

Abstract
We have previously developed a method to purify recombinant proteins, termed inverse transition cycling (ITC) that eliminates the need for column chromatography. ITC exploits the inverse solubility phase transition of an elastin-like polypeptide (ELP) that is fused to a protein of interest. In ITC, a recombinant ELP fusion protein is cycled through its phase transition, resulting in separation of the ELP fusion protein from other Escherichia coli contaminants. Herein, we examine the role of the position of the ELP in the fusion protein on the expression levels and yields of purified protein for four recombinant ELP fusion proteins. Placing the ELP at the C-terminus of the target protein (protein-ELP) results in a higher expression level for the four ELP fusion proteins, which also translates to a greater yield of purified protein. The position of the fusion protein also has a significant impact on its specific activity, as ELP-protein constructs have a lower specific activity than protein-ELP constructs for three out of the four proteins. Our results show no difference in mRNA levels between protein-ELP and ELP-protein fusion constructs. Instead, we suggest two possible explanations for these results: first, the translational efficiency of mRNA may differ between the fusion protein in the two orientations and second, the lower level of protein expression and lower specific activity is consistent with a scenario that placement of the ELP at the N-terminus of the fusion protein increases the fraction of misfolded, and less active conformers, which are also preferentially degraded compared to fusion proteins in which the ELP is present at the C-terminal end of the protein.
Keywords: elastin-like polypeptides, fusion proteins, fusion order, inverse transition cycling, protein yields, specific activities
Introduction
We have previously developed a method to purify recombinant proteins, termed inverse transition cycling (ITC) that eliminates the need for column chromatography. ITC exploits the inverse solubility phase transition of an elastin-like polypeptide (ELP) that is fused to a protein of interest.1–4 Herein, we examine the role of the fusion order (protein-ELP or ELP-protein) on the expression level, yield and specific activity of purified protein for a set of recombinant ELP fusion proteins.
ELPs are artificial, repetitive protein polymers that consist of repeats of the pentapeptide Val-Pro-Gly-Xaa-Gly, where the guest residue Xaa can be any amino acid residue except Pro.5–7 Upon heating, an aqueous solution of ELP undergoes an inverse phase transition within a very narrow temperature range (∼2–3°C); below its inverse transition temperature (Tt), the ELP is soluble in aqueous solution, but when the solution temperature is raised above its Tt, the ELP becomes insoluble and forms micron sized aggregates. The aggregation of an ELP is a reversible process, so that the ELP is completely resolubilized in buffer when the solution temperature is reduced below the transition temperature of the ELP. The inverse phase transition of ELPs can also be isothermally triggered by depressing their Tt below solution temperature by the addition of kosmotropes from the Hofmeister series.8–11
At the molecular design level, the Tt of an ELP can be tuned by two orthogonal parameters: the identity and mole fraction of the “guest” residue in the fourth position (Xaa), and the chain length of the ELP; recombinant synthesis provides complete control over these two variables, so that its is possible to synthesize ELPs with exquisite control of their Tt, which is important for different applications of these stimulus responsive polypeptides.12,13
We discovered that the phase transition behavior of ELPs is retained when ELPs are fused to soluble proteins,1 and we exploited this observation to develop a non-chromatographic method—ITC—for purification of recombinant proteins.1,3,4,12,14,15 In ITC, a recombinant ELP fusion protein is cycled through its phase transition, resulting in separation of the ELP fusion protein from other Escherichia coli contaminants. Importantly, the target protein is active in the fusion construct and its activity is retained through multiple rounds of ITC.1 ITC is highly efficient, as it enables the facile purification of soluble proteins even at low expression levels.1,3,4,12,14,15
Motivated by the goal of optimization of ITC, we have sought to examine how the design of an ELP fusion protein impacts ITC. Some of the design parameters of ELP fusion proteins are: (1) composition of the ELP; (2) its chain length; and (3) the position of the ELP relative to the target protein. In previous studies, we have investigated the effect of two of these variables—ELP chain length and composition—on ITC.1,3,4,12,15 However, the proteins in those studies were expressed as a C-terminal ELP fusion (i.e., protein-ELP). Here, we systematically examine the role of the fusion order of a set of ELP fusion proteins (i.e., N- and C-terminal fusions) on their purification by ITC, and demonstrate that the position of the ELP relative to the target protein has profound consequences on their expression level and activity.
Results
Fusion protein constructs
Four target proteins were fused to the N- or C-termini of an ELP in this study: blue fluorescent protein (BFP), chloramphenicol acetyltransferase (CAT), thioredoxin (Trx), and interleukin-1 receptor antagonist (IL1Ra). We have previously shown that BFP, CAT, and Trx express with high yields and in an active conformation when the ELP is fused to the C-terminus of the protein (protein-ELP).3,16 IL1Ra has been fused to ELPs at the N- and C- terminus in previous studies16,17 but the expression levels and yields of the purified proteins were not directly compared in these studies. The ELP in this study is a 90 pentapeptide repeat in which the guest residue is a mixture of Val, Ala, and Gly in a 5:2:3 ratio, respectively; this ELP is termed ELP[Val5Ala2Gly3-90] using the lexicographical notation that we have previously developed to describe ELPs.12
Fusion protein purification by inverse transition cycling
BLR(DE3) cells were separately transformed with the plasmids encoding all eight ELP fusion proteins, grown in triplicate in 50 mL TB media and purified by ITC.1–4
A schematic of the ITC purification method is shown in Figure 1. The phase transition of an ELP fusion protein in its soluble cell lysate is isothermally triggered by adding NaCl to the lysate at a concentration that depresses the Tt of the ELP fusion protein below the solution temperature (typically ambient). When the expression level of an ELP fusion protein is high enough, the aggregates of the ELP fusion protein that are formed cause the cell lysate to visibly turn turbid, thereby providing a convenient visual confirmation that the phase transition has occurred in the cell lysate. The turbid suspension is then centrifuged at room temperature (termed “hot spin”). The pellet, containing the fusion protein, is retained, while the supernatant is discarded. The pellet is then redissolved in low ionic strength buffer at a temperature below the Tt of the fusion protein. At this point, a centrifugation step at 4°C (termed “cold spin”) is carried out to remove contaminants that may have been physically trapped in the ELP fusion protein pellet during centrifugation. In the cold spin step, the fusion protein stays in solution, whereas denatured contaminants aggregate and are separated in the pellet fraction. The supernatant from the cold spin is retained, and the pellet is discarded. This process constitutes one round of ITC.
