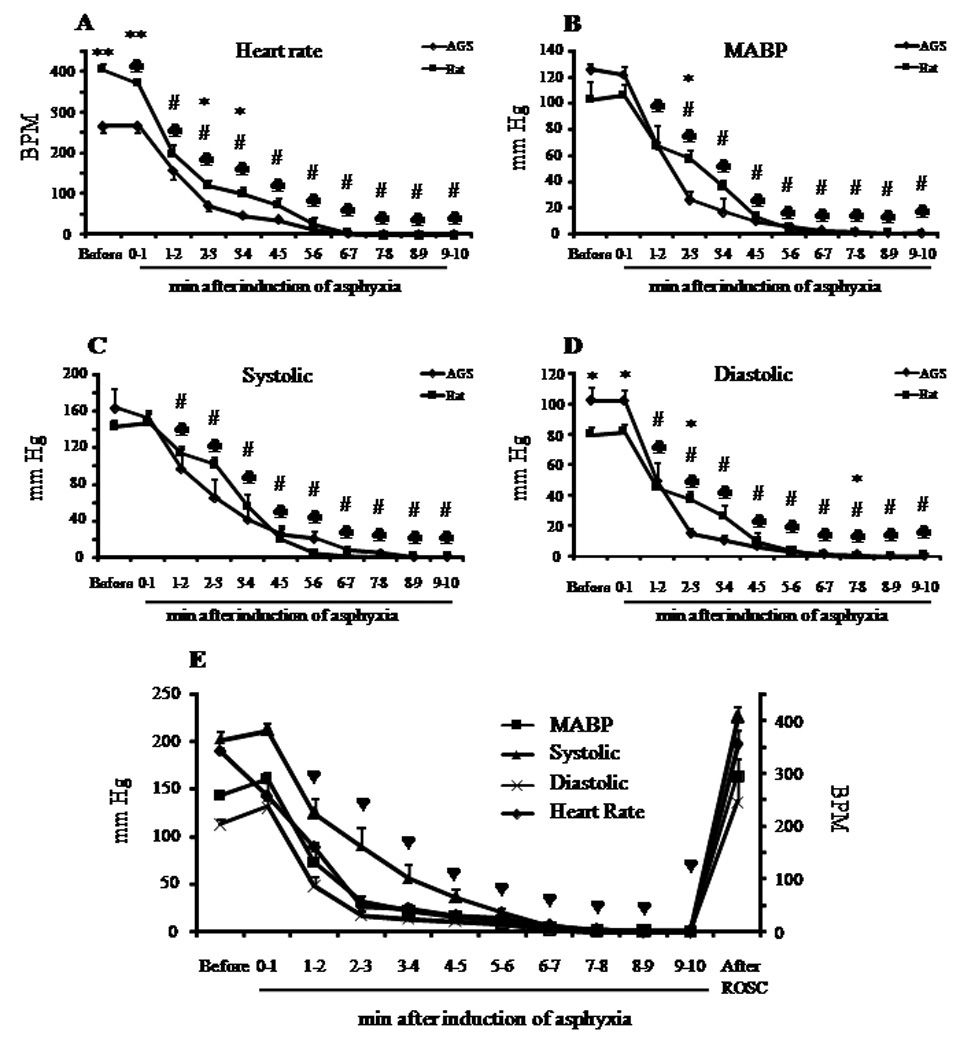
The average (A) heart rate (BPM), (B) mean arterial blood pressure (MABP), (C) systolic and (D) diastolic pressure before and during cardiac arrest (CA) in rats and AGS for in vivo ID experiments. (E) Mean heart rate, MABP, systolic and diastolic pressure before, during and after CA in AGS for histology experiments. Left ordinate represents data for MABP, systolic and diastolic pressure; right ordinate represents data for heart rate. *, p<0.05; **, p<0.001 rat vs AGS. # and ♣, p<0.05 vs baseline for AGS and rat, respectively. ♥, p<0.05 vs baseline for all parameters in (E).
Figure 2. Histopathology in CA1 hippocampus following 10 minutes of asphyxia cardiac arrest in AGS.
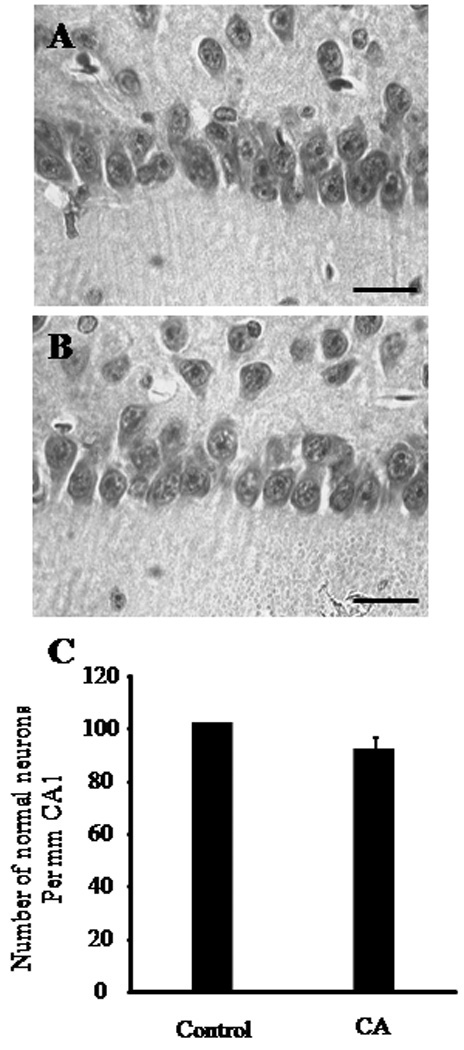
Representative histological images of hippocampal CA1 region in (A) naïve control AGS or in (B) AGS after 7 days of reperfusion following 10 minutes of cardiac arrest (CA) (n = 5). All images were captured at 40X magnification. (C) The AGS brains were examined by an investigator blinded to the experimental conditions. The normal neurons were counted in CA1 region of hippocampus. The number of normal neurons in the CA1 region (which includes the middle, medial and lateral sub-region) of naïve AGS hippocampus or AGS hippocampus 7 days after 10 minutes of CA are presented in a graphical form. Scale bar indicates 30 µm.
