

Abstract
Background
Prior loss-of-function analyses revealed that ATP8B1 (FIC1) post-translationally activated the Farnesoid X-Receptor (FXR).
Methods
Mechanisms underlying this regulation are elaborated upon by these gain-of-function studies in UPS cells, which lack endogenous FIC1 expression. FXR function was assayed in response to wild type and mutated FIC1 expression constructs using a human bile salt export pump (BSEP) promoter and a variety of cellular localization techniques.
Results
FIC1 overexpression led to enhanced phosphorylation and nuclear localization of FXR that was associated with FXR-dependent activation of the BSEP promoter. The FIC1 effect was lost after mutation of the FXR response element in the BSEP promoter. Despite similar levels of FIC1 protein expression, Byler-disease FIC1 mutants did not activate BSEP, while benign recurrent intrahepatic cholestasis mutants partially activated BSEP. The FIC1 effect was dependent upon the presence of the FXR ligand, chenodeoxycholic acid. The FIC1 effect on FXR phosphorylation and nuclear localization and its effects on BSEP promoter activity could be blocked with protein kinase C (PKC) ζ inhibitors (pseudosubstrate or siRNA silencing). Recombinant PKCζ directly phosphorylated immunoprecipitated FXR. Mutation of threonine 442 of FXR to alanine yielded a dominant negative protein, while the phosphomimetic conversion to glutamate resulted in FXR with enhanced activity and nuclear localization. Inhibition of PKCζ in Caco-2 cells resulted in activation of the human apical sodium dependent bile acid transporter promoter.
Conclusion
These results demonstrate that FIC1 signals to FXR via PKCζ. FIC1-related liver disease is likely related to downstream effects of FXR on bile acid homeostasis. BRIC emanates from a partially functional FIC1 protein. Phosphorylation of FXR is an important mechanism for regulating its activity.
Keywords: nuclear receptor, cholestasis, liver, ileum, bile acid
Mutations in ATP8B1 (Familial Intrahepatic Cholestasis 1, FIC1) lead to a spectrum of liver diseases (1–4). The more mild end of the spectrum of FIC1 disease is termed benign recurrent intrahepatic cholestasis (BRIC) (5), while the more severe disease is known as Byler disease or PFIC1 (6). The range of liver disease is presumed in large part to be related to the severity of the functional defect associated with the specific mutation in ATP8B1, although this has not been formally assessed (4). The liver disease may be accompanied by extrahepatic manifestations. These problems do not improve after liver transplantation; the diarrhea may worsen considerably and steatohepatitis may develop as a new problem after liver replacement (7). FIC1 is expressed broadly amongst tissues in the body, accounting in part for its varied extrahepatic manifestations (1, 8, 9).
The precise function of FIC1 and the pathophysiology of its variable disease manifestations are not well understood. Nucleotide homology analysis suggests that FIC1 could be a phospholipid flippase, potentially transferring aminophospholipids from the outer to inner hemi-leaflet of the lipid bilayer (1, 10). A chinese hamster ovary cell line that lacks FIC1 has impaired lipid transport capacity (8, 11). Expression of FIC1 in this cell line enhances phosphatidylserine transport (8, 12). Analysis of a limited number of human ileal tissue samples suggested that FIC1 might signal through the Farnesoid X-Receptor (FXR) (13). Confirmation of these findings using human liver tissue has been controversial and problematic due to the limited number of samples analyzed and the potential effects of the intrinsic liver disease on gene expression (14, 15). In vitro studies revealed that nuclear localization of FXR was diminished when FIC1 was knocked-down (13). Overexpression of FXR after FIC1 silencing did not rescue the effect, suggesting that post-transcriptional regulation was operative. FXR plays a key role in a variety of biologically important processes (16–23). FXR-mediated transcriptional effects are of fundamental relevance in bile acid homeostasis including regulation of ileal bile acid uptake by the apical sodium-dependent bile acid transporter (ASBT) and canalicular bile acid excretion via the bile salt excretory pump (BSEP) (24–29). The following studies were performed using a gain-of-function model to further assess the potential role that FIC1 may play in modifying FXR function.
EXPERIMENTAL PROCEDURES
Cells and Cell Culture
UPS cells (generously provided by Dr. Richard Pagano, Mayo Medical Center, Rochester, MN) were grown and maintained in Ham’s F-12 medium supplemented with 10% fetal calf serum (FCS). CV-1 (monkey kidney) (29), Caco-2 and HEK-293 cells (CRL-1573 ATCC, Rockville, MD) were grown and maintained in Dulbecco’s modified Eagle’s medium containing 10% FCS. UPS cells were cultured at 33°C, while CV-1 and HEK-293 cells were cultured at 37°C, both in 5% CO2. The effect of the FXR ligand, chenodeoxycholic acid (CDCA), was investigated by incubating cells in 0.5% charcoal treated fetal calf serum (CTFCS, Cocalico Biological, Inc, Reamstown PA) with or without additional CDCA. Concentrations of serum total bile acid (TBA), and the principal individual bile acids, chenodeoxycholic acid (CDCA), cholic acid (CA), deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA) were measured in undiluted FCS and CTFCS by stable-isotope dilution selected ion monitoring gas chromatography-mass spectrometry using previously described and validated methods (30–32).
