

To the Editor
In general, classic acrocephalosyndactyly syndromes are characterized by coronal craniosynostosis, particular hand and foot malformations, and facial dysmorphology including hypertelorism, down-slanting palpebral fissures, and midface hypoplasia. The most recently described of these syndromes is craniosynostosis, Philadelphia type, whose distinct clinical manifestations include sagittal craniosynostosis, rather than coronal craniosynostosis, and complete soft-tissue syndactyly of the fingers and toes (Fig. 1) with a relatively normal facial appearance [Robin et al., 1996]. Pedigree analysis of a five generation kindred indicates that the syndrome is inherited in an autosomal dominant fashion, with variable expression of the hand findings, and incomplete penetrance of the sagittal craniosynostosis [Robin et al., 1996]. Previous studies have excluded FGFR1 (implicated in Pfeiffer syndrome), FGFR2 (implicated in Pfeiffer, Apert, Crouzon, and Beare-Stevenson syndromes), FGFR3 (implicated in Muenke syndrome), and FGF8 where a rare 18 bp duplication of exon 1c was identified in two affected members of the family but did not segregate with the trait [Yoshiura et al., 1997; Muenke and Wilkie, 2001]. Without any evidence of previous mutations, we performed linkage analysis on a single family, the only described family with this disorder to date. Since craniosynostosis, Philadelphia type, is a clinically distinct craniosynostosis, with apparently a separate genetic etiology, characterizing the molecular basis for this disorder will point to a novel mechanism important in craniofacial and limb development. Specifically this would be the first gene that is involved primarily in sagittal craniosynostosis.
Fig. 1.

Hands and feet of an individual with craniosynostosis Philadelphia type.
We performed linkage analysis using microsatellite markers genotyped at deCODE genetics (1.5 cM average spacing, 2000 markers). For all markers, Mendelian errors were examined using PEDCHECK and when Mendelian errors were detected, the markers were removed from the analysis [O’Connell and Weeks, 1998]. Autosomal parametric and non-parametric linkage analysis was performed using GENEHUNTER-PLUS [Kruglyak et al., 1996; Kong and Cox, 1997]. An autosomal dominant model was tested with the allele frequency of the disease causing mutation set at 0.001. For regions of linkage haplotyping was performed using MERLIN’s-best function [Abecasis et al., 2002]. Haplotypes in regions of linkage were evaluated using HAPLOPAINTER in order to define minimal critical genomic intervals by examining recombination events [Thiele and Nurnberg, 2005].
Parametric linkage analysis identified four suggestive regions of linkage for craniosynostosis Philadelphia type on 2q35-36.3, 12q14.1-21.2, 20q11.2-13.13, and 22q12.1-13.1 with a LOD = 2.40 and multipoint NPL = 8.50. A review of the literature identified an overlapping locus for a phenotypically similar disorder, syndactyly type 1A or complete soft tissue syndactyly of digits two through five [Bosse et al., 2000; Ghadami et al., 2001]. Given the presence of complete soft tissue syndactyly as the major finding in craniosynostosis Philadelphia type, the phenotypic similarity and overlapping regions of linkage for the two disorders suggest they have the same etiology. Syndactyly type 1A has been demonstrated to be inherited in an autosomal dominant fashion with linkage to 2q35 in two families [Bosse et al., 2000; Ghadami et al., 2001]. The minimal critical interval for syndactyly 1A spans from D2S2319 to D2S344 (214.5–221.3 MB) [Bosse et al., 2000]. The overlapping minimal critical interval for craniosynostosis Philadelphia type spans from D2S2361 to D2S2297 (216.2–230.2 MB, Figs. 2 and 3). Thus, narrowing the interval using both phenotypes, gives a minimal critical interval spans from 216.2 to 221.3 MB on chromosome 2.
Fig. 2.
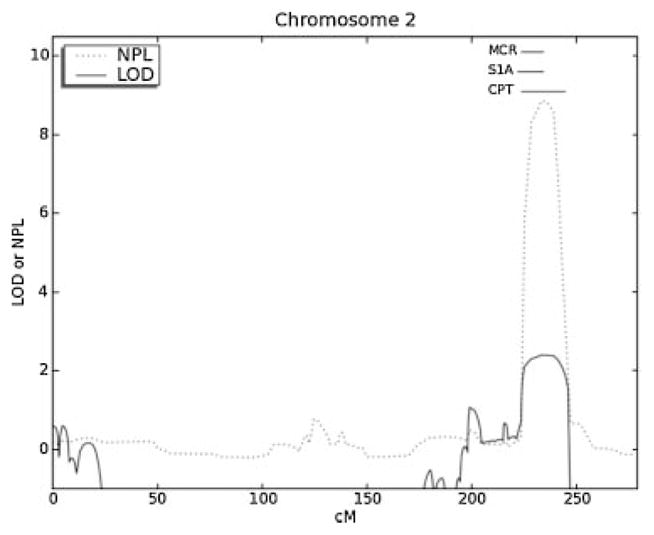
Linkage results on chromosome 2 indicate a region of linkage to chromosome 2q35-36.3 for craniosynostosis Philadelphia type (CPT; D2S2361 to D2S2297 or 216.2–230.2 MB). This overlaps with a previously described linkage peak for syndactyly 1A (S1A, D2S2319 to D2S344 or 214.5 MB to 221.3 MB) suggesting the disorders share the same etiology. Using information for both disorders narrows the minimal critical region of linkage (MCR) to 216.2–221.3 MB on chromosome 2.
Fig. 3.

