

Abstract
We describe a method for studying quantitative changes in accessibility of surface lysine residues of the PB1 subunit of the influenza RNA polymerase as a result of association with the PA subunit to form a PB1-PA heterodimer. Our method combines two established methods: (i) the chemical modification of surface lysine residues of native proteins by N-hydroxysuccinimidobiotin (NHS-biotin) and (ii) the stable isotope labeling of amino acids in cell culture (SILAC) followed by tryptic digestion and mass spectrometry. By linking the chemical modification with the SILAC methodology for the first time, we obtain quantitative data on chemical modification allowing subtle changes in accessibility to be described. Five regions in the PB1 monomer showed altered reactivity to NHS-biotin when compared with the [PB1-PA] heterodimer. Mutational analysis of residues in two such regions—at K265 and K481 of PB1, which were about three- and twofold, respectively, less accessible to biotinylation in the PB1-PA heterodimer compared with the PB1 monomer, demonstrated that both K265 and K481 were crucial for polymerase function. This novel assay of quantitative profiling of biotinylation patterns (Q-POP assay) highlights likely conformational changes at important functional sites, as observed here for PB1, and may provide information on protein–protein interaction interfaces. The Q-POP assay should be a generally applicable approach and may detect novel functional sites suitable for targeting by drugs.
Keywords: Q-POP assay, influenza virus polymerase, PB1-PA heterodimer, SILAC, NHS-biotin, LC-MS/MS
Introduction
Many biological functions are performed by multimers of different protein subunits. To fully describe and even to begin to understand the function of such multimeric complexes, high-resolution X-ray crystallography is required. However, for many protein complexes, such as the influenza RNA polymerase—a heterotrimer of PB1, PB2, and PA subunit—such data has not yet been obtained. Furthermore, crystallographic data may not necessarily reflect the functional states of proteins in solution. In the absence of such high resolution data on the heterotrimeric complex, it is of interest to explore biochemical methods of obtaining information on the topology of PB1 in a monomeric state, which in this article we compare with that of PB1 in the well-characterized PB1-PA heterodimer.1–6
Among biochemical methods to identify functionally important features of protein topology, the specific chemical modification of functional groups of amino acid residues exposed at the surfaces of proteins is a well-established approach.7 One particularly informative approach is the biotinylation of solvent-accessible lysine residues using amine-specific biotinylated derivatives of NHS-(N-hydroxysuccinimide) esters as labeling reagents.8 Lysine residues may be accessible to NHS-biotin (the biotinylation reagent) if a protein is in a monomeric state, but may be shielded if the protein is bound to an interaction partner. Sequencing of biotinylated peptides generated through proteolytic cleavage of the protein—typically by mass spectrometry—is required to assess differences in the extent of biotinylation of the monomeric protein, compared with the monomer bound to an interaction partner. Ideally, a biotinylated peptide would be present in the monomer, whereas the same peptide would be absent from the bound sample. A major drawback of this approach is that more subtle, but still potentially important, changes in accessibility may be missed.
Quantitative proteomics techniques such as stable isotope labeling of amino acids in cell culture (SILAC)9 can detect subtle changes of peptide abundances, but have not yet been exploited to address the effects of protein–protein interaction on protein conformation. To close this gap in the analytical toolbox, we report here a novel strategy of quantitative profiling of biotinylation patterns (Q-POP-assay). Our method involves the specific biotinylation of solvent-accessible lysine residues combined with SILAC using 12C6-lysine versus 13C6-lysine as the stable isotope label. Specifically, the Q-POP assay can yield quantitative information on surface residues that have altered reactivity as a result of protein–protein interactions. This can form the basis of further studies on a given protein using molecular biology-based techniques.
To develop and demonstrate the analytical power of our approach, we compared the biotinylation pattern of the polymerase basic protein 1 (PB1) subunit of the influenza A virus RNA polymerase in its monomeric form with the biotinylation pattern of PB1 when bound to the polymerase acidic protein (PA) subunit of the polymerase as a heterodimer. Influenza A viruses are a major threat to human health.10 In particular, H5N1 avian virus strains are of growing concern in this respect because they have the potential to cause a human “bird flu” pandemic in the future. Very recently, in April 2009, evidence of a new H1N1 human pandemic (swine flu) is emerging—emanating from Mexico. This may be the first pandemic of the 21st century. Resistance to existing antivirals, such as oseltamivir (Tamiflu), has already been reported in H5N1 strains, which have infected people giving urgency to the need to develop new antivirals.10 The influenza A virus RNA polymerase is a possible new target for antiviral drugs.11–13 For this purpose, a detailed understanding of structure-function relationships in polymerase subunits is of major interest.