Plasmid Constructs
231 base pairs of the BSEP promoter (−145 to +86) linked to a luciferase expression vector pSV0ALΔ5′ (p-145/Luc) (29) was used as a read-out of the FXR activity. For analyses in Caco-2 cells, the wild type human apical sodium dependent bile acid transporter promoter (hASBT) and the retinoic acid receptor cis-element mutant (hASBTμ) were utilized as previously described (33). pEF-FIC1, encoding a full length wild type human FIC1 coding region was used to express FIC1 in UPS cells (8). pEF-FIC1/G308V and pEF-FIC1/δ795-7 contained mutant FIC1 genes that have been characterized in the Byler and BRIC diseases, respectively (34). Four additional human FIC1 mutant constructs, pEF-FIC1/D554N (1660 G>A ccaggcagcctctcccaatgaaggtgccctgg – mutated nucleotide in bold italics), pEF-FIC1/G1040R (3118 G>A gcttgttgcatagggtcc) (both described in Byler disease), pEF-FIC1/R600W (1798 C>T ggacttcaacagtgactcgaagcg) and pEF-FIC1/R600Q (1799 G>A ggacttcaacagtgaccagaagcg) (both described in BRIC) (4) were prepared using the Quick-Change site directed mutagenesis kit (Stratagene, La Jolla, CA) (28). These specific mutations were chosen because their clinical phenotypes are relatively well-described. The point mutations were confirmed by DNA sequencing. FIC1 silencing was accomplished with a previously described construct, siFIC1, while a scrambled anti-sense construct, siScr, was utilized as a control (13).
A PKC ζ siRNA expression construct was synthesized (GenScript Corporation Piscataway, New Jersey) using PKC ζ cDNA sequence derived from UPS cells. The coding sequence for hamster PKC ζ cDNA was determined by RT/PCR of the hamster gene using the following oligonucleotide primers - PKC ζ 5′-ACCTCGTCCCGCTGACCTGCAGG-3′ and 5′-ACTCTGCCTCTGCATGTGGAAC-3′ (nucleotides 504-525 and 1000-1028 respectively, in the human PKC ζ gene, GenBank™ accession number Z15108 – hamster partial PKC ζ gene, GenBank™ accession number DQ913741). The entire region was utilized for northern analyses of the expression of PKCζ in UPS cells. The selected sequence for the PKC ζ anti-sense was 5′-AUGAUCAGAUCUAUGCCAUGA-3′. The siRNA expression construct was co-transfected into UPS cells along with BSEP-promoter luciferase construct and FIC1 expression plasmid as a measure of FIC1 activity. A scrambled RNA antisense construct was used as a negative control. FXR was silenced using a previously described construct (35).
A fragment of human FXR (hFXR) (accession number U68233), inserted into pCMX, (resulting in pCMX-hFXR) (24) was utilized in transfections to express FXR in CV-1 cells. Mutant pCMX-hFXR constructs were generated using QuickChange mutagenesis kit (Stratagene). The T442A mutant was synthesized by a point mutation, ACA --> GCA, using the following forward primer (mutated nucleotides in bold italics): 5 ′-cgcctgactgaattacgggcattcaatcatcacc-3′. The T442E mutant was synthesized by a point mutation from the T442 A mutant, ACA --> GAA, using the following forward primer, 5′-cgcctgactgaattacgggaattcaatcatcacc-3′.
Transient Transfection Assays
The procedures for transient transfection and luciferase analysis of cells were performed as described previously (36). Confluent cells (5×106)were transfected with 3μg of human BSEP-luciferase reporter and 0.3μg of a control plasmid (pRL-TK; Promega). Luciferase activities were determined by the dual luciferase reporter assay system (Promega) using a Turner 20/20 Luminometer (Turner BioSystems). All transfections were performed in triplicate and repeated in two separate sets of experiments.
FIC1 Anti-fusion Protein Antibodies
A fusion protein of human FIC1 (aa 748-851) coupled to maltose binding protein was generated by amplification of the corresponding cDNA from human intestinal cDNA and ligation into the vector pmalE C2 (New England Biolabs, Beverly, MA). Anti-FIC1 antibodies were generated in rabbits (Covance Research Products, Denver, PA). Antibodies were assessed by Western blotting of protein extracts from cells shown to either express or not express FIC1.
Protein Kinase Inhibitor Treatments
Staurosporine (100 nM) (37, 38), Bisindolylmaleimide I (10μM) (39, 40), Wortmannin (25 nM) (41), Protein Kinase C ζ Pseudosubstrate Inhibitor (PKC ζ PS) (Myristoylated) (100 μM) (39, 40), Gö6976 (10 μM) (39) were all purchased from EMD Biosciences, Inc (San Diego, CA). These inhibitors were added at the stated concentrations to the cells 38 hours after transfection, and the cells were harvested two hours after treatment was added to medium.
Generation of GFP Fused Human FXR (hFXR-GFP)
The full-length wild type hFXR cDNA was inserted downstream in-frame into the Bgl II and Xba I sites of an ecdysone-inducible expression vector, pIND (Invitrogen, Carlsbad, CA), containing the expression sequence of a green fluorescent protein (GFP), to produce the GFP-fused hFXR plasmid construct.
hFXR-GFP Transient Transfection and Fluorescence Microscopy
HEK-293 cells were transiently transfected with hFXR-GFP cDNA using Lipofectamine™ reagent (GIBCO/BRL) as describe previously (29). Briefly, cells were co-transfected for 3 hours with 0.5 μg/well of both hFXR-GFP and pVgRXR (Invitrogen) using Lipofectin at a DNA/lipid ratio of 1:3. After 48 hours growth in serum containing media cells were treated for 24 hours with inducer, 5μM Ponasterone A (Invitrogen) (dissolved in 100% ETOH). Cells were then untreated or treated with 100μM PKC ζ PS inhibitor for 4 hours before they were fixed for fluorescent microscopy of confluent cells.
Protein Preparation, Immunoprecipitation and Western Blotting
Cytoplasmic and nuclear extracts were prepared from Caco-2 and/or UPS cells co-transfected with FIC1 and/or siPKC ζ expression constructs using NE-PER™ (Pierce, Rockford, IL). Control untransfected cells were left in minimum essential medium. The immunoprecipitation method used was described by Anderson and Blobel (42). Total nuclear, cytoplasmic or FXR antibody immunoprecipitated proteins were analyzed by western blot analysis (36). FIC1 and FXR proteins were detected with the antibodies described above, while the IP FXR was blotted with an anti-phosphothreonine antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Sample loading was examined with a mouse monoclonal anti-actin or anti histone H1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Protein Kinase C Zeta Assay
Whole cell extracts were prepared from 5 × 107 UPS cells that were transfected with a wild type FXR construct and siPKCζ. FXR was immunoprecipitated and then treated directly with either purified recombinant PKCζ (Upstate, Temecula, CA) or bovine serum albumin as a negative control. Dephosphorylated myelin basic protein was used as a positive control for PKCζ mediated phosphorylation. Phosphorylation of either FXR or myelin basic protein was assessed by Western blot analysis with a phosphothreonine antibody.
