

Abstract
Objectives
Fetal hemoglobin (HbF) induction involves NO-cGMP signaling pathways. L-arginine, an NO precursor, and the phosphodiesterase (PDE) 5 inhibitor sildenafil, which potentiates cGMP, were studied in adults with sickle cell disease (SCD) who were stably on HU.
Methods
24 courses of L-arginine (0.1–0.2 g/Kg divided TID) or sildenafil (25–100 mg TID), assigned based on gender due to concerns about sildenafil-related priapism, were successfully completed. Biochemical assays, pulmonary pressures, and cardiopulmonary exercise capacity are reported from patients in whom serial values are available. Hematologic responses are reported in 14 subjects with HbSS who had stable baseline HbF levels.
Results and Conclusions
L-arginine increased plasma arginine and ornithine, but not citrulline, suggesting diversion by plasma arginase from NO, and citrulline, generation. Glutathione (GSH) increased only in patients on L-arginine. Sildenafil increased plasma cGMP and citrulline, but not other amino acids. Pulmonary pressures and 6-minute walk distances improved only in patients on sildenafil.
In subjects with stable baseline HbF levels, HbF levels changed little from a normalized baseline on L-arginine, decreasing by 2.9±16.1%, n=6; p=n.s., but increased on sildenafil, by 7.5±11.7%, n=8, p<.05. Absolute reticulocyte counts initially decreased in patients on sildenafil.
L-arginine, at doses that increase plasma arginine levels, altered redox potential in red cells. The lack of clinically detectable efficacy of L-arginine may be due to increased arginine metabolism in SCD patients.
In vivo augmentation of the cyclic nucleotide pathway by PDE inhibition may induce HbF slightly, but strikingly improves hemodynamic and functional status in SCD
Introduction
Sickle cell disease (SCD) is an inherited hematologic disease, characterized by recurrent vaso-occlusive crises and widespread vasculopathy, strikingly in the cerebral, pulmonary, and renal vasculature. Sickle hemoglobin (HbS) arises from a single base-pair mutation, A to T, in the adult β-globin gene, with a glutamine (GAG) to valine (GTG) substitution in the β–globin polypeptide chain. Deoxygenated HbS can polymerize in the red cell, causing vaso-occlusion and hemolysis. Sickle cell syndromes encompass both homozygous sickle mutations in the β–globin gene (HbSS), which predominate, and compound heterozygous mutations (HbSC and HbSβ-thalassemia).
The β-globin gene locus is developmentally regulated. Fetal hemoglobin (HbF), made from alpha (α) and gamma (γ) globin polypeptides, is the major hemoglobin in late fetal and early neonatal life, and is gradually supplanted, to over 98% total hemoglobin after birth, by adult hemoglobin (HbA), α- and beta- (β) globin polypeptides.
In a randomized, multi-centered trial, hydroxyurea (HU) increased the number of HbF-containing red cells and diminished the frequency of pain crises, acute chest syndromes, the need for transfusions (1) and, likely, mortality (2). HU increases HbF, decreases the white cell count, and alters vaso-active molecules. HU can be metabolized to nitric oxide (NO) (3). SCD patients on HU have elevated levels of NO metabolites, which activate soluble guanylate cyclase (sGC) and produce cGMP (4–6). Both cGMP and cAMP have been implicated in the pharmacologic up-regulation of HbF (4–7). In vitro models of erythropoiesis treated with NO-donors such as HU, have shown elevated γ-globin mRNA, HbF, and sGC (4). It is possible that augmentation of NO-, cGMP- or cAMP-related pathways, and/or modulation of PDEs, can induce fetal hemoglobin.
Sildenafil, an inhibitor of PDE 5 (which degrades cGMP) and a treatment for pulmonary hypertension, can potentiate cGMP-dependent signaling; Erythrocyte cGMP levels correlate with HbF levels in some patients on HU (8).