Ischemic depolarization is delayed in AGS during CA
Based on hypotheses regarding ion channel arrest and enhanced ion homeostasis in torpid hibernators, we next asked if ion homeostasis was preserved during global cerebral ischemia associated with CA in AGS. Loss of ion homeostasis was indicated by the ID in cerebral cortex during cardiac arrest. The brain temperature was maintained at 37°C in both experimental groups throughout the experiment. We observed that the onset of ID occurred at 1.9±0.22 minutes (n = 8) in rats and at 3.1±0.52 minutes (n=5) in AGS, a delay of 1.23 minutes, (p<0.05) (Figure 3). The time for the ID to peak in amplitude was likewise delayed in AGS. The interval between initiation of CA and peak amplitude in ID was 2.2±0.18, (n = 8) in rats and 3.8±0.41 minutes (n = 5) in AGS (p<0.01). These data suggest that preservation of ion homeostasis contributes to tolerance to CA in AGS.
Figure 3. Ischemic depolarization (ID) in rat and AGS brains following cardiac arrest.
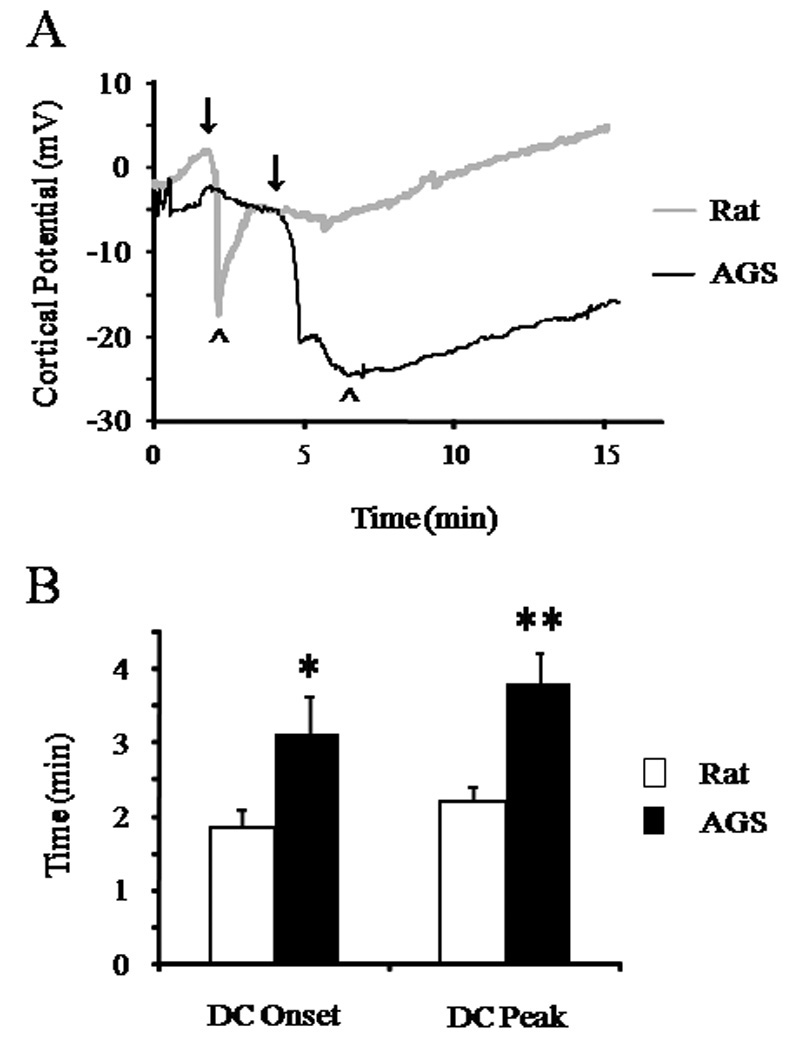
A) Representative traces of ID in AGS and rat brains indicating that time to onset of ID is delayed. Zero second indicates onset of asphyxia. Onset of ID is indicated by ↓ while time - to -peak amplitude is indicated by ^. B) Bar graphs showing time to onset of ID and time for the ID to peak in amplitude. *, p<0.05 and **, p<0.005 rat vs AGS.