The influenza virus RNA-dependent RNA polymerase is a heterotrimeric complex catalyzing both viral RNA transcription (vRNA → mRNA) and replication (vRNA → cRNA; cRNA → vRNA) in the nucleus of infected cells.14 The polymerase complex is composed of three subunits, PB1, polymerase basic protein 2 (PB2), and PA, and all three subunits are generally believed to be essential for polymerase function.14 The PB1 subunit plays a central role in the catalytic activity of the viral polymerase. The PB2 subunit binds the cap structure of host mRNAs, which is required for the initiation of viral RNA transcription. The PA subunit is known to be involved in a number of functions including promoter binding, cap binding, endonuclease activity, and chain elongation, amongst others.14–16 Recently, structural and functional studies have demonstrated that the N-terminal domain of PA is an endonuclease,17,18 crucial for the function of the polymerase complex in “stealing” cap structures from host mRNAs.18
During viral infection, the PB1, PB2, and PA subunits of the influenza RNA polymerase are synthesized in the cytoplasm of infected cells as independent protein subunits. They must be transported into nucleus and assembled into a trimeric complex before further assembly with influenza nucleoprotein and viral RNA into a enzymatically active, ribonucleoprotein complex.14 Recent work has suggested that the trimeric complex is assembled by first forming a PB1-PA heterodimer in the cytoplasm.1–3 The heterodimer is then targeted to the nucleus where the heterodimer complexes with PB2 to form the trimeric complex.1 The PB1-PA heterodimer is capable of binding to the viral RNA promoter.2,5
No detailed structural data of the PB1-PA heterodimer or of the PB1-PA-PB2 heterotrimeric polymerase complex is available yet, except for low-resolution electron microscopy data of the heterotrimer suggesting a relatively compact structure.19 However, some of the protein–protein interactions between the individual polymerase subunits are known from biochemical studies, suggesting that PB1 is the core of the polymerase complex, because it interacts with both PA and PB2.4,6,20 A short peptide at the N-terminus of PB1 is known from biochemical and recent X-ray structural studies to interact with the C-terminal region of PA,11–13,21,22 whereas no direct interaction has been detected between PB2 and PA. High-resolution X-ray structural data are also available on short regions near the C-terminus of PB223,24 and on the cap-binding domain of PB2.25
Here, using the novel Q-POP assay, we have identified sites in the PB1 subunit that undergo changes in chemical reactivity presumably because of conformational changes upon binding to the PA subunit. We show that two of these sites, which are novel in the influenza polymerase literature, are crucial for polymerase function.
Results
Isolation of equal amounts of PB1 monomer and PB1-PA heterodimers from SILAC-labeled transfected cells
Transient transfection of human embryonic kidney 293T cells has proved to be a suitable system for expression and partial purification (see Materials and Methods section) of individual influenza polymerase monomers, heterodimers, and heterotrimeric complexes of the PB1, PB2, and PA.2 Figure 1 illustrates our new Q-POP approach to study the interaction of two proteins, exemplified by the study of the interaction of the PB1 and PA subunits of the influenza A virus A/WSN/33 RNA polymerase. We anticipated that differences in the PB1 biotinylation patterns between the PB1 monomer and the PB1-PA heterodimer may be more subtle than all-or-nothing effects, and thus applied SILAC9 to introduce stable isotope labels into the proteins, in order to be able to obtain quantitative abundance ratios of biotinylated peptides from the two samples. In addition, the introduction of SILAC into the experimental workflow enabled us to mix both samples before LC-MS/MS analysis and analyze them in one run per experiment (see Fig. 1). Among the various SILAC labeling systems available, we chose the 12C6-lysine versus 13C6-lysine system because it directly reports the abundance ratios of biotinylated peptides.
Figure 1.

