

Abstract
Overproduction of nitric oxide by neuronal nitric oxide synthase (nNOS) has been linked to several neurodegenerative diseases. We have recently designed potent and isoform selective inhibitors of nNOS, but the lead compound contains several basic functional groups. A large number of charges and hydrogen bond donors can impede the ability of molecules to cross the blood brain barrier and thereby limit the effectiveness of potential neurological therapeutics. Replacement of secondary amines in our lead compound with neutral ether and amide groups was made to increase bioavailability and to determine if the potency and selectivity of the inhibitor would be impacted. An ether analogue has been identified that retains a similar potency and selectivity to that of the lead compound, and shows increased ability to penetrate the blood brain barrier.
Keywords: neuronal nitric oxide synthase, neuronal nitric oxide synthase inhibitors, ether, bioavailability, blood-brain barrier, aminopyridine
Introduction
Neuronal nitric oxide synthase (nNOS) has been implicated in various neurodegenerative diseases, including Parkinson’s disease 1 and neuronal damage resulting from stroke.2 Inhibition of nNOS could have therapeutic benefit in these and other diseases, but this inhibition must be achieved without effect on the other isoforms of NOS, endothelial (eNOS) and inducible (iNOS); inhibition of eNOS could lead to side effects such as hypertension,3 and inhibition of iNOS could result in a higher probability of Alzheimer’s disease.4
We have recently shown that a highly potent and isoform selective nNOS inhibitor (1) affects the extent of neurological damage suffered by rabbit fetuses under hypoxic conditions when administered to the dam intravenously.5 Although this in vivo efficacy was encouraging, we were concerned that the efficacy of the compound in adult disease models may be limited by unfavorable pharmacokinetics.6 Specifically, the high number of basic groups leads to significant positive charge at physiological pH, which could impede the ability of the compound to cross a fully formed blood brain barrier (BBB).7
Systematic replacement of basic functional groups in the lead with non-basic ones was carried out. The goal was to identify molecules that inhibited nNOS with a potency and selectivity comparable to 1, but which carried less charge, or at least had fewer hydrogen bond donors, and therefore could show increased uptake into the CNS.
The secondary amine of the pyrrolidine ring of 1 interacts with a key aspartate residue in nNOS.8 This is critical for nNOS over eNOS selectivity, so this group was not altered. The secondary amine adjacent to the pyrrolidine was replaced with an ether linkage to form 2 and an amide linkage to form 3. Replacing the secondary amine in the chain with an ether linkage gave 4. These three compounds were tested for activity against the NOS isoforms, and it was discovered that having an ether next to the pyrrolidine was a beneficial modification, but that having an ether in the chain resulted in a significant decrease in potency. The ether modification was incorporated into the design of 5 and 6, which had amides in the chain, with the carbonyls on either side of the nitrogen.
Chemistry
The synthesis of alcohol 7 has been described previously. The carbamate proton adjacent to the pyridine ring is more acidic than the hydroxyl group proton, allowing selective alkylation of the nitrogen preferentially to the oxygen (Scheme 1). Protection of this nitrogen with a benzyl group is necessary for both the Mitsunobu reaction9 and ether formation. A Mitsunobu reaction using acetic acid followed by hydrolysis of the resulting ester converted the trans-alcohol (8) to the desired cis-alcohol (10). Allyl bromide proved to be the best alkylating agent for ether formation. Alkylating reagents such as bromoacetonitrile and other alkylbromides and tosylates gave poor yields, most likely because of the high basicity of the secondary alkoxide. The allyl ether could be converted to aldehyde 12 under ozonolysis conditions.
Scheme 1.

Performing the Mitsunobu reaction on 8 using DPPA as a source of azide gave 13, which could be reduced to amine 14 with concomitant removal of the benzyl group (Scheme 2).
Scheme 2.

3-Fluorophenethylamine was alkylated with ethyl bromoacetate, and the resulting secondary amine was Boc protected (Scheme 3). The ester was hydrolyzed to give acid 16. 3-Fluorophenethanol was alkylated with allyl bromide and converted to aldehyde 18.
Scheme 3.

Compound 12 and 3-fluorophenethylamine were mixed to form the imine, which was then reduced to the secondary amine using sodium triacetoxyborohydride (Scheme 4). Simultaneous removal of the benzyl group and the Boc groups was achieved using high-pressure hydrogenation and strong acid to give 2 in a high yield.
Scheme 4.

Amine 14 was coupled to 16 using standard peptide coupling reagents to give the amide. This was treated with acid to remove the Boc groups, producing 3. A second portion of amine 14 was mixed with 18 then reduced with sodium triacetoxyborohydride. The Boc groups were removed with acid to give 4.
Aldehyde 12 was oxidized with oxone,10 and the resulting acid was coupled to 3-fluorophenethylamine (Scheme 5). Deprotection gave 5. Aldehyde 12 and benzylamine were mixed and reduced to the secondary amine. The two benzyl protecting groups were removed under hydrogenation conditions. The primary amine was coupled to 3-fluorophenylacetic acid, and the Boc groups were removed to give 6.
Scheme 5.

Results and Discussion
In vitro NOS inhibition assay
The compounds were tested for their ability to inhibit the three NOS isoforms using an established in vitro assay.11 The results are shown in Table 1.
Table 1.
Inhibition of the Three Isozymes of NOS by 1-6.
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Ki (nNOS) (μM) | 0.014 | 0.015 | 0.053 | 0.4 | 2.3 | 5.0 |
| Ki (eNOS) (μM) | 28 | 31 | 27 | 33 | 145 | 107 |
| Ki (iNOS) (μM) | 4 | 9.5 | 5.4 | 4 | 52 | 77 |
Racemic lead compound 1 is a competitive inhibitor of nNOS with low nanomolar potency. It shows 2000-fold selectivity for nNOS over eNOS, which is critical for this class of compounds to be therapeutically useful. Ether 2 has the same potency with nNOS as the analogous amine compound 1. The potencies for 2 with eNOS and iNOS are also comparable with those for 1, so the selectivity for nNOS over the other isoforms is excellent (~2000-fold over eNOS, ~600-fold over iNOS). These results indicate that the secondary amine adjacent to the pyrrolidine ring in 1 is either not contributing to binding to the active site, or that the ether linkage contributes equally as an amine to binding, possibly as a hydrogen bond acceptor. Crystal structures of analogues of 1 in the active site of nNOS indicate that this amine interacts with a heme propionate group via an active site water molecule. It is possible that an ether could also hydrogen bond to the same water molecule. Replacing the amine with an amide to give 3 resulted in a slight loss of binding affinity (3-4 fold), perhaps the result of increased steric hindrance. However, replacing the amine in the chain with an ether linkage as in 4, or with an amide as in 5 and 6 resulted in a significant loss in potency with nNOS. In the case of 4, the ether linkage is not only neutral, but a hydrogen bond donor has been replaced with a hydrogen bond acceptor. In the case of the amides, the NH is a potential H-bond donor, but the potency is not recovered. This suggests that this amine is involved in a charge-charge interaction that is important for binding, not a simple hydrogen bonding interaction.