Figure 1.
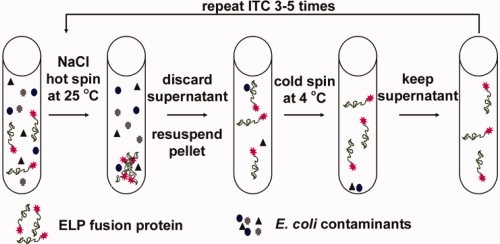
Schematic of the ITC protein purification method. First, cells are lysed and the cell debris is removed by centrifugation. NaCl is added to isothermally trigger the phase transition and the fusion protein is collected by centrifugation (hot spin). The supernatant is discarded and the pellet containing the fusion protein is resuspended in cold, low-ionic strength buffer. Some denatured contaminants may be trapped in the pellet, and therefore, a cold centrifugation step (cold spin) is carried out to remove the insoluble denatured contaminants; the fusion protein stays in solution during this step because the solution temperature is lower than the Tt of the fusion protein. The ITC process is repeated 3–5 times to obtain pure fusion protein.
All fusion proteins in this study were purified by ITC using the same basic protocol with some important exceptions; higher NaCl concentrations (5M in the first two rounds of ITC) were used to trigger the phase transition of ELP-proteins compared to the 3M NaCl used for the protein-ELP fusions; 3M NaCl is not sufficient to lower the Tt of the ELP-protein fusions below room temperature because these proteins express at lower levels than the protein-ELP fusions. As the Tt of an ELP fusion protein is inversely proportional to its concentration, dilute ELP fusion proteins have a higher Tt, so that a higher concentration of NaCl is required to depress their Tt below the ambient temperature at which the aggregation of the ELP fusion protein is carried out. Five rounds of ITC were carried out for Trx, BFP, and CAT constructs to obtain pure fusion protein as assessed by SDS-PAGE, while three rounds of ITC were sufficient to purify both IL1Ra constructs to homogeneity, as evaluated by SDS-PAGE.
The purification steps for each fusion protein were visualized by Coomassie blue stained SDS-PAGE; Figure 2 shows typical SDS-PAGE gels for Trx-ELP and ELP-Trx. After each round of ITC, the pellets were resuspended in smaller volumes to concentrate the fusion protein; for example the volume of the soluble cell lysate was 2 mL and pellets from the first and final hot spins were resuspended in 500 and 100 μL buffer, respectively. The volumes loaded on the gels at different stages of ITC for Trx-ELP and ELP-Trx in Figure 2 were proportional to the resuspended pellet volumes; the volume loaded of soluble cell lysate was 10 μL (Lane 2), the volumes after the first round of ITC in Lanes 3 and 4 were 2 μL, and the volume loaded in Lane 5 after the final cold spin was 0.4 μL. Therefore, the expression levels of the two constructs can be visually compared by comparing the intensities of the fusion constructs in the soluble cell lysates. It is clear from visual inspection of the intensities of the bands that the concentration of ELP-Trx in the soluble cell lysate was significantly lower than that of Trx-ELP, indicating that the level of expression of Trx is highly dependent on its position in the fusion protein. The resuspended pellets from the first NaCl hot spin in Lane 3 in both panels of Figure 2 and the supernatants from the first cold spin in Lane 4 showed some impurities, however, after the final (fifth) cold spin, no impurities were visible on the gel for either Trx fusion construct [Lane 5; Fig. 2(A,B)].
Figure 2.
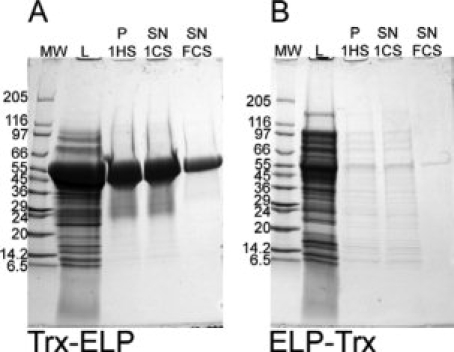
SDS-PAGE following the purification of (A) Trx-ELP and (B) ELP-Trx stained with Coomassie blue. Lane 1 on both gels: molecular weight markers (MW); Lane 2: lane soluble cell lysate (L); Lane 3: the resuspended pellet from the first hot spin (P 1HS); Lane 4: the supernatant from the first cold spin (SN 1CS); Lane 5: the supernatant from the final, fifth cold spin (SN, FCS). The molecular weights of Trx-ELP and ELP-Trx are 50.0 kDa for both proteins.
SDS-PAGE gels that document the purification of the all eight fusion protein constructs are shown in Supporting Information (Fig. S1). Bands corresponding to the fusion proteins indicate that the concentrations of ELP-protein constructs were lower in the soluble cell lysate compared to the protein-ELP constructs in all cases, similar to the expression of Trx. The SDS-PAGE data also show that ITC purification did not result in significant losses of any fusion protein during purification. The low concentrations of some the ELP-protein fusions made it difficult to visualize the progress in purification on Coomassie blue stained SDS-PAGE gels. Hence, the gels for ELP-proteins in Figure S1 were stained with silver; though we note that even the silver stained SDS-PAGE gels showed very faint bands for ELP-IL1Ra, ELP-CAT, and ELP-BFP fusion constructs. Weak bands corresponding to contaminants were observed in the silver stained gels, but the concentration of those contaminants was low. The concentration of ELP-BFP after the final cold spin was so low that the protein was not visible on the silver stained gel after the final round of ITC using the concentration as described earlier; this protein was visualized by loading an aliquot with 10-fold greater concentration (Lane 6, Fig. S1).