The amino acid L-arginine is metabolized to NO and citrulline, unless diverted to ornithine by arginase. Arginine is depleted in SCD patients at baseline, and more so in patients with additional complications (9). Plasma arginase, which degrades L-arginine, may serve as a marker for poor outcome in sickle cell disease (10–12). L-arginine supplementation, in mice and in humans (12,13), has been reported to ameliorate some clinical sequelae of HbSS. This amelioration may be mediated by decreasing red cell density (mean corpuscular hemoglobin concentration (MCHC) or by increasing red-cell glutathione (GSH) levels, both of which have been reported following L-arginine supplementation in transgenic mouse models of sickle cell disease(13,14).
Here, we report on an open-label phase I-II study in which L-arginine, at high doses, and sildenafil were examined for safety, impact on cardiopulmonary function (pulmonary artery pressures and six-minute walk distance (6MWD)), and impact on HbF in patients with sickle syndromes who were already on HU. Disease severity was estimated by indirectly assessing intravascular hemolysis (increased lactate dehydrogenase (LDH) and reticulocyte count), endothelial activation (increased serum vascular cell adhesion molecule (VCAM) levels), and risk of sickle polymer formation (increased intracellular hemoglobin concentration (MCHC)). Clinical events and hospitalizations, but not outpatient pain symptoms, during therapy were analyzed.
We describe the largest number of adult SCD patients so far reported who have received extended courses of both HU and L-arginine or sildenafil; and this is the only published description of prolonged L-arginine dosing at levels sufficient to alter plasma amino acid concentrations.
Methods
Design
Enroller patients were maintained on a non-toxic, non-escalating dose of HU until hematologic stability could be confirmed. They were then treated for 12 weeks with either L-arginine or sildenafil, non-randomly assigned by gender due to concerns, not subsequently borne out (15,16), that high-dose sildenafil could cause priapism in males with SCD. Serial measurements were made of HbF, plasma amino acids, total erythrocyte GSH, VCAM, arginase, blood chemistry, complete blood counts, 6MWD, and tricuspid regurgitant (TR) jet velocity. HbF-containing red cells and reticulocytes (F-cells and F-reticulocytes respectively) were also measured. MCHC in g/dL [Hb (g/dL) × 1000)/(mean corpuscular volume (fL) × red cell count (per liter)] was calculated for each time point. cGMP was measured in a subset of patients.
The protocol was approved by the Institutional Review Board of the NHLBI and is registered at clinicaltrials.gov as protocol NCT00056433.
Assays and functional tests
Plasma cGMP and VCAM levels were measured by kits (cGMP immunoassay and solid phase ELISA respectively, R&D Systems, Minn., MN, USA).
Arginase activity (12, 17), HbF, F-cells and F-reticulocytes (18), and total erythrocyte GSH levels (19) were measured as published in detail previously.
TR jet velocity, by echocardiogram, and 6MWD were evaluated serially on therapy, per institutional guidelines, published previously (20).
Statistics
Mean ± standard deviation was calculated for evaluable data points. The 1-way repeated measures ANOVA (RMA) was used to compare serial results within a treatment group; unless otherwise noted, all statistical results are 1-way RMA analyses. The 2-way RMA was used to compare serial results between treatment groups. The students’ t-test was used to compare initial values between groups (Table 1), limited time interval within groups, and episodes of toxicity.
Table 1. Patient Demographics.
Shown are data for all patients who were stabilized on HU and randomized to second drug. Data are from week 0 of second drug, following stabilization on HU.
| Patient Demographics† | Sildenfil, n=14*, (mean ± S.D.) | L-arginine, n=13 (mean ± S.D.) | p Value (t-test) |
|---|---|---|---|
| Gender | 13 F, 1 M | 3 F, 10 M | p<0.001 |
| Genotype | HbSS, n=14 | HbSS, n=13; HbSC, n=1 | n.s. |
| Age, years | 39 ± 9.7 | 37.67 ± 9.3 | n.s. |
| Hb, g/dl | 8.9 ± 1.4 | 9.4 ± 1.95 | n.s. |
| MCV, fL | 110.2 ± 10.6 | 110.25 ± 2.73 | n.s. |
| Hb F, % | 12.1 ± 3.5 | 12.98 ± 7.12 | n.s. |
| LDH, IU/L | 340 ± 133 | 320 ± 85 | n.s. |
| Arginine, mM/L | 35.6 ± 18.1 | 50.1 ± 17.0 | 0.07 |
| Ornithine, mM/L | 71.3 ± 10.3 | 76.7 ± 12.3 | n.s. |
| Citrulline, mM/L, | 16.5 ± 5.7 | 20.4 ± 13.4 | n.s. |
| TR Jet, M2/sec, | 2.72 ± 0.43 | 2.53 ± 0.3 | n.s. |
| 6 Minute Walk, M | 433 ± 104 | 485 ± 105 | 0.07 |
| Arginase, mM/L | 2.19 ± 1.65 | 1.15 ± 0.60 | n.s. |
| VCAM, (ng/ml) | 1132.5 ± 250.3 | 895.5 ± 204.2 | 0.04 |
| Total RBC GSH (μM) | 1090.1 ± 342.8 | 1222.0 ± 424.9 | n.s. |
6 patients reported previously (31).
