

Abstract
Colon tumors expressing high levels of β‐catenin and c‐myc have been reported in male F344 rats given three short cycles of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP) alternating with a high‐fat (HF) diet. Using the same experimental protocol, rats were euthanized 24 h after the last dose of PhIP so as to examine early changes in colonic crypt homeostasis and β‐catenin expression, before the onset of frank tumors. PhIP/HF dosing caused a significant increase in the bromodeoxyuridine labeling index throughout the entire colon, and within the colonic crypt column cleaved caspase‐3 was elevated in the basal and central zones, but reduced in the luminal region. In vehicle/HF controls, β‐catenin was immunolocalized primarily at the border between cells at the top of the crypt, whereas in rats given PhIP/HF diet there was strong cytoplasmic staining, which appeared as a gradient of increased β‐catenin extending from the base of the crypt column to the luminal region. Quantitative real‐time PCR and immunoblot analyses confirmed that β‐catenin and c‐myc were increased significantly in the colonic mucosa of rats given PhIP/HF diet. Collectively, these findings suggest that PhIP/HF cycling alters β‐catenin and c‐myc expression in the colonic mucosa, resulting in expansion of the proliferative zone and redistribution of apoptotic cells from the lumen to the central and basal regions of the colonic crypt. Thus, during the early stages of colon carcinogenesis, alternating exposure to heterocyclic amines and a high‐fat diet might facilitate molecular changes resulting in dysregulated β‐catenin and c‐myc expression. (Cancer Sci 2008; 99: 1754–1759)
High‐temperature cooking of meat and fish generates heterocyclic amine mutagens,( 1 ) including the compound 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP).( 2 ) PhIP is a multiorgan carcinogen in the rat, inducing tumors of the colon, prostate, mammary gland and other sites.( 3 ) These tumors are characterized by high expression levels of β‐catenin and β‐catenin/T‐cell factor (Tcf) target genes,( 4 , 5 , 6 , 7 ) as seen in primary human colon cancers and human colorectal cancer cell lines.( 8 , 9 , 10 )
Recently, it was reported that three short cycles of PhIP alternating with a high‐fat (HF) diet was as effective for the induction of colon tumors as continuous dietary treatment with the carcinogen.( 11 , 12 ) It was speculated that with continuous PhIP administration, multiple disadvantageous mutations might interfere with the growth of focal populations, resulting in cell death, so that only a subset of transformed cells survive and proliferate. It was suggested that intervals between PhIP exposure might produce ‘less chance of lethal mutations occurring [and] high‐fat diet could directly promote cell growth’.( 11 ) This selection process might be further influenced by postinitiation exposure to dietary phytochemicals, so that certain oncogenic mutants progress in preference to others, such as those affecting β‐catenin stability.( 7 , 12 )
To provide insight into the possible selection pressures during early stages of colon carcinogenesis, we examined the effects of short‐term intermittent PhIP/HF diet exposure on colonic cell homeostasis in the rat, before the onset of frank colon tumors. We report here on the induction of cell proliferation and apoptosis within the proliferative zone of the colonic crypt, with strongly elevated levels of cytoplasmic and nuclear β‐catenin. There was a concomitant increase in β‐catenin and c‐myc mRNA and protein expression, consistent with results obtained in colon tumors at 1 year.( 12 )
Materials and Methods
Animals and treatment. Prior approval was obtained from the Institutional Animal Care and Use Committee. The treatment protocol is illustrated in Fig. 1, and full details were reported recently by Wang et al.( 12 ) In brief, male F344 rats obtained at 4–5 weeks of age from the US National Cancer Institute were housed two per cage and given AIN93G diet (Dyets Inc., Bethlehem, PA) and water ad libitum. After 1 week acclimatization to the cages, PhIP (Toronto Research Chemicals, Ontario, Canada) was administered to each rat by daily oral gavage at a dose of 50 mg/kg body weight, which approximates the daily exposure in rats given 400 parts per million (p.p.m.) PhIP in the diet.( 3 ) Controls received vehicle alone (0.8% dimethyl sulfoxide [DMSO] in ultra‐pure water, adjusted to pH 3.5 with 0.1 N HCl). Following 2 weeks of PhIP or vehicle treatment, rats were placed on AIN93G diet supplemented with Primex hydrogenated vegetable oil, comprising 23% fat‐derived calories, and referred to hereafter as high‐fat (HF) diet. This diet was obtained from a commercial vendor (Dyets Inc., Bethlehem, PA, USA), and the composition is shown in Table 1. After 4 weeks on HF diet, animals were returned to standard AIN93G diet and treated again with PhIP or vehicle for 2 weeks, followed by an additional 4 weeks on HF diet. A third cycle of PhIP or vehicle then was completed, and 24 h later the rats were euthanized by CO2 inhalation. Colons from four PhIP‐treated rats and three vehicle‐treated rats were Swiss‐rolled and fixed in 10% formalin for immunohistochemical analyses. Colons from five additional PhIP‐treated rats and six vehicle‐treated rats were opened longitudinally, and the mucosa was scraped and frozen in liquid nitrogen, then stored at –80°C for subsequent molecular work.