The yields of the purified protein-ELP fusions were greater than the ELP-protein fusions for the BFP, CAT, and Trx constructs (P < 0.01, Student's t-test) whereas the difference in the yield for IL1Ra in the two positions verge on significance (P = 0.06, Student's t-test; Fig. 3 and Table S1 in Supporting Information). From SDS-PAGE gels of samples collected at different stages of ITC, we found that the concentration of ELP-protein fusions were significantly lower in the soluble cell lysate compared to protein-ELPs (Fig. 2 and Supporting Information Fig. S1). In addition, the SDS-PAGE gels showed no significant loss of protein during purification by ITC.
Figure 3.
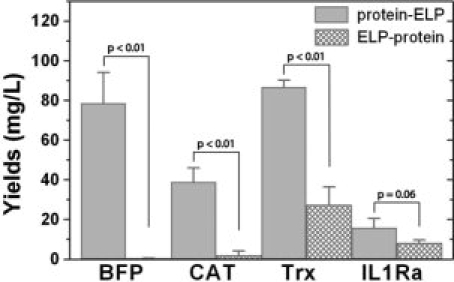
Yields obtained after purification of each fusion protein in triplicate. The proteins were grown in 50 mL cultures and the yields were extrapolated to 1 L. The raw data are reported in Table S1 in Supporting Information.
Inclusion body formation
We hypothesized that the preferential formation of inclusion bodies during expression is a possible reason for the lower expression level observed of the ELP-protein construct relative to the protein-ELP fusion in the soluble lysate. If a significant fraction of the expressed protein is shuttled into inclusion bodies, the fraction of soluble protein in the cell lysate is likely to be lower, leading to a lower level of expressed protein in the soluble cell lysate. To examine this possibility, SDS-PAGE gels that compare the soluble cell lysate with the insoluble cell lysate (resuspended in either PBS or in 6M guanidinium chloride) are shown in Figure 4. Contrary to our hypothesis, the BFP-ELP, CAT-ELP, and IL1Ra-ELP fusions, which have a high level of expression in the soluble fraction of the cell lysate as compared to constructs in the opposite order, also exhibited a significant concentration of fusion protein in the insoluble fraction of the cell lysate. In contrast, ELP-BFP, ELP-CAT, and ELP-IL1Ra, which exhibited lower levels of soluble expression, did only form small fractions of inclusion bodies of the fusion protein. The Trx fusions showed no significant formation of inclusion bodies in either fusion construct. Hence, these results clearly suggest that that shuttling of the ELP fusion proteins into insoluble inclusion bodies is not the major factor that could account for the lower expression level of soluble ELP-protein fusions as compared to the protein-ELP constructs.
Figure 4.
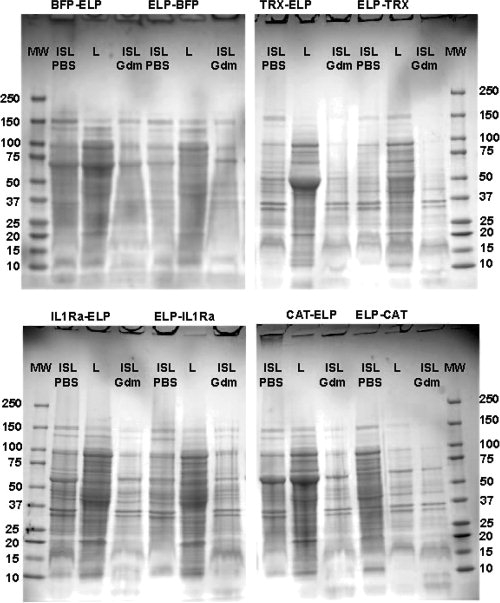
SDS-PAGE gels showing the soluble and insoluble cell lysate of BFP-ELP and ELP-BFP, Trx-ELP and ELP-Trx, IL1Ra-ELP and ELP-IL1Ra, and CAT-ELP and ELP-CAT. The insoluble cell lysate has been resuspended in either PBS or 6M guanidinium chloride. The lanes on each gel represents: the molecular weights marker (MW), the soluble cell lysate (L), the insoluble cell lysate resuspended in PBS (ISL), and the insoluble cell lysate resuspended in 6M guanidinium chloride (ISL Gdm). The molecular weights of the eight fusion proteins are: Trx-ELP, 50.0 kDa; BFP-ELP, 65.5 kDa; CAT-ELP, 62.6 kDa; IL1Ra-ELP, 54.2 kDa, ELP-Trx, 50.0 kDa, ELP-BFP, 63.7 kDa, ELP-CAT, 63.7 kDa; ELP-IL1Ra, 55.1 kDa.
mRNA levels during transcription
We next examined whether lower transcription levels are a possible cause for the consistently lower expression level of the ELP-protein construct compared to the protein-ELP construct. We quantified the mRNA levels during transcription for the two BFP constructs, which were chosen because these constructs showed the largest difference in yields compared to the other proteins. The two BFP fusion proteins were grown in TB media as described previously, and aliquots of the culture were taken for mRNA analysis at six time points from 8 to 24 h before harvesting the cells. In addition, aliquots of bacteria from the cultures were cultured on agar plates and the live bacteria were counted. No difference in bacteria levels are found between the two cultures (data not shown), suggesting that the total density of viable cells could not account for the observed difference in the expression level of the proteins. The total RNA was isolated from both cultures, followed by one-step reverse transcription and relative real-time PCR18 using BFP and ribosomal RNA specific primers (refer Fig. 5). rRNA was chosen as an internal reference because its level should be constant both cultures, so that the BFP transcript levels normalized to rRNA can be compared between the two fusion proteins. Throughout the log phase (8–16 h), mRNA levels were statistically identical, but as the bacteria entered the late stationary phase (21–24 h) greater variability was observed in BFP-ELP mRNA levels, which is probably due to accumulation of fusion protein insides the cells or stress induced nutrient depletion.19 In all cases, however, the levels of BFP-ELP mRNA were not significantly greater than that of ELP-BFP mRNA. These results lead us to conclude that differences in mRNA levels are most likely not the cause of the major differences in protein yield.