Results
Enrollment
Patients were offered study medication after stabilization on HU. 13 courses of L-arginine (10 males, 3 females) and 14 courses of sildenafil (13 females, 1 male) were given; 3 patients were treated sequentially with both medications after a 12-week wash-out period, for a total of 27 evaluable courses in 24 patients. Therapy was terminated early in 3 patients, due to headaches (n=2, on sildenafil) or anxiety (n=1, on L-arginine). 15 additional patients were never offered study medication, due to poor-compliance (n=4), medication intolerance (n=6), relocation (n=2), or clinical deterioration (n=3).
Hematologic stability, defined as a <10% change in HbF over 6 weeks, with ≥2 serial measurements, was not strictly achieved in all patients, due to delayed HU effects or red cell transfusions. Primary hematologic endpoints are reported only in patients with HbSS who achieved hematologic stability as stringently applied; that is, 8 patients on sildenafil and 6 patients on L-arginine. We report the percent change from the normalized baseline that was induced by a given treatment, as baseline HbF values varied widely, from 3.7 to 27.5 % of total Hgb.
Non-hematologic endpoints are reported on all evaluable patients for whom sufficient data are available, irrespective of baseline HbF levels. The total number of reported values varies between analyses due to missed data points. In a single male patient with HbSC, treated with L-arginine, only toxicity and biochemical data were analyzed.
Patient Demographics
Total hemoglobin (Hb), MCV, white blood cell count (WBC), HbF, F-cells, F-reticulocytes, and lactate dehydrogenase (LDH, Table 1 and not shown), did not differ at baseline between L-arginine- and sildenafil-treated patients. Physiologic and laboratory parameters suggested more active disease in the, predominantly female, patients assigned to sildenafil; in these patients, VCAM levels(p=.04), arginase levels, and TR jet velocities tended to be higher, while 6-MW distances and arginine levels (p=.07) tended to be lower.
Pharmacology
In all evaluable patients on therapy, plasma concentrations [mM/L] of arginine and its metabolite ornithine approximately doubled (figure 1A & B) with L-arginine (n=8), but did not change on sildenafil (n=7, p<.005 and p<.03, respectively, 2-way RMA between therapies). Strikingly, concentrations of the NO-related metabolite citrulline rose by one-third in patients on sildenafil, and did not change in patients on L-arginine (figure 1C, p=.47 and n.s., respectively).
Figure 1. Plasma Arginine, Ornithine, and Citrulline levels.

(A) Arginine and (B) ornithine levels rose with L-arginine supplementation (n=8 all evaluable, from 47±16 to 96±58, p<.005, and from 74±13 to 128±77, p=.03, respectively, 2-way RMA relative to sildenafil) but not with sildenafil therapy (n=7 all evaluable, from 35.0±15.6 to 28.6±11.3, p=n.s., and from 67.9±10.6 to 61.2±7.0, p=n.s., respectively). (C) Citrulline did not change in patients on L-arginine (from 22.3±11.0 to 23.4±12.8, p=n.s.), but rose in patients on sildenafil (from 15.7±4.0 to 21.1±4.7, p=.047).
The monitored first oral dose of sildenafil, 25 mg, in 5 patients was associated with a <10% change in mean and systolic arterial pressure and a minimal increase in heart rate (p=n.s. for all, not shown). Plasma cGMP was measured in 3 of these patients; it increased by an average of 2 pM/ml at hour 3 and returned to baseline by hour 5 (not shown).