Figure 1.
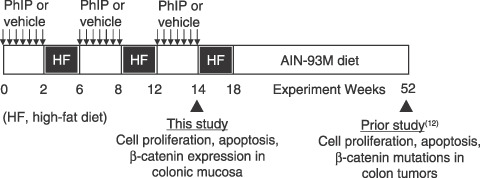
Experimental protocol for short‐term intermittent 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP) treatment alternating with high‐fat diet. This figure was modified from a recent report by Wang et al., which provided full details of the treatment protocol.( 12 ) In brief, male F344 rats were given PhIP by daily oral gavage at a dose of 50 mg/kg body weight, whereas controls received vehicle alone (0.8% dimethyl sulfoxide [DMSO] in ultra‐pure water, adjusted to pH 3.5 with 0.1 N HCl). Following 2 weeks of PhIP or vehicle treatment, rats were placed on AIN93G diet supplemented with Primex hydrogenated vegetable oil, comprising 23% fat‐derived calories (high‐fat [HF] diet). After 4 weeks on HF diet, animals were returned to standard AIN93G diet and treated again with PhIP or vehicle for 2 weeks, followed by an additional 4 weeks on HF diet. A third cycle of PhIP or vehicle then was completed, and 24 h later the rats were euthanized by CO2 inhalation. Note: in our prior study, which lasted 1 year,( 12 ) rats were switched as recommended from AIN93G (growth) diet to AIN94M (maintenance) diet at week 18.
Table 1.
Composition of AIN93G and high‐fat (HF) AIN93G diets
| Ingredient | AIN93G (Dyets #110700) g/kg | HF AIN93G (Dyets #181059) g/kg |
|---|---|---|
| Casein | 200 | 200 |
| L‐Cystine | 3 | 3 |
| Sucrose | 100 | 63.5 |
| Cornstarch | 397.486 | 252.186 |
| Dyetrose | 132 | 83.8 |
| Soybean oil | 70 | 70 |
| t‐Butylhydroquinone | 0.014 | 0.014 |
| Primex (hydrogenated vegetable.oil) | 0 | 230 |
| Cellulose | 50 | 50 |
| Mineral mix #210025 | 35 | 35 |
| Vitamin mix #310025 | 10 | 10 |
| Choline bitartrate | 2.5 | 2.5 |
| Total | 1000 | 1000 |
Scoring of cell proliferation and apoptosis indices. Details of the methods used for tissue isolation and immunohistochemistry were reported elsewhere.( 12 ) In brief, 5–6 rats in each group were selected at random and injected i.p. with bromodeoxyuridine (BrdU, 200 µmol/kg body wt), 1 h before sacrifice. Colons were removed and processed for immunostaining using the BrdU in situ detection kit (BDBiosciences, San Jose, CA, USA). In addition, cleaved caspase‐3 was immunolocalized using the EnVision+System‐HRP Kit (Dako, Carpinteria, CA, USA) in conjunction with a polyclonal rabbit anticleaved caspase‐3 antibody, which detects the endogenous large fragment of cleaved caspase‐3 resulting from cleavage adjacent to Asp175 (Cell Signaling Inc., Danvers, MA, USA). Labeling indices were calculated as the number of positive cells divided by the total number of cells in the basal, central and apical regions of the colonic crypt column, for a minimum of 15 crypts in each of the distal, middle and proximal regions of the colon (=45 crypts scored per colon). Only complete, well‐oriented, longitudinally sectioned crypts were evaluated.
