

Abstract
Hsp31 is a stress-inducible molecular chaperone involved in the management of protein misfolding at high temperatures and in the development of acid resistance in starved E. coli. Each subunit of the Hsp31 homodimer consists of two structural domains connected by a flexible linker that sits atop a continuous tract of nonpolar residues adjacent to a hydrophobic bowl defined by the dimerization interface. Previously, we proposed that while the bowl serves as a binding site for partially folded species at physiological temperatures, chaperone function under heat shock conditions requires that folding intermediates further anneal to high-affinity binding sites that become uncovered upon thermally induced motion of the linker. In support of a mechanism requiring that client proteins first bind to the bowl, we show here that fusion of a 20-residue-long hexahistidine tag to the N-termini of Hsp31 abolishes chaperone activity at all temperatures by inducing reversible structural changes that interfere with substrate binding. We further demonstrate that extending the C-termini of Hsp31 with short His tags selectively suppresses chaperone function at high temperatures by interfering with linker movement. The structural and functional sensitivity of Hsp31 to lengthening is consistent with the high degree of conservation of class I Hsp31 orthologs and will serve as a cautionary tale on the implications of affinity tagging.
Keywords: molecular chaperone, heat shock protein, DJ-1, His-tag, affinity tag, polyhistidine
Introduction
Molecular chaperones and heat-shock proteases are a class of proteins that rely on conformational changes fueled by ATP hydrolysis or triggered by physicochemical changes in the cellular environment (e.g., temperature, pH, redox state, etc.) to transiently bind, unfold, refold, or degrade partially folded polypeptides.1–5 E. coli Hsp31—a member of the DJ-1 protein superfamily6—is a stress-inducible molecular chaperone and weak aminopeptidase that helps exponentially growing cells manage severe thermal stress and stationary phase cells survive the deleterious effects of acidification by acting as a holdase.7–13 Highly conserved Hsp31 orthologs are present in a number of pathogenic bacteria,13 and more distant relatives have been identified on the basis of structure-guided alignments.14 These include archaebacterial Pfp-I type peptidases,15,16 and human DJ-1, a protein implicated in familial Parkinson's disease.17
E. coli Hsp31 is organized as a homodimer of 31-kDa subunits that each contain a Cys-His-Asp catalytic triad responsible for its peptidase activity.8 Each protomer consists of two α-β domains termed A and P that are connected by a 22-residue-long flexible linker.14,18 The P domain, which exhibits no homology to other folds, is responsible for dimer formation via extensive interactions with the A′ and P′ domains of the neighboring subunit. Furthermore, the P domain dimerization interface defines a shallow hydrophobic bowl ∼20 Å in diameter that has been proposed to serve as a binding site for partially folded proteins.19 Upon temperature increase, the linker (Gln-28 to His-49), and a neighboring loop extending from Met-103 to Lys-115, move to expose normally shielded, nonpolar residues to the solvent.18,19 These amino acids form continuous hydrophobic tracts on either side of the bowl that are thought to allow high-affinity substrate binding under heat-shock conditions. Substrate ejection is believed to be driven by the return of the linker-loop “gate” to its original position as thermal stress subsides. Fine-tuning of Hsp31 mode of action may involve additional mechanisms since chaperone activity is negatively regulated by ATP binding at high temperatures,13 and each subunit contains a Zn(II) binding site in the vicinity of the linker-loop region.20
In this report, we show that fusion of His6 tags to either the N- or C-termini of E. coli Hsp31 affects both its structure and function. While N-terminal His-tagging induced major conformational changes that abolished chaperone activity at both low and high temperatures, addition of C-terminal extensions predominantly affected Hsp31 structure and activity at elevated temperatures. The mechanistic implications of these results are discussed.