The quantitative profiling of biotinylation patterns method (Q-POP)—an experimental strategy for studying interactions between influenza virus polymerase subunits PB1 and PA. The PB1-TAP subunit was expressed in 293T cells cultured in 12C6-lysine-containing medium, and the PB1-TAP-PA heterodimer was expressed in 13C6-lysine-containing medium. They were separately partially purified, followed by in vitro biotinylation with NHS-biotin. They were then mixed at ∼1:1 ratio and concentrated by precipitation and separated on SDS-PAGE. The band containing both 12C6-lysine- and 13C6-lysine-labeled PB1 was excised followed by in-gel trypsin digestion. The biotinylated peptides were purified and finally analyzed by LC-MS/MS.
Briefly, 293T cells that had been passaged in 12C6-lysine (light lysine)-containing medium were transfected with an expression plasmid for PB1-TAP, that is, PB1 tagged with a C-terminal tandem affinity purification (TAP) tag. In parallel, 293T cells passaged in 13C6-lysine (heavy lysine)-containing medium were transfected with plasmids expressing both PB1-TAP and PA. At 40-h post-transfection, the recombinant PB1-TAP monomers and PB1-TAP-PA dimers were separately purified from cell lysates by IgG Sepharose affinity chromatography, a method that relies on the affinity of the IgG constant region with the protein A domains in the TAP tag (see Materials and Methods section). The PB1-TAP monomers and PB1-TAP-PA heterodimers were separately biotinylated and then mixed in approximately equal amounts before concentration and separation of PB1-TAP by SDS-PAGE from PA and other proteins, for example, Hsp90. PB1-TAP and PA were present in comparable amounts (see Fig. 2) as expected from their known association in dimers, in line with our previous findings.2 Moreover, the yield of PB1-TAP monomer was similar to that of the PB1-TAP in the PB1-TAP-PA heterodimer. This confirmed that the samples were suitable for subsequent analysis.
Figure 2.

PB1 and PA polymerase subunits before and after biotinylation and mixing (A) partially purified PB1-TAP (L) and PB1-TAP-PA (H) before biotinylation, silver-stained, (B) mixture (L + H) of light PB1-TAP and heavy PB1-TAP-PA after biotinylation and concentration by precipitation, coomassie blue-stained. The positions of the PB1-TAP, PA, and Hsp90 bands are indicated on 7% SDS-PAGE.
Identification of SILAC pairs corresponding to biotinylated peptides
After in-gel trypsin digestion followed by extraction, peptides were analyzed by LC-MALDI MS/MS (see Fig. 1). The assignment of biotinylated peptides was guided by the observation of characteristic biotinyl-lysine-derived immonium ions present in the MS/MS spectra (see Fig. 3). In the MS/MS spectra shown, these ions showed up at m/z = 310.253 [Fig. 3(A)] and m/z = 326.250 [Fig. 3(B)]. These corresponded to fragment ions of the calculated mass of 310.159 Da for the light biotinylated lysine immonium ion after a loss of ammonia,26 and an additional species most likely due to oxidation of the biotinylated lysine yielding an ion of calculated mass of 326.154 Da for the light version. Heavy counterparts of these ions are also annotated in Figure 3. The calculated masses for the heavy species are 315.176 Da for the heavy biotinylated lysine immonium ion after loss of ammonia and 331.171 Da for the heavy oxidized species (see also Supporting Information Figure). The mass difference (5 Da) in these pairs of ions further confirms their assignment as immonium ions, because they lack the carbonyl carbon atom. Their high intensities are consistent with the presence of a fixed positive charge on a nitrogen atom,26 and references therein.
Figure 3.

Characteristic fragment ions derived from immonium ions of biotinylated lysine. (A) MS/MS spectra of peptide 119VDKLTQR126 of PB1 with biotinylated K121 (MH+ = 1142.599 Da, SILAC light), (B) MS/MS spectrum of the corresponding peptide with an oxidized biotinylated K121 (MH+ = 1158.594 Da). b-ions are derived from the peptide N-termini, y-ions are derived from the C-termini.
We were able to assign 40 SILAC pairs to biotinylated peptides derived from PB1. These covered a total of 23 lysine residues out of 48 within the PB1 amino acid sequence. The majority of lysine residues in the SILAC pairs observed showed no significant change of accessibility to biotinylation. However, seven lysine residues in SILAC pairs displayed significant abundance changes between the PB1 monomer and PB1-PA heterodimer sample. Figure 4 shows three examples of SILAC pairs, specifically K265 that displayed decreased accessibility to biotinylation in the PB1-PA heterodimer [Fig. 4(A)], specifically K121 [Fig. 4(B), showing increased accessibility in the heterodimer], and K737 [Fig. 4(C), no change in accessibility between monomer and heterodimer]. A comprehensive list of SILAC pairs of biotinylated peptides and light/heavy ratios is given in the Supporting Information Table. We only included peptides for which we also obtained unambiguous MS/MS sequencing data. No peptide assignment was allowed on the basis of peptide mass alone. This full MS/MS data for one experiment are given in the Supporting Information Figure.