Immunohistochemical detection of β‐catenin. The basic method was identical to that used for cleaved caspase‐3, but with polyclonal rabbit antiβ‐catenin (Abcam, ab2982, Cambridge, MA, USA) at 500‐fold dilution in phosphate‐buffered saline.
Quantitative real‐time PCR (qPCR). Ctnnb1 (β‐catenin), cyclin D1, c‐myc and c‐jun mRNA levels were determined by quantitative (q)PCR and normalized to glyceraldehyde‐3‐phosphate dehydrogenase (Gapdh), exactly as reported.( 5 , 12 ) In brief, frozen samples of colonic mucosa (5–6 rats per group) were thawed and the mRNA was extracted using the RNeasy kit (Qiagen, Valencia, CA, USA). RNA was reverse‐transcribed using Omniscript Reverse Transcriptase (Qiagen) and random hexamers (Invitrogen). PCR was conducted on an Opticon Monitor 2 system (Finnzymes, Finland), in 20 µL total reaction volume containing cDNAs, SYBR Green I dye (DyNAmo master solution, Finnzymes) and gene‐specific primers. The amount of specific mRNA was quantified by determining the point at which the fluorescence accumulation entered the exponential phase (Ct), and the Ct ratio of the target gene to Gapdh was calculated for each sample. Three separate experiments were performed for each sample, and the corresponding results were expressed as mean ± SE.
Immunoblotting. β‐Catenin, c‐myc, c‐jun and cyclin D1 were examined using the immunoblotting methodology reported elsewhere, with β‐actin as loading control.( 6 , 7 )
Statistics. Data were plotted as mean ± SE and compared using Students t‐test. In the figures, significant P‐values are shown as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
Results
Cycling of PhIP/HF diet strongly induces cell proliferation throughout the colon. Immunohistochemical staining for BrdU incorporation revealed a marked difference between vehicle‐ and PhIP‐treated rats (Fig. 2). When quantified for the entire crypt column (Fig. 2a), the BrdU labeling index showed a highly significant increase in rats given PhIP in the proximal, middle and distal regions of the colon. Specifically, 6–9% of the cells were BrdU‐positive in vehicle controls, compared with 14–19% in rats given PhIP. BrdU‐positive cells also were quantified for different regions within the crypt column (Fig. 2b–d); increased labeling was seen in the central and basal regions, with little or no change in the luminal zone (upper‐third).
Figure 2.
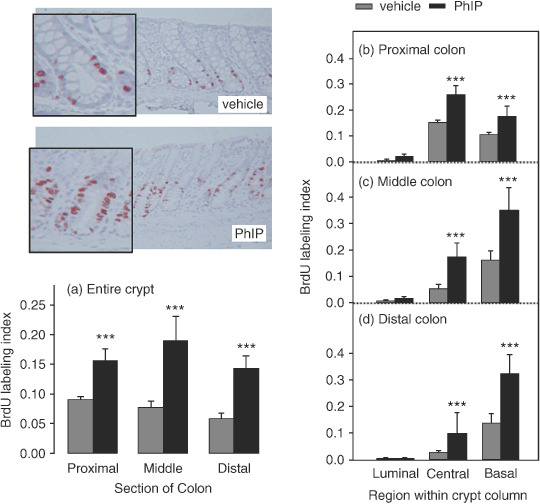
Cycling of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP)/high‐fat diet induces cell proliferation in the colon. Rats were treated with PhIP and a high‐fat diet (see Fig. 1) and the bromodeoxyuridine (BrdU) labeling index was determined as described in Materials and Methods. Data bars, mean ± SE; ***P < 0.001, by Students t‐test, PhIP (n = 4) versus corresponding vehicle controls (n = 3).