Inhibition of εPKC during ischemia blocks delayed ion homeostasis collapse in AGS
εPKC is a key protective pathway activated in different models of brain ischemia tolerance (Di-Capua et al. 2003; Raval et al. 2003; Lange-Asschenfeldt et al. 2004; Bright and Mochly-Rosen 2005; Chou and Messing 2005; Li et al. 2005; Perez-Pinzon et al. 2005; Long et al. 2006). We tested the hypothesis that activation of εPKC is necessary for the delayed collapse of ion homeostasis. We mimicked cerebral ischemia in acute hippocampal slices by depriving slices of oxygen and glucose (oxygen glucose deprivation or OGD). We observed that in TAT treated rat and AGS hippocampal slices the onset of ID occurred at 2.8±0.23 (n = 4) and 6.6±1.60 (n = 4) minutes after onset of OGD, respectively. Similar to observations made during CA in vivo, we observed that onset and peak amplitude of OGD-induced ID were significantly delayed by 3.8 minutes (p<0.01, n = 4) and 4 minutes (p<0.01, n = 4) in AGS hippocampal slices compared to rat hippocampal slices (Figure 4). Moreover, evoked field potential persisted in slices from AGS but not in slices from rat during OGD and reperfusion (Figure 4). Pre-treating AGS hippocampal slices with a selective TAT-conjugated εPKC inhibitor peptide (εV1–2; 200 nM) for 30 minutes prior to OGD accelerated onset of the ID by ~ 2 minutes when compared with slices treated with TAT carrier (vehicle) (p<0.05). However, pre-treatment with εPKC inhibitor had no effect on ID and time-to-peak in hippocampal slices harvested from rats (Figure 4D). These results demonstrate that εPKC activation is required for the robust ion homeostasis in AGS brain tissue during experimental ischemia.
Figure 4. Ischemic depolarization (ID) in rat and AGS hippocampal slices following oxygen glucose deprivation and the effect of εPKC activation on ID.
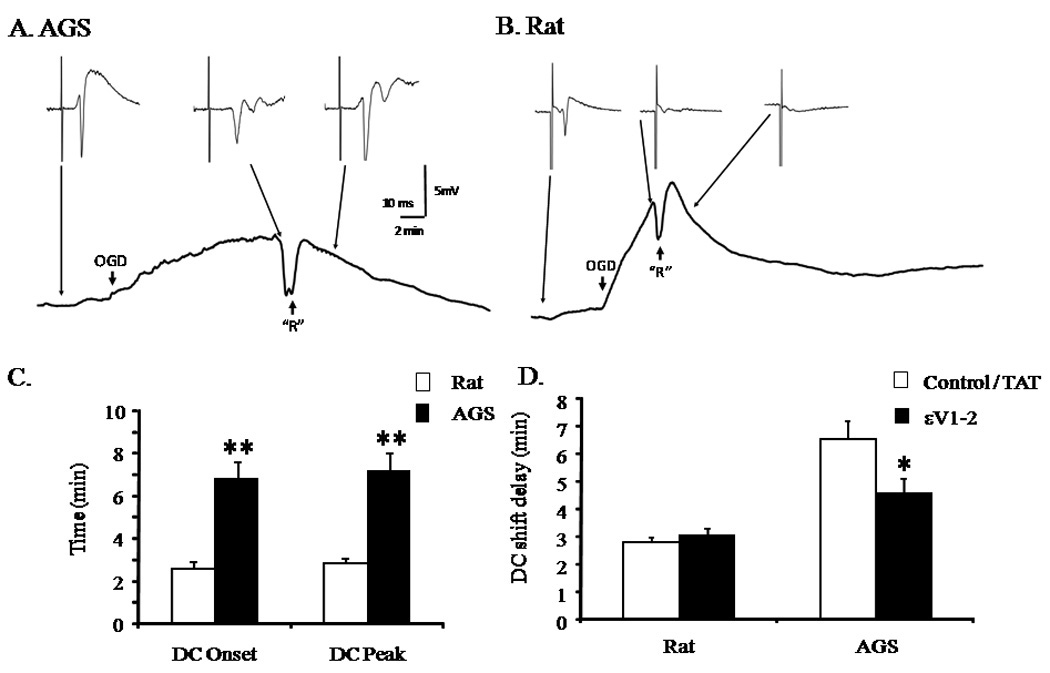
Representative traces of ID in (A) AGS and (B) rat hippocampal slices indicating that time to onset of ID is delayed. ↓ OGD indicates onset of OGD, while onset of reperfusion is indicated by ↑ “R”. C) Bar graphs showing time to onset of ID and time for the ID to peak in amplitude. D) Bar graphs showing the effect of εPKC selective TAT-conjugated εPKC inhibitor peptide (εV1–2) or TAT carrier (vehicle) (200 nM each) on onset of ID in hippocampal slices harvested from rat or AGS. *, p<0.05 TAT vs εV1–2. **, p<0.001 rat vs AGS.
εPKC inhibition prevents OGD- induced εPKC activation
To confirm that εV1–2 inhibited εPKC translocation during OGD in rat and AGS hippocampal slices, we next measured levels of εPKC in soluble and particulate (membrane) fractions in OGD + TAT carrier peptide and OGD + εV1–2 - treated slices (Figure 5) (Gray et al. 1997). The εPKC inhibitor (εV1–2) increased the amount of εPKC remaining in the soluble fraction after OGD, confirming that εV1–2 blocked εPKC activation during OGD in both rat and AGS slices.
Figure 5. εPKC inhibition prevents OGD- induced εPKC activation in AGS and rat.