Figure 5.
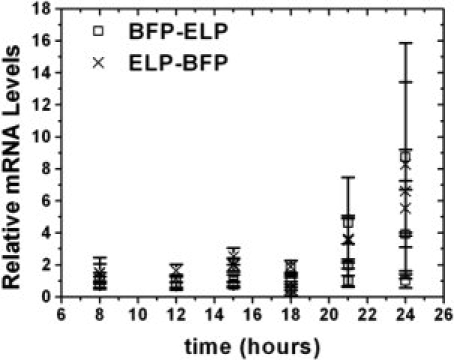
Relative mRNA levels of BFP-ELP and ELP-BFP. For each time point and construct, mRNA has been isolated from three flasks, and RT-PCR run in triplicates. Red points represent relative mRNA levels of ELP-BFP constructs, blue data point represent relative mRNA levels of BFP-ELP constructs. Bars represent a 95% confidence interval, calculated by the relative quantitation (RQ) study software (Applied Biosystems).
Specific activity of fusion proteins
An important factor to consider is the activity of the target proteins in the two positions relative to the ELP. Indeed, the specific activities of the fusion proteins are a more important measure of the yields compared to measures of the yields of purified ELP fusions determined by UV-vis spectrophotometry, because the latter may include soluble, but misfolded conformers with little or no activity. This is an important consideration for ELP fusion proteins as ELPs are highly soluble with a solubility limit of hundreds of milligrams per mL in aqueous solutions,2,3,20 so that ELP tags may allow misfolded proteins to remain soluble, whereas in the absence of the fused ELP, the misfolded conformers would most likely become insoluble and aggregate.
Previous studies have shown all ELP fusion proteins studies to date are active when fused as protein-ELP. No significant changes in activity were observed for Trx-ELP and BFP-ELP compared to the free target protein1,4 whereas CAT-ELP showed a small, ∼15% decrease in activity compared to free CAT.4 In contrast, we found that the IL1Ra-ELP activity was ∼100-fold lower17 than that of IL1Ra while ELP-IL1Ra's activity was 500-fold lower compared to the free IL1Ra, which is the largest difference observed to date for the specific activity of ELP fusion proteins compared to the native protein.16 Nevertheless, the significantly lower specific activity of IL1ra fusions compared to the native protein suggest that the IL1Ra potency is dependent upon the position in the fusion construct, and provide a rationale for these measurements.
With the exception of BFP, all ELP-protein fusions in this study had a lower specific activity compared to their protein-ELP counterpart (P < 0.01, Students t-test; refer Fig. 6). The specific activity of ELP-Trx was ∼60% of the Trx-ELP specific activity and that of the ELP-CAT construct is ∼37% of the specific activity of the ELP-CAT construct (Fig. 6 and Table S1 Supporting Information). Similar to CAT and Trx, ELP-IL1Ra also exhibited ∼25% of the specific activity of IL1Ra-ELP.
Figure 6.
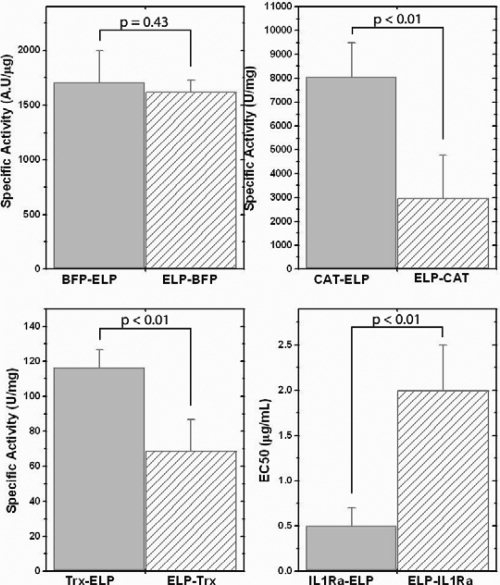
Bar graphs comparing the activities for the four target proteins fused in two directions compared to the ELP. BFP is not a biologically active molecule and the activity is measured by its fluorescence, CAT activity is measured by a 1-deoxychloramphenicol acetylation assay, Trx activity is measured by insulin reduction assay, and IL1Ra potency is measured by a lymphocyte proliferation assay.
BFP is the only exception to this trend. BFP is not a biologically active protein but fluoresces in the near-UV region and its fluorescence is sensitive to changes in the tertiary structure that may be indicative of misfolding; for example, even slight misfolding of GFP (the native wild-type protein from which BFP is derived) can limit the autooxidation of the residues that create the endogeneous fluorophore in the protein.21–23 We have, therefore, used the intrinsic fluorescence of BFP as a surrogate of its activity. No significant differences were observed in the mass normalized fluorescence of BFP-ELP and ELP-BFP.
Discussion
The development of high throughput and scalable protein purification methods is in high demand for fundamental protein interaction studies in the post-genomic era, and for the production of proteins as therapeutics. Our laboratory has developed ITC, a protein purification method that eliminates the need for column chromatography. Chromatography, despite its wide-spread use has significant limitations; capital costs are high, resins are often expensive, yields are limited by the loading capacity of the resin,24 elution conditions are specific to the tag and can be detrimental to the stability of the target protein, and the method can be time consuming. In contrast, protein purification by ITC is technologically simple, as it can be performed with widely available centrifuges or filtration devices, and can provide >95% purity in a few rounds of ITC.
In an ongoing effort to optimize ITC, we have characterized the expression and purification, by ITC, of fusion proteins where the ELP tag is fused to either the C- or the N-terminus of the target protein. Four target proteins were fused to either the N- or the C-terminus of an ELP in this study. In all cases we find that protein-ELP fusions have a greater level of expression than ELP-protein fusions (refer Fig. 3). We have also observed that concentrations of all ELP-protein fusions are lower in the soluble cell lysate compared to protein-ELP fusions (Fig. 2 and Supporting Information). Furthermore, these differences are exacerbated when the metric used for comparison is functional activity rather than protein concentration, as three out of four ELP-protein fusions have significantly lower specific activity as compared to their corresponding protein-ELP construct.