Cycling of PhIP/HF diet alters the distribution of apoptotic cells within the colonic crypt. Immunohistochemical staining for cleaved caspase‐3, a marker of apoptosis, identified positive cells along the uppermost region of the crypt in vehicle controls, and more generalized staining throughout the crypt column in rats given PhIP (Fig. 3). Quantification of the labeling index for the entire crypt column showed no significant differences in the proximal, middle or distal regions of the colon (Fig. 3a). However, when the cleaved caspase‐3 labeling index was quantified for regions within the crypt column (Fig. 3b–d), a highly significant difference was seen between vehicle controls and PhIP‐treated rats. In the latter case, cleaved caspase‐3‐positive cells were more‐or‐less evenly distributed throughout the luminal, central and basal crypt regions, whereas in vehicle controls the labeling was almost exclusively in the luminal region of the crypt. We interpret these data as evidence that PhIP/HF cycling increased cell proliferation in the lower two‐thirds of the crypt (Fig. 2), giving rise to more apoptotic cells in the same region and a concomitant reduction in cell death at the top of the crypt.
Figure 3.
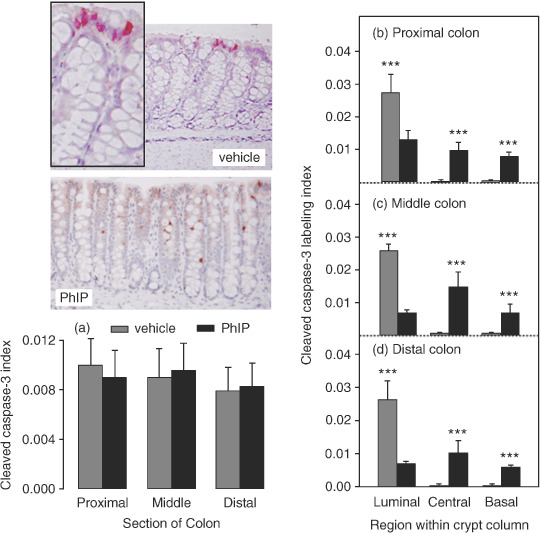
Cycling of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP)/high‐fat diet increases cleaved caspase‐3 labeling in the central and basal regions of the colonic crypt column and reduces it in the luminal region. The cleaved caspase‐3 labeling index was determined as described in Materials and Methods. Data bars, mean ± SE; ***P < 0.001, by Students t‐test, PhIP (n = 4) versus corresponding vehicle controls (n = 3).
Cycling of PhIP/HF diet increases β‐catenin staining in the colonic crypt. An important regulator of cell proliferation and apoptosis in the colon is β‐catenin and immunohistochemical staining revealed a marked difference in β‐catenin expression within the crypt column of rats given PhIP compared with vehicle (Fig. 4). Thus, in vehicle controls, histological studies showed there was a generalized low level of β‐catenin expression that was restricted to the borders between cells in the upper region of the crypt column (arrows), with no cytoplasmic or nuclear staining detected. In marked contrast, colons from rats given PhIP had widespread labeling throughout the lower half to two‐thirds of the crypt column and β‐catenin was strongly expressed in the cytoplasm and sometimes also in the nucleus (Fig. 4, open arrowheads).
Figure 4.
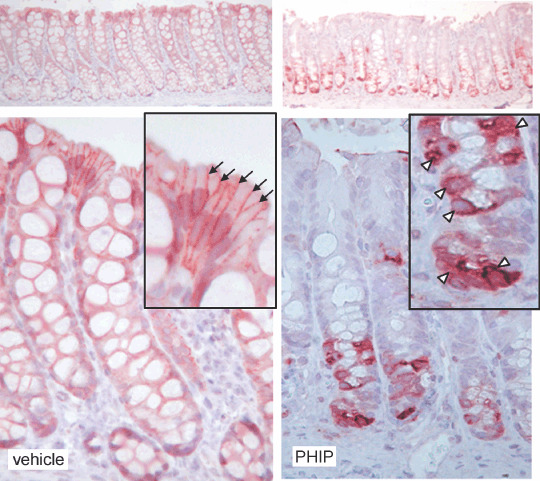
Cycling of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP)/high‐fat diet increases β‐catenin expression in the colon. Immunohistochemistry revealed low overall expression levels of β‐catenin in vehicle controls, but high levels in PhIP‐treated rats. In vehicle controls, β‐catenin was restricted to the borders between cells (arrows), with little or no cytoplasmic and nuclear staining, whereas in PhIP‐treated rats there was strong cytoplasmic β‐catenin expression (arrow heads).