In vivo brain uptake assay
To determine the extent to which 1 and 2 cross the blood brain barrier, the compounds were administered to mice at 3.7 mg/kg (10 nmol/g) via intraperitoneal injection. At 5, 10, 20, and 40 minutes after administration, five mice were sacrificed, and blood and brain samples were taken. The amount of compound in each sample was determined by LCMS and quantified by comparison to a standard curve. The assay was performed on 1 and 2 separately and as a co-administration experiment. The plasma and brain concentrations of 1 and 2 calculated in the co-administration experiment are shown in Fig 2 and Fig 3. The shapes of the curves and relative values did not differ significantly between the co-administration and separate administrations, suggesting that the two compounds do not interfere with one another. The values were initially quantified in μmol/mL of plasma and μmol/g of brain tissue. The brain concentrations were converted to μM by assuming the density of brain tissue is approximately 1 g/mL.
Figure 2.
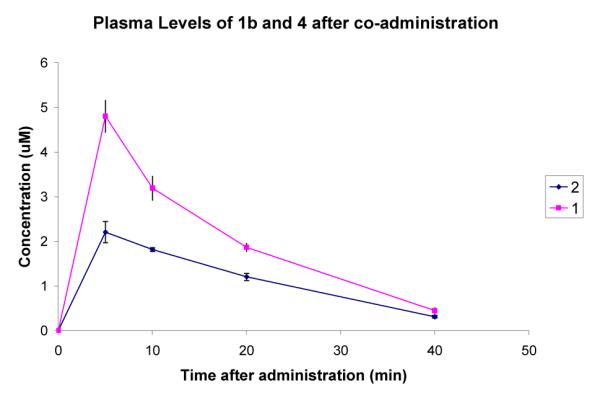
Comparison of the plasma concentrations of 1 and 2 after ip administration at 3.7 mg/kg. Error bars show the standard error mean.
Figure 3.
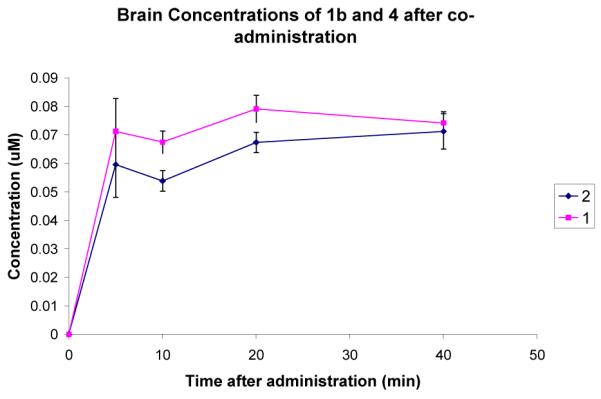
Comparison of the brain concentrations of 1 and 2 after ip administration at 3.7 mg/kg. Error bars show the standard error mean.
A comparison of the plasma concentrations gives a surprising result. The plasma concentration of 1 is significantly higher than that of 2 throughout the 40 min experiment. The brain level of 1 is also slightly higher than that of 2. Fig 4 shows the ratio of brain to blood concentration for the two compounds.
Figure 4.
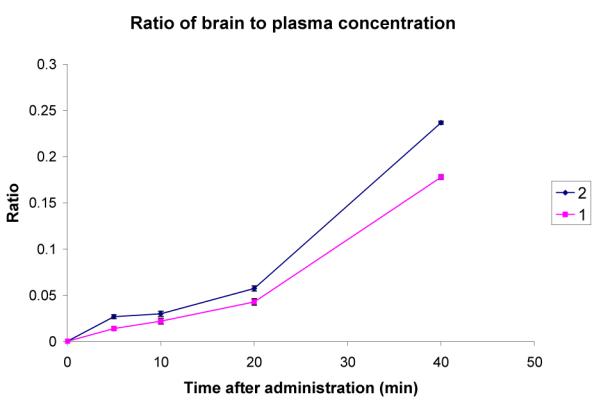
Comparison of the ratio of brain to blood concentrations of 1 and 2 after ip administration at 3.7 mg/kg. Error bars show the standard error mean.
The brain/blood ratio for 2 is consistently higher than that of 1, indicating that it does indeed penetrate the BBB more efficiently.
The secondary amine in 1 that is adjacent to the pyrrolidine ring is probably not protonated at physiological pH, as the presence of two secondary amines two carbons away will lower the pKa significantly.12 This may explain why there is little loss in binding affinity when this amine is replaced with an ether linkage. The ether modification in 2 is therefore not useful in reducing the overall charge on the molecule, which may explain why there is only a small difference in BBB penetration between 1 and 2. However, the ether modification does reduce the number of H-bond donors. Furthermore, according to molecular properties calculations, 2 has a slightly higher calculated logD (0.11) and lower polar surface area (81 Å2) than 1 (−0.09 and 84 Å2, respectively).13 All of these subtle changes may be responsible for the modest increase in the ability of the compound to cross the BBB.
A cursory inspection of the in vivo results suggests that although 2 crosses the BBB better than 1, there has been no improvement as there is no positive increase in final brain concentration. However, the concentration of 2 is above 60 nM throughout the 40 min experiment. This is four times the Ki with nNOS, and could be enough to produce a therapeutic response. In contrast, the plasma levels are less than 2 μM, which is significantly less than the Ki with eNOS. Although the brain to blood ratio is low, when administered at this dose, the compound should reach sufficient levels in the brain to have a therapeutic effect while not affecting eNOS, and therefore not causing a hypertensive side effect. The plasma concentration of 1 is considerably higher than that of 2, although still insufficient to effectively inhibit eNOS. In contrast, the brain concentrations of the two compounds are approximately the same. Since the two compounds have the same ability to inhibit nNOS, compound 2 could potentially have the same therapeutic effect as 1 while at a lower systemic level, and therefore have a lower potential for side effects. Compound 1 was found to be highly effective in a cerebral palsy rabbit model and exhibited no cardiovascular effects.5
Conclusions
The replacement of secondary amino groups in a potent and selective nNOS inhibitor with ether or amide linkages had varied effects on potency. In one case, replacement with an ether resulted in a negligible difference in potency and selectivity. In other cases, the replacement resulted in dramatic losses in potency. The ether analogue that was as potent as the lead compound showed a modest increase in brain uptake over the lead compound. These results are consistent with the notion that hydrogen bond donor groups are more detrimental to BBB penetration than hydrogen bond acceptor groups.
Experimental Procedures
General Methods
Proton nuclear magnetic resonances (1H NMR) were recorded in deuterated solvents on a Varian Inova 500 (500 MHz) spectrometer. Chemical shifts are reported in parts per million (ppm, δ) relative to tetramethylsilane (δ 0.00). 1H NMR splitting patterns are designated as singlet (s), doublet (d), triplet (t), or quartet (q). Splitting patterns that could not be interpreted or easily visualized were recorded as multiplet (m) or broad (br). Coupling constants are reported in Hertz (Hz). Proton-decoupled carbon (13C-NMR) spectra were recorded on a Varian Inova 500 (125 MHz) spectrometer and are reported in ppm using the solvent as an internal standard (CDCl3, δ 77.23). NMR spectra recorded in D2O were not normalized. In many cases, the presence of rotamers made the NMR spectra complex. In the case of two peaks that are clearly a pair of rotamers, but are too far apart for an average to accurately represent the spectrum, the pair is written enclosed in parentheses, or the presence of rotamers is indicated. Electrospray mass spectra (ESMS) were obtained using an LCQ-Advantage with methanol as the solvent in positive ion mode, unless otherwise stated. For most compounds, 1H and 13C NMR and ESMS data are presented. Final compounds were analyzed for purity using multiple HPLC conditions (see Supplementary Information). Analysis of biological samples was carried out using an API 3000 liquid chromatography-tandem mass spectrometry system (Applied Biosystems, Foster City, CA) equipped with an Agilent 1100 series HPLC system (Agilent Technologies, Wilmington, DE).