We have further examined possible causes for the significant difference between the expression levels of fusing the protein in the two different positions compared to the ELP. Important variables that can impact the observed yield of a protein are its transcriptional and translational levels, formation of inclusion bodies and misfolding coupled to intracellular degradation.
The preferential formation of insoluble inclusion bodies is one possible reason for the difference in the observed levels of soluble protein between the two ELP-fusion constructs. Although we found small fractions of some fusion proteins in the insoluble fraction of the cell lysate, significant quantities of ELP-protein fusion were not found in the insoluble cell lysate fraction, and therefore, we conclude that the low expression level of the ELP-protein constructs is not due to the preferential formation of inclusion bodies of the ELP-protein fusions (refer Fig. 4).
Another possible explanation for the lower expression levels is the incomplete transcription of the ELP-proteins. Measurement of the mRNA levels for the two BFP constructs that exhibited the largest difference in expression level and purified yield (refer Fig. 5) showed no significant difference in the mRNA levels, so that it is unlikely that the lower yields of ELP-protein fusions are due to their lower transcriptional levels as compared to their corresponding protein-ELP fusion.
Although we do not have direct evidence to support this assertion—by a process of elimination of other possible reasons—we suggest two possible explanations for the lower yields of ELP-protein fusions: (1) lower translational levels of the mRNA in the ELP-protein direction; and (2) intracellular degradation of the translated fusion ELP-proteins. One explanation for the lower yields of ELP-protein fusions is lower translational levels of the mRNA in the ELP-protein direction. Pulse-chase experiments may determine if translational differences are the cause for the lower yield of ELP protein fusion, but they are beyond the scope of this article, however this possibility has not been ruled out at the present time. The second, equally likely possibility is intracellular degradation. Degradation of a polypeptide during or immediately following translation can occur via several pathways within a cell. The most common degradation pathways are: (1) N-terminal degradation, (2) C-terminal degradation, and (3) degradation of misfolded proteins. We suggest that ELP-protein constructs may be preferentially marked for degradation, either by the addition of adding a tag that targets it for intracellular degradation or due to misfolding which is recognized by proteases,25 so that ELP-protein fusions are simply degraded while they are being translated or shortly thereafter leaving behind no detectable truncated products in the cell that are visible on SDS-PAGE of the soluble cell extract. Of the possible degradation routes, we can exclude N-terminal degradation as a potential cause for the observed difference in yields between protein-ELPs and ELP-proteins because none of the fusion proteins have residues at the N-terminus that are required for this degradation pathway (Phe, Leu, Trp, Tyr, Arg, or Lys).26–28 Intracellular C-terminal degradation can also occur in E. coli and is a possible cause for the differences in yield; this mechanism involves stalling of the mRNA during translation followed by a 10 amino acid addition encoded by SsrA RNA which marks the protein for degradation.29–32 Because the C-terminus of all the ELP-protein constructs are dissimilar, and because the ELP on the N-terminal end is identical in these fusion proteins, we believe it is unlikely, that C-terminal degradation is responsible for the differences in the observed yield and specific activity of the ELP-protein constructs relative to their protein-ELP counterparts.
Instead, we believe that the most likely reason is that the ELP-protein constructs have a larger fraction of misfolded conformers compared to protein-ELP constructs, which targets them preferentially for intracellular degradation. This assertion is supported by the fact that all the fusion proteins with a measured biological activity (i.e., Trx, CAT, and IL1Ra) show lower specific activity in the ELP-protein position as compared to the protein-ELP position, which is a strong indicator of misfolding. Nevertheless, one cannot rule out that the bulky ELP domain, accounting for a significant mass fraction of the fusion protein, could sterically hinder association of the protein domain with a target receptor. Such effects would be most important for the ELP fusions with IL1Ra, CAT, and Trx, where bimolecular interaction with a substrate is required for activity. If so, then the ideal ELP position may depend on the nature of the fusion domain interaction with its therapeutic target. Misfolded proteins are more likely to be digested by E. coli proteases during translation, therefore, digestion is one of the two most plausible explanations for the lower yields of the ELP-proteins. In addition, we have also observed a lower specific activity of the purified ELP-protein fusions relative to the protein-ELP fusion, which we believe is due to a larger fraction of soluble, but misfolded conformers of the ELP-protein fusion compared to fusion proteins expressed in the opposite direction that avoid intracellular degradation.
Materials and Methods
Materials
Restriction endonucleases were purchased from New England Biolabs (Beverly, MA), DNA plasmids were purified using the spin miniprep system from QIAGEN, (Valencia, CA), the expression vectors pET25b, pET24d, pET32b, and the BLR(DE3) E. coli bacteria strain were from Novagen, (Milwaukee, WI), SuperScript® III Platinum® SYBR® Green One-Step qPCR Kit w/ROX was from Invitrogen, and RNAeasy kit was from Qiagen. The cultures were grown in Terrific Broth (TB) media from Mo Bio Laboratories (Carlsbad, CA), silver staining kit and precast SDS-PAGE Mini-PROTEAN 4–20% Tris HCl gels were from Bio-Rad (Hercules, CA), FAST CAT Green assay kit was from Invitrogen (Carlsbad, CA), coenzyme A (95% purity) was from Sigma-Aldrich (St. Louis, MO), CellTiter-Glo luminescent cell viability assay was from Promega (Madison, WI), human peripheral blood leukocytes RPMI 1788 were obtained from ATCC (Rockville, MD), interleukin 1 was from Pierce (Rockford, IL) and human IL1Ra was from R & D Systems (Minneapolis, MN).
Expression constructs
The gene synthesis of BFP-ELP and Trx-ELP fusions was published previously.1,3,4 The CAT gene (from Invitrogen) was retrieved by PCR and TA cloning as described earlier.4 The ELP fusion proteins were cloned as protein-ELP or ELP-protein fusions in pET24 (CAT-ELP, ELP-CAT, ELP-Trx), pET25 (BFP-ELP, ELP-BFP, IL1Ra-ELP, ELP-IL1Ra), or pET32a (Trx-ELP) expression vectors using standard molecular biology procedures. The synthesis of protein-ELP and ELP-protein constructs of the same proteins were carried out in pET24d and pET25b vectors using the same procedures as described previously. The ELP used in all fusions is a 36 kDa peptide with 90 repeats of the pentapeptide having 50% valine, 20% alanine, and 30% glycine in the guest residue position; using our notation system this ELP is ELP[V5A2G3]-90.