Quantification of β‐catenin‐positive cells revealed significant differences between vehicle‐ and PhIP‐treated rats. Specifically, in vehicle controls, <0.2% of the cells stained positive for nuclear and/or cytoplasmic β‐catenin (grey bars, Fig. 5a), but ~78% of the cells were positive for membrane‐associated β‐catenin (grey bars, Fig. 5b), and this was seen in the proximal, middle and distal regions of the colon. In the proximal colon of PhIP‐treated rats, 4% of the cells stained positive for nuclear/cytoplasmic β‐catenin (solid bar, Fig. 5a) and ~25% for membrane‐associated β‐catenin (solid bar, Fig. 5b). In the middle colon, the corresponding data were 8–9% for nuclear/cytoplasmic β‐catenin and 18.5% for membrane‐associated β‐catenin. In the distal colon of both PhIP‐ and vehicle‐treated rats, ~75% of the cells stained positive for membrane‐associated β‐catenin (Fig. 5b), but few cells had nuclear/cytoplasmic staining.
Figure 5.
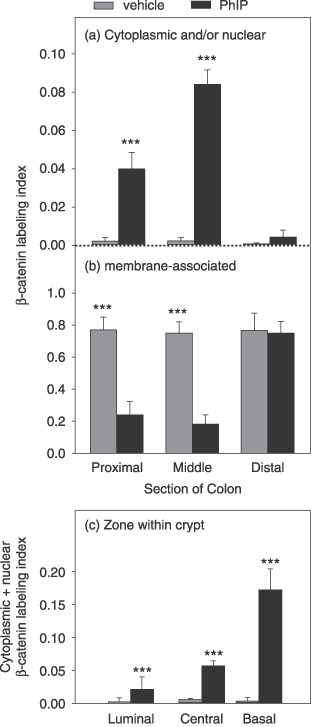
Cycling of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP)/high‐fat diet increases cytoplasmic β‐catenin expression in the colon. The β‐catenin labeling index was scored as the number of cells stained positively for cytoplasmic (and/or nuclear) versus membrane‐associated expression, divided by the total number of cells in that region of the colon. Data bars, mean ± SE; PhIP n = 4, vehicle controls n = 3. **P < 0.01; ***P < 0.001, PhIP group significantly different from the corresponding vehicle controls.
Nuclear/cytoplasmic β‐catenin labeling was further examined according to region within the colonic crypt column (Fig. 5c). There was evidence for a gradient of increased β‐catenin expression in the colonic crypt of PhIP‐treated rats compared with vehicle controls, with higher β‐catenin levels in the basal zone than in the central or luminal zones. The data presented in Fig. 5(c) for PhIP‐treated rats (solid bars) revealed 2%, 5.6% and 17.2% of the cells stained positive for nuclear/cytoplasmic β‐catenin in the luminal, central and basal regions of the crypt, respectively (P < 0.001, each region versus the corresponding vehicle control).
Cycling of PhIP/HF diet elevates β‐catenin and c‐myc mRNA and protein expression. We next examined Ctnnb1 (β‐catenin) and three β‐catenin/Tcf target genes, c‐myc, c‐jun and cyclin D1, for changes in mRNA expression (Fig. 6). Colonic mucosa from PhIP‐treated rats had 30% higher levels of Ctnnb1 and 65% higher levels of c‐myc mRNA compared with vehicle controls (P < 0.05). No difference in c‐jun or cyclin D1 expression was observed after normalizing to Gapdh.
Figure 6.