All chemical reagents were purchased from Aldrich and were used without further purification. NADPH, calmodulin, and human ferrous hemoglobin were obtained from Sigma-Aldrich. Tetrahydrobiopterin (H4B) was purchased from Alexis Biochemicals. HEPES, DTT, and some conventional organic solvents were purchased from Fisher Scientific.
Tetrahydrofuran (THF) was distilled from sodium and benzophenone prior to use. Methylene chloride (CH2Cl2) was distilled from calcium hydride prior to use, if dry solvent were required. Dimethylformamide (DMF) was purchased as an anhydrous solvent and was used directly.
General alkylation procedure
To a solution of the compound in anhydrous DMF (3 - 10 mL) at 0 °C was added NaH (1.1 equiv). The mixture was allowed to warm to room temperature and stirred for 30 min. The alkylating agent (1.1 equiv) was added dropwise, and the solution was stirred for 16 h. The solvent was removed in vacuo, and the residue was dissolved in EtOAc, washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo.
General Mitsunobu procedure
To a solution of the alcohol in anhydrous THF (5 mL) were added PPh3, DIAD and the nucleophile. The mixture was stirred for 16 h. The mixture was poured into brine and extracted with EtOAc (3 × 10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo.
General reductive amination procedure
A solution of the aldehyde and the amine in CH2Cl2 (5 mL) was stirred for 10 min. NaHB(OAc)3 was added, and the solution was stirred for 1 h.
General procedure for amide formation
To a solution of the acid and the amine in CH2Cl2 (3 mL) were added TEA, EDC, and HOBt. The mixture was stirred for 16 h. The solution was diluted with CH2Cl2 (20 mL) and washed with 1N HCl (2 × 15 mL), sat NaHCO3 (15 mL), and brine (15 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo.
General procedure for benzyl group removal
To a solution of the compound in EtOH (5 mL) was added Pd(OH)2/C (~5 mg). The mixture was stirred under a hydrogen atmosphere at 50 °C for 48 h. The mixture was filtered through Celite, and the solvent was removed in vacuo.
General procedure for Boc group removal
A solution of the compound in HCl in dioxanes (4 N, 3 mL) was stirred for 16 h. The solvent was removed under a stream of nitrogen, and the residue was dissolved in water (10 mL). The solution was washed with EtOAc, and the solvent was removed in vacuo. The resulting salt was dissolved in the minimum amount of MeOH, and anhydrous ether was added causing the salt to precipitate. The ether layer was decanted to leave the salt as a white solid.
trans-tert-Butyl 3-((6-(benzyl(tert-butoxycarbonyl)amino)-4-methylpyridin-2-yl)methyl)-4-hydroxypyrrolidine-1-carboxylate (8)
The general alkylation procedure was carried out on 7 (491 mg, 1.21 mmol) using benzyl bromide (148 μL, 1.25 mmol) as the alkylating agent. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 2:3) to afford 8 as a white solid (402 mg, 0.81 mmol, 67%). 1H NMR (500 MHz, CDCl3) δ 7.34 – 7.18 (m, 6H), 6.70 (m, 1H), 5.12 (s, 2H), 4.05 (m, 1H), 3.74 – 3.55 (m, 2H), 3.19 (m, 1H), 3.08 (m, 1H), 2.81 – 2.63 (m, 2H), 2.40 (m, 1H), 2.29 (s, 3H), 1.45 (s, 9H), 1.42 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 157.5, 154.8, 154.5, 154.1, 149.4, 139.6, 128.4, 127.3, 127.0, 120.8, 118.4, 81.6, 79.5, (75.3 + 74.6), (52.9 + 52.5), 50.6, (49.9 + 49.5), (45.6 + 44.8), 39.4, 28.7, 28.4, 21.3; ESMS m/z = 498 (M + H)+.
tert-Butyl 3-acetoxy-4-((6-(benzyl(tert-butoxycarbonyl)-amino)-4-methylpyridin-2-yl)methyl)pyrrolidine-1-carboxylate (9)
The general Mitsunobu procedure was carried out on 8 (402 mg, 0.81 mmol) using PPh3 (262 mg, 1 mmol), DIAD (188 μL, 1 mmol) and acetic acid (86 μL, 1.5 mmol) as the nucleophile. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 1:3) to afford 9 as a white solid (216 mg, 0.40 mmol, 49%). Unreacted 8 was also recovered. 1H NMR (500 MHz, CDCl3) δ 7.44 (m, 1H), 7.29 – 7.18 (m, 5H), 6.62 (s, 1H), 5.16 (s, 2H), 4.11 (m, 1H), 3.43 (m, 3H), 3.10 (m, 1H), 2.85 (m, 1H), 2.66 (m, 2H), 2.29 (s, 3H), 2.05 (s, 3H), 1.45 (s, 9H), 1.41 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.7, 156.9, 154.5, 154.2, 148.9, 140.1, 128.3, 127.2, 126.8, 126.7, 119.9, 117.5, 81.4, 79.6, (75.0 + 74.1), 60.6, (53.0 + 52.6), 50.2, (49.6 + 49.1), (42.0 + 41.4), 35.0, 28.7, 28.4, 21.3; ESMS m/z = 540 (M + H)+, 562 (M + Na)+.
cis-tert-Butyl 3-((6-(benzyl(tert-butoxycarbonyl)amino)-4-methylpyridin-2-yl)methyl)-4-hydroxypyrrolidine-1-carboxylate (10)
To a solution of 9 (216 mg, 0.40 mmol) in MeOH (3 mL) was added aqueous NaOH (1 N, 3 mL). The mixture was stirred at room temperature overnight. The pH of the solution was adjusted to ~8, and the product was extracted with EtOAc (3 × 15 mL). The organic layers were combined, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 2:3) to afford 10 as a white solid (184 mg, 0.37 mmol, 93%). 1H NMR (500 MHz, CDCl3) δ 7.36 (d, 1H), 7.28 – 7.20 (m, 5H), 6.74 (d, 1H), 5.09 (s, 2H), 3.95 (m, 1H), 3.59 – 3.36 (m, 4H), 3.14 (m, 1H), 2.86 (m, 1H), 2.78 (m, 1H), 2.31 (s, 3H), 1.44 (s, 9H), 1.41 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 157.8, 154.5, 154.2, 149.8, 139.2, 128.5, 127.0, 120.7, 118.7, 81.7, 79.3, (71.2 + 70.4), (53.9 + 53.6), 50.6, (49.6 + 49.2), (45.2 + 44.6), 35.3, 28.8, 28.4, 21.4; ESMS m/z = 498 (M + H)+, 520 (M + Na)+.