Results
Influence of His-tagging on Hsp31 chaperone activity
Two His6-tagged Hsp31 variants were constructed as described in the Materials and Methods section. Each monomer of the N-terminally tagged protein contains about 2 kDa, 20-amino acid-long extension (MGSSHHHHHHSSGLVPAGSH), specifying a thrombin recognition site. Cleavage of the tag yields an Hsp31 derivative (cl-Hsp31) with 3-amino acid-long N-terminal scars (GSH) that behaves indistinguishably from authentic Hsp31 in structural and functional assays.19 The C-terminally His-tagged variant (Hsp31-His6) contains seven extra amino acids (EHHHHHH) per monomer that add ≈9 kDa to the molecular mass of native Hsp31. Consistent with the fact that about 1900 Å2 of surface area are buried upon dimer formation,14 all Hsp31 variants assembled as homodimers as judged by size exclusion chromatography and native polyacrylamide gel electrophoresis (Fig. 1). Nevertheless, the presence of a trailing edge in the elution profile of His6-Hsp31 [Fig. 1(A)] suggests that N-terminal His-tagging alters the protein hydrodynamic radius.
Figure 1.

Purification and quaternary structure of Hsp31 variants. (A) The indicated proteins (1.2 μg) were injected on a Biosep S2000 column developed at 1 mL/min in 150 mM Tris-HCl, pH 7.4. Under these conditions, the calibration proteins albumin (67 kDa) and chymotrypsinogen A (25 kDa) elute at 6.7 and 8.7 mL, respectively (arrows). Absorbance was monitored at 280 nm and is reported in arbitrary units (A.U.) (B) Hsp31 variants were mixed with Laemmli sample buffer lacking (native lanes) or containing SDS and DTT. SDS/DTT-treated samples were heated at 95°C for 5 min before loading. Proteins were fractionated on a 10% polyacrylamide gel under nondenaturing conditions.
In light of preliminary experiments indicating that both N- and C-terminal tags influenced Hsp31 chaperone activity, we first examined the functional consequences of His-tagging on the low-temperature, housekeeping chaperone activity of Hsp31, which has been proposed to involve the binding of partially folded substrates to the Hsp31 hydrophobic bowl on the basis of mutagenesis experiments.19 For these experiments, guanidine hydrochloride-unfolded malate dehydrogenase (MDH) or urea-unfolded citrate synthase (CS) were diluted into refolding buffer containing no additive or supplemented with a sixfold molar excess of cl-Hsp31, His6-Hsp31, or Hsp31-His6 at 23°C. Like native Hsp31,13,19 cl-Hsp31 improved the recovery of active MDH by twofold and that of CS by sevenfold [Fig. 2(A)]. By contrast, Hsp31-His6 was less than half as efficient at improving MDH and CS refolding yields, and His6-Hsp31 was completely incapable of enhancing the reactivation of either substrate [Fig. 2(A)]. Because the N-termini of Hsp31 are proximal to the bowl (see Fig. 7), we considered the possibility that the N-terminal hexahistidine extensions might occupy the substrate binding site. To test this idea, His6-Hsp31 was cleaved with immobilized thrombin and the 17-residue-long peptide MGSSHHHHHHSSGLVPA was recovered. Figure 2(B) shows that, even when added to the refolding cocktail at a fivefold molar excess over Hsp31, the purified tag did not influence Hsp31′s ability to support the refolding of chemically unfolded MDH and CS. Thus, the lack of His6-Hsp31 chaperone activity cannot be attributed to the N-terminal hexahistidine extensions acting as pseudosubstrate.
Figure 2.
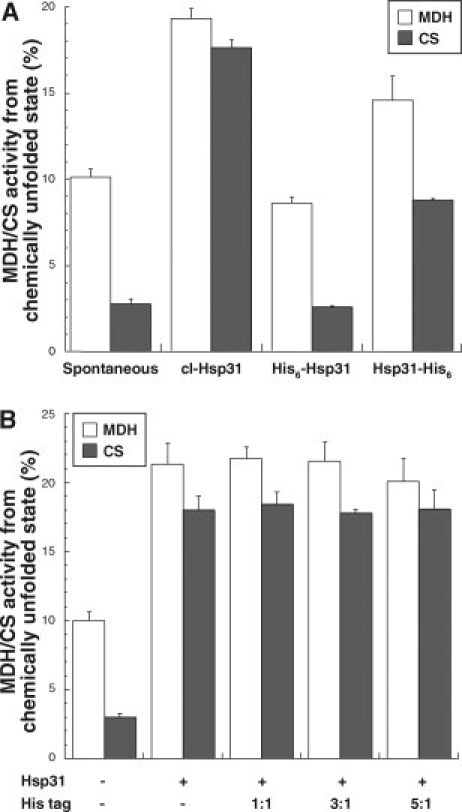
Influence of His-tagging on Hsp31 housekeeping chaperone activity. (A) Guanidine hydrochloride-unfolded MDH (open bars) or urea-unfolded CS (filled bars) were rapidly diluted to a 0.4 μM final concentration in refolding buffer supplemented with no additives or with a sixfold molar excess (based on protomers) of cl-Hsp31, His6-Hsp31, or Hsp31-His6. Enzymatic activities were assayed after 3h (MDH) or 30 min (CS) of incubation at 23°C. There was no change in activity after these time periods. (B) MDH (open bars) and CS (filled bars) were unfolded as above and diluted into refolding buffer containing a sixfold molar excess of native Hsp31 and the indicated molar excess of the MGSSHHHHHHSSGLVPA peptide (His tag) over Hsp31 monomers. All samples were assayed in triplicate and error bars were obtained for three independent experiments.