Adverse Events
There was no significant difference between therapies in the number of admissions (n=4 on sildenafil and 7 on L-arginine), of episodes of leg edema (4 each), or of ocular complications (n=5 on sildenafil and 2 on L-arginine). Headaches were more common in patients on sildenafil, 9/14, vs. 0/13 with L-arginine (p=.005, t-test); these headaches resolved with conservative management in most patients in spite of continued therapy, but prompted termination of sildenafil therapy in 2 patients. Four patients on sildenafil, and none on L-arginine (p<.05), developed facial or periorbital swelling, but no medication changes were prompted by these self-limited events.
Hematologic Responses and Markers of Disease Activity
After 10–12 weeks on sildenafil, relative HbF levels rose by 7.5±11.7% from a normalized baseline (figure 2A, n=8, p<.01) and relative F-cell levels also increased, by 6.8±13.1% (figure 2B, n=7, p=n.s.), while total Hgb dropped, from 8.8±1.6 to 8.4±1.6 g/dL (p=n.s., not shown). On L-arginine, baseline-normalized HbF declined slightly in HbSS patients on L-arginine, by 2.9±16% (n=6, p=n.s.), as did F-cells, by 2.6±15.7% (n=6, p=n.s.), while total Hgb rose slightly, from 9.3±0.9 to 9.4±0.8 g/dL (n=6, p=n.s.). Calculated absolute HbF levels, in g/dL were not different between groups.
Figure 2. Relative HbF and F-cell levels in patients with a stable baseline HbF.

(A) Percent change in HbF %, from week 0 set at 0 %. In patients who were treated with sildenafil, rising by 7.5±11.7% at week 10 (n=8, plotted from week -6, solid black line, p<.05), but not in patients treated with L-arginine (solid grey line, n=6, dropping by 2.9±16% at week 12, p=n.s.). (B) Percent change in F-cell % from week 0, set at 0 %. In patients on sildenafil rising by 6.8±13.1% at week 12 (n=7, plotted from week 0, solid black line, p=n.s.) and in patients on L-arginine diminishing by 2.6±15.7% at week 12 (n=6, solid grey line, p=n.s.).
The absolute reticulocyte count (x103/μL, not shown) dropped rapidly in patients on sildenafil, from 240±110 at baseline to 199±106 at 4 weeks (n=8, p=.03, t-test, weeks 0–4) not seen in patients on L-arginine, from 187±41 week 0 to 202±34 week 4 (n=6, p=n.s. t-test, weeks 0–4). This difference was no longer seen at 12 weeks (weeks 0–12, p=n.s.).
Arginase levels tended to be higher at week 0 and diminished by week 12, especially in patients on sildenafil (p=n.s., not shown). LDH and VCAM levels did not change during therapy with either agent (not shown). MCHC did not change in HbSS patients with a stable baseline HbF after either sildenafil or L-arginine treatment (n=8 and 6, respectively, p=n.s., not shown).
Total erythrocyte GSH levels at week 8 were increased with L-arginine treatment (n=8, p=.03), but not with sildenafil treatment (n=6, p=n.s., student’s t-test).
Physiologic and Hemodynamic Responses
In all HbSS patients, irrespective of HbF stability at baseline, TR jet velocity dropped on sildenafil (n=13, from a mean of 2.79±0.38 to 2.68±0.32 meters/sec, p<.001), but not on L-arginine (n=11, from a mean of 2.57±0.34 to 2.69±0.41 m/s, p=n.s.). The 6-MW distances increased after sildenafil therapy (n=13, from a mean of 399.8±88.5 meters to 491.3±109.5 meters, p<.05), but did not change with L-arginine therapy (n=9, from 494.3±108.5 meters to 511.0±115.9 meters, p=n.s.). A direct comparison of therapeutic effect between therapies is not possible due to higher TR jet velocities at entry in the sildenafil group compared with the L-arginine group.
Discussion
This study was designed to evaluate, in SCD patients on HU, whether therapy that supplements the NO pathway (L-arginine) or enhances guanylate cyclase effects (sildenafil) has an impact on HbF, on markers of disease severity, or on cardiopulmonary function.