Figure 4.
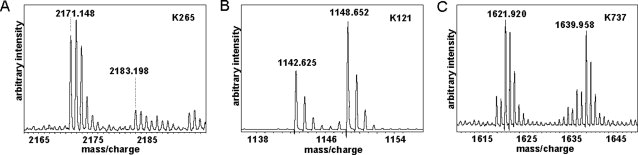
Examples of SILAC pairs with (A) decreased accessibility, that is, peptide SICEKLEQSGLPVGGNEK modified at K265, (B) increased accessibility, that is, peptide VDKLTQGR modified at K121, and (C) similar accessibility, that is, peptide IKKEEFTEIMK modified at K737 to biotinylation in PB1-PA heterodimers compared with monomeric PB1.
Lysine residues 121, 265, 288, 471, and 479–481 display altered accessibility to biotinylation upon PB1-PA heterodimerization
Table I summarizes the light to heavy abundance ratios of 10 biotinylated peptides from PB1 that displayed a significant change in abundance in the comparison of the PB1 monomer with the PB1-PA heterodimer. The mean abundance ratio of all observable SILAC pairs was 1.25 (±0.4) as determined by the quantitation software (see Materials and Methods section). All the peptides listed in Table I had an abundance ratio statistically different (confidence interval > 95%) from the mean value. The abundance of the peptides that contained K265 was reduced more than threefold in the heterodimer samples compared with the PB1 monomers. A similar drastic change was observed for the peptide that contained biotinylated K471. A less extensive, but still significant reduction of abundance was observed for peptides that contained biotinylated lysine residues 288 and 479. The biotinylated peptide in which both K480 and K481 were modified displayed a similar (about twofold) significant reduction in accessibility to biotinylation in the heterodimer. On the other hand, the peptides containing the biotinylated K121 were significantly more abundant in the PB1-PA heterodimer than in the PB1 monomer. Overall, these results suggested that four regions of PB1, that is, near K265, near K288, near K471, and near K479–K481 were less accessible to biotinylation in the heterodimer than in the monomer. A fifth region, the 121 region of PB1, by contrast, became more accessible in the heterodimer (see Discussion section).
Table I.
PB1 Peptides Containing Biotinylated Lysine Residues with Significantly Altered Ratios in the PB1 Monomer (Light Sample) Compared with the PB1-PA Heterodimer (Heavy Sample)
| Peptide sequence | Mass (MH+) of SILAC pair | aa range | Modified residue | Ratio light/ heavya |
|---|---|---|---|---|
| VDKLTQGR | 1142.625/1148.652 | 119–126 | K121 | 0.52 |
| VDKbLTQGR | 1158.625/1164.651 | 119–126 | K121 | 0.44 |
| SICEKLEQSGLPVGGNEK | 2171.148/2183.198 | 261–278 | K265 | 4.38 |
| SICEKbLEQSGLPVGGNEK | 2187.027/2199.042 | 261–278 | K265 | 4.48 |
| KMMTNSQDTEISFTITGDNTK | 2588.280/2600.443 | 288–308 | K288 | 2.24 |
| KMMTNSQDTEISFTITGDNTK | 2604.414/2616.209 | 288–308 | K288 | 2.63 |
| TCKbLLGINMSK | 1522.797/1534.838 | 469–479 | K471 | 4.07 |
| LLGINMSKK | 1229.746/1241.786 | 472–480 | K479 | 2.35 |
| LLGINMSKK | 1245.730/1257.75 | 472–480 | K479 | 2.35 |
| KKSYINR | 1376.720/1388.749 | 480–486 | K480 + 481c | 2.61 |
The mean abundance ratio of all observable SILAC pairs was 1.25 ± 0.4. The data in this table were derived from a single experiment. Further details are given in the Supporting Information Table and Figure.
Lysine biotinylated and oxidized.