Figure 7.

“Bottom” and “side” views of the Hsp31 dimer. One of the monomers is color-coded: the P1, P2, and P3 segments of the P domain are shown in different shades of green, the flexible linker is in red, and the A domain is in blue. The N-terminal Thr-5 and C-terminal Gly-283 are labeled N and C, respectively. Trp-107 (W107), Trp-173 (W173) and Trp-229 (W-229) are shown as balls and sticks. The hydrophobic bowl is bounded by an ellipse. Molecular surfaces were calculated and rendered using 1n57 coordinates14 and the Swiss-Pdb viewer package.22
We have previously proposed that high-temperature chaperoning by Hsp31 requires that folding intermediates first interact with the bowl and next anneal to adjacent hydrophobic regions that become solvent-exposed as a result of heat-induced linker-loop movement.18,19 Such thermal activation of chaperone activity can be tested by assaying the recovery of MDH or CS activity at 23°C following 30-min incubation at 45°C, or by measuring the ability of Hsp31 to suppress the aggregation of model substrates at high temperature.13,19 Figure 3(A) shows that, although cl-Hsp31 promoted the recovery of 30% (MDH) or 45% (CS) of the original enzymatic activity following heat treatment, neither His6-Hsp31 nor Hsp31-His6 were able to support MDH or CS reactivation. In agreement with these results, a sixfold molar excess of Hsp31-His6 was no more effective than the control protein BSA at suppressing the heat-induced aggregation of CS [Fig. 3(B)] or alcohol dehydrogenase [ADH; Fig. 3(C)]. In the same functional test, His6-Hsp31 aggravated the aggregation of both model substrates compared to control experiments conducted with the same concentration of BSA or in the absence of additives [Fig. 3(B,C)]. Taken together, the above results show that, while N-terminal extensions abolish the ability of Hsp31 to function as a chaperone at both low and high temperatures, C-terminal His-tagging primarily interferes with Hsp31 chaperone activity under heat-shock conditions.
Figure 3.

His6-Hsp31 and Hsp31-His6 chaperone activity is lost at high temperatures. (A) MDH (open bars) or CS (filled bars) were injected to a final concentration of 0.4 μM in refolding buffer held at 45°C and supplemented with no additive or with a sixfold molar excess of the indicated Hsp31 variants. Mixtures were incubated for 30 min at 45°C and assayed after 30 min of further incubation at 23°C. Reactivation yields did not change after this time. All samples were assayed in triplicate and error bars were obtained for three independent experiments. (B) CS was injected at a 0.4 μM final concentration into buffer held at 45°C and supplemented with no additive, a sixfold molar excess of BSA, His6-Hsp31 or Hsp31-His6, or a threefold molar excess of cl-Hsp31 (all based on protomers). Samples were transferred to a fluorescence spectrophotometer, and aggregation was monitored at 45°C with excitation and emission wavelengths set at 500 nm. (C) ADH aggregation experiments were conducted as above except that the final ADH concentration was 1.6 μM and the incubation temperature was 41.5°C. AU, arbitrary units.