Statistical Analysis
Data was expressed as the means ± SD from at least six experimental measurements. Differences between experimental groups were evaluated for statistical significance using Student’s t test, where p < 0.05 were considered to be statistically significant (GraphPad Software Inc., San Diego CA).
RESULTS
Gain of Function Studies in UPS Cells
UPS cells were selected because of their defective non-endocytic uptake of fluorescent phosphatidylserine analogs (11). UPS cells lack endogenous expression of FIC1, unlike the parent CHO cell line (8, 11). Western blot utilizing the FIC1 anti fusion antibody (Figure 1) detected a 140 kDa protein in wild type Caco-2 cells, wild type CHO cells and in UPS cells transfected with the wild type, G308V Byler and δ795-7 BRIC FIC1 constructs. The FIC1 antibody did not detect antigen in FIC1 antisense treated Caco-2 cells nor in untransfected UPS cells. Preincubation of the anti-fusion FIC1 antibody with GST-FIC1 fusion peptide abolished the antibody reactivity to FIC1 protein (data not shown). Comparison of the anti fusion FIC1 antibody with the antibody previously described by Ujhazy et al (8) revealed that proteins of identical mobility were detected by both antibodies (data not shown).
Figure 1. Western Blot of FIC1 Protein Expression in Whole Cellular Extracts.

FIC1 protein expression was detected by anti-fusion FIC1 antibody generated from immunization of rabbits as described in materials and methods. FIC1 protein was endogenously expressed in Caco-2 cells (lane 1) and in Chinese Hamster Ovary cells (wild type CHO in lane 3) and in UPS cells transfected with wild type FIC1 (lane 5), with G308V, a Byler’s construct (lane 6) and with δ795-7, a BRIC construct (lane 7). FIC1 protein expression was not found in Caco-2 cells treated with FIC1 anti-sense (lane 2) and in non-transfected UPS cells (lane 4).
The human BSEP promoter is activated by FXR and its activity is directly relevant to bile flow (29, 43, 44). As such, a human BSEP promoter fragment linked to a luciferase reporter gene (p-145/Luc) was used as a read-out of FXR activity. This BSEP promoter is known to be positively regulated by FXR, and contains one FXR cis element (IR-1 at bp −50/−63) (29). BSEP promoter activity was diminished by 72% in UPS cells in comparison to the parent CHO cells (UPS 100 ± 8; CHO 384 ± 22%, p < 0.0001). FXR and short heterodimer partner (SHP) protein expression were reduced in UPS cells compared to CHO cells (Figure 2). FIC1 transfection of UPS cells led to a 6-fold activation of the BSEP promoter, which was abrogated when the FXR-response element was mutated (Supplemental Table 1). None of the Byler disease constructs activated BSEP, while the BRIC disease constructs yielded a 2-fold activation (Supplemental Table 1 and Figure 3A). Equivalent amounts of the appropriate sized FIC1 protein were generated by each of the mutant constructs (Figure 3B). FXR and SHP protein expression were increased in cells transfected with wild type FIC1, while minimal induction of these proteins was seen after transfection with the Byler disease constructs (Figure 3B). Intermediate FXR and SHP protein induction was observed after transfection with the BRIC disease constructs. The effects of FIC1 were dependent upon the presence of either 10% fetal calf serum (FCS) or the addition of 50 μM CDCA to CTFCS (Figure 3C). Total bile acid concentration in FCS was 6.7 μM, while it was 0.7 μM in the CTFCS (Supplemental Table 2).
Figure 2. Western Blot of FXR Signaling in Whole Cellular Extracts.

There sets of UPS (U) or CHO (C) cells were analyzed by western blot of total cellular proteins. FIC1 protein was repeatedly not detectable in UPS cells. FXR and SHP protein expression were reduced in UPS cells relative to CHO cells. The actin signal shows equivalent loading.
Figure 3.



3A. Bile Salt Excretory Pump (BSEP) Promoter Activity in UPS Transfected Cells. UPS cells were transfected with wild type (wt) FIC1, or one of three Byler constructs (G308V, D554N and G1040R), or one of three BRIC constructs (R600W, ∂795-7 and R600Q). 6-fold activation of the BSEP promoter was observed with wt FIC1 compared to untransfected UPS cells. None of the Byler disease constructs activated the BSEP promoter, while all the BRIC disease constructs yielded a 2-fold activation (mean of 236%). The Renilla activity was the same in all transfected cells (not shown). 3B. Western analysis of protein expression after FIC1 transfection. Equivalent expression and size of the translated FIC1 protein is observed. FXR and SHP expression is minimal in the untransfected cells and those cells transfected with Byler disease constructs. Transfection with the BRIC disease constructs yield a level of expression of FXR and SHP which is intermediate between the Byler disease and wild type constructs. 3C. Analysis of the effect of bile acids on the activation of BSEP promoter activity by FIC1 in UPS cells. BSEP promoter activity is minimal in the cells cultured in 0.5% charcoal treated fetal calf serum (CTFCS). Promoter activity for the various FIC1 constructs was markedly increased in the presence of either 10% FCS or supplementation with 50 μM CDCA.
Effect of Protein Kinase Inhibitors on FIC1 Mediated Increase of BSEP Promoter Activity in UPS cells
PK and PKC inhibitors were tested on UPS cells co-transfected with FIC1 plasmid and the BSEP promoter as a read-out of FXR activity. All results are expressed relative to BSEP promoter activity in untreated UPS cells (Table 1). FIC1 activation of BSEP was inhibited by wortmannin, staurosporine, and Bisindolylmaleimide I but not Gö 6976. PKC ζ pseudosubstrate (PS), a more specific inhibitor of PKC ζ also inhibited FIC1 stimulation of BSEP activity (Table 1).