Protein expression and purification
Each fusion construct was expressed in E. coli BLR(DE3) cells and purified in triplicate from 50 mL TB, supplemented by either 100 μg/mL ampicillin or 15 μg/mL kanamycin. The 50 mL cultures were each inoculated by 0.5 mL of a 2.5 mL overnight starter culture [overnight starter cultures were inoculated from frozen DMSO stocks kept at −80°C except for IL1Ra-ELP and ELP-IL1Ra; starter cultures were inoculated from newly transformed BLR(DE3) cells from plates] and grown for 24 h at 37°C without the induction because ELP fusion proteins express with higher yields without induction.3 The cells were harvested by centrifugation for 15 min at 4°C. The pellets were each resuspended in 2 mL PBS buffer and frozen at −80°C. The resuspended pellets were thawed and lysed by ultrasonic disruption on ice. Poly(ethyleneimine) (100 μL of 10%) was added to each lysed pellet suspension before centrifugation at 16120g (15 min) to separate insoluble cell debris from the soluble cell lysate.
All eight constructs were purified by ITC using NaCl to trigger the phase transition at room temperature. Typically, in the first two rounds of ITC, the NaCl concentrations were 3 or 5M for the protein-ELP and ELP-protein constructs, respectively, except for ILRa-ELP where the salt concentrations during purification were identical to those of ELP-IL1Ra. In the following rounds of ITC, the NaCl concentrations were 1.5 or 3M for protein-ELP and ELP-protein, respectively. After triggering the phase transition, the solutions were centrifuged and the pellets were resuspended in cold PBS, followed by a cold spin to remove denatured contaminants trapped in the pellets. Three to five rounds of ITC were carried out for each construct. In the final round of ITC, the pellets were resuspended in 100 μL PBS. Concentration of the purified fusion proteins were measured on a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE) spectrophotometer (corresponding to absorbance at 280 nm) using the extinction coefficients 1.975 × 104 M−1 cm−1, 2.418 × 104 M−1 cm−1, 4.95 × 104 M−1 cm−1, and 2.231 × 104 M−1 cm−1 for Trx-ELP, BFP-ELP, CAT-ELP, and IL1Ra-ELP, respectively. The extinction coefficients for ELP-Trx, ELP-BFP, ELP-CAT, and ELP-IL1Ra were 2.231 × 104 M−1 cm−1, 2.674 × 104 M−1 cm−1, 5.078 × 104 M−1 cm−1, and 2.359 × 104 M−1 cm−1, respectively. The extinction coefficients were estimated from the amino acid sequence using the method of Gill and von Hippel.33 The number of disulfide bonds were found in PDB files; 2trx (Trx), 1bfp (BFP), 1pd5 (CAT), and 1ilr (IL1Ra). The purification was followed and visualized by SDS-PAGE and stained with Coomassie blue or silver stain. Molecular weights of the eight fusion proteins: Trx-ELP, 50.0 kDa; BFP-ELP, 65.5 kDa; CAT-ELP, 62.6 kDa; IL1Ra-ELP, 54.2 kDa, ELP-Trx, 50.0 kDa, ELP-BFP, 63.7 kDa, ELP-CAT, 63.7 kDa; ELP-IL1Ra, 55.1 kDa. SDS-PAGE gels showing the soluble and insoluble fractions were generated by resuspending the cell debris in the same volume as the removed soluble cell lysate in either PBS buffer or 6M guanidinium hydrochloride in water and 4 μL of each fraction was loaded onto the gel.
Activity assays
Trx activity was measured by the insulin disulfide reduction assay, as described by Holmgren.34,35 The assay was carried out in a cuvette using a total volume of 600 μL; 50 mM phosphate buffer containing 1 mM EDTA at pH 7.0, 400 μM initial concentrations of NADPH, 107 μM insulin, 0.06 μM thioredoxin reductase, and 3.2–6.2 μM Trx-ELP or ELP-Trx. The enzymatic reactions were initiated by adding insulin and the decrease in NADPH absorbance at 340 nm was monitored spectrophotometrically on a Cary 300 spectrophotometer (Varian, Palo Alto, CA). The rate was measured by the initial slopes of the curves. For blank measurements, PBS buffer was added instead of Trx. The extinction coefficient for NADPH is 6200 M−1 cm−1 and reduction of 1 μM disulfide corresponds to a decrease in absorbance of 0.0062. The specific activity of Trx was determined as U/μg fusion construct where 1 U represents the conversion of 1 μM substrate per minute. The activity of each of the 50 mL purified protein was measured three times.
BFP fluorescence was measured on a Cary Eclipse fluorescence spectrophotometer (Varian, Palo Alto, CA). The excitation wavelength was 385 nm and the emission spectra were recorded from 430 to 600 nm. The fluorescence of three purified batches of BFP-ELP were measured (n = 3), while the fluorescence from two purified batches of ELP-BFP were measured (n = 2 for the first batch and n = 1 for the second batch). The concentrations of BFP-ELP were 0.72–0.73 μM and of ELP-BFP 0.60–0.93 μM. All volumes for fluorescence measurements were 600 μL. A blank spectrum of PBS buffer was subtracted from each BFP spectrum and the curves were integrated from 430 to 600 nm. The obtained areas were normalized by the BFP concentration to determine the specific activity of the BFP fusion proteins.