In this study, three months of sildenafil with HU therapy slightly increased relative %HbF in SCD patients; f-cells also trended higher in these patients, most of whom were female. X-linked polymorphisms have been associated with higher basal HbF levels in SCD(21), but gender differences have not predicted response to pharmacologic augmentation of HbF (22). Single-nucleotide polymorphisms in the PDE7B gene have been associated both with high basal levels of HbF and with increased HbF responsiveness to HU in SCD (23, 24), and both cAMP and cGMP have been implicated in the pharmacologic up-regulation of HbF. PDE5’s substrate is cGMP and PDE7B’s substrate is cAMP(25); cross-reactions between PDE inhibitors in bone marrow, described in other tissues, is feasible. Ongoing studies of sildenafil as therapy for pulmonary hypertension in sickle cell disease should confirm or refute this observation in a larger mixed gender population.
Red cell density did not change in our patients, in spite of reports of L-arginine decreasing MCHC in experimental murine models of SCD (13). This may relate to a species-specific release of red cell arginase into plasma during hemolysis, less in mice than in man (11, 26, 27), and an acute rise in plasma arginase activity has been seen in patients with SCD after co-administration of L-arginine and Hydroxyurea (28). Arginase metabolizes L-arginine to ornithine, and diverts it from the citrulline-NO pathway. This would be consistent with amino acid profiles in our patients, in which L-arginine supplementation increased ornithine, but not citrulline, while sildenafil therapy increased only citrulline.
Of note, L-arginine increased GSH levels in our patients, suggesting that L-arginine increases antioxidants, as described in murine models of SCD (14). Erythrocyte GSH depletion has been linked to hemolysis and to oxidative stress (29, 30). Aberrations in the glutathione pathway are well established in SCD (19 and references therein).
As expected, given the known vasodilatory effects of PDE5 inhibition on the pulmonary vasculature (31), TR jet velocities dropped, and 6-MW distances rose, with sildenafil.
This is the largest study published to date on the chronic use of L-arginine or sildenafil in adult patients with SCD. In an earlier randomized study, L-arginine supplementation had no effect on NO levels, red cell Gardos channels, or MCHC in 48 children with SCD. However, in sharp distinction with our current report, plasma arginine levels did not detectably increase in that earlier study (32). Also in contrast with earlier reports (12), L-arginine did not improve TR jet velocities in our patients. However, our patients had less cardiovascular compromise than described earlier, and further improvement with pharmacological therapy may not have been possible. Serum arginine and total red cell glutathione levels rose in our patients on L-arginine, but without hematologic or physiologic improvement, nor notable toxicity.
We recognize the many limitations of this non-randomized phase I/II trial. However, this study represents the largest and most comprehensive assessment of NO donor therapies in a complex therapeutic and phenotypic background. The results clearly indicate a number of potential benefits of augmenting the cGMP pathway in sickle cell disease, while raising questions about the efficacy of L-arginine therapy in this population. Randomized studies in a larger patient population will be needed to confirm these intriguing preliminary results.
Figure 3. Total erythrocyte GSH levels.
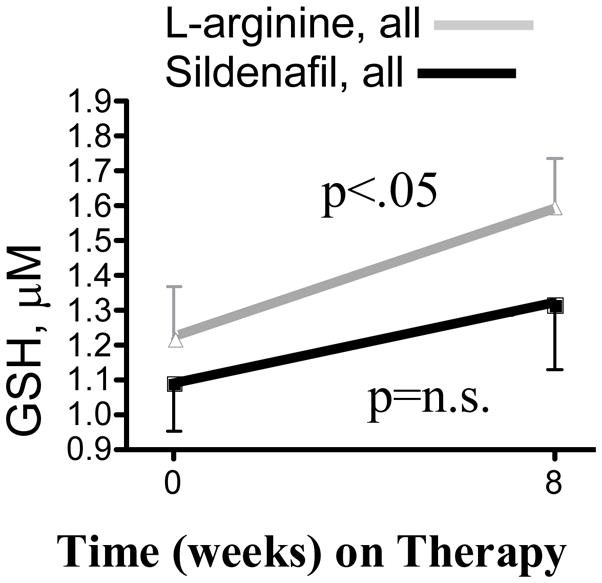
Red cell GSH levels rose in patients on L-arginine (all evaluable, grey line, from 1221.9±424.9 μM week 0 to 1593.4±405.9 μM week 8, n=8, p=0.03), but rose insignificantly in patients on sildenafil (all evaluable, black line, from 1090.1±324.8 μM week 0 to 1318±459.0 μM week 8, n=6, p=n.s.).