Influence of His-tagging on Hsp31 structure
In an effort to correlate the loss of function of His-tagged variants with structural changes, we first compared the secondary structures of His6-Hsp31 and Hsp31-His6 to that of cl-Hsp31 using far-UV CD spectroscopy. Figure 4 and Table I show that, while His6-Hsp31 exhibits an increase in α-helical content and a concomitant decrease in random coils relative to cl-Hsp31 (or native Hsp31), Hsp31-His6 contains fewer α-helices and a higher amount of β- structure than cl-Hsp31. On the other hand, CD spectra and predicted α-helix and β-sheet content21 were virtually identical at 23°C (Fig. 4, closed symbols) and 45°C (open symbols) for each variant, indicating that changes in secondary structure induced by His tagging are stable.
Figure 4.

Influence of His-tagging on the secondary structure of Hsp31. Far-UV CD spectra of cl-Hsp31 (circles), His6-Hsp31 (diamonds) and Hsp31-His6 (squares) were recorded at 23°C (closed symbols) and 45°C (open symbols). The CD spectrum of native Hsp31 (not shown) is indistinguishable from that of cl-Hsp31.
Table I.
Secondary Structure Content of Hsp31 Variantsa
| Secondary structure content (%) |
||||||||
|---|---|---|---|---|---|---|---|---|
| 23°C |
45°C |
|||||||
| Hsp31 | cl-Hsp31 | His6-Hsp31 | Hsp31-His6 | Hsp31 | cl-Hsp31 | His6-Hsp31 | Hsp31-His6 | |
| Helical | 37 | 37 | 54 | 26 | 37 | 36 | 58 | 27 |
| Sheet | 14 | 13 | 18 | 25 | 15 | 14 | 11 | 25 |
| Random | 49 | 50 | 28 | 49 | 49 | 49 | 31 | 49 |
Hsp31 contains three tryptophan residues at positions 107, 173, and 229 that are suitable for probing the protein's tertiary structure by intrinsic fluorescence spectroscopy. We recorded the emission spectra of cl-Hsp31, His6-Hsp31, and Hsp31-His6 at 23°C and 45°C following excitation at 295 nm. In all cases, the maximal emission wavelength was found to be 348 ± 1 nm, indicating that tryptophan residues experience a hydrophilic environment, irrespective of the presence of an extension or of a change in the incubation temperature. However, the maximal emission intensity of His6-Hsp31 was more than sixfold higher than that of cl-Hsp31 at 23°C, and Hsp31-His6 exhibited a small increase in fluorescence emission intensity relative to the control (Fig. 5, closed symbols). In addition, while raising the temperature to 45°C did not alter the intrinsic tryptophan fluorescence spectrum of cl-Hsp31, it led to decreased emission intensities for both His6-Hsp31 and Hsp31-His6 (Fig. 5, open symbols). Thus, both N- and C-terminally tagged variants undergo high-temperature conformational rearrangements in nonsecondary structural elements that directly or indirectly quench tryptophan fluorescence.
Figure 5.

Influence of His-tagging on the tertiary structure of Hsp31. Intrinsic tryptophan fluorescence spectra of cl-Hsp31 (circles), His6-Hsp31 (diamonds), and Hsp31-His6 (squares) were recorded at 23°C (closed symbols) and 45°C (open symbols). Fluorescence intensities are reported in arbitrary units (AU).
To confirm these results, we made use of bis-1-anilino-8-naphthalene sulfonate (bis-ANS), a small reporter molecule that exhibits little fluorescence in its free state but becomes highly fluorescent when bound to solvent-exposed hydrophobic domains. Consistent with intrinsic tryptophan fluorescence data, the bis-ANS fluorescence of His6-Hsp31 was 3.8-fold higher than that of cl-Hsp31 at 23°C, while that of Hsp31-His6 was only marginally larger (20%) than the control. At 45°C, however, bis-ANS-treated His6-Hsp31 and Hsp31-His6 were 4- and 4.6-fold more fluorescent than cl-Hsp31, respectively. We conclude that, while N-terminal His-tagging affects the tertiary structure of His6-Hsp31 at both low and high temperatures, the destabilizing effect of the C-terminal extension is most significant upon exposure to heat.