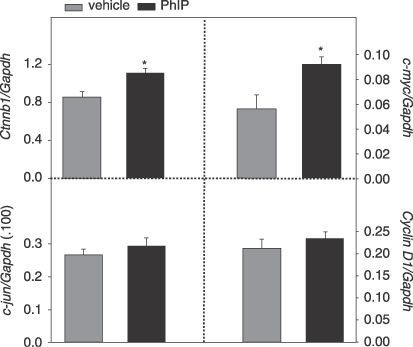
Cycling of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP)/HF diet increases β‐catenin and c‐myc mRNA expression in the rat colon. Quantitative real‐time PCR was performed as described in Materials and Methods, for β‐catenin (Ctnnb1) and three β‐catenin/Tcf target genes, namely c‐myc, c‐jun and cyclin D1, normalized to Gapdh. Data bars, mean ± SE, PhIP n = 5, vehicle controls n = 6. *P < 0.05, PhIP group significantly different from corresponding vehicle controls.
Immunoblotting revealed higher expression levels of β‐catenin and c‐myc proteins in colonic mucosa of PhIP‐treated rats, compared with vehicle controls (Fig. 7). No differences were seen among the treatment groups for cyclin D1 and c‐jun, after normalizing to β‐actin (data not shown).
Figure 7.
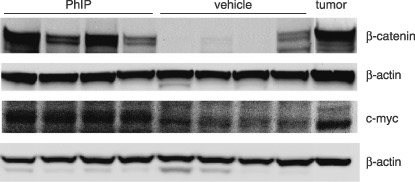
Immunodetection of increased β‐catenin and c‐myc in the rat colon before the onset of frank tumors. Representative immunoblots of β‐catenin and c‐myc in colonic mucosa from rats treated with three cycles of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP) (or vehicle) alternating with high‐fat diet. A PhIP‐induced colon tumor from the corresponding 1‐year carcinogenicity study,( 12 ) is shown in the furthest right lane as a positive control. β‐Actin, loading control.
Discussion
Ubagai et al. studied the combined effects of PhIP and a HF diet on colon carcinogenesis in the F344 rat.( 11 ) Animals were given 400 p.p.m. PhIP in the basal diet for 2 weeks, followed by HF diet for up to 110 weeks, and this resulted in 3 of 19 rats with large intestinal tumors (19% incidence). Alternatively, 400 p.p.m. dietary PhIP was given for 2 weeks, followed by 4 weeks on HF diet, and the PhIP/HF cycling was repeated three times, ending with continuous HF diet for up to 60 weeks. Large intestinal tumors were seen in 9 out of 20 rats (45% incidence). The latter result compares favorably with the 43% incidence observed in rats treated continuously with 100 p.p.m. dietary PhIP for 104 weeks,( 3 ) in which a ~10‐fold greater total amount of PhIP was administered per animal. Thus, PhIP/HF cycling represents an efficient means of inducing colon tumors, using a fraction of the amount of total carcinogen. We further modified the experimental protocol by giving PhIP via oral gavage, at a dose (50 mg/kg body wt per day) that was matched to the daily carcinogen intake from 400 p.p.m. PhIP in the diet,( 3 ) and after three cycles of PhIP/HF treatment, standard AIN93M diet rather than HF diet was administered. Using this modified protocol, the colon tumor incidence after 1 year was 41.6%.( 12 )
In the current investigation, rats were euthanized immediately after completing the third and final cycle of PhIP dosing, so as to examine changes in colonic crypt homeostasis before the onset of frank tumors. Under these conditions, there was a striking increase in BrdU‐positive cells throughout the entire length of the colon, with particularly high staining in the lower two‐thirds of the crypt column. Moreover, there was an increase in cleaved caspase‐3‐positive cells in the proliferative zone of each crypt (central and basal regions), coupled with a reduction of apoptotic cells in the luminal region. The present study was not designed to specifically compare intermittent PhIP/HF dosing with continuous dietary PhIP treatment, but this might be interesting in a follow up investigation. Previously, a modest 1.5‐fold increase in colonic BrdU labeling was seen at 8 weeks in male (but not female) rats fed continuously with 400 p.p.m. PhIP in the diet, and no significant changes were seen in cell proliferation rates for male or female rats at 4 or 12 weeks.( 13 ) However, the latter study did not assess changes in apoptosis or β‐catenin expression. In general, we observed that cleaved caspase‐3 did not show as good a concordance as did BrdU labeling for the corresponding β‐catenin positive cells.