tert-Butyl 3-(allyloxy)-4-((6-(benzyl(tert-butoxy-carbonyl)amino)-4-methylpyridin-2-yl)methyl)pyrrolidine-1-carboxylate (11)
To a solution of 10 (994 mg, 2.0 mmol) in DMF (10 mL) was added NaH (60% in mineral oil, 120 mg, 3.0 mmol) at 0 °C. After being stirred for 15 min, allyl bromide (360 μL, 3.0 mmol) was added. The reaction mixture was allowed to stir at room temperature for 30 min, then quenched by H2O (1.0 mL). The solvent was removed in vacuo, and the resulting material was purified by flash column chromatography (silica gel, EtOAc / hexanes, 1:9-1:4) to yield 11 (1.07 g, 99%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.40 (m, 1H), 7.27 – 7.17 (m, 5H), 6.69 (s, 1H), 5.80 (m, 1H), 5.22 – 5.11 (m, 4H), 3.96 (m, 1H), 3.72 – 3.64 (m, 2H), 3.55 – 3.37 (m, 2H), 3.21 – 3.09 (m, 2H), 2.93 (m, 1H), 2.72 (m, 1H), 2.59 (m, 1H), 2.29 (m, 3H), 1.45 – 1.41 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 158.0, 154.9, 154.1, 148.7, 140.1, 134.9, 128.3, 127.2, 126.7, 120.4, 117.3, 116.8, 81.4, 79.3, 78.0, 70.4, (51.2 + 50.7), 50.1, (49.6 + 49.2), (43.1 + 42.4), 34.8, 28.8, 28.4, 21.4; ESMS m/z = 538 (M + H)+, 560 (M + Na)+.
tert-Butyl 3-((6-(benzyl(tert-butoxycarbonyl)amino)-4-methylpyridin-2-yl)methyl)-4-(2-oxoethoxy)-pyrrolidine-1-carboxylate (12)
A solution of 11 (535 mg, 1.0 mmol) in CH2Cl2 (20 mL) was cooled to −78 °C, to which O3 was charged until the reaction solution turned purple (~20 min). The O3 flow was stopped, and the reaction was allowed to stir at the same temperature for 1.5 h. To the resulting solution was added Zn (195 mg, 3.0 mmol) and 50% aqueous AcOH (20 mL). The reaction mixture was then warmed to room temperature over 45 min. The solvent was removed by rotary evaporation and the resulting material was purified by flash column chromatography (silica gel, ethyl acetate/hexanes, 1:4-1:1) to yield 12 (405 mg, 75%) as an oily white solid. 1H NMR (500 MHz, CDCl3) δ 9.56 (s, 1H), 7.44 (m, 1H), 7.28 – 7.16 (m, 5H), 6.69 (s, 1H), 5.17 (s, 2H), 4.06 – 3.94 (m, 1H), 3.81 – 3.63 (m, 2H), 3.54 – 3.41 (m, 2H), 3.22 – 3.11 (m, 2H), 2.98 (m, 1H), 2.77 (m, 1H), 2.71 – 2.53 (m, 1H), 2.30 (s, 3H), 1.46 (s, 9H), 1.42 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 202.4, 157.6, 154.7, 154.1, 148.9, 140.0, 128.4, 127.1, 120.2, 117.2, 81.5, 80.7, 79.7, 74.7, (51.1 + 50.5), 50.1, (49.4 + 49.0), (42.9 + 42.3), 34.4, 28.7, 28.4, 21.3; ESMS m/z = 572 (M + MeOH)+.
tert-Butyl 3-azido-4-((6-(benzyl(tert-butoxycarbonyl)-amino)-4-methylpyridin-2-yl)methyl)pyrrolidine-1-carboxylate (13)
The general Mitsunobu procedure was carried out on 8 (519 mg, 1.04 mmol) using PPh3 (342 mg, 1.3 mmol), DIAD (255 μL, 1.35 mmol) and DPPA (281 μL, 1.30 mmol) as the nucleophile source. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 1:4) to afford 13 as a white solid (461 mg, 0.88 mmol, 85%) 1H NMR (500 MHz, CDCl3) δ 7.47 (m, 1H), 7.37 (m, 1H), 7.24 (m, 4H), 6.69 (s, 1H), 5.17 (s, 2H), 3.76 (m, 1H), 3.58 – 3.37 (m, 2H), 3.31 (m, 1H), 3.00 (q, J = 11 Hz, 1H), 2.85 (m, 1H), 2.72 (m, 1H), 2.62 (m, 1H), 2.30 (m, 3H), 1.45 (s, 9H), 1.42 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 156.8, 154.6, 154.2, 149.0, 140.0, 130.3, 128.3, 127.0, 126.8, 126.4, 120.5, 117.4, 81.5, 79.8, (63.5 + 62.7), 51.6, 50.2, 48.9, (42.8 + 42.2), 35.2, 28.7, 28.4, 21.4; ESMS m/z = 523 (M + H)+, 545 (M + Na)+.
tert-Butyl 3-amino-4-((6-(tert-butoxycarbonylamino)-4-methylpyridin-2-yl)methyl)pyrrolidine-1-carboxylate (14)
The general procedure for the removal of benzyl groups was carried out on 13 (461 mg, 0.88 mmol). The crude product was purified using flash column chromatography (silica gel, EtOAc / methanol, 9:1) to afford 14 as a white solid (170 mg, 0.42 mmol, 48%). 1H NMR (500 MHz, CDCl3) δ 7.62 (s, 1H), 6.64 (s, 1H), 3.56 – 3.14 (m, 4H), 2.78 – 2.61 (m, 3H), 2.46 (m, 1H), 2.30 (m, 3H), 1.52 (s, 9H), 1.44 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 161.4, 158.1, 155.0, 152.6, 151.6, 150.4, 119.4, 110.5, 81.0, 79.4, (71.8 + 70.7), (61.3 + 60.4), (50.0 + 49.6), (49.0 + 48.5), (35.3 + 34.7) , 28.7, 28.4, 21.5; ESMS m/z = 407 (M + H)+.
Ethyl 2-(tert-butoxycarbonyl(3-fluorophenethyl)-amino)-acetate (15)
A solution of ethyl bromoacetate (119 μL, 1 mmol) and 3-fluorophenethylamine (130 μL, 1.5 mmol) in ethanol was stirred for 16 h. Boc2O (327 mg, 1.5 mmol) was added, and the solution was stirred a further 2 h. The mixture was poured into brine and extracted with EtOAc (3 × 15 mL). The organic layers were combined, dried over Na2SO4, and concentrated in vacuo. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 1:4) to afford 15 as a colorless oil (146 mg, 0.45 mmol, 45%). 1H NMR (500 MHz, CDCl3) δ 7.24 (m, 1H), 6.99 – 6.88 (m, 3H), 4.18 (q, J = 7 Hz, 2H), 3.88 (s, 1H), 3.76 (s, 1H), 3.49 (m, 2H), 2.84 (m, 2H), 1.44 (s, 9H), 1.27 (t, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.2, (164.7 + 162.8), 155.8, 141.9, 130.2, 124.8, 115.9, 113.5, 80.6, 61.3, 50.4, 49.4, 35.0, 28.5, 14.5; ESMS m/z = 326 (M + H)+.
2-(tert-Butoxycarbonyl(3-fluorophenethyl)amino)acetic acid (16)
To a solution of 15 (146 mg, 0.45 mmol) in MeOH (3 mL) was added aqueous NaOH (1 N, 3 mL) dropwise. The solution was stirred for 16 h. Aqueous HCl (1N, 4 mL) was added dropwise. The mixture was diluted with brine (5 mL) and extracted with ethyl acetate (3 × 15 mL). The organic layers were combined, dried over Na2SO4, and concentrated in vacuo to afford 16 as a white solid (133 mg, 0.45 mmol, quant). 1H NMR (500 MHz, CDCl3) δ 10.22 (br, 1H), 7.24 (m, 1H), 6.98 – 6.86 (m, 3H), (3.93 + 3.81) (s, rotamers, 2H), 3.49 (m, 2H), 2.84 (m, 2H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ (175.9 + 175.4), (164.1 + 162.2), 156.1, 141.7, 130.3, 124.8, 116.0, 113.5, 81.2, 50.5, 49.3, 34.8, 28.5; ESMS (negative ion mode) m/z = 296 (M - H)—.