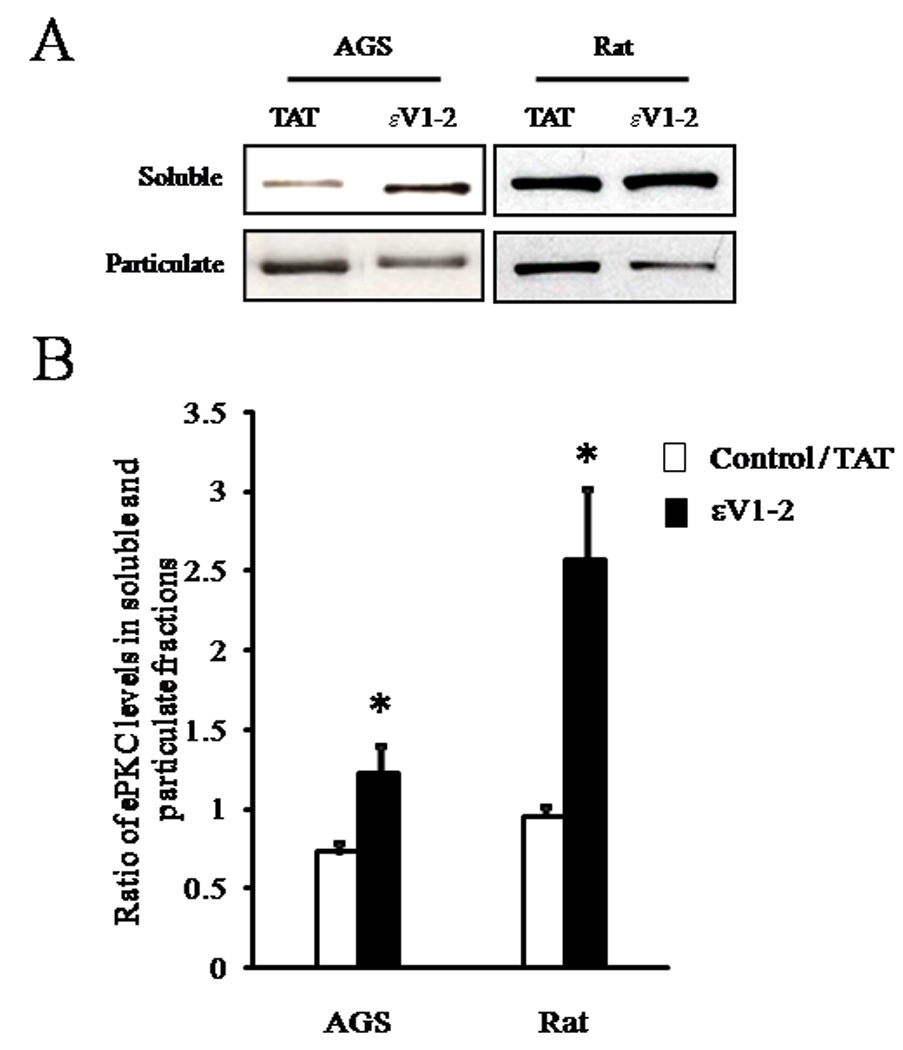
Slices collected at the end of the electrophysiology experiments were frozen in liquid nitrogen. Particulate and soluble fractions were prepared as described in the methods. (A) Representative immunoblot of εPKC isozyme in rat or AGS hippocampal slices subjected to OGD following treatment with εPKC selective TAT-conjugated εPKC inhibitor peptide (εV1–2) or TAT carrier (vehicle) (200 nM each). (B) Immunoblots as represented in (A) were subjected to densitometric analysis and percent translocation from soluble to particulate fraction was determined. Results are expressed as ratio of εPKC levels in soluble and particulate fractions for each sample. *, p < 0.05 TAT vs εV1–2.
Discussion
The ischemic tolerance of AGS is independent of hibernation status (Dave et al. 2006). Here, we show that ischemic-tolerance was associated with robust maintenance of ion homeostasis during ischemia, as indicated by a delay in ID in vivo and in vitro. Moreover, the delay in the collapse of ion homeostasis required activation of protein kinase C epsilon (εPKC) in AGS.
Ion homeostasis and ischemia / anoxia tolerance
Neuronal depolarization during anoxia or ischemia represents a collapse in ion homeostasis. Without reperfusion, this “ischemic” or “anoxic” depolarization leads to acute neuronal death and represents a reliable correlate of ensuing brain damage (Kaminogo et al. 1998). If oxygen and glucose are re-introduced immediately after ID, slices generally recover and evoked potentials return to pre-OGD amplitude (Perez-Pinzon et al. 1998). Blocking or delaying the ID can significantly improve recovery (Takeda et al. 2003; Anderson et al. 2005). In hypoxia-tolerant species such as the fresh water turtle, persistent ion homeostasis diminishes metabolic demand during periods of limited oxygen availability (Hochachka 1986; Perez-Pinzon et al. 1992; Bickler et al. 2000). During hibernation, AGS experience ischemic-like levels of blood flow but lack neuropathology due to decreased energy demand coupled to decreased energy supply (Lust et al. 1989; Frerichs et al. 1994; Frerichs et al. 1995). Downregulation of ion channels, termed “ion channel arrest”, is hypothesized to occur in torpid hibernators (Hochachka 1986). Ion channel arrest slows the collapse of ion gradients by preventing the flow of ions in and out of neurons.