Broad specificity proteases such as proteinase K (PK) are useful tools to probe overall protein conformation since structural changes or packing defects induced by mutations, ligand binding, or temperature are often accompanied by an increase in the exposure of cleavage sites (aliphatic, aromatic, or hydrophobic amino acids in the case of PK). In agreement with crystallographic and biochemical data showing that Hsp31 relies on structural rearrangements as part of its mechanism of action at high temperatures,18,19 we found that, although native Hsp31 was fairly resistant to degradation by PK at 37°C, its susceptibility to proteolysis was greatly increased at 45°C [Fig. 6(A)]. We took advantage of this observation to compare the effect of His tags at 45°C. Not unexpectedly, both N- and C-terminal extensions increased the initial rate and the extent of Hsp31 degradation to a comparable extent [Fig. 6(B)].
Figure 6.

Temperature and His-tagging increase the proteolytic susceptibility of Hsp31. (A) Native Hsp31 was incubated with proteinase K (PK) at 37 or 45°C. Samples collected at the indicated time points were fractionated by SDS-PAGE (inset) and band intensities were quantified by videodensitometric analysis of the gels. An arbitrary value of 100% was assigned to samples collected at time zero. (B) Side-by-side PK digestions of cl-Hsp31 (circles), His6-Hsp31 (diamonds) and Hsp31-His6 (squares) were conducted as above at 45°C.
Discussion
Hsp31 orthologs have been identified in all kingdoms on the basis of structural homology in their A domains, and have been divided into three classes based on homologs variations in their catalytic triad, P domain, and oligomeric status. Whereas class I homologs (exemplified by E. coli Hsp31) have a complete P domain and form dimers, class II homologs have a truncated P domain and unknown quaternary structure, and class III homologs (e.g., members of the PfpI family of intracellular proteases) completely lack the P domain and are hexameric.14 Among class I members, the N-terminal segment of the P domain (known as P1 and extending from residues 1 to 31 in E. coli Hsp31) is well conserved in both length and sequence and its dimerization interface defines the shallow hydrophobic bowl (see Fig. 7). Previously, we reported that glutamate substitution in four nonpolar residues located in the P1 segment (Phe-19, Phe-20, Tyr-24, and Leu-26) reduced the ability of Hsp31 to support the reactivation of chemically denatured and heat-inactivated MDH and CS by 20–70%, and proposed that the bowl serves as a substrate binding site. In this study, we found that extending the N-termini of Hsp31 by 20 amino acids completely abolished chaperone activity at both high and low temperatures, but that removal of 17 of these residues via thrombin digestion led to full recovery of function (Figs. 2–3). Considering that the N-termini of Hsp31 flank the bowl (see Fig. 7), these results bring strong support to the proposal that binding of partially folded client proteins to the bowl is a required initial step under both low- and high-temperature conditions, and explain why P1 segments only range between 31 and 34 amino acids in length among the highly homologous (>55% sequence identity) class I Hsp31 homologues.14
There are several possible mechanisms by which N-terminal extensions could interfere with substrate binding: they may act as pseudosubstrates that occupy the bowl, they may alter its architecture, or they may physically block substrate access. Based on the observation that a purified peptide encompassing 17 residues of the N-terminal extension does not interfere with Hsp31 chaperone activity [Fig. 2(B)], the first scenario is unlikely. Indeed, His tags are hydrophilic and unlikely substrates for a chaperone. On the other hand, N-terminal extensions led to significant changes in Hsp31 secondary structure and to an increase in fluorescence emission and hydrophobicity at 23°C. Trp173 and 229 are unlikely to be responsible for the higher fluorescence since they are located away from mobile elements. However, Trp107/Trp107′ are proximal to the bowl (see Fig. 7), and the observed increase in intrinsic tryptophan fluorescence is well explained by a reduction in fluorescence quenching associated with tag-induced deformation of the P1 segments. Structural rearrangements in the P1/P1′ region could also account for the increase in Hsp31 hydrophobicity. Whether these structural changes alone account for the loss of chaperone activity remains an open question. However, based on our observations that the N-terminal tags are fully accessible to immobilized thrombin, that they alter the hydrodynamic radius of Hsp31, and that their structural and functional impact can be fully reversed by shortening their length to three amino acids, it is likely that they project into solution. Thus, N-terminal His tags may directly contribute to the loss of chaperone function by acting as steric brushes that prevent folding intermediates from interacting with the bowl.