Table 1. Effect of protein kinase inhibition on FIC1 mediated activation of the BSEP promoter.
UPS cells were transfected with human BSEP promoter as a reporter for FXR activity. Activity is reported relative to BSEP promoter activity in UPS cells without co-transfection with the FIC1 expression construct (row 1 of the table). Various protein kinase inhibitors and the substrates of the inhibitor are displayed in the table. The Renilla activity was the same in all transfected cells (not shown).
| + FIC1 | INHIBITOR | SUBSTRATE | % BSEP PROMOTER ACTIVITY | |
|---|---|---|---|---|
| MEAN | STANDARD DEVIATION | |||
| no | None | None | 100 | 11 |
| yes | None | None | 560 | 74 |
| yes | Wortmannin | PI3 Kinase | 103 | 12 |
| yes | Staurosporine | General protein kinase | 93 | 9 |
| yes | Bisindolylmaleimide I | Protein kinase C | 97 | 10 |
| yes | Gö 6976 | Classical protein kinase C | 572 | 31 |
| yes | Myristoylated PKC ζ pseudosubstrate | PKC ζ | 91 | 44 |
| yes | PKC ζ siRNA | PKC ζ | 112 | 5 |
| yes | Scrambled siRNA | None | 605 | 39 |
siPKC ζ Effects on FIC1 Mediated Induction of Bsep Promoter Activity in UPS Cells
To determine the role of PKC ζ specifically, an siRNA hamster PKC ζ expression construct was synthesized using the hamster PKC ζ as template. PKC ζ mRNA was knocked-down by 87% in UPS cells after siRNA treatment (northern analysis of PKC ζ mRNA levels: control 9400 ± 300, siRNA 1000 ± 100, scrambled siRNA 9400 ± 100, p <0.0001 for siRNA vs either control or scrambled). siPKC ζ treatment yielded a reduction in basal BSEP promoter activity and FIC1 induction of BSEP activity was abrogated (Table 1).
Effect of PKCζ Inhibition on Human ASBT Promoter Activity
siRNA mediated silencing and pseudosubstrate inhibition led to a marked increase in human ASBT promoter activity in Caco-2 cells (Supplemental Table 3). Promoter activity was also enhanced after siFIC1 treatment. Basal activity of the retinoic acid receptor mutant promoter was reduced and this promoter construct was not activated after inhibition of PKCζ or silencing of FIC1.
Effects of FIC1 and PKC ζ Knock-down of FXR Phosphorylation and Localization
Under basal conditions, which included the bile acids found in FCS, total FXR and phosphorylated FXR were distributed between cytoplasm and nucleus (Figure 4A). FIC1 overexpression led to an increase in FXR phosphorylation and nuclear translocation. In contrast, FIC1 overexpression in the context of PKC ζ knock-down, showed a marked decrease in FXR protein phosphorylation and nuclear localization (Figure 4A).
Figure 4.

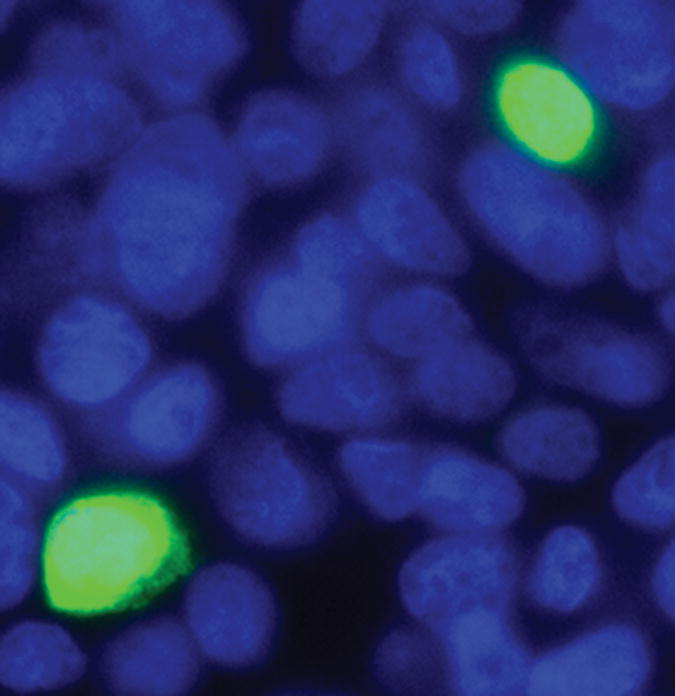
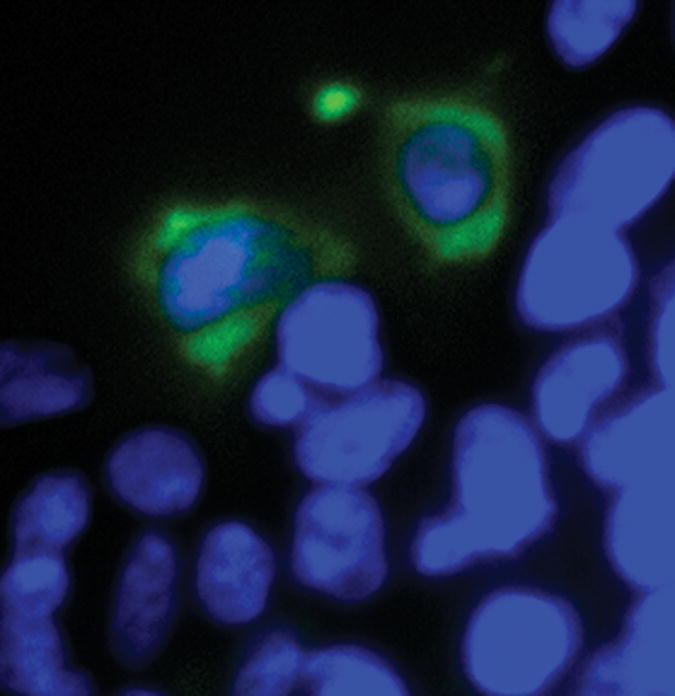
Figure 4A. Effect of siPKC ζ on nuclear localization and phosphorylation of FXR in UPS cells. UPS cells were transfected with a human FIC1 expression construct and an siPKC ζ antisense construct. Cytosolic and nuclear proteins were analyzed by western blot. Actin and histone H1 antibodies serve as loading controls for cytosolic and nuclear proteins, respectively. In the basal state (FIC1 −, siPKC ζ −) FXR is found in both the cytosol and nucleus. FIC1 transfection (FIC1 +, siPKC ζ −) led to nuclear localization of FXR, while siPKC ζ treatment of the FIC1 transfected cells (FIC1 +, siPKC ζ +) blocked translocation to the nucleus. FXR antibodies were used to immunoprecipitate FXR, which was then blotted for phosphothreonine (P-FXR). FXR phosphorylation is increased after FIC1 transfection, which is blocked by siPKC ζ co-treatment.