Peptide contained both biotinylated lysine and lysine biotinylated and oxidized. Modified lysine residues are given in bold font. M is oxidized methionine.
Mutagenesis indicated that the K265 and K481 regions of PB1 are crucial for polymerase activity
We first selected the K265 region of PB1 for further functional analysis because, of all the peptides characterized in Table I, the peptide with the biotinylated K265 showed the largest difference in accessibility between monomer and dimer. Thus, mutation of K265 and adjacent residues might disrupt the interaction of PB1 and PA. Alternatively, the K265 region of PB1 may be a region where there is a marked conformational change on interaction with PA, thus reducing the accessibility of lysine residues, without disrupting the interaction between PB1 and PA. To distinguish between these hypotheses, we constructed a point mutant at position K265 (K → A), a double mutant at positions 264–265 (EK → AA), and a triple mutant at positions 263–265 (CEK → AAA) in the pcDNA-PB1-TAP expression plasmid. An alignment of part of the PB1 sequences of the related influenza A, B, and C viruses in the K265 region shows that K265 is conserved in influenza A and C viruses, whereas the adjacent residues, E264 and C263, are conserved in all three virus types [Fig. 5(A)]. Using these mutants, we initially tested heterodimer (PB1-PA) formation, which might be compromised if K265 was critical for PB1-PA interaction. It can be seen that the three PB1-TAP mutants pulled down nontagged PA as efficiently as wild-type PB1-TAP [Fig. 5(B), upper panel], suggesting that the mutations at these positions did not disrupt PB1-PA dimerization. This result was confirmed in the purified 3P complexes (see Materials and Methods section), because PA and comigrating PB2 were also present in similar amounts in the mutants and wild type [Fig. 5(B), lower panel].
Figure 5.
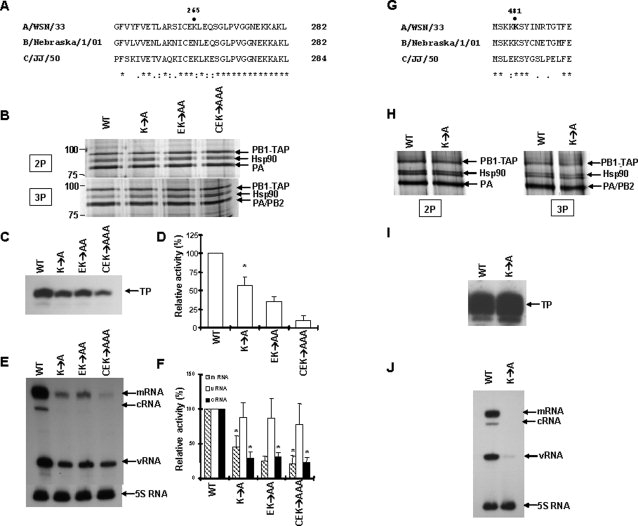
Functional effects of mutations at positions 265 and 481 of PB1 upon PA binding and polymerase activity. (A) Alignment of amino acid sequences of PB1 subunits of influenza A, B, and C viruses near the K265 position. (B) Effects of the single, double, and triple mutant PB1 (see text) on 2P (PB1-PA) dimerization and 3P formation. 293T cells were transfected with plasmids expressing either wild-type PB1-TAP or the three mutant PB1-TAP, together with either a plasmid expressing PA for 2P heterodimerization (upper panel) or plasmids expressing PA and PB2 for 3P formation (lower panel). Protein size markers and positions of PB1-TAP, PA, PB2, and Hsp90 are shown. (C) In vitro ApG-primed transcription mediated by the wild type (WT), using samples from Figure 5(B) (lower panel), and the 3 indicated mutant PB1 polymerases. TP, transcription product. (D) Quantitation of four independent ApG-primed transcription reactions. Values are % of wild-type levels. Error bars = S.D; asterisks (*) indicate the values significantly different from 100% (P < 0.01, Student's t-test). (E) Primer extension assays of influenza vRNA, mRNA, and cRNA isolated from 293T cells expressing either wild-type RNPs or the indicated mutant RNPs (see Experimental Procedures). The positions of transcription products derived from NA reporter mRNA, cRNA, vRNA, and 5S RNA, used as a control, are indicated. (F) Quantitation of four independent primer extension assays. Values were expressed as % of the wild-type values set to 100%. Values significantly different from 100% (P < 0.01, Student's t-test) are indicated by an asterisk (*). (G) Alignment of amino acid sequences of PB1 subunits of influenza A, B, and C viruses near the K481 position. (H) Effect of the single mutant PB1 (K481A, see text) on 2P (PB1-PA) dimerization and 3P formation. A doublet of Hsp90α and β is visible. Additionally, the yields of the Hsp90 doublet appear to be high with respect to the PB1-TAP band, possibly because of partial degradation of PB1-TAP for unknown reasons. (I) In vitro ApG-primed transcription mediated by the wild-type (WT) and indicated mutant PB1 polymerases. (J) In vivo primer extension assay with wild-type and indicated mutant RNPs.
