

Abstract
The inflammatory response plays out over time in a reproducible and organized manner after an initiating stimulus. Here we showed that the genes activated in cultured mouse fibroblasts in response to the cytokine tumor necrosis factor (TNF) can be divided roughly into three groups, each with different induction kinetics. Whereas differential transcription is important in determining the grouping of these genes, differential mRNA stability also exerted a strong influence—in some cases overriding that of transcriptional control elements—on the temporal order of gene expression. mRNA transcripts expressed early have abundant AU-rich elements in their 3′-untranslated regions whereas those expressed later contain fewer. Thus mRNA stability and transcriptional control, two intrinsic characteristics of genes, control the kinetics of proinflammatory cytokine-induced gene expression.
Introduction
As long ago as the first century BC, the inflammatory process was recognized as evolving in stereotyped stages 1. No matter what invading organism or tissue injury incites the response, the temporal order of inflammatory events is quite similar. From a cellular point of view, neutrophils are the first to arrive at a site of inflammation and they stay for only a short time (first 24 hours). Monocytes arrive somewhat later but stay for days. Tissue remodeling processes take over during the late stages of inflammation 2.
There are two possibilities for how this stereotypical process could be orchestrated: each stage could induce the next or the temporal order of events could be `encoded' intrinsically in the genes involved in the inflammatory process. By analyzing events in cultured cells, we have come to realize that much of the time-course of the response program is encoded in the genes themselves. The sequential order of gene expression and the relative duration of the various inflammatory events appear to be pre-set as a part of the gene activation program. The process is likely the result of an interplay between elements regulating transcriptional induction, transcriptional repression and mRNA stability. Here we focus on the role of mRNA stability.
Tumor necrosis factor (TNF) (http://www.signaling-gateway.org/molecule/query?afcsid=A002291) is an important inflammatory cytokine involved in both acute and chronic inflammation 3. The effects of TNF are mediated by a group of transcription factors including NF-κB, AP-1 and interferon regulatory factors (IRFs). Among these, NF-κB is a key factor involved critically in most of the effects of TNF 4-6. Global gene activation induced by TNF and other proinflammatory cytokines6-9 has been studied extensively. These studies mainly focused on identifying genes induced in response to a particular stimulus 8, in a particular cell type 7, or during a particular disease 9. These genes have been grouped according to their functions 10, and transcriptional regulation modes 5-9,11. However, these studies did not concentrate on examining properties common to genes induced in a particular temporal order.
To understand the molecular processes underlying the temporal order of gene expression after TNF stimulation, we studied cultured fibroblasts, which are representative of cells at the site of an injury. At a site of injury, the resident as well as the incoming cells produce TNF 3; thus all cells in the vicinity are bathed in TNF throughout the response. We therefore examined cells continuously exposed to TNF. We found that the activated genes fell into three kinetic categories. Whereas transcriptional control was certainly a determinant of the kinetics of gene expression, stability of the mRNAs encoded by the three groups of genes played an unexpectedly key role.
Results
Temporal gene induction patterns in fibroblasts
We started our study by examining the timing of gene activation induced by TNF. To this end, we performed a microarray analysis of gene expression in mouse embryonic 3T3 fibroblasts at three times (0.5, 2, 12 hours) following addition of TNF. Using a criterion of a greater than 2-fold induction with a P < 0.01, approximately 250 genes were up-regulated or down-regulated at some or all of these time points (Fig.1a). The induced genes were clustered with 2D clustering wizard and the Agglomerative Clustering Algorithm 12 provided in the software. We then further investigated only those genes (180 genes) that were upregulated after TNF stimulation.
Figure 1.

TNF-activated genes fall into three kinetically different groups. (a) Microarray analysis of gene expression in 3T3 fibroblasts stimulated with recombinant mouse TNF (10 ng/ml) for 0.5, 2, and 12 hours. Activated genes, defined by an induction ratio equal to or greater than 2-fold (P < 0.01), are shown in green and were clustered with an Agglomerative Clustering Algorithm into three groups (I, II, III). Genes suppressed by TNF treatment are shown in red. The number of genes in each group is shown in parentheses. (b). Representative kinetics of expression of genes in each group.
As we had seen in an earlier, more limited study 5, the kinetics of gene induction by TNF varies considerably from gene to gene. However, within this larger set of upregulated genes, we could discern three broad classes of induction kinetics, each of which is most directly characterized by the time of peak mRNA expression. Group I mRNAs peaked at 0.5 hour, Group II at 2 hours and Group III at 12 hours (Fig. 1b). By examining more time points, individual variations could be found within the groups 5, but the representative kinetics of individual genes analyzed by RT-qPCR illustrated the strength of the generalization (Fig. 2 and Supplementary Fig. 1, online). These three groups could be called early, intermediate and late genes and, in fact, the Group I mRNAs predominated early, the Group II mRNAs predominated at around 2 hours and the Group III mRNAs predominated at later time points after TNF stimulation (data not shown and Supplementary Table 1).
Figure 2.