Compared to N-terminal His-tagging, addition of 7-residue-long hexahistidine tails to the C-termini of Hsp31 had a more modest impact on the secondary and tertiary structure of the protein at 23°C (Figs. 4–5). Yet, these minor structural changes had significant functional consequences, as the ability of Hsp31-His6 to improve the spontaneous refolding of MDH or CS from the chemically denatured state was half that of cl-Hsp31 or native Hsp31 [Fig. 2(A)]. Because the C-termini of Hsp31 are located away from the bowl and the tag is short (see Fig. 7), physical blocking of substrate binding is unlikely to account for the suboptimal performance of Hsp31-His6 at 23°C. On the other hand, the attachment points of the His tags (Gly-283 and Gly-283′) are proximal to the end of the flexible linker, and long-range structural constraints, which may propagate along the linker, could alter the architecture of the bowl and reduce substrate binding efficiency.
Bis-ANS fluorescence measurements and PK accessibility experiments (see Fig. 6) show that the C-terminal His-tags exert a more pronounced effect on Hsp31 tertiary structure at 45°C. In fact, we found that like His6-Hsp31, Hsp31-His6 exhibits no chaperone activity at elevated temperatures (see Fig. 3). Considering the proximity of the Hsp31 C-terminus to the linker and given the fact that a similar loss of chaperone function occurs when this flexible segment is immobilized onto the core of the protein via disulfide crosslinking,19 the most likely explanation for our results is that the C-terminal extensions interfere with linker movement and precludes the exposure of underlying hydrophobic residues.
Taken together, our results indicate that substrate binding to the hydrophobic bowl is a first and necessary step for Hsp31 chaperone function at all temperatures and bring additional support to the proposal that efficient stabilization of partially folded substrates under heat-shock conditions requires that they further anneal to the normally shielded hydrophobic patches flanking the bowl.19 From a more pragmatic standpoint, the structural and functional sensitivity of Hsp31 to both N- or C-terminal His-tagging was rather unexpected as affinity tags are generally considered innocuous and often left uncleaved based on the premise that they will not affect protein structure, function, or ability to crystallize.23–27 We note that although the original Hsp31 structure was solved using the C-terminally His-tagged version of the protein,14 its Cα trace is superimposable with those of subsequent structures obtained with untagged Hsp31.7,20 While this is consistent with our observation that C-terminal tags have little influence on Hsp31 conformation at low temperatures, the story may have been different if crystallization trials had been conducted with His6-Hsp31. In this respect, it is worth pointing out that a search for the hexahistidine (HHHHHH) motif in the Protein Data Bank reveals that more than 5800 structures have been solved using His-tagged proteins. How many of these structures are impacted by the presence of His tags remains unknown.
Materials and Methods
DNA manipulations
Plamid pNT-hchA, a pET28+ (Novagen) derivative encoding His6-Hsp31, was described elsewhere.19 To construct Hsp31-His6, the hchA gene was amplified using primers 5′-TAATACGACTCACTATAGGG-3′ and 5′-CCACTCGAGACCCGCGTAAGCTGC-3′ and pKV111 plasmid DNA13 as a template. The PCR-amplified fragment was digested with NdeI and XhoI and cloned in the same sites of pET22b+ (Novagen) to yield pCT-hchA. Gene integrity was verified by DNA sequencing.
Protein purification
BL21(DE3) cells (Novagen) harboring the various Hsp31 expression plasmids were grown at 37°C in 250 mL of LB medium supplemented with 50 μg/mL neomycin or 100 μg/mL carbenicillin. Cultures were induced with 1 mM IPTG at A600 ≈ 0.4 and incubated to A600 ≈ 4.0. Cells were sedimented by centrifugation at 8000g for 10 min, resuspended in 50 mM sodium phosphate pH 8.0, and disrupted with a French press at 10,000 psi. Insoluble material was removed by centrifugation at 15,000g for 15 min. Imidazole (35 mM) and NaCl (300 mM) were added to the supernatant, which was loaded onto a 10 mL Ni2+-NTA column (Qiagen). Proteins were eluted in 30 mL of phosphate buffer containing 300 mM NaCl and 250 mM imidazole. Samples were dialyzed for 24 h at 4°C against 100 mM Tris-HCl, pH 7.5. To prepare cl-Hsp31, purified His6-Hsp31 was incubated with 300 μL thrombin-agarose (Sigma) for 6 h at room temperature followed by extensive dialysis against 100 mM Tris-HCl, pH 7.5. Complete removal of the tag was confirmed by immunoblotting with anti-His6 antibodies (Novagen). To prepare the MGSSHHHHHHSSGLVPA peptide, His6-Hsp31 was digested with thrombin-agarose, and the supernatant was loaded onto a Ni2+-NTA column. The purified tag was eluted as above and desalted on a G-50 column. In all cases, purity was greater than 97%. Protein concentrations were determined using the Coomassie Dye Binding Protein Assay Kit (Sigma).