Preservation of ion homeostasis contributes to tolerance to CA in AGS
Our results demonstrate robust ion homeostasis during ischemia in euthermic AGS. Persistent ion homeostasis in euthermic AGS brain results in a delay of membrane depolarization, one of the first steps in the ischemic cascade resulting from a disruption in energy supply that likely contributes to ischemia tolerance observed in this species. A delay of more than a minute in ID is significant given the current 5 minute window for successful resuscitation from CA in ischemia - vulnerable species such as rats and humans (Takeda et al. 2003; Anderson et al. 2005). Indeed, neuronal cells can withstand 2.9 times longer duration of ischemia if ID is delayed by 1.1 minutes (Takeda et al. 2003). Nonetheless, persistent ion homeostasis may be only one component of the endogenous tolerance of ischemia in AGS. Other mechanisms thought to play a role downstream to ID include differences in NMDA receptor expression and enhanced Ca2+ homeostasis (Zhao et al. 2006). Moreover, euthermic AGS have chronically high HIF 1α protein levels in brain that may be due to mild, chronic hypoxemia associated with a low respiratory drive (Ma et al. 2005). In rats, hypoxia-induced preconditioning is linked to HIF 1α regulated gene expression (Bergeron et al. 2000; Jones and Bergeron 2001; Bernaudin et al. 2002; Prass et al. 2003) and enhanced tolerance to subsequent ischemic events. Protective effects of hypoxic preconditioning are reported using in vivo and in vitro models of cerebral ischemia (Gidday et al. 1994; Bruer et al. 1997; Miller et al. 2001). Thus, increased levels of HIF 1α may also, in part, be responsible for neuroprotection that we observed in the present study. In addition, the persistence of robust ion homeostasis in acutely prepared brain slices from AGS also supports the hypothesis that ischemic tolerance (as measured by delay to ID) is an intrinsic property of neurons and glia, rather than a property of the cerebral vasculature of the AGS brain.
εPKC activation delays collapse of homeostasis in AGS brain during experimental ischemia
This is the first report that a neuronal signaling pathway, in this case εPKC, is implicated in ischemic tolerance during the euthermic state of a hibernating species. Ischemia tolerance in AGS provides an example of a chronic and persistent state of neuroprotection that shares many similarities with ischemic preconditioning (IPC). IPC is prophylactic against lethal ischemic insults in many organs including brain and heart via mechanisms involving εPKC (Perez-Pinzon 2007; Savitz and Fisher 2007). In the brain, IPC induces robust neuroprotection against ischemia in the CA1 region of the hippocampus in a variety of in vivo and in vitro models (Perez-Pinzon et al. 1997; Raval et al. 2003; Lange-Asschenfeldt et al. 2004; Dave et al. 2006; Kim et al. 2007). The εPKC isozyme plays an essential role in the neuroprotective effects of IPC (Raval et al. 2003).
εPKC activation inhibits Na+/K+-ATPase and VGSC (Nowak et al. 2004; Chen et al. 2005), both key players in ion homeostasis and its collapse during ischemia. VGSCs drive action potentials in neurons. A barrage of action potentials increases intracellular sodium, which in turn requires extrusion via the ATP-driven Na+/K+-ATPase. We hypothesize that εPKC is poised to regulate this balance by decreasing VGSC and Na+/K+-ATPase function. εPKC reduces peak Na+ currents by ~30% via phosphorylation of sodium channel subunits (Chen et al. 2005). After oxidant injury in renal proximal tubular cells, activation of εPKC decreases the activity of Na+/K+-ATPase by 60% (Nowak et al. 2004). Inhibition of VGSC could be sufficient, on its own, to delay loss of ion homeostasis. Alternatively, since εPKC activation does not inhibit VGSC completely (Chen et al., 2005) simultaneous inhibition of VGSC and Na+/K+-ATPase may act synergistically to decrease the rate of Na+ influx and decrease the rate of ATP hydrolysis ultimately resulting in the persistence of ion homeostasis. PKC is considered a target for modulating metabolic suppression in hibernating animals (Mehrani and Storey 1997; Lee et al. 2002; Eddy et al. 2005). In support of this hypothesis, Na+/K+-ATPase activity is decreased during hibernation (MacDonald and Storey 1999) and this down regulation of Na+/K+-ATPase contributes to ischemia tolerance (Ross et al. 2006). The present results show that εPKC activation preserved ion homeostasis during experimental ischemia in AGS brain. It is noted that εPKC activation had no effect on ion homeostasis during experimental ischemia in rat brain (Figure 3D). This evidence suggests that physiological differences between rat and AGS may contribute to this effect, and warrant further investigation.
Contribution of other physiological variables in neuroprotection against ischemia in AGS
AGS show differences in physiological parameters when compared to rats and other mammals. AGS maintain higher levels of blood glucose after similar periods of fasting when compared to rats. Higher blood glucose may contribute to hypoxia tolerance in AGS, but would not be expected to contribute to tolerance to cerebral ischemia. Glucose, supplied via blood flow to the brain can contribute to glycolysis and ATP production. Despite the fact that plasma glucose levels before inducing ischemia delays the decline in ATP (Folbergrova et al. 1997) and delays the onset of ID (Li et al. 1996), an essential influence of glucose can be ruled out as glucose concentration used in ACSF for in vitro studies was the same for slices obtained from both species and was constant throughout the experiment. Moreover, hyperglycemia during reperfusion actually worsens outcome following stroke (Gilmore and Stead 2006). In vivo, AGS maintain chronically low PaO2 and chronically high PaCO2 levels as reported here and previously (Ma et al. 2005; Dave et al. 2006). It has yet to be determined what physiological mechanisms are responsible for this respiratory status or if chronic hypoxemia contributes to ischemia tolerance. High blood CO2 levels were not associated with lowered blood pH. Indeed arterial HCO3− concentrations were approximately twice as high in AGS compared with in rat. Enhanced pH buffering capacity may contribute to ischemia tolerance in AGS, but also requires further investigation.