Gene expression after TNF withdrawal (a) 3T3 fibroblasts were exposed to TNF (10 ng/ml) continuously for 10 hours (TNF; red line), or were exposed to TNF for 6 hours, after which TNF was washed away (TNF withdrawal; blue line). Expression of genes in each group was measured by qRT-PCR at indicated time points. The data are mean ± s.d. of triplicate samples and are representative of three to six independent experiments with similar results. (b). 3T3 fibroblasts were stimulated with medium alone (Medium) or with TNF for 6 hours (TNF), or were exposed to TNF for 6 hours after which TNF was washed away and cells were incubated in medium for an additional 18 hours (TNF withdrawal). CCL2 and CCL5 protein in supernatants were measured by ELISA. Note that the production of CCL2 and CCL5 in medium alone was too low (approximately 20 pg/ml) to be shown and CCL5 production is delayed in kinetics and thus lower comparing with CCL2. The data are mean ± s.d. of triplicate samples and representative of three independent experiments with similar results.
We then examined whether the genes in one group share common molecular properties that distinguish them from genes in the other groups. We first extensively compared the function, subcellular localization, expression, and fold induction after TNF stimulation of each gene product None of these criteria mapped cleanly onto the groups (Supplementary Table 1). What accurately characterized the groups was solely the kinetics of induction. However, although we initially used the peak expression as the determining factor, we noted more similarities in the patterns of genes in each group (Fig. 1b). For example, Group I genes peak at 0.5 hours, and then quickly decrease in expression, often to base line or below, by 2 hours. The Group II mRNAs continued to increase in expression until 2 hours after TNF stimulation, and thereafter their expression was sustained; many genes were expressed in elevated amounts throughout the course of our observation (up to 24 hours). The mRNAs of Group III genes gradually increased in abundance but with delayed kinetics, often not reaching peak expression even at the end of a 24 hour observation period.
The expression patterns of these genes were reproducible in various different derivatives of 3T3 fibroblasts (with a few exceptions as discussed below). To compare a quite disparate cell, we studied human primary dermal fibroblasts and found similar patterns, with a few differences. For example, the basal amounts of many genes were low, and some Group I genes did not sharply go down after peaking at 0.5 hour (Supplementary Fig. 2).
Ccl20, Ccl2 and Mmp13 are three exceptions to these patterns. The Group II gene Ccl20 (and less frequently, Ccl2), peaked at 0.5 hour like a Group I gene in some experiments (Supplementary Figure 3 and data not shown). However their expression then remained elevated throughout the time course like Group II genes. Thus, these two genes may be borderline members of Group I and Group II. Similarly, the pattern of Mmp13 seemed to incorporate elements of Group II and Group III (data not shown). However, most genes fell consistently within one group.
Response to inducer removal
The patterns of mRNA accumulation are influenced by ongoing transcription and mRNA degradation. To distinguish the influence of each of these variables we first examined gene expression changes in response to TNF removal. We removed TNF at 6 hours after its addition and extensively washed the cells. Most Group I mRNA transcripts were expressed in such low amounts that removal of TNF had little discernable effect; detectable Group I mRNA transcripts rapidly returned to base line following TNF removal (Fig.2a). Accumulation of Group II mRNA stopped abruptly and quickly returned to base line (Fig. 2a). This suggested that the washing process was effective in removing TNF and that continual stimulation is essential for maintaining the sustained phase of these genes. In contrast, Group III mRNA transcripts were resistant to the withdrawal of TNF. Although further accumulation ceased, Group III mRNA transcript expression did not significantly decrease even 4 hours after removal of TNF (Fig. 2a). This result was further confirmed by studying additional group III genes in mouse fibroblasts and the same genes in human primary dermal fibroblasts. Expression of the Group III genes Ccl5 and C3 were still high after removal of TNF (Supplementary Fig. 2). Similarly, CCL5 protein was still robustly secreted after TNF removal, whereas CCL2, a protein encoded by a Group II gene, decreased in expression following TNF removal (Fig. 2b).
These results imply that the three groups of genes are controlled via quite different mechanisms. Group I genes are transcribed quickly after TNF addition but then turn off after half an hour. Group I mRNAs must therefore be very unstable, and their transcription must be inhibited because even in the presence of TNF, their expression fell to base line. Many but not all Group II genes were also induced quickly after TNF stimulation, but their accumulation continued until about 2 hours when it reached a plateau. As removal of TNF quickly reduced Group II mRNA expression at 6 hours, these mRNAs are continually transcribed but are also quite unstable. Group III genes expression was induced more slowly but once transcription was under way, these mRNAs are very stable.
One trivial explanation of the data might be that different genes react differently to a particular concentration of inducer. Thus we varied the concentration of TNF (Supplementary Fig. 3 and data not shown). We did see some prolongation of induction at higher concentrations but no change in the kinetic pattern of gene expression.
mRNA stability influences induction patterns
To directly investigate the role played by mRNA stability, we measured mRNA expession changes over time by RT-qPCR after blocking transcription with actinomycin D (ActD). We measured base line (uninduced) mRNA stability and mRNA stability after various times of induction, normalized to the relatively stable Rpl32 (L32) mRNA. It is well known that some Group I mRNA transcripts such as c-fos are very unstable 13. We tested 15 genes in Group I, and all behaved similarly (Fig. 3, Table 1 and data not shown). Group I mRNA transcript half lives varied from 0.2 to 0.9 hours, with some tendency to be shorter before or just after induction (Table 1). Group II mRNAs were more stable, with half lives varying from 1 to 8 hours or more. Again, the base line stability was somewhat less than the stability after induction. For the Group III genes, we could not measure a half life because we saw no consistent decrease in expression up to 4 hours after ActD treatment. The relative values of many Group III mRNA transcripts actually increased after normalizing to Rpl32 (Fig. 3). The concentration (10μg/ml) of ActD used here acts quickly and completely 14 so the reason for this latter observation may be that Rpl32 mRNA does show some instability and the Group III mRNAs are even more stable than Rpl32. Modulation of mRNA stability by extracellular signals does occur through the induction of Zfp36 (or Tristetraprolin, TTP) 15-18 and other factors 19. Zfp36 expression was indeed highly induced by TNF in 3T3 fibroblasts (Supplementary Fig. 1) and may be responsible for the small half life decrease of some Group I genes immediately after TNF stimulation. However, TNF did not significantly alter mRNA stability and the relative differences of mRNA stability between the three groups of genes appear to be intrinsic to the genes themselves.