The activity of CAT fusion proteins was measured by the green FAST CAT assay using the protocol published by Molecular Probes/Invitrogen. A 9 mM coenzyme A solution was prepared in water. CAT-ELP and ELP-CAT were diluted 300,000 and 3000 times in PBS buffer, respectively; 60 μL CAT fusion protein, 10 μL 1-deoxy-chlorampenicol, and 2.5 μL H2O was incubated for 10 min at 37°C before adding 7.5 μL Coenzyme A which incubated for 30 min at 37°C. After separating the remaining substrate from the product by thin layer chromatography, the extracted samples were diluted 10 times before the fluorescence of both substrate and product were measured on a Victor31420 multilabel counter plate reader using the fluorescein (FITC) bandpass filters (Perkin Elmer, Shelton, CT).
IL-1Ra activity was determined by a cell proliferation assay using human peripheral blood leukocytes RPMI 1788. The cells were grown in suspension in a 75 cm2 tissue culture flask in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 50 mM β-mercaptoethanol, 10 mM HEPES buffer, 50 U/mL penicillin, and 50 μg/mL streptomycin. The cells were grown in suspension with an initial density of 1000 cells/well in a 96-well plate in 80 μL of assay medium (culture medium with 3% heat-inactivated bovine serum). The cells were stimulated with 10 μL of a 29 pM IL1 solution and treated with 10 μL IL1Ra-ELP, ELP-IL1Ra, or human IL1Ra. Eight serial dilutions were used for each of IL1Ra-ELP, ELP-IL1Ra, and commercial IL1Ra, and the cells were incubated for 72 h at 37°C. At that point, the number of surviving cells was quantified by the CellTiter Glo luminescence assay. The data was fit using non-linear least squares regression (MATLAB CurveFit Toolbox) to a sigmoidal dose-response curve to derive EC50 parameters.
RT-PCR
Total RNA was isolated from cultures at different time points ranging from 8 to 24 h. Total RNA was purified using the RNAeasy kit (Qiagen). One step reverse transcription and relative real-time PCR was performed using SuperScript III Platinum SYBR Green One-Step qPCR Kit w/ROX (Invitrogen). The RNA template was diluted 100-fold and amplified in a real-time PCR cycler with fluorescence detection (ABI 7300 sequence detection system, Applied Biosystems). Primers for BFP mRNA amplification segment (230 bp) were:
Forward—GGC AAC TAC AAG ACA CGT GCT G
Reverse—GGG CCA TCG CCA ATT GGA GTA T
Detection of the housekeeping gene 16S rRNA was performed with the following universal primers: 8F—GGA TCC AGA CTT TGA TYM TGG CTC AG
314R—CTGCTGCCTCCCGTAGG (306 bp segment).
PCR conditions were: 5 min at 50°C, 5 min at 95°C, then 40 cycles of 15 s at 95°C and 1 min at 60°C. Single product amplification was verified by dissociation analysis as well as by visualization by agarose gel electrophoresis. Real-time PCR was performed without reverse transcriptase as a negative control to ensure that contaminating residual DNA was at least 210-fold lower than mRNA levels (a difference of at least 10 cycles). The relative transcription levels were analyzed with the ABI Prism 700 SDS software (Applied Biosystems). For each of the six flasks (three expressing BFP-ELP and three expressing ELP-BFP), the levels of BFP mRNA were calculated relative to the levels of 16s rRNA to achieve a measure of BFP transcripts per cell.18 Further, the levels of BFP to rRNA transcripts of the first BFP-ELP flask was calibrated to one for each time point and all other values for this time point were normalized accordingly. Aliquots from each culture were grown on agar plates, incubated at 37°C overnight, and the live bacteria counted the following day.
Conclusions
The position of the target protein relative to the ELP tag in ELP fusion proteins is an important variable in controlling the expression level and specific activity of ELP fusion proteins. Placing the ELP at the C-terminus of the target protein (protein-ELP) result in a higher expression level for the four proteins studied herein. The position of the fusion protein also has a significant impact on the specific activity, as ELP-protein constructs have a lower specific activity than protein-ELP constructs in three out of the four proteins studied herein. Our results suggest that transcriptional differences are not responsible for the difference in protein expression between the two positions of an ELP in the fusion protein. We believe that two possible scenarios can explain the lower expression levels and lower specific activity of ELP-protein constructs compared to protein-ELP constructs: First, placement of the ELP at the N-terminus of the fusion protein increases the fraction of misfolded protein that are more likely to be targeted for degradation, but that not all of the misfolded conformers are degraded, so that the misfolded conformers that evade degradation decrease the measured specific activity of the fusion protein. Second, differences in translation may provide another, complementary possibility that could account for these observations. We conclude that these studies provide a strong rationale to consider the position in the design of an ELP fusion protein. We end with the caveat that although the results are unequivocal, they are based on a small set of four proteins and a single ELP. Because protein expression is a complex process and is highly protein-dependent, it is possible that in some instances an ELP-protein construct may express better or display higher specific activity than the equivalent protein-ELP construct, so that it may be prudent in to synthesize and express both variants of a protein, if possible.
Glossary
Abbreviations:
- BPP
blue fluorescent protein
- CAT
chloramphenicol transferase
- ELP
elastin-like polypeptide
- ITC
inverse transition cycling
- IL1
interleukin 1
- IL1Ra
interleukin 1 receptor antagonist
- Trx
thioredoxin;Tt, transition temperature.