1-(2-(Allyloxy)ethyl)-3-fluorobenzene (17)
The general alkylating procedure was carried out on 3-fluorophenethanol (127 μL, 1 mmol) using allyl bromide (130 μL, 1.5 mmol) as the alkylating agent and THF as the solvent. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 1:4) to afford 17 (143 mg, 0.79 mmol, 79%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.23 (m, 1H), 7.00 (d, J = 7.5 Hz, 1H), 6.91 (m, 2H), 5.89 (m, 1H), 5.25 (dd, J = 1, 17 Hz, 1H), 5.17 (d, J = 10 Hz, 1H), 3.98 (d, J = 5.5 Hz, 2H), 3.64 (t, J = 7 Hz, 2H), 2.89 (t, J = 7 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ162.1, 141.9, 134.9, 129.9, 124.8, 117.2, 116.0, 113.3, 72.2, 70.9, 36.3.
2-(3-Fluorophenethoxy)acetaldehyde (18)
A solution of 17 (143 mg, 0.79 mmol) in CH2Cl2 (5 mL) was cooled to —78 °C. Ozone was passed through the solution for 1 h. Zn powder (104 mg, 1.6 mmol) and 50% aqueous acetic acid (5 mL) were added, and the mixture was allowed to warm to room temperature. The mixture was stirred a further 1 h. The mixture was poured into NaHCO3 (aq), and the product was extracted with CH2Cl2 (3 × 20 mL). The organic layers were combined, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 1:3) to afford 18 as a white solid (110 mg, 0.61 mmol, 80%). 1H NMR (500 MHz, CDCl3) δ 9.69 (s, 1H), 7.25 (m, 1H), 6.94 (m, 3H), 4.07 – 3.43 (m, 4H), 2.90 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 200.1, 163.0, 141.2, 130.1, 124.8, 116.1, 113.6, 73.3, 72.6, 36.1.
tert-Butyl 3-((6-(benzyl(tert-butoxycarbonyl)amino)-4-methylpyridin-2-yl)methyl)-4-(2-(tert-butoxycarbonyl(3-fluorophenethyl)-amino)ethoxy) pyrrolidine-1-carboxylate (19)
The general reductive amination procedure was carried out using aldehyde 12 (463 mg, 0.86 mmol), 3-fluorophenethylamine (224 μL, 1.72 mmol), and NaHB(OAc)3 (212 mg, 1 mmol). To the crude mixture was added DIEA (200 μL, 1.39 mmol) and Boc2O (218 mg, 1 mmol), and the solution was stirred for 2 h. The mixture was poured into brine (15 mL) and extracted with EtOAc (3 × 15 mL). The organic layers were combined, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 1:4) to afford 19 as a white solid (249 mg, 0.33 mmol, 38%). 1H NMR (500 MHz, CDCl3) δ 7.42 (m, 1H), 7.24 – 7.17 (m, 6H), 6.94 – 6.86 (m, 3H), 6.62 (m, 1H), 5.17 (s, 2H), 3.60 (m, 1H), 3.52 – 3.17 (m, 9H), 3.06 (q, J = 11 Hz, 1H), 2.88 – 2.79 (m, 3H), 2.68 (m, 1H), 2.58 (m, 1H), 2.28 (m, 3H), 1.43 (m, 27H); 13C NMR (125 MHz, CDCl3) δ 164.1, 162.1, 157.9, 155.5, 154.8, 154.6, 154.1, 148.8, 142.2, 140.1, 130.1, 128.3, 127.2, 126.7, 124.8, 120.2, 117.3, 115.9, 113.5, 81.4, 79.8, 79.3, 68.3, 64.5, 60.6, 50.3, 50.2, 49.5, 47.6, 43.0, 35.0, 34.5, 28.7, 28.4, 21.4; ESMS m/z = 763 (M + H)+.
6-((4-(2-(3-Fluorophenethylamino)-ethoxy)-pyrrolidin-3-yl)methyl)-4-methyl-pyridin-2-amine (2)
To a solution of 19 (190 mg, 0.25 mmol) in EtOH (5.0 mL) was added a 1:1 mixture of EtOH / concentrated HCl (10 mL) and Pd(OH)2/C (20%, 150 mg). The mixture was charged with H2 at 575 psi. The reaction was allowed to stir at room temperature for 48 h. The catalyst was removed by filtration through Celite, and the resulting Celite cake was washed with EtOH (4 × 3 mL) and 2 N HCl (3 mL). The combined solution was concentrated in vacuo to yield 2 as a white trihydrochloride salt (84 mg, 0.23 mmol, 90%). 1H NMR (500 MHz, D2O) δ 7.24 (q, J = 7 Hz, 1H), 7.02 (d, J = 8 Hz, 1H), 6.97 (d, J = 10 Hz, 1H), 6.90 (t, J = 9 Hz), 6.57 (s, 1H), 6.45 (s, 1H), 4.10 (m, 1H), 3.75 (m, 1H), 3.70 (m, 1H), 3.63 – 3.53 (m, 4H), 3.38 (m, 1H), 3.29 – 3.20 (m, 4H), 3.04 (m, 1H), 2.96 (m, 1H), 2.84 (m, 1H), 2.68 (m, 1H), 2.21 (s, 3H); 13C NMR (125 MHz, D2O) δ 163.8, 161.8, 158.4, 154.0, 145.9, 138.9, 130.8, 124.9, 115.6, 114.4, 110.6, 78.4, 64.0, 60.4, 49.5, 48.4, 47.2, 47.1, 41.7, 31.4, 29.3, 21.3; ESMS m/z = 373 (M + H)+. HRMS calcd 373.24036 found 373.24036 (M + H)+.
tert-Butyl 3-(2-(tert-butoxycarbonyl-(3-fluorophenethyl)amino)acetamido)-4-((6-(tert-butoxycarbonylamino)-4-methylpyridin-2-yl)methyl)pyrrol-idine-1-carboxylate (20)
The general procedure for amide formation was carried out on 16 (76 mg, 0.26 mmol) and 14 (106 mg, 0.25 mmol) using EDC (54 mg, 0.28 mmol), HOBt (38 mg, 0.28 mmol), and DIEA (52 μL, 0.28 mmol). The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 2:1) to afford 20 (151 mg, 0.22 mmol, 88%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.62 (s, 1H), 7.24 (m, 1H), 6.91 (m, 3H), 6.60 (m, 1H), 4.57 (m, 1H), 3.81 (m, 2H), 3.56 – 3.39 (m, 4H), 3.25 – 3.06 (m, 2H), 2.86 – 2.60 (m, 5H), 2.27 (s, 3H), 1.52 (s, 9H), 1.42 (s, 9H), 1.38 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 171.3, 164.1, 162.1, 157.2, 156.5, 154.4, 152.7, 151.6, 150.3, 141.2, 130.3, 124.8, 119.4, 116.0, 113.7, 110.7, 81.4, 80.9, 79.6, 52.4, 51.1, 50.5, 49.2, 49.0, 40.9, 36.3, 34.7, 28.6, 21.5; ESMS m/z = 686 (M + H)+, 708 (M + Na)+.