Figure 3.
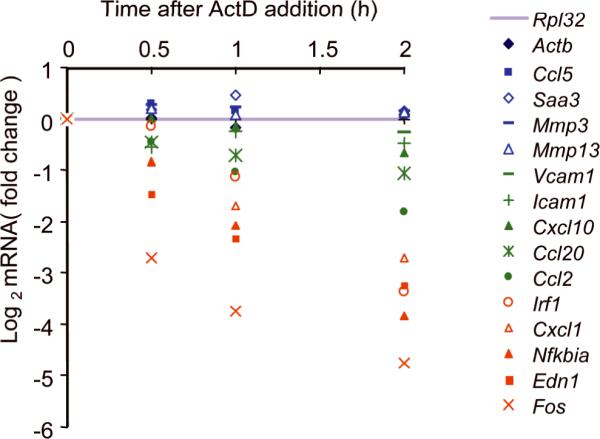
mRNA stability of genes in each group. mRNA transcripts encoding the indicated genes were measured by qRT-PCR at indicated times after addition of the transcription inhibitor actinomycin D (ActD) to 3T3 fibroblasts. The results are shown as a ratio to the base line value measured just before ActD addition (in log base 2 scale). Group I, II, and III genes are shown in red, green and blue, respectively. All values were normalized to Rpl32. The data are an average of triplicate samples with variations less than 20% and representative of three independent experiments with similar results.
Table 1.
mRNA half life in mouse cells
| Genes | AREs in 3'UTR | mRNA half life ( hour, mean ± s.d., n=3 ) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 3T3 | BMDM | ||||||||
|
|
|||||||||
| Basal | TNF (0.5h) | TNF (6h) | Basal | TNF (0.5h) | TNF (6h) | LPS (0.5h) | LPS(6h) | ||
| Group I | |||||||||
| Fos | 10 | 0.22 ± 0.04 | 0.17 ± 0.02 | TLTD | 0.14 ± 0.01 | 0.08 ± 0.02 | 0.16 ± 0.05 | 0.29 ± 0.02 | 0.20 ± 0.04 |
| Tnf | 10 | TLTD | TLTD | TLTD | 0.17 ± 0.05 | 0.11 ± 0.01 | 0.13 ± 0.08 | 1.45 ± 0.17 | 0.47 ± 0.04 |
| Zfp36 | 8 | 0.28 ± 0.03 | 0.18 ± 0.05 | 0.28 ± 0.02 | 0.23 ± 0.02 | 0.10 ± 0.01 | 0.20 ± 0.06 | 3.70 ± 0.10 | 0.45 ± 0.04 |
| Edn1 | 6 | 0.34 ± 0.12 | 0.31 ± 0.03 | 0.50 ± 0.10 | 0.46 ± 0.07 | 0.40 ± 0.11 | 0.38 ± 0.16 | stable | 0.31 ± 0.05 |
| Tnfaip3 | 6 | 0.28 ± 0.05 | 0.32 ± 0.08 | 0.32 ± 0.05 | 0.23 ± 0.05 | 0.12 ± 0.01 | 0.19 ± 0.07 | 0.55 ± 0.19 | 0.27 ± 0.04 |
| Nfkbia | 5 | 0.53 ± 0.06 | 0.48 ± 0.09 | 0.49 ± 0.12 | 0.28 ± 0.04 | 0.19 ± 0.02 | 0.21 ± 0.02 | 0.52 ± 0.12 | 0.29 ± 0.06 |
| Il1b | 5 | TLTD | TLTD | TLTD | 0.26 ± 0.10 | 0.27 ± 0.13 | 0.75 ± 1.17 | stable | stable |
| Cxcl1 | 7 | 0.64 ± 0.08 | 0.51 ± 0.07 | 0.94 ± 0.26 | 0.16 ± 0.06 | 0.15 ± 0.06 | 0.25 ± 0.10 | stable | 1.74 ± 1.11 |
| Cxcl2 | 7 | 0.54 ± 0.17 | 0.50 ± 0.11 | 0.68 ± 0.31 | 0.16 ± 0.04 | 0.17 ± 0.05 | 0.19 ± 0.01 | stable | 5.31 ± 4.36 |
| Irf1 | 4 | 0.74 ± 0.20 | 0.73 ± 0.09 | 0.68 ± 0.09 | 0.47 ± 0.12 | 0.40 ± 0.03 | 0.38 ± 0.09 | 4.16 ± 4.09 | 0.49 ± 0.11 |
| Group II | |||||||||
| Ccl20 | 4 | 1.46 ± 0.37 | 2.28 ± 0.93 | 2.78 ± 1.20 | TLTD | TLTD | TLTD | TLTD | TLTD |
| Ccl2 | 2 | 1.04 ± 0.08 | 1.37 ± 0.30 | 1.29 ± 0.26 | 0.39 ± 0.08 | 0.36 ± 0.04 | 0.61 ± 0.38 | stable | 1.09 ± 0.69 |
| Cxcl10 | 2 | 4.84 ± 2.59 | 5.06 ± 4.63 | 3.85 ± 0.31 | 0.77 ± 0.38 | 1.16 ± 0.70 | 1.03 ± 0.78 | 2.03 ± 1.83 | 2.06 ± 0.38 |
| Icam1 | 2 | 4.19 ± 1.90 | > 8.0 | 5.37 ± 1.83 | 1.52 ± 0.89 | 6.56 ± 1.33 | 2.94 ± 1.53 | stable | 6.25 ± 2.41 |
| Nfkbie | 1 | 4.48 ± 0.74 | > 8.0 | 2.88 ± 1.17 | |||||
| Vcam1 | 2 | 7.69 ± 1.04 | > 8.0 | > 8.0 | |||||
| Mmp10 | 1 | > 8.0 | > 8.0 | > 8.0 | |||||
| Group III | |||||||||
| Mmp13 | 2 | stable | stable | stable | 2.92 ± 2.56 | 5.71 ± 3.30 | stable | stable | stable |
| C3 | 0 | stable | stable | stable | |||||
| Mmp3 | 1 | stable | stable | stable | TLTD | TLTD | TLTD | TLTD | TLTD |
| Ccl5 | 0 | stable | stable | stable | stable | stable | stable | stable | stable |
| Saa3 | 0 | stable | stable | stable | stable | stable | stable | stable | stable |
| Serpina3g | 0 | stable | stable | stable | |||||
| House-keeping | |||||||||
| Actb | stable | stable | stable | ||||||
| Rpl32 | stable | stable | stable | ||||||
| Rpl13 | stable | stable | stable | ||||||
mRNA half lives are presented, determined as described in Methods. TLTD means too low to be determined