Activity assays and aggregation suppression experiments
The ability of Hsp31 variants (2.4 μM based on protomers) to support the reactivation of MDH (0.4 μM) from the GuHCl-unfolded state or that of CS (0.4 μM) from the urea-unfolded state was determined after 3-h (MDH) or 30-min (CS) incubation at room temperature as previously described.13,19 Reactivation yields do not increase after these time periods. The ability of Hsp31 variants (2.4 μM) to support the reactivation of MDH (0.4 μM) or CS (0.4 μM) denatured by 30-min incubation at 45°C was determined 30 min after transfer of the reaction mixtures to 23°C as described.13,19 All samples were assayed in triplicate, and error bars were obtained for three independent experiments. ADH (0.4 μM) aggregation suppression experiments by Hsp31 variants (1.2 μM) at 41.5°C were conducted as described.13
Spectroscopy
CD spectra of Hsp31 variants (10 μM in 10 mM sodium phosphate, pH 7.5) were recorded on a thermostated Jasco J-720 spectropolarimeter at 23 and 45°C using a 1-mm pathlength cuvette. Intrinsic tryptophan fluorescence experiments were conducted with 0.4 μM of Hsp31 in 1 mL of 100 mM sodium phosphate, pH 7.5 supplemented with 5 mM MgCl2. The buffer was preincubated at 45°C for high-temperature experiments. Fluorescence emission spectra were collected after 15-min incubation in a thermostated Hitachi F4500 spectrophotometer with an excitation wavelength of 295 nm and slit widths set at 5 nm. For bis-ANS fluorescence experiments, Hsp31 variants (0.8 μM) in 1 mL of 150 mM Tris-HCl, pH 7.5, 10 mM KCl, and 5 mM MgCl2 were held at 23°C or 45°C for 5 min and supplemented with 12 μM of bis-ANS. Samples were transferred to a thermostated Hitachi F4500 spectrophotometer and emission intensities at 477 nm following excitation at 340 nm were measured after 15-min incubation. Readings were taken in triplicates and data corrected for the emission of buffer supplemented with 12 μM bis-ANS.
PK digestion
Hsp31 variants (15 μg) were incubated at 37 or 45°C with 5 μg of PK in 50 μL of 100 mM Tris-HCl, pH 7.5. Aliquots (8 μL) were removed at the indicated time points and immediately heated at 95°C for 5 min in SDS-PAGE loading buffer. Samples were fractionated on 12% minigels, and the intensity of the Hsp31 bands was quantified by videodensitometric analysis using Image J.
References
- 1.Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22:1399–1408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- 2.Dougan DA, Mogk A, Bukau B. Protein folding and degradation in bacteria: to degrade or not to degrade? That is the question. Cell Mol Life Sci. 2002;59:1607–1616. doi: 10.1007/PL00012487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 4.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 5.Winter J, Jakob U. Beyond transcription: new mechanisms for the regulation of molecular chaperones. Crit Rev Biochem Mol Biol. 2004;39:297–317. doi: 10.1080/10409230490900658. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Ringe D, Wilson MA, Ondrechen MJ. Identification of functional subclasses in the DJ-1 superfamily proteins. PLoS Comput Biol. 2007;3:e10. doi: 10.1371/journal.pcbi.0030010. [DOI] [PubMed] [Google Scholar]
- 7.Lee SJ, Kim SJ, Kim IK, Ko J, Jeong CS, Kim GH, Park C, Kang SO, Suh PG, Lee HS, Cha SS. Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J Biol Chem. 2003;278:44552–44559. doi: 10.1074/jbc.M304517200. [DOI] [PubMed] [Google Scholar]
- 8.Malki A, Caldas T, Abdallah J, Kern R, Eckey V, Kim SJ, Cha SS, Mori H, Richarme G. Peptidase activity of the Escherichia coli Hsp31 chaperone. J Biol Chem. 2005;280:14420–14426. doi: 10.1074/jbc.M408296200. [DOI] [PubMed] [Google Scholar]
- 9.Malki A, Kern R, Abdallah J, Richarme G. Characterization of the Escherichia coli YedU protein as a molecular chaperone. Biochem Biophys Res Commun. 2003;301:430–436. doi: 10.1016/s0006-291x(02)03053-x. [DOI] [PubMed] [Google Scholar]