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, Critical Care Medicine Department and The National Heart, Lung and Blood Institute. We acknowledge, with much gratitude, the patients who participated in this study. The contributions made by the clinical team and staff of the Sickle Cell Disease clinic at the National Institutes of Health were invaluable, and this study would not have been possible without their able assistance.
References
- 1.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia [see comments] N Engl J Med. 1995;332(20):1317–22. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. Jama. 2003;289(13):1645–51. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 3.King SB. N-hydroxyurea and acyl nitroso compounds as nitroxyl (HNO) and nitric oxide (NO) donors. Curr Top Med Chem. 2005;5(7):665–73. doi: 10.2174/1568026054679362. [DOI] [PubMed] [Google Scholar]
- 4.Cokic VP, Smith RD, Beleslin-Cokic BB, Njoroge JM, Miller JL, Gladwin MT, et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Invest. 2003;111(2):231–9. doi: 10.1172/JCI16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keefer JR, Schneidereith TA, Mays A, Purvis SH, Dover GJ, Smith KD. Role of cyclic nucleotides in fetal hemoglobin induction in cultured CD34+ cells. Exp Hematol. 2006;34(9):1151–61. doi: 10.1016/j.exphem.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 6.Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98(4):1847–52. doi: 10.1073/pnas.041599798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue A, Kuroyanagi Y, Terui K, Moi P, Ikuta T. Negative regulation of gamma-globin gene expression by cyclic AMP-dependent pathway in erythroid cells. Exp Hematol. 2004;32(3):244–53. doi: 10.1016/j.exphem.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Conran N, Fattori A, Saad ST, Costa FF. Increased levels of soluble ICAM-1 in the plasma of sickle cell patients are reversed by hydroxyurea. Am J Hematol. 2004;76(4):343–7. doi: 10.1002/ajh.20129. [DOI] [PubMed] [Google Scholar]
- 9.Morris CR, Kuypers FA, Larkin S, Vichinsky EP, Styles LA. Patterns of arginine and nitric oxide in patients with sickle cell disease with vaso-occlusive crisis and acute chest syndrome. J Pediatr Hematol Oncol. 2000;22(6):515–20. doi: 10.1097/00043426-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Schnog JJ, Jager EH, van der Dijs FP, Duits AJ, Moshage H, Muskiet FD, et al. Evidence for a metabolic shift of arginine metabolism in sickle cell disease. Ann Hematol. 2004;83(6):371–5. doi: 10.1007/s00277-004-0856-9. [DOI] [PubMed] [Google Scholar]
- 11.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. Jama. 2005;294(1):81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris CR, Morris SM, Hagar W, Van Warmerdam J, Claster S, Kepka-Lenhart D, et al. Arginine Therapy: A New Treatment for Pulmonary Hypertension in Sickle Cell Disease? Am J Respir Crit Care Med. 2003 doi: 10.1164/rccm.200208-967OC. [DOI] [PubMed] [Google Scholar]
- 13.Romero JR, Suzuka SM, Nagel RL, Fabry ME. Arginine supplementation of sickle transgenic mice reduces red cell density and Gardos channel activity. Blood. 2002;99(4):1103–8. doi: 10.1182/blood.v99.4.1103. [DOI] [PubMed] [Google Scholar]
- 14.Dasgupta T, Hebbel RP, Kaul DK. Protective effect of arginine on oxidative stress in transgenic sickle mouse models. Free Radic Biol Med. 2006;41(12):1771–80. doi: 10.1016/j.freeradbiomed.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bialecki ES, Bridges KR. Sildenafil relieves priapism in patients with sickle cell disease. Am J Med. 2002;113(3):252. doi: 10.1016/s0002-9343(02)01165-8. [DOI] [PubMed] [Google Scholar]