Although ActD may have side effects, the results from the ActD experiments strongly agree with those of the TNF withdrawal analysis (Fig. 2a, Supplementary Table 2). Jointly these results are very supportive of the view that mRNA stability is a key determinant controlling the kinetic patterns of TNF-induced gene expression.
Gene induction patterns in primary macrophages
To examine TNF-induced kinetic patterns of gene expression in another cell type, we examined mouse bone marrow-derived primary macrophages. Most genes activated by TNF in fibroblasts were also activated by TNF in macrophages; in addition, most activated genes fell into the same groups as in fibroblasts, when classified by kinetics and half-life. (Fig. 4a and Table 1).
Figure 4.

Kinetic patterns of gene expression in macrophages. Bone marrow-derived macrophages were stimulated with TNF (left) or LPS (right) for the indicated times and gene expression was measured by qRT-PCR. (a) The expression of one representative gene in each group is shown; additional genes are shown in Table 1. (b) Altered expression patterns of Ccl2 and Il1b induced by LPS compared to TNF. The data are mean ± s.d. of triplicate samples and representative of three independent experiments with similar results.
We noted a few outstanding exceptions. First, Ccl2, which in fibroblasts is borderline between Group I and Group II, showed clear Group I kinetics in macrophages (Fig. 4b). Mmp13 -- borderline between Group II and Group III in fibroblasts -- cleanly fell into Group II in macrophages (data not shown). In both cases these discrepancies were due to decreased half life of mRNA (Table 1). In fact, overall, mRNA transcripts in macrophages had shorter half lives than in fibroblasts (Table 1). Thus, relative mRNA stability appears to be a major determinant of activation kinetics in both fibroblasts and macrophages. Interestingly, although Tnf and Il1b were highly induced by TNF in macrophages, their induction was almost undetectable in fibroblasts (Table 1). This cell-specific difference may represent an important mechanism for rapid propagation of inflammatory signals; its basis is unclear.
LPS-induced mRNA stabilization
To examine whether another proinflammatory stimulus incites the same kinetic patterns of gene expression, we used the bacterial cell wall product lipopolysaccharide (LPS), which stabilizes Tnf and Il1b mRNA 20,21. Many genes were expressed with similar kinetics in TNF- and LPS-stimulated macrophages (Fig. 4a). However, LPS, unlike TNF, stabilized Tnf and Il1b mRNA (Table 1). Interestingly, LPS also stabilized mRNA transcripts encoding the neutrophil attracting chemokines CXCL1 and CXCL2 and the monocyte/macrophage chemokine CCL2, but had less effect on the T cell attracting chemokine CXCL10 (IP-10), Fos and IκBα (Table 1). The stabilization effect of LPS was transient, being stronger at 0.5 hour than at 6 hour (Table 1). However, gene activation induced by LPS involves complications not presented by TNF, possibly because LPS potentiates NF-κB activation 22 and induces production of autocrine factors such as TNF itself 23, and interferon-β 24. LPS-induced alterations in mRNA stabilization produced predictable effects on the overall induction kinetics of these mRNA transcripts (Fig. 4b and data not shown), further supporting the notion that mRNA stability exerts a strong influence over gene expression patterns.