- 10.Mujacic M, Bader MW, Baneyx F. Escherichia coli Hsp31 functions as a holding chaperone that cooperates with the DnaK-DnaJ-GrpE system in the management of protein misfolding under severe stress conditions. Mol Microbiol. 2004;51:849–859. doi: 10.1046/j.1365-2958.2003.03871.x. [DOI] [PubMed] [Google Scholar]
- 11.Mujacic M, Baneyx F. Regulation of Escherichia coli hchA, a stress-inducible gene encoding molecular chaperone Hsp31. Mol Microbiol. 2006;60:1576–1589. doi: 10.1111/j.1365-2958.2006.05207.x. [DOI] [PubMed] [Google Scholar]
- 12.Mujacic M, Baneyx F. Chaperone Hsp31 contributes to acid resistance in stationary phase Escherichia coli. Appl Environ Microbiol. 2007;73:1014–1018. doi: 10.1128/AEM.02429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sastry MSR, Korotkov K, Brodsky Y, Baneyx F. Hsp31, the Escherichia coli yedU gene product, is a molecular chaperone whose activity is inhibited by ATP at high temperatures. J Biol Chem. 2002;277:46026–46034. doi: 10.1074/jbc.M205800200. [DOI] [PubMed] [Google Scholar]
- 14.Quigley PM, Korotkov K, Baneyx F, Hol WGJ. The 1.6-Å crystal structure of the class of chaperones represented by Escherichia coli Hsp31 reveals a putative catalytic triad. Proc Natl Acad Sci USA. 2003;100:3137–3142. doi: 10.1073/pnas.0530312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du X, Choi I-G, Kim R, Wang W, Jancarik J, Yokota H, Kim S-H. Crystal structure of an intracellular protease from Pyrococcus horikoshii at 2-Å resolution. Proc Natl Acad Sci USA. 2000;97:14079–14084. doi: 10.1073/pnas.260503597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halio SB, Blumentals II, Short SA, Merrill BM, Kelly RM. Sequence, expression in Escherichia coli, and analysis of the gene encoding a novel intracellular protease (PfpI) from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol. 1996;178:2605–2612. doi: 10.1128/jb.178.9.2605-2612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 18.Quigley PM, Korotkov K, Baneyx F, Hol WGJ. A new native EcHsp31 structure suggests a key role of structural flexibility for chaperone function. Protein Sci. 2004;13:269–277. doi: 10.1110/ps.03399604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sastry MSR, Quigley PM, Hol WGJ, Baneyx F. The linker-loop region of Escherichia coli chaperone Hsp31 functions as a gate that modulates high-affinity substrate binding at elevated temperatures. Proc Natl Acad Sci USA. 2004;101:8587–8592. doi: 10.1073/pnas.0403033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, Liu D, Kaluarachchi WD, Bellamy HD, White MA, Fox RO. The crystal structure of Escherichia coli protein YedU reveals three potential catalytic active sites. Protein Sci. 2003;12:2303–2311. doi: 10.1110/ps.03121403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrade MA, Chacón P, Merelo JJ, Morán F. Evaluation of secondary structure of proteins from UV circular dichroism using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 22.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 23.Bucher MH, Evdokimov AG, Waugh DS. Differential effects of short affinity tags on the crystallization of Pyrococcus furiosus maltodextrin-binding protein. Acta Crystallogr D Biol Crystallogr. 2002;58:392–397. doi: 10.1107/s0907444901021187. [DOI] [PubMed] [Google Scholar]
- 24.Jenny RJ, Mann KG, Lundblad RL. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 25.Mohanty A, Wiener MC. Membrane protein expression and production: effects of polyhistidine tag length and position. Protein Expr Purif. 2004;33:311–325. doi: 10.1016/j.pep.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 26.Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 27.Waugh DS. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]