Haplotype analysis in the studied pedigree demarcates a minimal critical interval for craniosynostosis Philadelphia type between D2S2361 and D2S2297.
This 5.1 MB interval contains 67 predicted or verified protein coding genes and three microRNAs. Transcripts were prioritized based on known function, expression and/or interactions in the literature. In addition we evaluated a published expression dataset consisting of murine forelimb RNA run on the Affymetrix Murnie Gene Chip (MOE-430) at embryonic day 12.5, just prior to the induction of apoptosis in the mouse inter-digital areas [Shou et al., 2005]. We sequenced coding exons of four protein coding genes (TMBIM1, RQCD1, RNF25, ATG9A) and one microRNA hairpin precursor (hsa-mir-153-1 predicted to regulate FGFR2) [Lewis et al., 2003, 2005] in two individuals with craniosynostosis Philadelphia type and two individuals with syndactyly 1A from the German and Iranian family, respectively. To date, there have been no mutations found co-segregating within families for this disorder; however, we have not excluded the involvement of mutations in untranslated regions or regulatory regions. Future studies will focus on other genes within the minimal critical interval including, IHH, IGFBP2, and IGFBP5.
Although initial linkage results identified four regions of linkage that were completely segregating with the trait, we chose to focus on chromosome 2q35 given the linkage to syndactyly type 1A [Bosse et al., 2000; Ghadami et al., 2001]. The similar limb finding in craniosynostosis, Philadelphia type is consistent with the notion that these disorders share a common etiology [Robin et al., 1996]. The assertion that these two disorders share a common genetic etiology could only be made after linkage analysis narrowed potential regions of linkage for craniosynostosis Philadelphia type.
Syndromic forms of craniosynostosis often manifest limb anomalies as associated findings [Wilkie et al., 2001]. One classic example is Apert syndrome, which is caused by mutations in FGFR2 and is characterized by coronal craniosynostosis and tissue and/or boney syndactyly of the hands and feet [Wilkie et al., 1995]. More recently, a specific mutation in FGFR3 has been shown to cause Muenke syndrome, which is characterized by coronal craniosynostosis and brachydactyly [Muenke et al., 1997]. The documented pleiotropic effect of genes involved in craniofacial and limb development [Wilkie et al., 2001], and the linkage evidence, strongly suggests that sagittal craniosynostosis and syndactyly 1A share a common genetic etiology.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Bosse K, Betz RC, Lee YA, Wienker TF, Reis A, Kleen H, Propping P, Cichon S, Nothen MM. Localization of a gene for syndactyly type 1 to chromosome 2q34-q36. Am J Hum Genet. 2000;67:492–497. doi: 10.1086/303028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadami M, Majidzadeh AK, Haerian BS, Damavandi E, Yamada K, Pasallar P, Najafi MT, Nishimura G, Tomita HA, Yoshiura KI, Niikawa N. Confirmation of genetic homogeneity of syndactyly type 1 in an Iranian family. Am J Med Genet. 2001;104:147–151. doi: 10.1002/ajmg.10061. [DOI] [PubMed] [Google Scholar]
- Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: A unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Muenke M, Wilkie AO. Craniosynostosis syndromes. In: Scriver CR, et al., editors. The metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill Companies, Inc; 2001. pp. 6117–6146. [Google Scholar]
- Muenke M, Gripp KW, McDonald-McGinn DM, Gaudenz K, Whitaker LA, Bartlett SP, Markowitz RI, Robin NH, Nwokoro N, Mulvihill JJ, Losken HW, Mulliken JB, Guttmacher AE, Wilroy RS, Clarke LA, Hollway G, Ades LC, Haan EA, Mulley JC, Cohen MM, Jr, Bellus GA, Francomano CA, Moloney DM, Wall SA, Wilkie AO. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet. 1997;60:555–564. [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. PedCheck: A program for identifying genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin NH, Segel B, Carpenter G, Muenke M. Craniosynostosis, Philadelphia type: A new autosomal dominant syndrome with sagittal craniosynostosis and syndactyly of the fingers and toes. Am J Med Genet. 1996;62:184–191. doi: 10.1002/(SICI)1096-8628(19960315)62:2<184::AID-AJMG13>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Shou S, Scott V, Reed C, Hitzemann R, Stadler HS. Transcriptome analysis of the murine forelimb and hindlimb autopod. Dev Dyn. 2005;234:74–89. doi: 10.1002/dvdy.20514. [DOI] [PubMed] [Google Scholar]
- Thiele H, Nurnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Oldridge M, Tang Z, Maxson RE., Jr Craniosynostosis and related limb anomalies. Novartis Found Symp. 2001;232:122–133. doi: 10.1002/0470846658.ch9. [DOI] [PubMed] [Google Scholar]
- Yoshiura K, Leysens NJ, Chang J, Ward D, Murray JC, Muenke M. Genomic structure, sequence, and mapping of human FGF8 with no evidence for its role in craniosynostosis/limb defect syndromes. Am J Med Genet. 1997;72:354–362. [PubMed] [Google Scholar]