Relative mRNA stability is encoded in the genome
The stability of mRNAs is at least partly determined by AU-rich elements (ARE) in the 3′UTR. The classical ARE sequence was originally recognized in the 3 ′UTR of Tnf 25,26, and Fos 13,27. Recently, other types of AU-rich elements have been found that are also involved in regulation of mRNA stability 19,28. To search widely for AU-rich elements, we used the sequence ATTT embedded in a string of at least 7 A and T residues as a core element to scan 3′UTRs in the mouse genome (www.ensembl.org). We found that the number of possible AREs is highest in the 3′UTRs of Group I mRNA transcripts (4-10), intermediate in Group II mRNA transcripts (2 to 4), and lowest in Group III mRNA transcripts (0 to 1) (Table 1). This finding strongly suggests that the relative stability of the mRNAs is determined by ARE content.
To examine directly whether the 3′ UTRs of these genes are the determining factor influencing their expression pattern, we created a series of GFP transgenes whose expression was controlled by either the Cxcl2 (Group I), Cxcl10 (Group II) or Ccl5 (Group III) promoter in combination with the 3′UTR of either Fos (Group I) or Ccl5 (Supplementary Fig. 4). As expected, GFP mRNA became unstable (although somewhat less unstable than actual Fos mRNA) when linked to the Fos 3′UTR, but remained stable with the 3′ UTR of Ccl5; these differences in stability were maintained independently of the promoter linked to the GFP transgene (Fig. 5a and data not shown). Basal amounts of transgenes linked to Group III 3′UTRs were high, presumably because the control mechanisms that ordinarily keep Group III genes repressed are not effective within the transgene context. The 3′ UTR of Fos resulted in Group I kinetics, regardless of whether it was linked to a Group I, II or III promoter; GFP transgenes linked to the 3′ UTR of Ccl5 showed Group III kinetics, even when driven by Group I and Group II promoters (Fig. 5b). These results strongly confirm that the 3′UTR is the main determining factor of gene expression patterns in this system.
Figure 5.

Gene expression patterns are determined by the 3′UTR of a gene. (a) 3T3 fibroblasts were infected with lentiviruses encoding GFP transgenes linked to the promoter of Cxcl10 and the 3′UTR of Ccl5 or Fos. Cells were pre-treated with TNF for 30 minutes before the addition of actinomycin D (ActD). mRNA transcripts encoding GFP, and endogenous Fos and Rpl32, were measured by qRT-PCR at indicated times after addition of ActD. The data are an average of triplicate samples with variations less than 20% and representative of three independent experiments. (b) 3T3 fibroblasts infected with the indicated lentiviruses were stimulated with TNF (20ng/ml). GFP expression was measured by qRT-PCR at indicated time points after the addition of TNF. The data are mean ± s.d. of triplicate samples and representative of three independent experiments with similar results.
Discussion
It is puzzling that a single cytokine, TNF, is able to induce a complex, dynamic process such as the inflammatory response. It is even more puzzling that TNF does this largely through activation of NF-κB; the effects of TNF are severely blunted in cells lacking NF-κB 4-6. A highly integrated set of events must ensue after the activation of the TNF receptor, and these events must influence a wide range of genes that are `programmed' to react to receptor activation in varied ways. We have been trying to understand these events and the genes they influence 4,5,23,29,30. Here we show how the genes that are activated by continuous exposure to TNF are expressed in patterns determined to a major extent by the stability of the mRNA encoded by each gene.
We classified TNF-induced genes into three groups according to their kinetics of expression. mRNA transcripts in each group showed distinct stability. Group I mRNA genes encoded highly unstable mRNA transcripts; thus when the transcription of these genes is reduced as it is within the first hour after TNF treatment, the amounts of the corresponding mRNA transcripts fall rapidly. The rapid decrease of Group I gene transcription after 30 minutes of TNF stimulation is most likely due to post-induction transcriptional suppression, because it correlates temporally with the synthesis of some negative regulators, such as IκBα and A20, and a sharp decrease in IKK activity 31-34. The great instability of Group I mRNA transcripts was not conferred on them by TNF treatment, as they were equally unstable in TNF-induced and resting cells. Thus instability is an intrinsic property of the gene structure. Instability correlated with a large number of AU-rich tracts, which are known to destabilize mRNA. Furthermore, a Group I 3′UTR appended to a transgene linked to a Group II or III promoter resulted in adoption of a Group I accumulation pattern.
Group II mRNA transcripts are more stable and contain fewer AU-rich tracts. Their transcription does not decrease as abruptly as that of the Group I genes. The combination of continued transcription and moderate stability means that their mRNAs plateau in amounts distinctly higher than amounts in resting cells.
Group III genes are the most remarkable. Their mRNA, whether in induced cells or uninduced cells, is so stable that over a 4 hour period we could not discern any instability. Group III mRNA transcript 3′ UTRs contain virtually no AU-rich regions. Their transcription is delayed but once started causes an accumulation that shows no drop, even over 12 hours. The 3′UTR from a Group III gene completely converted the accumulation pattern of a transgene linked to a Group I or II promoter to that of a Group III mRNA.
Although all of the Group III mRNAs showed the same high stability, by another criterion Group III genes are quite different. Some, like Ccl5, are induced with the same kinetics regardless whether or not cells are treated with cycloheximide to stop protein synthesis at the time of induction (35 and our unpublished data). Others are blocked by cycloheximide indicating that their induction requires a new protein to be made. Other investigators have considered this a key characteristic with which to categorize TNF- or growth factor-induced genes 35,36.