The current findings show that ischemia tolerance in AGS compared with rat does not depend on gender or the hibernation season. Our study does not rule out influences of these factors since neither variable was systematically studied in the present set of experiments. Gender in rats and humans influence outcome after cerebral ischemia (Hurn and Macrae 2000; Bramlett 2005). Phenotype expressed during the hibernation season is necessary for resistance to ischemia/reperfusion injury in the intestine (Kurtz et al. 2006). However, the maximum duration of global ischemia that AGS can tolerate without neuropathology must be defined to address the role of gender and hibernation season in tolerance to cerebral ischemia during cardiac arrest. This limit remains unknown and is beyond the scope of the present study. Such studies would be complicated by the properties of cardiac tissue that limit capacity to achieve ROSC as opposed to the resistance of neurons to global cerebral ischemia. In the present study, male AGS were tested in the spring just after the hibernation season. These animals tolerated 10 minutes of CA. Similarly, in a previous study we observed that female AGS tested during the pre-hibernation season tolerated 8 minutes of CA. Tolerating 8 or 10 minutes of CA without neuronal cell death contrasts substantially from the massive cell loss in the hippocampus CA1 region and severe neurological deficits experienced by rats of either sex subjected to 8 min of CA (Raval et al. 2005; Dave et al. 2006). Thus, in comparison to rat both male and female AGS resist neuronal cell death induced by extended periods of CA.
Summary
Minimizing time to treatment is a significant challenge for treating ischemic brain injury in humans. During the initial chaos that ensues during cerebral blood flow deficits in brain, the narrow therapeutic window for CA and stroke patients strongly limits the capacity to translate preclinical neuroprotection strategies to the clinic. For people at risk for stroke and cardiac arrest, prophylactic treatments may extend the therapeutic window and improve clinical outcome (Savitz and Fisher 2007). In our study, the chronic neuroprotection in AGS was associated with enhanced ion homeostasis regulated by εPKC. We conclude that loss of ion homeostasis is delayed in AGS brain and that this delay requires εPKC activation. If these results are translated to a prophylactic treatment for patients at risk for CA or stroke, this delay could significantly increase the therapeutic window. Other factors downstream to loss of ion homeostasis also contribute to the remarkable ischemia tolerance of AGS and require further investigation.
Supplementary Material
Acknowledgement
This study was supported by NIH grants NS34773, NS05820, NS045676, NS054147, NS041069-06 (NINDS and NIMH) and US Army Medical Research and Materiel Command grant # 05178001. We thank Velva Combs, Heather McFarland and Dr. Maritza Martinez for technical assistance. TAT and εV1–2 were purchased from KAI Pharmaceuticals Inc., South San Francisco, CA.
References
- Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD. Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. J Neurophysiol. 2005;93:963–979. doi: 10.1152/jn.00654.2004. [DOI] [PubMed] [Google Scholar]
- Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–296. [PubMed] [Google Scholar]
- Bernaudin M, Nedelec AS, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab. 2002;22:393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Donohoe PH, Buck LT. Hypoxia-induced silencing of NMDA receptors in turtle neurons. J Neurosci. 2000;20:3522–3528. doi: 10.1523/JNEUROSCI.20-10-03522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlett HM. Sex differences and the effect of hormonal therapy on ischemic brain injury. Pathophysiology. 2005;12:17–27. doi: 10.1016/j.pathophys.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Brierley JB, Cooper JE. Cerebral complications of hypotensive anaesthesia in a healthy adult. J Neurol Neurosurg Psychiatry. 1962;25:24–30. doi: 10.1136/jnnp.25.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright R, Mochly-Rosen D. The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke. 2005;36:2781–2790. doi: 10.1161/01.STR.0000189996.71237.f7. [DOI] [PubMed] [Google Scholar]
- Bruer U, Weih MK, Isaev NK, Meisel A, Ruscher K, Bergk A, Trendelenburg G, Wiegand F, Victorov IV, Dirnagl U. Induction of tolerance in rat cortical neurons: hypoxic preconditioning. FEBS Lett. 1997;414:117–121. doi: 10.1016/s0014-5793(97)00954-x. [DOI] [PubMed] [Google Scholar]
- Buck LT, Hochachka PW. Anoxic suppression of Na(+)-K(+)-ATPase and constant membrane potential in hepatocytes: support for channel arrest. Am J Physiol. 1993;265:R1020–R1025. doi: 10.1152/ajpregu.1993.265.5.R1020. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cantrell AR, Messing RO, Scheuer T, Catterall WA. Specific modulation of Na+ channels in hippocampal neurons by protein kinase C epsilon. J Neurosci. 2005;25:507–513. doi: 10.1523/JNEUROSCI.4089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WH, Messing RO. Protein kinase C isozymes in stroke. Trends Cardiovasc Med. 2005;15:47–51. doi: 10.1016/j.tcm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Dave KR, Prado R, Raval AP, Drew KL, Perez-Pinzon MA. The arctic ground squirrel brain is resistant to injury from cardiac arrest during euthermia. Stroke. 2006;37:1261–1265. doi: 10.1161/01.STR.0000217409.60731.38. [DOI] [PubMed] [Google Scholar]