Figures 4B and C. Effects of PKC ζ pseudosubstrate inhibitor treatment on cellular distribution of FXR-GFP in transfected HEK-293 cells. HEK-293 cells were grown on glass coverslips and transiently transfected with hFXR-GFP cDNA. The transfected HEK-293 cells were untreated (top panel – Figure 4B) or treated (bottom panel – Figure 4C) with 100 μM PKC ζ pseudosubstrate inhibitor for 4 hours before they were fixed for fluorescent microscopy. The fluorescent pictures were merged from blue (DAPI stained nucleus) and green (FXR-GFP) images.
These findings were confirmed by transfecting a GFP-tagged FXR protein expression construct into HEK-293 cell, and treating these cells with or without the PKC ζ PS inhibitor. As shown in Figure 4B, in the cells untreated with the PKC ζ inhibitor, the vast majority of the GFP-tagged FXR is found in the nucleus. In contrast, addition of PKC ζ PS inhibitor redistributes the FXR to the cytoplasm (Figure 4C). PKC ζ PS inhibitor treatment did not lead to an alteration in Histone H1 distribution (data not shown). Treatment of immunopurified FXR with recombinant PKCζ resulted in threonine based phosphorylation (Figure 5).
Figure 5. PKCζ treatment of immunopurified FXR.

FXR was immunopurified from UPS cells that were transfected with both wild type FXR (to enhance expression) and siPKCζ (to minimize endogenous phosphorylation). FXR or myelin basic protein (MBP) were treated with PKCζ (+) or bovine serum albumin (−) and then were analyzed by western blot using a phosphothreonine antibody. PKCζ but not albumin treatment yielded phosphorylated FXR (P-FXR) or myelin basic protein (P-MBP).
Effects of T442 mutations on FXR function and localization
Site directed mutagenesis was performed to assess the importance of FXR phosphorylation. Studies were performed in CV-1 cells, which lack endogenous FXR, and readout of FXR activity was assessed using the BSEP promoter. T442, one of the predicted threonine phosphorylation sites, was chosen for initial analysis based upon the Scansite 2.0 algorithm (http://scansite.mit.edu/). Alanine substitution was used to prevent phosphorylation, while glutamate was used to simulate phosphorylation. T442A was associated with diminished baseline activity, while T442E was associated with enhanced activation of the BSEP promoter (Table 2). Activation of BSEP by T442E was dependent upon the presence of CDCA as a ligand. The wild type and two mutant FXR proteins were equally expressed in the CV-1 cells (Supplement Figure 1). Cellular localization of the mutated FXR constructs was performed by Western blot of cytoplasmic or nuclear proteins. The T442A mutant construct was localized to either the cytosol or nucleus in proportions similar to wild type FXR (Supplement Figure 2). In contrast, the T442E mutant was localized exclusively to the nucleus as might be predicted if phosphorylation of this residue controls nuclear localization (Figure 6).
Table 2. Effect of Threonine 442 Mutations on FXR Activity and Dependence Upon the FXR Ligand CDCA.
CV-1 cells were transfected with human BSEP promoter as a reporter for FXR activity. Activity is reported relative to BSEP promoter activity in CV-1 cells in 10% fetal calf serum with no exogenous FXR (row 1 of the table). The effects of various changes in media and FXR expression constructs are displayed in the table. The Renilla activity was the same in all transfected cells (not shown).
| FXR CONSTRUCT | MEDIA | % BSEP PROMOTER ACTIVITY | |
|---|---|---|---|
| mean | standard deviation | ||
| none | FCS | 100 | 17 |
| wild type | FCS | 603 | 110 |
| wild type | CTFCS | 118 | 22 |
| wild type | CTFCS + 100 μM CDCA | 545 | 81 |
| T442A | FCS | 26 | 3 |
| T442E | FCS | 1460 | 230 |
| T442E | CTFCS | 113 | 2 |
| T442E | CTFCS + 100 μM CDCA | 1212 | 87 |
Figure 6. Cellular localization of T442E FXR.

CV-1 cells were untransfected (n), transfected with a wild type FXR (wt) or a T442E FXR mutant (m+). FXR was found in both the cytoplasmic and nuclear extracts in wild type transfected cells, while FXR was found only in the nucleus in the T442E transfected cells.
DISCUSSION
Prior loss of function studies using siRNA-mediated knock-down of FIC1 in Caco-2 cells demonstrated diminished FXR function that was associated with reduction of its nuclear localization (13). Overexpression of FXR did not rescue the effect, suggesting that the effect was not simply on the basis of FXR abundance but instead involved posttranslational modification. The current studies extend these observations to a gain-of-function model, where exogenous overexpression of FIC1 results in enhanced FXR activity that is associated with its increased phosphorylation and nuclear localization. FXR-mediated downstream activation of the BSEP promoter is absent in cells lacking FIC1 (UPS), cells transfected with FIC1 constructs that harbor Byler-type mutations and in UPS cells co-transfected with wild type FIC1 and an FXR-response element mutated BSEP promoter. FXR activity is present in cells that endogenously express FIC1 and is reconstituted after FIC1 transfection. Interestingly, the BRIC-type mutant partially activated BSEP, indicating diminished but not absent functional activity of the FIC1 gene in BRIC. This novel functional assay of FIC1 activity corresponds with previously reported FIC1 disease phenotypes (1, 4).