Thus the behavior of the three groups of genes can be explained in terms of three parameters: induction of gene transcription, secondary induced transcriptional silencing and intrinsic stability. These parameters interact to lead to three phases of the inflammatory response to TNF. An early phase is dominated by Group I genes, a middle phase is dominated by Group II genes and, during a late phase, Group III genes become increasingly important. This response is largely encoded in the genome and therefore its temporal order is virtually unaltered in mouse versus human cells or fibroblasts versus macrophages. While the patterns within a particular group are quite stereotyped, the amounts of the mRNAs vary enormously (see the scales on the various Figures), the fold-inductions vary significantly and the genes in each groups exert a wide range of functions.
Gene activation kinetics induced by various stimuli have been described previously. Ten different gene activation patterns were identified in an early study of human primary fibroblasts responding to serum treatment 37. More recently, activation patterns were recorded for cells treated with growth factors 18, LPS 24,38 or TNF 35. These studies were mainly focused on analysis of transcriptional regulation modes for these patterns. For example, Yarilina et al. 35 described a similar three-class activation pattern in macrophages stimulated by TNF to what we have described here. However, their grouping criterion was based on the distinct behaviors of gene activation in the presence of cycloheximide, and thus their gene classifications differed somewhat from ours. For example, they grouped Ccl5 and Ccl10 together but we grouped them separately, a grouping that is justified by their different mRNA stabilities. In addition, they did not study an early time point (0.5h), and therefore did not put the highly unstable Cxcl1 in the early category. However, the major difference from our work is our focus on the common characteristic of mRNA stability underlying the groupings.
Another important perspective can be derived from our work. The Group I and II genes, being induced rapidly after TNF addition, will be induced even by a short pulse of inducer. A pulse as short as 15 minutes will lead to full induction of a Group I gene (30 and data not shown). This means that the response to a transient stimulus will be qualitatively different than that to a prolonged stimulus. There may be an evolutionary importance to this phenomenon, as it may allow minor stimuli to elicit only an early, neutrophil-dominated response. The transcription factors encoded by Group I genes (e.g. Irf1, Fos, Junb) will activate pathways that involve other genes, so the response to a pulse could nevertheless constitute a rich program of events. It is significant that Fos is a potential oncogene. Restricting Group I expression to a short pulse may be a mechanism to reduce the time window during which oncogenes could initiate cellular transformation 39,40. It is notable that although we grouped the genes entirely on the basis of their expression kinetics, many of these genes have functional roles that predominate during the temporal phase associated with the group in which we classified them. Group I genes such as Cxcl1 and Cxcl2 encode neutrophil chemoattractants that can initiate the response to infection. Some chemokines encoded by Group II genes, such as Ccl2 41 and Ccl7 42, recruit monocytes, which become macrophages and ingest foreign matter and dying cells. Other Group II genes, Vcam1 43 and Csf1 44, help to tether these cells in the inflamed tissue and drive their differentiation into macrophages. Similarly, a number of tissue remodeling and wound repairing genes are clustered in Group III (e.g. those encoding MMP3, MMP13, PEDF 45, and CyCAP 46). Group III gene products MMP3 and MMP13 also play a role in inactivating the chemokines MCP-1 47 and MCP-3 48, which are encoded by Group II genes. In this way, the temporal ordering also facilitates switching from recruiting monocytes to the ensuing tissue remodeling/healing process. This correlation between gene expression kinetics and function suggests that the timing of many of the events of inflammation is built into the sequence of the genes that control these events.
In this study, we focused on fibroblasts, which support tissues and are actively involved in the evolution of the inflammatory response in tissues. Fibroblasts produce chemokines and cytokines in a pre-programmed fashion when stimulated by the TNF released locally from resident hematopoietic cells like mast cells or macrophages that have encountered stimuli such as LPS. Several genes (e.g. Mmp3 and Ccl20) are expressed in fibroblasts but not in macrophages, supporting the notion that they play necessary roles in inflammation. MMP3 modulates the extracellular matrix and inactivates some chemokines 47,48. CCL20 is a very potent chemoattractant for immature dendritic cells 49. Fibroblasts have other features that make them a good inflammatory model. In macrophages, TNF can induce the release of TNF itself and IL-1β. Although an important mechanism of rapidly propagating inflammatory signals, this secondary TNF and IL-1β production can obscure analysis of gene expression induced by a primary TNF stimulus. Fibroblasts, in contrast, do not express TNF or IL-1β in response to TNF. This property facilitated the global analysis of gene activation profiles in this study.
Methods
Reagents
Recombinant human and mouse TNF and M-CSF were from R&D Systems. E. Coli LPS (Cat#: L6529) and Actinomycin D (A9415) were from Sigma-Aldrich.
Cell culture and mice
Mouse embryonic immortal 3T3 fibroblast cell lines were generated as previously described 5 and maintained at less than 90% confluence in DMEM medium supplemented with 10% bovine calf serum (Hyclone) and penicillin and streptomycin. For experiments, cells were cultured for 24 hours in low density with normal culture medium and were then maintained overnight with 0.5% serum before being stimulated with recombinant mouse TNF for indicated time periods. The cells were then rapidly harvested for RNA extraction. The cell medium was also saved for ELISA. Human dermal fibroblasts-adult (Cat# 2320) were purchased from ScienCell Research Laboratories and cultured in complete Fibroblast Medium (Cat# 2310) from the same company, as suggested by the manufacturer. The cells were seeded in low density, incubated with a 1:10 dilution of culture medium with basal medium and stimulated similarly to the 3T3 fibroblasts. Mouse bone marrow-derived macrophages were obtained as described 50 from C57Bl/6 mice (from the Jackson Laboratory) and were maintained in DMEM supplemented with 10% FBS (Hyclone) and 20 ng/ml mouse M-CSF for 7 days. The medium was changed to fresh culture medium without M-CSF two hours before addition of stimulants.