N-(4-((6-Amino-4-methylpyridin-2-yl)-methyl)-pyrrolidin-3-yl)-2-(3-fluorophenethylamino)-acetamide (3)
The general procedure for the removal of Boc groups was carried out on 20 (151 mg, 0.22 mmol) to give 3 (94 mg, 0.19 mmol, 86%) as a pale yellow trihydrochloride salt. 1H NMR (500 MHz, D2O) δ 7.24 (q, J = 7.5 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.92 (m, 2H), 6.50 (s, 2H), 4.55 (m, 1H), 3.89 (s, 2H), 3.59 (m, 1H), 3.46 (m, 1H), 3.32 (m, 1H), 3.22 (m, 2H), 3.10 (m, 1H), 2.93 (t, J = 8 Hz, 2H), 2.83 (m, 1H), 2.73 (t, J = 7.5 Hz, 2H), 2.15 (s, 3H); 13C NMR (125 MHz, D2O) δ 166.3, 163.8, 161.9, 158.3, 154.0, 145.2, 138.8, 130.9, 124.9, 115.8, 114.4, 110.7, 50.5, 50.4, 48.6, 48.1, 48.0, 40.4, 31.5, 29.4, 21.4; ESMS m/z = 386 (M + H)+. HRMS calcd 386.23561, found 386.23616 (M + H)+.
tert-Butyl 3-(tert-butoxycarbonyl(2-(3-fluorophenethoxy)ethyl)amino)-4-((6-(tert-butoxycarbonylamino)-4-methylpyridin-2-yl)methyl)pyrrolidine-1-carboxylate (21)
The general reductive amination procedure was carried out using amine 14 (69 mg, 0.17 mmol) and aldehyde 18 (27 mg, 0.15 mmol) with NaHB(OAc)3 (42 mg, 0.2 mmol). DIEA (100 μL, 0.7 mmol) and Boc2O (50 mg, 0.23 mmol) were added to the crude product, and the solution was stirred another 2 h. The mixture was poured into NaHCO3 (aq), and the product was extracted with CH2Cl2 (3 × 20 mL). The organic layers were combined, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 1:3) to afford 21 as a white solid (81 mg, 0.12 mmol, 80%). 1H NMR (500 MHz, CDCl3) δ 7.60 (m, 1H), 7.31 – 7.23 (m, 2H), 6.93 (m, 2H), 6.61 (m, 1H), 3.64 – 3.48 (m, 5H), 3.42 – 3.04 (m, 5H), 2.87 (m, 4H), 2.62 (m, 1H), 2.44 (m, 1H), 2.29 (m, 3H), 1.52 (s, 9H), 1.44 (s, 18H); ESMS m/z = 673 (M + H)+, 695 (M + Na)+.
6-((4-(2-(3-Fluorophenethoxy)ethylamino)pyrrolidin-3-yl)methyl)-4-methyl-pyridin-2-amine (4)
The general procedure for the removal of Boc groups was carried out on 21 (81 mg, 0.12 mmol) to give 4 (54 mg, 0.11 mmol, 92%) as a white trihydrochloride salt. 1H NMR (500 MHz, D2O) δ 7.12 (m, 1H), 6.97 – 6.91 (m, 2H), 6.73 (m, 1H), 6.61 – 6.53 (m, 2H), 4.24 (m, 1H), 4.12 (m, 1H), 3.85 – 3.45 (m, 6H), 3.34 – 3.17 (m, 3H), 3.10 – 2.99 (m, 2H), 2.82 – 2.69 (m, 2H), 2.54 (m, 1H), 2.20 (s, 3H); ESMS m/z = 373 (M + H)+. HRMS calcd 373.24036, found 373.24082 (M + H)+.
2-(4-((6-(Benzyl(tert-butoxycarbonyl)amino)-4-methyl-pyridin-2-yl)methyl)-1-(tert-butoxycarbonyl)-pyrrolidin-3-yloxy)acetic acid (22)
To a solution of 12 (166 mg, 0.31 mmol) in anhydrous DMF (3 mL) was added oxone (190 mg, 0.31 mmol). The mixture was stirred at room temperature for 3 h, and the solvent was removed in vacuo. The residue was dissolved in 1N HCl (aq) and extracted with EtOAc (5 × 15 mL). The organic layers were combined, dried over anhydrous Na2SO4, and concentrated in vacuo to afford 22 as a white solid (143 mg, 0.258 mmol, 83%). 1H NMR (500 MHz, CDCl3) δ 7.25 (m, 6H), 6.92 – 6.70 (m, 1H), 5.18 (m, 2H), 4.12 (m, 1H), 3.89 (m, 2H), 3.48 (m, 2H), 3.17 (m, 2H), 2.92 (m, 2H), 2.59 (m, 1H), 2.32 (m, 3H), 1.44 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 172.4, 158.4, 157.0, 155.1, 154.4, 151.5, 139.3, 128.5, 127.2, 120.7, 87.9, 82.1, 79.8, 74.8, 66.6, 51.1, 50.4, 49.4, 42.9, 34.8, 28.7, 28.4, 21.6; ESMS m/z = 556 (M + H)+, ESMS (negative ion mode) m/z = 554 (M — H)+.
tert-Butyl 3-((6-(benzyl(tert-butoxycarbonyl)-amino)-4-methylpyridin-2-yl)methyl)-4-(2-(3-fluorophenethylamino)-2-oxoethoxy)pyrrolidine-1-carboxylate (23)
The general procedure for amide formation was carried out on 22 (143 mg, 0.258 mmol) and 3-fluorophenethylamine (40 μL, 0.3 mmol) using TEA (43 μL, 0.3 mmol, EDC (50 mg, 0.26 mmol), and HOBt (35 mg, 0.26 mmol). The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 2:1) to afford 23 as a white solid (35 mg, 0.05 mmol, 20%). 1H NMR (500 MHz, CDCl3) δ 7.44 (m, 1H), 7.23 (m, 6H), 6.95 (m, 1H), 6.89 (m, 2H), 6.52 (m, 1H), 5.14 (s, 2H), 3.87 (m, 1H), 3.67 – 3.36 (m, 6H), 3.13 (m, 1H), 2.99 (m, 1H), 2.79 (m, 2H), 2.69 (m, 1H), 2.54 (m, 2H), 2.30 (m, 3H), 1.45 (s, 9H), 1.40 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 169.5, 164.2, 157.2, 154.3, 149.0, 140.0, 130.4, 128.4, 127.0, 126.9, 124.6, 120.0, 117.5, 115.9, 113.8, 83.7, 81.6, 80.4, 79.7, 68.9, (50.8 + 50.5), 50.2, (49.3 + 49.0), (42.7 + 42.2), 39.8, 35.6, 34.5, 28.7, 28.4, 21.4; ESMS m/z = 677 (M + H)+.
tert-Butyl 3-((6-(tert-butoxycarbonylamino)-4-methylpyridin-2-yl)methyl)-4-(2-(3-fluorophenethylamino)-2-oxoethoxy)pyrrolidine-1-carboxylate (24)
The general procedure for the removal of benzyl groups was carried out on 23 (34 mg, 0.05 mmol). The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 2:1) to afford 24 as a yellow oil (21 mg, 0.036 mmol, 72%). 1H NMR (500 MHz, CDCl3) δ 7.61 (m, 1H), 7.05 (s, 1H), 6.98 (m, 1H), 6.89 (m, 2H), 6.71 (m, 1H), 6.53 (s, 1H), 4.01 (m, 1H), 3.84 (m, 2H), 3.65 – 3.46 (m, 4H), 3.29 (m, 1H), 3.07 (m, H), 2.84 (m, 2H), 2.71 (m, 1H), 2.57 (m, 2H), 2.30 (s, 3H), 1.52 (s, 9H), 1.46 (s, 9H); ESMS m/z = 587 (M + H)+.