The effects of FIC1 on FXR function leads to a plausible hypothesis of the pathophysiology of FIC1 related liver disease. Severe cholestasis may result from a combination of diminished canalicular excretion and enhanced intestinal reabsorption of bile acids (13). FXR transcriptionally activates BSEP, thus FIC1 deficiency would be expected to lead to reduced canalicular bile acid excretion. ASBT (SLC10A2) expression was increased in human ileal samples from patients with FIC1 disease (13). Negative feedback regulation of human ASBT by bile acids is mediated via the FXR-SHP cascade on the retinoic acid receptor response element (33). Thus in FIC1 deficiency there is reduced inhibition of ASBT resulting in induction of its expression and enhanced intestinal reclamation of bile acids. The more “benign” disease seen in BRIC appears to be related to incomplete inactivation of FIC1. The reason for the periodic nature of BRIC remains unknown, although this novel model system may be useful in examining the effects of exogenous factors on FIC1 activity.
FIC1 has been proposed and experimentally shown to function as an aminophospholipid flippase/transporter (1, 8, 12). Since phosphatidylserine may activate protein kinases initial experiments focused upon this signaling pathway (45, 46). PKC ζ was of particular interest since the atypical protein kinases can be activated directly by phosphatidylserine. PKC ζ is expressed in liver and has been implicated in mediating other bile acid related processes (39, 47, 48). PKCζ is downstream of phosphatidyl inositol 3-kinase (PI3K), and its activation depends of PI3K products (49) and phosphorylation by phosphoinositide-dependent protein kinase (PDK1) followed by auto-phosphorylation in some cells (50). Wortmannin (a general PI3K inhibitor), staurosporine (a general protein kinase inhibitor) and bisindolylmaleimide I (a protein kinase C inhibitor) all abrogated the activation of BSEP by FIC1 implicating the aforementioned signaling pathway. These inhibitors also reduced basal BSEP promoter activity, suggesting an element of basal induction of BSEP through this pathway. Gö 6976, a classical PKC inhibitor, had no effect on either basal activity or FIC1 mediated induction of BSEP, suggesting involvement of an atypical PKC. The specificity of the response via PKC ζ was determined using a PKC ζ pseudosubstrate and siRNA mediated silencing, both of which reduced basal BSEP promoter activity and eliminated activation by FIC1. In light of known nonspecific effects of PKC inhibitors, the siRNA studies provide the strongest specific support for the role of PKC ζ in mediating the FIC1 effect. Pseudosubstrate inhibition of PKC ζ markedly reduced nuclear targeting of a GFP tagged FXR molecule.
Human ASBT is under negative feedback regulation by bile acids primarily mediated by the FXR-SHP cascade acting upon the retinoic acid receptor (33). Inhibition of PKCζ in Caco-2 cells leads to activation of the human ASBT promoter in a manner akin to that observed with FIC1 silencing (13). This activation is dependent upon transcriptional effects mediated by the retinoic acid receptor cis element.
Recombinant PKC ζ directly phosphorylated FXR, supporting the potential functional importance of FXR phosphorylation in mediating the FIC1 effect. FIC1 activity, manipulated either by knock-down or exogenous overexpression, correlates positively with FXR phosphorylation and nuclear localization. There are multiple potential phosphorylation sites in the FXR protein. Initial studies focused on threonine sites as the immunoprecipitation studies were positive with a phosphothreonine antibody. The importance of FXR phosphorylation was modeled by site directed mutagenesis of FXR at one of the predicted threonine phosphorylation sites, T442. Alanine substitution, which prevents phosphorylation yielded a dominant negative protein. This protein could still translocate to the nucleus presumably related to phosphorylation of other sites in FXR (e.g. T219 – unpublished data). Glutamate substitution yielding a phosphomimetic amino acid change generated an activated FXR molecule with accentuated nuclear localization. Activation was dependent upon the presence of FXR ligand, although the exact mechanism of activation deserves further study. T442 is conserved amongst the rat, mouse and human FXR genes and has an intriguing pattern of conservation and substitution amongst other nuclear receptors (51). Its position in helix 11 underscores the potential critical relevance of this residue in conferring agonist or antagonist properties upon FXR (51–54).
In summary, these studies suggest that FIC1 function leads to activation of PKC ζ with subsequent phosphorylation, nuclear localization and activation of FXR. Future studies are needed to determine if there is a direct link between the aminophospholipid flippase activity of FIC1 and the observed changes in the PKC ζ activity and the phosphorylation status of FXR. Effects on downstream targets of FXR, including the canalicular bile salt transporter and the apical sodium dependent bile acid transporter, may underlie the cholestasis observed in individuals with FIC1 deficiency. Milder FIC1 related disease is associated with partial functionality of this protein. Post-translational modifications may be an important mechanism for regulation of FXR function.
Supplementary Material
Acknowledgments
Financial support: This work was supported in part by grants from the National Institutes of Health, DK 54165 and 69942 (to BLS) and HD 20632 (to FJS).