GFP transgenes
The endogenous promoters of Cxcl2 (-1043 to +67 relative to transcription start site), Cxcl10 (-1000, +66), and Ccl5 (-1997,+48) were amplified by PCR using C57Bl/6 mouse genomic DNA as the template and subcloned into the third generation lentiviral vector pHAGE2-CMV-eGFP-W (A.B. Balazs) by replacing the original CMV promoter located between SpeI and NotI sites. The full length 3′UTRs of Fos and Ccl5 were cloned into the same vector after the stop codon of GFP by replacing a DNA region including the WPRE element between the BamHI and Acc65I sites. The new constructs were confirmed by functional assays (e.g. TNF inducibility) and DNA sequencing. These GFP constructs were stably integrated into the genome of polyclonal mouse 3T3 fibroblasts by infection with lentiviruses carrying the constructs. Briefly, the lentiviruses were made by co-expression of the lentiviral construct with packaging vectors (Tat, Rev, Gag/pol, VSV-G) in 293T cells and collection of the medium at 36 to 72 hours post transfection. The fresh virus-containing medium mixed with fresh medium (1:1) was added to the sparsely seeded 3T3 cells in the presence of polybrene (Chemicon, Cat#TR-1003-G, final conc. 10μg/ml) and incubated for 12 hours at 37°C before changing to fresh medium for an additional three days. The efficiency of viral infection was estimated by the fraction of cells expressing GFP and was about 50-80%.
mRNA analysis
For quantitative real time PCR, DNA-free RNA was extracted with RNeasy Mini kit (Qiagen) followed by DNase treatment. 0.4 μg total RNA was reverse transcribed using Invitrogen SuperScript II first-strand synthesis kit. qPCR was performed using SYBR GREEN PCR Master Mix (Applied Biosystems) and a 7300 Real-Time PCR machine. Samples were performed in triplicates. The results were normalized to Rpl32 (2-16 of the Rpl32 mRNA defined as 1 unit of activity). The sequences of most of primers were from Primer Bank (http://pga.mgh.harvard.edu/primerbank/index.html) and are listed in supplementary Table 3.
mRNA stability
10μg/ml actinomycin D (ActD) was directly added to cell cultures that were already treated with TNF or LPS for the indicated times without removing the original stimulant. The cells were harvested 0.5, 1, 2 and 4 hours after addition of ActD. mRNA amounts were measured by qRT-PCR as described above and normalized to Rpl32 before calculation of half lives. Because of the non-linearity of mRNA degradation kinetics, the half lives of labile mRNA (Group I mRNAs and most Group II mRNAs) were determined by a short treatment of Act D (0.5h), whereas the half lives of more stable mRNA (group III mRNAs and part of Group II mRNAs) were determined by longer ActD treatment (2 or 4 hours) for better estimation.
ELISA
Supernatants were collected from cell cultures at various times after TNF stimulation. The antibodies and recombinant proteins were from R&D Systems. ELISAs were done using R&D Systems ELISA kit as recommended by the manufacturer.
Microarray
For microarray hybridization, 2 μg RNA was labeled, fragmented and hybridized to an Affymetrix Mouse Genome 430 2.0 Array. After the arrays were scanned, the expression value for each gene was analyzed with Rosetta Resolver 7.1 Gene Expression Data Analysis System (Rosetta Inpharmatics LLC). Only those genes whose absolute fold change over a non-treatment control was equal to or greater than 2.0 and P < 0.01 were clustered using 2D Clustering Wizard and Agglomerative Hierarchical Cluster Algorithm. Microarray data were deposited in the GEO database under accession number GSE14071.
Supplementary Material
Acknowledgements
We thank M. Boldin, R. O'Connell, X. Luo, and K. Taganov for valuable comments on the manuscript and A. Balazs for kindly providing with pHAGE2-CMV-eGFP-W plasmid. This work was supported by a grant from National Institutes of Health to D.B (2R01GM039458). The microarray analysis was supported by the Millard and Muriel Jacobs Genetics and Genomics Laboratory at California Institute of Technology.