2-(4-((6-Amino-4-methylpyridin-2-yl)-methyl)-pyrrolidin-3-yloxy)-N-(3-fluorophenethyl)-acetamide (5)
The general procedure for the removal of Boc groups was carried out on 24 (0.036 mmol) to give 5 as a greasy solid (5.1 mg, 0.011 mmol, 31%). 1H NMR (500 MHz, D2O) δ 7.14 (q, J = 7 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 10 Hz, 1H), 6.75 (m, 1H), 6.55 (s, 1H), 6.26 (s, 1H), 4.00 (m, 1H), 3.86 (m, 1H), 3.70 (m, 1H), 3.42 (m, 6H), 3.13 (m, 1H), 3.01 (m, 1H), 2.75 – 2.58 (m, 3H), 2.16 (s, 3H); ESMS m/z = 387 (M + H)+. HRMS calcd 387.21963, found 387.22094 (M + H)+.
tert-Butyl 3-((6-(benzyl(tert-butoxycarbonyl)-amino)-4-methylpyridin-2-yl)methyl)-4-(2-(benzylamino)ethoxy)-pyrrolidine-1-carboxylate (25)
The general reductive amination procedure was carried out using aldehyde 12 (59 mg, 0.11 mmol), benzylamine (36 μL, 0.33 mmol), and NaHB(OAc)3 (32 mg, 0.15 mmol). The crude product was purified using flash column chromatography (silica gel, EtOAc) to afford 25 as a white solid (63 mg, 0.1 mmol, 91%). 1H NMR (500 MHz, CDCl3) δ 7.41 – 7.18 (m, 11H), 6.64 (s, 1H), 5.17 (s, 2H), 3.79 (s, 2H), 3.65 – 3.52 (m, 3H), 3.44 – 3.35 (m, 2H), 3.28 (m, 1H), 3.19 (m, 1H), 3.09 (m, 1H), 2.89 (m, 1H), 2.74 – 2.67 (m, 2H), 2.57 (m, 1H), 2.25 (s, 3H), 1.45 (s, 9H), 1.41 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 157.9, 155.0, 154.7, 154.1, 148.8, 140.6, 140.1, 128.6, 128.3, 127.3, 127.2, 126.8, 126.7, 120.2, 117.3, 81.4, 79.7, 79.4, 78.8, (68.9 + 68.8), 54.0, (51.1 + 50.6), 50.2, 49.6, (49.0, (43.0 + 42.4), 34.7, 28.8, 28.5, 21.4; ESMS m/z = 631 (M + H)+.
tert-Butyl 3-(2-aminoethoxy)-4-((6-(tert-butoxycarbonyl-amino)-4-methylpyridin-2-yl)methyl)-pyrrolidine-1-carboxylate (26)
The general procedure for the removal of benzyl groups was carried out on 25 (63 mg, 0.10 mmol). The crude product was purified using flash column chromatography (silica gel, EtOAc / MeOH, 9:1) to afford 26 as a white solid (13 mg, 0.03 mmol, 30%). 1H NMR (500 MHz, CDCl3) δ 7.55 (s, 1H), 7.20 (s, 1H), 6.58 (s, 1H), 3.73 (m, 1H), 3.58 – 3.20 (m, 6H), 3.08 (m, 1H), 2.82 (m, 2H), 2.61 (m, 1H), 2.46 (m, 1H), 2.22 (s, 3H), 1.45 (s, 9H), 1.39 (s, 9H); ESMS m/z = 451 (M + H)+.
N-(2-(4-((6-Amino-4-methylpyridin-2-yl)methyl)pyrrolidin-3-yloxy)ethyl)-2-(3-fluorophenyl)acetamide (27)
The general procedure for amide formation was carried out on 26 (13 mg, 0.03 mmol) and 3-fluorophenylacetic acid (5 mg, 0.03 mmol) using EDC (6 mg, 0.03 mmol), HOBt (5 mg, 0.03 mmol), and TEA (3 μL, 0.03 mmol). The crude product was purified using flash column chromatography (silica gel, EtOAc / hexanes, 3:1) to afford 27 as a white solid (9 mg, 0.015 mmol, 51%). 1H NMR (500 MHz, CDCl3) δ 7.63 (m, 1H), 7.23 (m, 2H), 7.01 – 6.88 (m, 3H), 6.58 (s, 1H), 6.42 (s, 0.5H), 6.30 (s, 0.5H), 3.71 (m, 1H), 3.66 – 3.52 (m, 4H), 3.50 – 3.34 (m, 4H), 3.24 (m, 1H), 3.06 (m, 1H), 2.70 (m, 1H), 2.49 (m, 2H), 2.31 (s, 3H), 1.53 (s, 9H), 1.46 (s, 9H); ESMS m/z = 587 (M + H)+, 609 (M + Na)+.
N-(2-(4-((6-Amino-4-methylpyridin-2-yl)-methyl)pyrrolidin-3-yloxy)ethyl)-2-(3-fluorophenyl)acetamide (6)
The general procedure for the removal of Boc groups was carried out on 27 (9 mg, 0.015 mmol) to give 6 as a greasy solid (3 mg, 0.0065 mmol, 44%). 1H NMR (500 MHz, D2O) δ 7.27 (s, 1H), 7.10 (q, J = 7.5 Hz, 1H), 6.97 (d, J = 7.5, 1H), 6.92 (d, J = 8Hz, 1H), 6.72 (m, 1H), 6.52 (s, 1H), 6.16 (s, 1H), 3.86 (m, 1H), 3.67 (m, 1H), 3.59 (m, 1H), 3.52 (m, 1H), 3.50 – 3.39 (m, 2H), 3.29 (m, 3H), 3.08 (dd, J = 3, 13 Hz, 1H), 2.89 (t, J = 11.5 Hz, 1H), 2.50 – 2.37 (m, 3H), 2.13 (s, 3H); ESMS m/z = 387 (M + H)+. HRMS calcd 387.21963, found 387.22097 (M + H)+.
In vitro enzyme assay
The NOS isoforms used were recombinant enzymes overexpressed in E. coli. Murine macrophage iNOS,14 rat nNOS,15 and bovine eNOS16 were overexpressed and isolated as reported. The formation of nitric oxide was monitored using a hemoglobin capture assay as described previously. Briefly, a solution of nNOS or eNOS containing 10 μM L-arginine, 1.6 mM CaCl2, 11.6 μg /mL calmodulin, 100 μM DTT, 100 μM NADPH, 6.5 μM H4B, 125 μg/mL oxyhemoglobin, and varying concentrations of inhibitor in 100 mM Hepes (pH 7.4) was monitored at 30 °C. For the determination of inhibition of iNOS, no additional Ca2+ or calmodulin were added. The assay was initiated by the addition of enzyme, and the absorption of UV light at 400 nm was recorded over one minute. As NO was evolved and coordinated to the hemoglobin, the absorption at 400 nm increased, producing a value for the enzyme velocity under these conditions. A value for the initial rate was obtained when no inhibitor was added (v0). The velocity of the enzyme (v) was then determined in the presence of varying concentrations of inhibitor, until a concentration of inhibitor that reduced the enzyme velocity to half its initial value (v/v0 ~ 0.5) was discovered. Concentrations of inhibitor above and below this value were tested and a graph of v/v0 versus inhibitor concentration ([I]) was plotted. Extrapolation of this graph allowed the determination of an IC50 value. Experiments were repeated at least three times or until graphs had R2 > 0.95. The Ki value can be estimated from the IC50 value if the Km for the substrate is known, using the equations below. The Km values used were: 1.3 μM (nNOS), 8.3 μM (iNOS) and 1.7 μM (eNOS).