List of abbreviations
- FIC1
familial intrahepatic cholestasis 1
- BRIC
benign recurrent intrahepatic cholestasis
- PFIC1
progressive familial intrahepatic cholestasis 1
- PS
phosphatidyl serine
- FXR
farnesoid x receptor
- BSEP
bile salt export pump
- PKC
protein kinase C
- CHO
Chinese hamster ovary
- CDCA
chenodeoxycholic acid
- ASBT
apical sodium dependent bile acid transporter
- SHP
short heterodimer partner
- FCS
fetal calf serum
- CTFCS
charcoal treated fetal calf serum
References
- 1.Bull LN, van Eijk MJT, Pawlikowska L, DeYoung JA, Juiun J, Liao M, Klomp LWJ, et al. Identification of a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 2.van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001;21:535–544. doi: 10.1055/s-2001-19034. [DOI] [PubMed] [Google Scholar]
- 3.van Ooteghem NA, Klomp LW, van Berge-Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol. 2002;36:439–443. doi: 10.1016/s0168-8278(01)00299-9. [DOI] [PubMed] [Google Scholar]
- 4.Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, Juijn JA, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27–38. doi: 10.1002/hep.20285. [DOI] [PubMed] [Google Scholar]
- 5.Summerskill WHJ, Walshe JM. Benign recurrent intrahepatic “obstructive” jaundice. Lancet. 1959;II:686–690. doi: 10.1016/s0140-6736(59)92128-2. [DOI] [PubMed] [Google Scholar]
- 6.Clayton RJ, Iber FL, Ruebner BH, McKusick VA. Byler disease. Fatal familial intrahepatic cholestasis in an Amish kindred. J Pediatr. 1965;67:1025–1028. [PubMed] [Google Scholar]
- 7.Carlton VE, Pawlikowska L, Bull LN. Molecular basis of intrahepatic cholestasis. Ann Med. 2004;36:606–617. doi: 10.1080/07853890410018916. [DOI] [PubMed] [Google Scholar]
- 8.Ujhazy P, Ortiz D, Misra S, Li S, Moseley J, Jones H, Arias I. Familial intrahepatic cholestasis 1: Studies of localization and function. Hepatology. 2001;34:768–775. doi: 10.1053/jhep.2001.27663. [DOI] [PubMed] [Google Scholar]
- 9.van Mil SW, van Oort MM, van den Berg IE, Berger R, Houwen RH, Klomp LW. Fic1 is expressed at apical membranes of different epithelial cells in the digestive tract and is induced in the small intestine during postnatal development of mice. Pediatr Res. 2004;56:981–987. doi: 10.1203/01.PDR.0000145564.06791.D1. [DOI] [PubMed] [Google Scholar]
- 10.Tang X, Halleck MS, Schlegel RA, Williamson P. A subfamily of P-type ATPases with aminophospholipid transporting activity. Science. 1996;272:1495–1497. doi: 10.1126/science.272.5267.1495. [DOI] [PubMed] [Google Scholar]
- 11.Hanada K, Pagano RE. A Chinese hamster ovary cell mutant defective in the non-endocytic uptake of fluorescent analogs of phosphatidylserine: isolation using a cytosol acidification protocol. J Cell Biol. 1995;128:793–804. doi: 10.1083/jcb.128.5.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paulusma CC, Folmer DE, Ho-Mok KS, de Waart DR, Hilarius PM, Verhoeven AJ, Oude Elferink RP. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47:268–278. doi: 10.1002/hep.21950. [DOI] [PubMed] [Google Scholar]
- 13.Chen F, Ananthanarayanan M, Emre S, Neimark E, Bull LN, Knisely AS, Strautnieks SS, et al. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology. 2004;126:756–764. doi: 10.1053/j.gastro.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 14.Alvarez L, Jara P, Sanchez-Sabate E, Hierro L, Larrauri J, Diaz MC, Camarena C, et al. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004;13:2451–2460. doi: 10.1093/hmg/ddh261. [DOI] [PubMed] [Google Scholar]
- 15.Demeilliers C, Jacquemin E, Barbu V, Mergey M, Paye F, Fouassier L, Chignard N, et al. Altered hepatobiliary gene expressions in PFIC1: ATP8B1 gene defect is associated with CFTR downregulation. Hepatology. 2006;43:1125–1134. doi: 10.1002/hep.21160. [DOI] [PubMed] [Google Scholar]
- 16.Lambert G, Amar MJ, Guo G, Brewer HB, Jr, Gonzalez FJ, Sinal CJ. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem. 2003;278:2563–2570. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Binz J, Numerick MJ, Dennis S, Luo G, Desai B, MacKenzie KI, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest. 2003;112:1678–1687. doi: 10.1172/JCI18945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace JL, Miller MJ. Nitric oxide in mucosal defense: a little goes a long way. Gastroenterology. 2000;119:512–520. doi: 10.1053/gast.2000.9304. [DOI] [PubMed] [Google Scholar]
- 20.Biet F, Locht C, Kremer L. Immunoregulatory functions of interleukin 18 and its role in defense against bacterial pathogens. J Mol Med. 2002;80:147–162. doi: 10.1007/s00109-001-0307-1. [DOI] [PubMed] [Google Scholar]
- 21.Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, Dong B, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–236. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 22.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 23.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makishima M, Okamoto A, Repa J, Tu H, Learned R, Luk A, Hull M, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 25.Parks D, Blanchard S, Bledsoe R, Chandra G, Consler T, Kliewer S, Stimmel J, et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Chen J, Hollister K, Sowers L, Forman B. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Molecular Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 27.Sinal C, Tohkin M, Miyata M, Ward J, Lambert G, Gonzalez F. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 28.Chen F, Ma L, Dawson P, Sinal C, Sehayek E, Gonzalez F, Breslow J, et al. Liver receptor homologue-1 mediated species- and cell line-specific bile acid dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chemistry. 2003;278:19909–19916. doi: 10.1074/jbc.M207903200. [DOI] [PubMed] [Google Scholar]