References
- 1.Majno G. The Healing Hand: Man and Wound in the Ancient World. Harvard University Press; Cambridge: 1975. [Google Scholar]
- 2.Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009–17. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- 3.Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149–60. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 4.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–4. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. Embo J. 2003;22:5530–9. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. 2005;280:17435–48. doi: 10.1074/jbc.M500437200. [DOI] [PubMed] [Google Scholar]
- 7.Viemann D, et al. TNF induces distinct gene expression programs in microvascular and macrovascular human endothelial cells. J Leukoc Biol. 2006;80:174–85. doi: 10.1189/jlb.0905530. [DOI] [PubMed] [Google Scholar]
- 8.Zhao B, Stavchansky SA, Bowden RA, Bowman PD. Effect of interleukin-1beta and tumor necrosis factor-alpha on gene expression in human endothelial cells. Am J Physiol Cell Physiol. 2003;284:C1577–83. doi: 10.1152/ajpcell.00243.2002. [DOI] [PubMed] [Google Scholar]
- 9.Schwamborn J, et al. Microarray analysis of tumor necrosis factor alpha induced gene expression in U373 human glioblastoma cells. BMC Genomics. 2003;4:46. doi: 10.1186/1471-2164-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 11.Li X, et al. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 2002;277:45129–40. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartigan JA. Clustering Algorithm. John Wiley and Sons; New Yock: 1975. [Google Scholar]
- 13.Wilson T, Treisman R. Removal of poly(A) and consequent degradation of c-fos mRNA facilitated by 3' AU-rich sequences. Nature. 1988;336:396–9. doi: 10.1038/336396a0. [DOI] [PubMed] [Google Scholar]
- 14.Franklin RM, Baltimore D. Patterns of macromolecular synthesis in normal and virus-infected mammalian cells. Cold Spring Harb Symp Quant Biol. 1962;27:175–98. doi: 10.1101/sqb.1962.027.001.019. [DOI] [PubMed] [Google Scholar]
- 15.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–5. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 16.Lai WS, et al. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol. 1999;19:4311–23. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauer I, et al. Interferons limit inflammatory responses by induction of tristetraprolin. Blood. 2006;107:4790–7. doi: 10.1182/blood-2005-07-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amit I, et al. A module of negative feedback regulators defines growth factor signaling. Nat Genet. 2007;39:503–12. doi: 10.1038/ng1987. [DOI] [PubMed] [Google Scholar]
- 19.Shim J, Karin M. The control of mRNA stability in response to extracellular stimuli. Mol Cells. 2002;14:323–31. [PubMed] [Google Scholar]
- 20.Han J, Brown T, Beutler B. Endotoxin-responsive sequences control cachectin/tumor necrosis factor biosynthesis at the translational level. J Exp Med. 1990;171:465–75. doi: 10.1084/jem.171.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YL, et al. Differential regulation of ARE-mediated TNFalpha and IL-1beta mRNA stability by lipopolysaccharide in RAW264.7 cells. Biochem Biophys Res Commun. 2006;346:160–8. doi: 10.1016/j.bbrc.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 22.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–61. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 23.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–7. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 24.Doyle S, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–63. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 25.Beutler B, Krochin N, Milsark IW, Luedke C, Cerami A. Control of cachectin (tumor necrosis factor) synthesis: mechanisms of endotoxin resistance. Science. 1986;232:977–80. doi: 10.1126/science.3754653. [DOI] [PubMed] [Google Scholar]
- 26.Caput D, et al. Identification of a common nucleotide sequence in the 3'-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci U S A. 1986;83:1670–4. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zubiaga AM, Belasco JG, Greenberg ME. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol. 1995;15:2219–30. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–50. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–64. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 31.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 32.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–10. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–5. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 35.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9:378–87. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 36.Tullai JW, et al. Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J Biol Chem. 2007;282:23981–95. doi: 10.1074/jbc.M702044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iyer VR, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–7. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 38.Gilchrist M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–8. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 39.Miller AD, Curran T, Verma IM. c-fos protein can induce cellular transformation: a novel mechanism of activation of a cellular oncogene. Cell. 1984;36:51–60. doi: 10.1016/0092-8674(84)90073-4. [DOI] [PubMed] [Google Scholar]
- 40.Lee WM, Lin C, Curran T. Activation of the transforming potential of the human fos proto-oncogene requires message stabilization and results in increased amounts of partially modified fos protein. Mol Cell Biol. 1988;8:5521–7. doi: 10.1128/mcb.8.12.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rollins BJ. JE/MCP-1: an early-response gene encodes a monocyte-specific cytokine. Cancer Cells. 1991;3:517–24. [PubMed] [Google Scholar]
- 42.Van Damme J, Proost P, Lenaerts JP, Opdenakker G. Structural and functional identification of two human, tumor-derived monocyte chemotactic proteins (MCP-2 and MCP-3) belonging to the chemokine family. J Exp Med. 1992;176:59–65. doi: 10.1084/jem.176.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neish AS, Williams AJ, Palmer HJ, Whitley MZ, Collins T. Functional analysis of the human vascular cell adhesion molecule 1 promoter. J Exp Med. 1992;176:1583–93. doi: 10.1084/jem.176.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker S, Warren MK, Haskill S. Colony-stimulating factor-induced monocyte survival and differentiation into macrophages in serum-free cultures. J Immunol. 1987;139:3703–9. [PubMed] [Google Scholar]
- 45.Sarojini H, et al. PEDF from mouse mesenchymal stem cell secretome attracts fibroblasts. J Cell Biochem. 2008;104:1793–802. doi: 10.1002/jcb.21748. [DOI] [PubMed] [Google Scholar]
- 46.Kong W, Li S, Longaker MT, Lorenz HP. Cyclophilin C-associated protein is up-regulated during wound healing. J Cell Physiol. 2007;210:153–60. doi: 10.1002/jcp.20830. [DOI] [PubMed] [Google Scholar]
- 47.McQuibban GA, et al. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–7. [PubMed] [Google Scholar]
- 48.McQuibban GA, et al. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–6. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 49.Dieu MC, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–86. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmitz J, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.