- Dave KR, Raval AP, Prado R, Katz LM, Sick TJ, Ginsberg MD, Busto R, Perez-Pinzon MA. Mild cardiopulmonary arrest promotes synaptic dysfunction in rat hippocampus. Brain Res. 2004;1024:89–96. doi: 10.1016/j.brainres.2004.07.050. [DOI] [PubMed] [Google Scholar]
- Di-Capua N, Sperling O, Zoref-Shani E. Protein kinase C-epsilon is involved in the adenosine-activated signal transduction pathway conferring protection against ischemia-reperfusion injury in primary rat neuronal cultures. J Neurochem. 2003;84:409–412. doi: 10.1046/j.1471-4159.2003.01563.x. [DOI] [PubMed] [Google Scholar]
- Eddy SF, Morin P, Jr, Storey KB. Cloning and expression of PPAR-gamma and PGC-1alpha from the hibernating ground squirrel, Spermophilus tridecemlineatus. Mol Cell Biochem. 2005;269:175–182. doi: 10.1007/s11010-005-3459-4. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Li PA, Uchino H, Smith ML, Siesjo BK. Changes in the bioenergetic state of rat hippocampus during 2.5 min of ischemia, and prevention of cell damage by cyclosporin A in hyperglycemic subjects. Exp Brain Res. 1997;114:44–50. doi: 10.1007/pl00005622. [DOI] [PubMed] [Google Scholar]
- Frerichs KU, Kennedy C, Sokoloff L, Hallenbeck JM. Local cerebral blood flow during hibernation, a model of natural tolerance to "cerebral ischemia". J Cereb Blood Flow Metab. 1994;14:193–205. doi: 10.1038/jcbfm.1994.26. [DOI] [PubMed] [Google Scholar]
- Frerichs KU, Dienel GA, Cruz NF, Sokoloff L, Hallenbeck JM. Rates of glucose utilization in brain of active and hibernating ground squirrels. Am J Physiol. 1995;268:R445–R453. doi: 10.1152/ajpregu.1995.268.2.R445. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Fitzgibbons JC, Shah AR, Park TS. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett. 1994;168:221–224. doi: 10.1016/0304-3940(94)90455-3. [DOI] [PubMed] [Google Scholar]
- Gilmore RM, Stead LG. The role of hyperglycemia in acute ischemic stroke. Neurocrit Care. 2006;5:153–158. doi: 10.1385/ncc:5:2:153. [DOI] [PubMed] [Google Scholar]
- Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J Biol Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- Hochachka PW. Defense strategies against hypoxia and hypothermia. Science. 1986;231:234–241. doi: 10.1126/science.2417316. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- Hypothermia-after-Cardiac-Arrest-Study-Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Jones NM, Bergeron M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab. 2001;21:1105–1114. doi: 10.1097/00004647-200109000-00008. [DOI] [PubMed] [Google Scholar]
- Kaminogo M, Suyama K, Ichikura A, Onizuka M, Shibata S. Anoxic depolarization determines ischemic brain injury. Neurol Res. 1998;20:343–348. doi: 10.1080/01616412.1998.11740529. [DOI] [PubMed] [Google Scholar]
- Kim E, Raval AP, Defazio RA, Perez-Pinzon MA. Ischemic preconditioning via epsilon protein kinase C activation requires cyclooxygenase-2 activation in vitro. Neuroscience. 2007;145:931–941. doi: 10.1016/j.neuroscience.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause GS, Kumar K, White BC, Aust SD, Wiegenstein JG. Ischemia, resuscitation, and reperfusion: mechanisms of tissue injury and prospects for protection. Am Heart J. 1986;111:768–780. doi: 10.1016/0002-8703(86)90114-6. [DOI] [PubMed] [Google Scholar]
- Kurtz CC, Lindell SL, Mangino MJ, Carey HV. Hibernation confers resistance to intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2006;291:G895–G901. doi: 10.1152/ajpgi.00155.2006. [DOI] [PubMed] [Google Scholar]
- Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab. 2004;24:636–645. doi: 10.1097/01.WCB.0000121235.42748.BF. [DOI] [PubMed] [Google Scholar]
- Lee M, Choi I, Park K. Activation of stress signaling molecules in bat brain during arousal from hibernation. J Neurochem. 2002;82:867–873. doi: 10.1046/j.1471-4159.2002.01022.x. [DOI] [PubMed] [Google Scholar]
- Li J, Niu C, Han S, Zu P, Li H, Xu Q, Fang L. Identification of protein kinase C isoforms involved in cerebral hypoxic preconditioning of mice. Brain Res. 2005;1060:62–72. doi: 10.1016/j.brainres.2005.08.047. [DOI] [PubMed] [Google Scholar]
- Li PA, Kristian T, Shamloo M, Siesjo K. Effects of preischemic hyperglycemia on brain damage incurred by rats subjected to 2.5 or 5 minutes of forebrain ischemia. Stroke. 1996;27:1592–1601. doi: 10.1161/01.str.27.9.1592. discussion 1601-1592. [DOI] [PubMed] [Google Scholar]
- Long C, Gao Y, Gao G, Han S, Zu P, Fang L, Li J. Decreased phosphorylation and protein expression of ERK1/2 in the brain of hypoxic preconditioned mice. Neurosci Lett. 2006;397:307–312. doi: 10.1016/j.neulet.2005.12.045. [DOI] [PubMed] [Google Scholar]
- Lust WD, Wheaton AB, Feussner G, Passonneau J. Metabolism in the hamster brain during hibernation and arousal. Brain Res. 1989;489:12–20. doi: 10.1016/0006-8993(89)90003-6. [DOI] [PubMed] [Google Scholar]
- Lutz PL, Milton SL. Negotiating brain anoxia survival in the turtle. J Exp Biol. 2004;207:3141–3147. doi: 10.1242/jeb.01056. [DOI] [PubMed] [Google Scholar]