- 29.Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf D, Suchy F. Human bile salt export pump promoter is transactivated by farsenoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. doi: 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- 30.Setchell KD, Matsui A. Serum bile acid analysis. Clin Chim Acta. 1983;127:1–17. doi: 10.1016/0009-8981(83)90070-0. [DOI] [PubMed] [Google Scholar]
- 31.Rodrigues CM, Setchell KD. Performance characteristics of reversed-phase bonded silica cartridges for serum bile acid extraction. Biomed Chromatogr. 1996;10:1–5. doi: 10.1002/(SICI)1099-0801(199601)10:1<1::AID-BMC536>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Lawson AM, Setchell KDR. Mass spectrometry of bile acids. In: setchell KDR, Kritchevsky D, Nair PP, editors. The bile acids, methods and applications. Vol. 4. New York: Plenum Press; 1988. pp. 167–267. [Google Scholar]
- 33.Neimark E, Chen F, Li X, Shneider BL. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40:149–156. doi: 10.1002/hep.20295. [DOI] [PubMed] [Google Scholar]
- 34.Harris MJ, Kagawa T, Dawson PA, Arias IM. Taurocholate transport by hepatic and intestinal bile acid transporters is independent of FIC1 overexpression in Madin-Darby canine kidney cells. J Gastroenterol Hepatol. 2004;19:819–825. doi: 10.1111/j.1440-1746.2004.03347.x. [DOI] [PubMed] [Google Scholar]
- 35.Frankenberg T, Rao A, Chen F, Haywood J, Shneider BL, Dawson PA. Regulation of the mouse organic solute transporter alpha-beta, Ostalpha-Ostbeta, by bile acids. Am J Physiol Gastrointest Liver Physiol. 2006;290:G912–922. doi: 10.1152/ajpgi.00479.2005. [DOI] [PubMed] [Google Scholar]
- 36.Chen F, Ma L, Al-Ansari N, Shneider B. The role of AP-1 in the transcriptional regulation of the rat apical sodium-dependent bile acid transporter. J Biol Chem. 2001;276:38703–38714. doi: 10.1074/jbc.M104511200. [DOI] [PubMed] [Google Scholar]
- 37.Simard M, Zhang W, Hinton DR, Chen TC, Weiss MH, Su YZ, Gopalakrishna R, et al. Tamoxifen-induced growth arrest and apoptosis in pituitary tumor cells in vitro via a protein kinase C-independent pathway. Cancer Lett. 2002;185:131–138. doi: 10.1016/s0304-3835(02)00261-6. [DOI] [PubMed] [Google Scholar]
- 38.Couldwell WT, Hinton DR, He S, Chen TC, Sebat I, Weiss MH, Law RE. Protein kinase C inhibitors induce apoptosis in human malignant glioma cell lines. FEBS Lett. 1994;345:43–46. doi: 10.1016/0014-5793(94)00415-3. [DOI] [PubMed] [Google Scholar]
- 39.McConkey M, Gillin H, Webster CR, Anwer MS. Cross-talk between protein kinases Czeta and B in cyclic AMP-mediated sodium taurocholate co-transporting polypeptide translocation in hepatocytes. J Biol Chem. 2004;279:20882–20888. doi: 10.1074/jbc.M309988200. [DOI] [PubMed] [Google Scholar]
- 40.Muscella A, Storelli C, Marsigliante S. Atypical PKC-zeta and PKC-iota mediate opposing effects on MCF-7 Na+/K+ATPase activity. J Cell Physiol. 2005;205:278–285. doi: 10.1002/jcp.20396. [DOI] [PubMed] [Google Scholar]
- 41.Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J. 1993;296 (Pt 2):297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson D, Blobel G. Immunoprecipitation of proteins from cell-free translations. Methods Enzymol. 1983;96:111–120. doi: 10.1016/s0076-6879(83)96012-3. [DOI] [PubMed] [Google Scholar]
- 43.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nature Genetics. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 44.Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, Hofmann AF, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of the mammalian liver. J Biol Chem. 1998;273:10046–10050. doi: 10.1074/jbc.273.16.10046. [DOI] [PubMed] [Google Scholar]
- 45.Ron D, Kazanietz MG. New insights into the regulation of protein kinase C and novel phorbol ester receptors. Faseb J. 1999;13:1658–1676. [PubMed] [Google Scholar]
- 46.Di Mari JF, Mifflin RC, Powell DW. The role of protein kinase C in gastrointestinal function and disease. Gastroenterology. 2005;128:2131–2146. doi: 10.1053/j.gastro.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 47.Rust C, Karnitz LM, Paya CV, Moscat J, Simari RD, Gores GJ. The bile acid taurochenodeoxycholate activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J Biol Chem. 2000;275:20210–20216. doi: 10.1074/jbc.M909992199. [DOI] [PubMed] [Google Scholar]
- 48.Sarkar S, Bananis E, Nath S, Anwer MS, Wolkoff AW, Murray JW. PKCzeta is required for microtubule-based motility of vesicles containing the ntcp transporter. Traffic. 2006;7:1078–1091. doi: 10.1111/j.1600-0854.2006.00447.x. [DOI] [PubMed] [Google Scholar]
- 49.Nakanishi H, Brewer KA, Exton JH. Activation of the zeta isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1993;268:13–16. [PubMed] [Google Scholar]
- 50.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mi LZ, Devarakonda S, Harp JM, Han Q, Pellicciari R, Willson TM, Khorasanizadeh S, et al. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol Cell. 2003;11:1093–1100. doi: 10.1016/s1097-2765(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 52.Nettles KW, Greene GL. Nuclear receptor ligands and cofactor recruitment: is there a coactivator “on deck”? Mol Cell. 2003;11:850–851. doi: 10.1016/s1097-2765(03)00133-3. [DOI] [PubMed] [Google Scholar]
- 53.Downes M, Verdecia MA, Roecker AJ, Hughes R, Hogenesch JB, Kast-Woelbern HR, Bowman ME, et al. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol Cell. 2003;11:1079–1092. doi: 10.1016/s1097-2765(03)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costantino G, Entrena-Guadix A, Macchiarulo A, Gioiello A, Pellicciari R. Molecular dynamics simulation of the ligand binding domain of farnesoid X receptor. Insights into helix-12 stability and coactivator peptide stabilization in response to agonist binding. J Med Chem. 2005;48:3251–3259. doi: 10.1021/jm049182o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.