The high cytoplasmic β‐catenin expression seen in the present study is noteworthy, because Ubagai et al.( 11 ) reported that all of the large intestinal tumors obtained following PhIP/HF treatment also had high accumulation of cytoplasmic and nuclear β‐catenin. Interestingly, only 55% of the tumors harbored a mutation in the Apc or Ctnnb1 (β‐catenin) genes, and it was suggested that PhIP/HF cycling produced ‘unknown genetic alterations’ in the Wnt–Apc–β‐catenin signaling pathway.( 11 ) Using the identical exposure protocol described here, colon tumors obtained at 1 year had a 36% frequency of β‐catenin mutations, although this was increased to 79% when rats were subjected to postinitiation treatment with caffeine, which resulted in tumor promotion.( 12 ) This clearly supports the notion that cells harboring specific genetic changes within a population can be influenced to progress (or not to progress) to neoplasia by external factors, such as phytochemicals and a HF diet. As discussed before,( 11 ) intermittent exposure to PhIP might allow populations of cells to survive and progress to tumors, when continuous carcinogen treatment might otherwise lead to removal of those cells via apoptosis. In the present study, there was increased apoptosis in the lower half of the colonic crypt following PhIP cycling, but this appeared to be a compensatory mechanism triggered by the greatly enhanced rate of cell proliferation, resulting in expansion of the crypt‐wide proliferative zone (Fig. 2).
Despite the high levels of β‐catenin detected in the present study following PhIP/HF treatment (Fig. 4), no β‐catenin mutations were observed in the colonic mucosa scrapings, using PCR‐based single strand conformation polymorphism (PCR‐SSCP) screening (data not presented). One interpretation is that β‐catenin mutations were indeed present in the colonic scrapings, but below the limit of detection of the PCR‐SSCP methodology, and that only after clonal expansion and tumor formation were the mutations readily detected owing to their enrichment relative to the wild type allele. Interestingly, among the three reported β‐catenin/Tcf target genes examined, only c‐myc was increased in the colonic mucosa of rats given PhIP/HF diet, as noted in the colon tumors obtained at 1 year.( 12 ) Interestingly, Ctnnb1 mRNA also was elevated in the colonic mucosa of rats given PhIP/HF diet (Fig. 6). Although this increase was modest, it is consistent with prior reports showing elevated levels of Ctnnb1 mRNA expression in PhIP‐ and 1,2‐dimethylhydrazine‐induced rat colon tumors,( 5 , 12 ) as well as in primary human colon carcinomas and their liver metastases.( 14 ) In some of the vehicle controls, low or undetectable levels of β‐catenin were seen via immunoblotting (Fig. 7), despite the presence of Ctnnb1 mRNA in most samples (Fig. 6) and membrane‐associated β‐catenin being detected in immunohistochemical analyses (Fig. 4). It is unclear whether this reflects the efficiency of the membrane extraction and/or immunoblot procedures used here, and further work is needed to clarify this question, perhaps on a larger subset of tissue samples from vehicle and PhIP‐treated animals. However, for the vehicle control samples shown here (Fig. 7), repeated immunoblotting confirmed the low or undetectable expression of β‐catenin, suggesting the data in this case were reproducible.
In summary, we have shown that in rats given three cycles of PhIP alternating with exposure to HF diet, there was an increase in colonic cell proliferation, elevated apoptosis in the proliferative zone of the colonic crypt and augmented expression of cytoplasmic β‐catenin. These results from the early stages of colon carcinogenesis suggested a possible independent dysregulation of β‐catenin and c‐myc expression, separate from the classical β‐catenin/Tcf pathway,( 15 ) in accordance with data obtained from the corresponding colon tumors at 1 year.( 12 ) Thus, early changes affecting colonic crypt homeostasis at the time of exposure to PhIP and HF diet may have persisted into much later stages of colon tumor formation.