References
- 1.Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat Biotechnol. 1999;17:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 2.Meyer DE, Trabbic-Carlson K, Chilkoti A. Protein purification by fusion with an environmentally responsive elastin-like polypeptide: effect of polypeptide length on the purification of thioredoxin. Biotechnol Prog. 2001;17:720–728. doi: 10.1021/bp010049o. [DOI] [PubMed] [Google Scholar]
- 3.Trabbic-Carlson K, Liu L, Kim B, Chilkoti A. Expression and purification of recombinant proteins from Escherichia coli: comparison of an elastin-like polypeptide fusion with an oligohistidine fusion. Protein Sci. 2004;13:3274–3284. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trabbic-Carlson K, Meyer DE, Liu L, Piervincenzi R, Nath N, LaBean T, Chilkoti A. Effect of protein fusion on the transition temperature of an environmentally responsive elastin-like polypeptide: a role for surface hydrophobicity? Protein Eng Des Sel. 2004;17:57–66. doi: 10.1093/protein/gzh006. [DOI] [PubMed] [Google Scholar]
- 5.Urry DW. Entropic elastic processes in protein mechanisms. I elastic structure due to and inverse temperature transition and elasticity due to internal chain dynamics. J Protein Chem. 1988;7:1–34. doi: 10.1007/BF01025411. [DOI] [PubMed] [Google Scholar]
- 6.Urry DW. Free energy transduction in polypeptides and proteins based on inverse temperature transitions. Prog Biophys Mol Biol. 1992;57:23–57. doi: 10.1016/0079-6107(92)90003-o. [DOI] [PubMed] [Google Scholar]
- 7.Urry DW. Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J Phys Chem B. 1997;101:11007–11028. [Google Scholar]
- 8.Luan C-h, Parker TM, Prasad KU, Urry DW. Differential scanning calorimetry studies of NaCl effect on the inverse temperature transition of some elastin-based polytetra-. polypenta-, and polynonapeptides. Biopolymers. 1991;31:465–475. doi: 10.1002/bip.360310502. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Furyk S, Bergbreiter DE, Cremer PS. Specific ion effects on the water solubility of macromolecules: PNIPAM and the hofmeister series. J Am Chem Soc. 2005;127:14505–14510. doi: 10.1021/ja0546424. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Furyk S, Sagle LB, Cho Y, Bergbreiter DE, Cremer PS. Effects of hofmeister anions on the LCST of PNIPAM as a function of molecular weight. J Phys Chem C. 2007;111:8916–8924. doi: 10.1021/jp0690603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho Y, Zhang Y, Christensen T, Sagle LB, Chilkoti A, Cremer PS. Effects of hofmeister anions on the phase transition temperature of elastin-like polypeptides. J Phys Chem B. 2008;112:13765–13771. doi: 10.1021/jp8062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer DE, Chilkoti A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: examples from the elastin-like polypeptide system. Biomacromolecules. 2002;3:357–367. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- 13.Meyer DE, Chilkoti A. Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules. 2004;5:846–851. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 14.Christensen T, Trabbic-Carlson K, Liu W, Chilkoti A. Purification of recombinant proteins from Escherichia coli at low expression levels by inverse transition cycling. Anal Biochem. 2007;360:166–168. doi: 10.1016/j.ab.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim DW, Trabbic-Carlson K, MacKay JA, Chilkoti A. Improved non-chromatographic purification of a recombinant protein by cationic elastin-like polypeptides. Biomacromolecules. 2007;8:1417–1424. doi: 10.1021/bm060849t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA. Development and characterization of a fusion protein between thermally responsive elastin-like polypeptide and interleukin-1 receptor antagonist: sustained release of a local antiinflammatory therapeutic. Arthritis Rheum. 2007;56:3650–3661. doi: 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- 17.Kim DH, Smith JT, Chilkoti A, Reichert WM. The effect of covalently immobilized rhIL-1ra-ELP fusion protein on the inflammatory profile of LPS-stimulated human monocytes. Biomaterials. 2007;28:3369–3377. doi: 10.1016/j.biomaterials.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Kolter R, Siegele DA, Tormo A. The stationary-phase of the bacterial life-cycle. Annu Rev Microbiol. 1993;47:855–874. doi: 10.1146/annurev.mi.47.100193.004231. [DOI] [PubMed] [Google Scholar]
- 20.Chow DC, Dreher MR, Trabbic-Carlson K, Chilkoti A. Ultra-high expression of a thermally responsive recombinant fusion protein in E coli. Biotechnol Prog. 2006;22:638–646. doi: 10.1021/bp0503742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rekas A, Alattia J-R, Nagai T, Miyawaki A, Ikura M. Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J Biol Chem. 2002;277:50573–50578. doi: 10.1074/jbc.M209524200. [DOI] [PubMed] [Google Scholar]
- 22.Paramban RI, Bugos RC, Su WW. Engineering green fluorescent protein as a dual functional tag. Biotechnol Bioeng. 2004;86:687–697. doi: 10.1002/bit.20077. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Patel HN, Lappe JW, Wachter RM. Reaction progress of chromophore biogenesis in green fluorescent protein. J Am Chem Soc. 2006;128:4766–4772. doi: 10.1021/ja0580439. [DOI] [PubMed] [Google Scholar]
- 24.Waugh DS. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 25.Rozkov A, Enfors S-O. Analysis and control of proteolysis of recombinant proteins in Escherichia coli. Adv Biochem Eng/Biotechnol. 2004;89:163–195. doi: 10.1007/b95567. [DOI] [PubMed] [Google Scholar]
- 26.Shrader TE, Tobias JW, Varshavsky A. The N-end rule in Escherichia coli: cloning and analysis of the leucyl, phenylalanyl-tRNA-protein transferase gene aat. J Bacteriol. 1993;175:4364–4374. doi: 10.1128/jb.175.14.4364-4374.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varshavsky A. The N-end rule pathway of protein degradation. Genes Cells. 1997;2:13–28. doi: 10.1046/j.1365-2443.1997.1020301.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang KH, Sauer RT, Baker TA. ClpS modulates but is not essential for bacterial N-end rule degradation. Genes Dev. 2007;21:403–408. doi: 10.1101/gad.1511907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keiler KC, Waller PRH, Sauer RT. Role of the peptide tagging system in detradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 30.Gottesman S, Roche E, Zhou Y, Sauer RT. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayes CS, Bose B, Sauer RT. Proline residues at the C terminus of nascent chains induce SsrA tagging during translation termination. J Biol Chem. 2002;277:33825–33832. doi: 10.1074/jbc.M205405200. [DOI] [PubMed] [Google Scholar]
- 32.Flynn JM, Neher SB, Kim Y-I, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 2003;11:671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 33.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 34.Holmgren A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitoland dihydrolipoamide. J Biol Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 35.Holmgren A, Bjornstedt M. Enzymatic reduction-oxidation of protein disulfides by thioredoxin. Methods Enzymol. 1984;107:295–300. doi: 10.1016/0076-6879(84)07019-1. [DOI] [PubMed] [Google Scholar]