- Ma YL, Zhu X, Rivera PM, Toien O, Barnes BM, LaManna JC, Smith MA, Drew KL. Absence of cellular stress in brain after hypoxia induced by arousal from hibernation in Arctic ground squirrels. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1297–R1306. doi: 10.1152/ajpregu.00260.2005. [DOI] [PubMed] [Google Scholar]
- MacDonald JA, Storey KB. Regulation of ground squirrel Na+K+-ATPase activity by reversible phosphorylation during hibernation. Biochem Biophys Res Commun. 1999;254:424–429. doi: 10.1006/bbrc.1998.9960. [DOI] [PubMed] [Google Scholar]
- Mehrani H, Storey KB. Protein kinase C from bat brain: the enzyme from a hibernating mammal. Neurochem Int. 1997;31:139–150. doi: 10.1016/s0197-0186(96)00130-1. [DOI] [PubMed] [Google Scholar]
- Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport. 2001;12:1663–1669. doi: 10.1097/00001756-200106130-00030. [DOI] [PubMed] [Google Scholar]
- Milton SL, Prentice HM. Beyond anoxia: the physiology of metabolic downregulation and recovery in the anoxia-tolerant turtle. Comp Biochem Physiol A Mol Integr Physiol. 2007;147:277–290. doi: 10.1016/j.cbpa.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, Rea T, Lowe R, Brown T, Dreyer J, Davis D, Idris A, Stiell I. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300:1423–1431. doi: 10.1001/jama.300.12.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak G, Bakajsova D, Clifton GL. Protein kinase C-epsilon modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. Am J Physiol Renal Physiol. 2004;286:F307–F316. doi: 10.1152/ajprenal.00275.2003. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA. Mechanisms of neuroprotection during ischemic preconditioning: lessons from anoxic tolerance. Comp Biochem Physiol A Mol Integr Physiol. 2007;147:291–299. doi: 10.1016/j.cbpa.2006.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Mumford PL, Sick TJ. Prolonged anoxic depolarization exacerbates NADH hyperoxidation and promotes poor electrical recovery after anoxia in hippocampal slices. Brain Res. 1998;786:165–170. doi: 10.1016/s0006-8993(97)01438-8. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Born JG, Centeno JM. Calcium and increase excitability promote tolerance against anoxia in hippocampal slices. Brain Res. 1999;833:20–26. doi: 10.1016/s0006-8993(99)01462-6. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Dave KR, Raval AP. Role of reactive oxygen species and protein kinase C in ischemic tolerance in the brain. Antioxid Redox Signal. 2005;7:1150–1157. doi: 10.1089/ars.2005.7.1150. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:175–182. doi: 10.1097/00004647-199702000-00007. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Rosenthal M, Sick TJ, Lutz PL, Pablo J, Mash D. Downregulation of sodium channels during anoxia: a putative survival strategy of turtle brain. Am J Physiol. 1992;262:R712–R715. doi: 10.1152/ajpregu.1992.262.4.R712. [DOI] [PubMed] [Google Scholar]
- Plum F. What causes infarction in ischemic brain?: The Robert Wartenberg Lecture. Neurology. 1983;33:222–233. doi: 10.1212/wnl.33.2.222. [DOI] [PubMed] [Google Scholar]
- Prass K, Scharff A, Ruscher K, Lowl D, Muselmann C, Victorov I, Kapinya K, Dirnagl U, Meisel A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke. 2003;34:1981–1986. doi: 10.1161/01.STR.0000080381.76409.B2. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci. 2003;23:384–391. doi: 10.1523/JNEUROSCI.23-02-00384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Prado R, Katz LM, Busto R, Sick TJ, Ginsberg MD, Mochly-Rosen D, Perez-Pinzon MA. Protein kinase C delta cleavage initiates an aberrant signal transduction pathway after cardiac arrest and oxygen glucose deprivation. J Cereb Blood Flow Metab. 2005;25:730–741. doi: 10.1038/sj.jcbfm.9600071. [DOI] [PubMed] [Google Scholar]
- Ross AP, Christian SL, Zhao HW, Drew KL. Persistent tolerance to oxygen and nutrient deprivation and N-methyl-D-aspartate in cultured hippocampal slices from hibernating Arctic ground squirrel. J Cereb Blood Flow Metab. 2006;26:1148–1156. doi: 10.1038/sj.jcbfm.9600271. [DOI] [PubMed] [Google Scholar]
- Savitz SI, Fisher M. Prophylactic neuroprotection. Curr Drug Targets. 2007;8:846–849. doi: 10.2174/138945007781077382. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- Stuart G. Voltage-activated sodium channels amplify inhibition in neocortical pyramidal neurons. Nat Neurosci. 1999;2:144–150. doi: 10.1038/5698. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Namba K, Higuchi T, Hagioka S, Takata K, Hirakawa M, Morita K. Quantitative evaluation of the neuroprotective effects of hypothermia ranging from 34 degrees C to 31 degrees C on brain ischemia in gerbils and determination of the mechanism of neuroprotection. Crit Care Med. 2003;31:255–260. doi: 10.1097/00003246-200301000-00040. [DOI] [PubMed] [Google Scholar]
- Weltzin MM, Zhao HW, Drew KL, Bucci DJ. Arousal from hibernation alters contextual learning and memory. Behav Brain Res. 2006;167:128–133. doi: 10.1016/j.bbr.2005.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HW, Ross AP, Christian SL, Buchholz JN, Drew KL. Decreased NR1 phosphorylation and decreased NMDAR function in hibernating Arctic ground squirrels. J Neurosci Res. 2006;84:291–298. doi: 10.1002/jnr.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.