Acknowledgments
We thank Mandy Louderback for assistance with oral gavage and necropsy. Laboratory Animal Service (LAR) staff are gratefully acknowledged for their help with animal care and maintenance. The experiments described here were supported in part by NIH grants CA90890, CA65525, CA90176 and CA122959, and by National Institute of Environmental Health Sciences Center grant P30 ES00210. Partial support for RHD was provided by the Foundation for Promotion of Cancer Research, Tokyo, Japan.
References
- 1. Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci 2004; 95: 290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Knize MG, Felton JS. Formation and human risk of carcinogenic heterocyclic amines formed from natural precursors in meat. Nutr Rev 2005; 63: 158–65. [DOI] [PubMed] [Google Scholar]
- 3. Ito N, Hasegawa R, Imaida K et al . Carcinogenicity of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP) in the rat. Mutat Res 1997; 376: 107–14. [DOI] [PubMed] [Google Scholar]
- 4. Dashwood RH, Suzui M, Nakagama H, Sugimura T, Nagao M. High frequency of β‐catenin (Ctnnb1) mutations in the colon tumors induced by two heterocyclic amines in the F344 rat. Cancer Res 1998; 58: 1127–9. [PubMed] [Google Scholar]
- 5. Wang R, Dashwood WM, Bailey GS, Williams DE, Dashwood RH. Tumors from rats given 1,2‐dimethylhydrazine plus chlorophyllin or indole‐3‐carbinol contain transcriptional changes in β‐catenin that are independent of β‐catenin mutation status. Mutat Res 2006; 601: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blum CA, Tanaka T, Zhong X et al . Mutational analysis of Ctnnb1 and Apc in tumors from rats given 1,2‐dimethylhydrazine or 2‐amino‐3‐methylimidazo[4,5‐f]quinoline: mutational ‘hotspots’ and the relative expression of β‐catenin and c‐jun. Mol Carcinogen 2003; 36: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blum CA, Xu M, Orner GA et al . β‐Catenin mutation in rat colon tumors initiated with 1,2‐dimethylhydrazine and 2‐amino‐3‐methylimidazo[4,5‐f]quinoline, and the effect of post‐initiation treatment with chlorophyllin and indole‐3‐carbinol. Carcinogenesis 2001; 22: 315–20. [DOI] [PubMed] [Google Scholar]
- 8. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006; 25: 7531–7. [DOI] [PubMed] [Google Scholar]
- 9. Gavert N, Ben‐Ze’ev A. beta‐Catenin signaling in biological control and cancer. J Cell Biochem 2007; 102: 820–8. [DOI] [PubMed] [Google Scholar]
- 10. Wood LD, Parsons DW, Jones S et al . The genomic landscapes of human breast and colorectal cancers. Science 2007; 318: 1108–13. [DOI] [PubMed] [Google Scholar]
- 11. Ubagai T, Ochiai M, Kawamori T et al . Efficient induction of rat large intestinal tumors with a new spectrum of mutations by intermittent administration of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine in combination with a high‐fat diet. Carcinogenesis 2002; 23: 197–200. [DOI] [PubMed] [Google Scholar]
- 12. Wang R, Dashwood WM, Löhr CV et al . Protective versus promotional effects of white tea and caffeine on PhIP‐induced tumorigenesis and β‐catenin expression in the rat. Carcinogenesis 2008; 29: 834–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ochiai M, Watanabe M, Kushida H, Wakabayashi K, Sugimura T, Nagao M. DNA adduct formation, cell proliferation, and aberrant crypt focus formation induced by PhIP in male and female rat colon with relevance to carcinogenesis. Carcinogenesis 1996; 17: 95–8. [DOI] [PubMed] [Google Scholar]
- 14. Mann B, Gelos M, Siedow A et al . Target genes of β‐catenin‐T cell‐factor/lymphoid‐enhancer‐factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 1999; 96: 1603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yekkala K, Baudino TA. Inhibition of intestinal polyposis with reduced angiogenesis in ApcMin/+ mice due to decreases in c‐Myc expression. Mol Cancer Res 2007; 5: 1296–303. [DOI] [PubMed] [Google Scholar]