- 16.Burnett AL, Bivalacqua TJ, Champion HC, Musicki B. Feasibility of the use of phosphodiesterase type 5 inhibitors in a pharmacologic prevention program for recurrent priapism. J Sex Med. 2006;3(6):1077–84. doi: 10.1111/j.1743-6109.2006.00333.x. [DOI] [PubMed] [Google Scholar]
- 17.Morris SM, Jr, Kepka-Lenhart D, Chen LC. Differential regulation of arginases and inducible nitric oxide synthase in murine macrophage cells. Am J Physiol. 1998;275(5 Pt 1):E740–7. doi: 10.1152/ajpendo.1998.275.5.E740. [DOI] [PubMed] [Google Scholar]
- 18.Little JA, McGowan VR, Kato GJ, Partovi KS, Feld JJ, Maric I, et al. Combination erythropoietin-hydroxyurea therapy in sickle cell disease: experience from the National Institutes of Health and a literature review. Haematologica. 2006;91(8):1076–83. [PMC free article] [PubMed] [Google Scholar]
- 19.Morris CR, Suh JH, Hagar W, Larkin S, Bland DA, Steinberg MH, et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood. 2008;111(1):402–10. doi: 10.1182/blood-2007-04-081703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anthi A, Machado RF, Jison ML, Taveira-Dasilva AM, Rubin LJ, Hunter L, et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med. 2007;175(12):1272–9. doi: 10.1164/rccm.200610-1498OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dover GJ, Smith KD, Chang YC, Purvis S, Mays A, Meyers DA, et al. Fetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2. Blood. 1992;80(3):816–24. [PubMed] [Google Scholar]
- 22.Maier-Redelsperger M, de Montalembert M, Flahault A, Neonato MG, Ducrocq R, Masson MP, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood. 1998;91(12):4472–9. [PubMed] [Google Scholar]
- 23.Wyszynski DF, Baldwin CT, Cleves MA, Amirault Y, Nolan VG, Farrell JJ, et al. Polymorphisms near a chromosome 6q QTL area are associated with modulation of fetal hemoglobin levels in sickle cell anemia. Cell Mol Biol (Noisy-le-grand) 2004;50(1):23–33. [PubMed] [Google Scholar]
- 24.Ma Q, Wyszynski DF, Farrell JJ, Kutlar A, Farrer LA, Baldwin CT, et al. Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea. Pharmacogenomics J. 2007 doi: 10.1038/sj.tpj.6500433. [DOI] [PubMed] [Google Scholar]
- 25.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58(3):488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 26.Spector EB, Rice SC, Kern RM, Hendrickson R, Cederbaum SD. Comparison of arginase activity in red blood cells of lower mammals, primates, and man: evolution to high activity in primates. Am J Hum Genet. 1985;37(6):1138–45. [PMC free article] [PubMed] [Google Scholar]
- 27.Morris CR, Kuypers FA, Kato GJ, Lavrisha L, Larkin S, Singer T, et al. Hemolysis-associated pulmonary hypertension in thalassemia. Ann N Y Acad Sci. 2005;1054:481–5. doi: 10.1196/annals.1345.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris CR, Vichinsky EP, van Warmerdam J, Machado L, Kepka-Lenhart D, Morris SM, Jr, et al. Hydroxyurea and arginine therapy: impact on nitric oxide production in sickle cell disease. J Pediatr Hematol Oncol. 2003;25(8):629–34. doi: 10.1097/00043426-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med. 1999;27(9–10):922–35. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 30.Wu G. Intestinal mucosal amino acid catabolism. J Nutr. 1998;128(8):1249–52. doi: 10.1093/jn/128.8.1249. [DOI] [PubMed] [Google Scholar]
- 31.Machado RF, Martyr S, Kato GJ, Barst RJ, Anthi A, Robinson MR, et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol. 2005;130(3):445–53. doi: 10.1111/j.1365-2141.2005.05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Styles L, Kuypers F, Kesler K, Reiss U, Lebeau P, Nagel R, et al. Arginine Therapy Does Not Benefit Children with Sickle Cell Anemia- Results of the CSCC Clinical Trial Consortium Multi-institutional Study. Blood. 2007 Abstract #2252. [Google Scholar]