In vivo brain uptake assay
1 and 2 were administered at 3.7 mg/kg to naive mice via ip injection as a 1 mM solution in phosphate buffered saline (PBS). At 5, 10, and 20 min after administration, mice were sacrificed, and blood was drawn by intracardiac puncture. The mice were perfused with PBS, and the brains were harvested.
Plasma and brain concentrations of 1 and 2 were quantified by liquid chromatography-tandem mass spectrometry after sample preparation by solid phase extraction. In brief, to an aliquot of plasma (100 μL) was added 3 or 4 (10 μL of 10 μM stock) as an internal standard. The mixture was diluted with 2% acetic acid (500 μL). Brains were weighed and homogenized in 2% formic acid / acetonitrile (2:1, 700 μL) and 3 or 4 (10 μL of 10 μM) was added. The homogenate was transferred to a centrifuge tube, and the homogenizer was washed with 2% formic acid in water / acetonitrile (2:1, 300 μL), which was also added to the centrifuge tube. The samples were centrifuged (10 krpm, 12 min), and the supernatant was transferred to a separate tube. Plasma and brain samples were loaded onto a preconditioned Oasis MCX micro-elution plate. The well was washed with 2% formic acid (500 μL) and methanol (500 μL), and the compound was eluted with 5% NH4OH in methanol (2 × 400 μL). The solvent was removed under a stream of nitrogen, and the residue was reconstituted in LCMS/MS mobile phase (200 μL). An aliquot (20 μL) was analyzed by LCMS and compared to standard curves. Standard curves for each compound in each matrix were linear between 0.005 and 5 μg/mL with R2 > 0.99.
Samples containing 2 and 4 were eluted isocratically from a Phenomenex MAX column (50 × 2.0 mm, Phenomenex, Torrance, CA) with a mobile phase consisting of water and methanol (80:20) containing 0.1 % TFA at a flow rate of 250 μL / min. The tandem mass spectrometer was operated with its electrospray source in the positive ionization mode. The mass to charge ratios of the precursor-to-product ion reactions monitored were 373.3→123.1 for 2 and 4. The retention time of 2 was 2.38 min; that of 4 was 4.74 min. Samples containing 1 and 4 were eluted isocratically from a Phenomenex MAX column (50 × 2.0 mm, Phenomenex, Torrance, CA) with a mobile phase consisting of water and methanol (75:25) containing 0.1 % TFA at a flow rate of 125 μL / min. The tandem mass spectrometer was operated with its electrospray source in the positive ionization mode. The mass to charge ratios of the precursor-to-product ion reactions monitored were 372.3→123.2 for 1. The retention time of 1 was 2.46 min; that of 4 was 4.74 min.
Supplementary Material
Figure 1.

nNOS-selective analogue inhibitors synthesized
Acknowledgements
The authors are grateful to the National Institutes of Health for financial support to R.B.S. (GM49725) and Dr. Bettie Sue Masters (GM52419, with whose laboratory P.M. and L.J.R. are affiliated), as well as the Robert A. Welch Foundation to Dr. Bettie Sue Masters (AQ1192). P.M. is supported by grants 0021620806 and 1M0520 from MSMT of the Czech Republic. The authors wish to thank Dr. Michael Avram and Lynn Luong of the Northwestern Clinical Pharmacology Core Facility and the Pharmaceutical Chemistry Translational Resource of the Center for Drug Discovery and Chemical Biology for carrying out the SPE and LCMS experiments.
Footnotes
Supporting Information Available: Chromatographic conditions and chromatograms for the determination of purity of compounds 2, 3, 4, 5 and 6. This material is available free of charge via the Internet at http://pubs.acs.org.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang L, Dawson VL, Dawson TM. Role of nitric oxide in Parkinson’s disease. Pharmacol. Therapeut. 2006;109:33–41. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 2002;40:511–526. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 3.Zicha J, Pechanova O, Dobesova Z, Kunes J. Hypertensive response to chronic N-ω-nitro-L-arginine methyl ester (L-NAME) treatment is similar in immature and adult Wistar rats. Clin. Sci. 2003;105:483–489. doi: 10.1042/CS20030078. [DOI] [PubMed] [Google Scholar]
- 4.Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer’s disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J. Neurosci. 2008;28:1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martásek P, Roman LJ, Vásquez-Vivar J, Poulos TL, Silverman RB. Selective neuronal nitric oxide synthase inhibitors for prevention of cerebral palsy. Ann. Neurol. doi: 10.1002/ana.21555. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adinolfi M, Beck Susan E., Haddad Susan A., Seller Mary J. Permeability of the blood-cerebrospinal fluid barrier to plasma proteins during foetal and perinatal life. Nature (London, United Kingdom) 1976;259:140–141. doi: 10.1038/259140a0. [DOI] [PubMed] [Google Scholar]
- 7.Seelig A. The role of size and charge for blood-brain barrier permeation of drugs and fatty acids. J. Mol. Neurosci. 2007;33:32–41. doi: 10.1007/s12031-007-0055-y. [DOI] [PubMed] [Google Scholar]
- 8.Ji H, Stanton BZ, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J. Am. Chem. Soc. 2008;130:3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawton GR, Ji H, Silverman RB. Remote protection prevents unwanted cyclizations with 2-aminopyridines. Tetrahedron Lett. 2006;47:6113–6115. [Google Scholar]
- 10.Travis BR, Sivakumar M, Hollist GO, Borhan B. Facile oxidation of aldehydes to acids and esters with oxone. Org. Lett. 2003;5:1031–1034. doi: 10.1021/ol0340078. [DOI] [PubMed] [Google Scholar]
- 11.Hevel JM, Marletta MA. Nitric-oxide synthase assays. Method Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 12.Dagnall SP, Hague DN, McAdam ME. C-13 Nuclear magnetic-resonance study of the protonation sequence of some linear aliphatic polyamines. J. Chem. Soc., Perkin Trans. 2. 1984:1111–1114. [Google Scholar]
- 13. Molecular properties were calculated using Molinspiration software ( www.molinspiration.com) on the expected form at pH 7.4.
- 14.Hevel JM, White KA, Marletta MA. Purification of the Inducible Murine Macrophage Nitric-Oxide Synthase - Identification as a Flavoprotein. J. Biol. Chem. 1991;266:22789–22791. [PubMed] [Google Scholar]
- 15.Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. High-Level Expression of Functional-Rat Neuronal Nitric-Oxide Synthase in Escherichia-Coli. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martasek P, Liu Q, Liu JW, Roman LJ, Gross SS, Sessa WC, Masters BSS. Characterization of bovine endothelial nitric oxide synthase expressed in E-coli. Biochem. Biophys. Res. Commun. 1996;219:359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.