We next analyzed the activity of the purified, mutant recombinant heterotrimeric polymerases in vitro by a standard (see Materials and Methods section) ApG-primed transcription assay resulting in the synthesis of a short, 14 nt run-off transcript from a model viral RNA promoter [Fig. 5(C)]. All three mutants around residue K265 showed significantly less activity than wild type—the triple mutant showing the greatest effect [Fig. 5(D)]. We also reconstituted recombinant ribonucleoprotein complexes from polymerase-expressing plasmids, a nucleoprotein-expressing plasmid, and a neuraminidase reporter in 293T cells and analyzed the activity of the resultant mutant polymerases in vivo compared with wild type by primer extension. This assay27 allows all three viral RNAs—mRNA, vRNA, and cRNA to be analyzed in the same experiment. Figure 5(E) (qualitative) and Figure 5(F) (quantitative) showed that all three PB1 mutants resulted in significantly reduced mRNA and cRNA levels compared with wild type, although vRNA levels remained unaffected for unknown reasons.
We next selected the K479, K480, and K481 region, rather than the other regions identified in Table I, for further functional analysis because this region was represented in three different peptides (Table I). In addition, compared with K265, the light to heavy abundance ratio differed from the mean value by a smaller yet still statistically significant difference (Table I). The K481 (K → A) mutant was selected, because within the K479–K481 region, lysine 481 is the most conserved residue between influenza A, B, and C strains [Fig. 5(G)]. Figure 5(H) showed that the K481A mutation did not disrupt PB1-PA dimerization or formation of the heterotrimeric complex. Although there was no inhibitory effect in the in vitro ApG-primed transcription assay [Fig. 5(I)], the recombinant mutant polymerase completely abolished the activity in the in vivo RNP reconstitution assay [Fig. 5(J)] (see Discussion section).
In summary, the functional data suggested that mutation of K265 and its surrounding amino acids in PB1, or mutation of K481, did not significantly disrupt PB1-PA heterodimerization. However, mutations in the K265 region significantly inhibited polymerase activity both in vitro and in vivo, whereas a mutation at K481 abolished polymerase activity completely in vivo, but not in vitro (see Discussion section).
Discussion
We report here the discovery of novel sites of potential functional importance in the influenza A virus polymerase PB1 subunit obtained by the Q-POP assay—a novel quantitative approach to screen for potential regions in a protein affected by protein–protein interactions. In particular, we demonstrate that the Q-POP assay can identify protein regions that undergo probable conformational changes induced by protein–protein interactions, even with low amounts (microgram quantities), of low-purity (≥20%) protein (see Experimental Procedures). This distinguishes the Q-POP assay not only from traditional protein surface modification assays28 but also from protein folding assays (e.g., hydrogen-deuterium (H/D) exchange).29 The traditional protein surface modification assays are more or less restricted to the detection of large effects on the accessibility of surface amino acid residues.28 This approach is problematic to quantify because an unmodified peptide of an assumed constant abundance is required as an internal reference.30 However, as shown here, induced conformational changes do not necessarily result in large effects, and are not reliably detectable without the introduction of a stable isotope label that permits direct quantitation of changes in accessibility of amino acids. The H/D exchange assay,29 in contrast to the Q-POP assay, requires higher amounts of high-purity protein. Moreover, it requires analysis at nonphysiological pH and gives no information about amino acid side chains.
Furthermore, another obvious advantage of the Q-POP assay is that this method allows an unbiased screening of the protein of interest without prior knowledge of which amino acid residues will undergo a presumed conformational change. This feature contrasts with more classical approaches to study conformational changes in proteins, such as spectroscopy-based approaches based on introduction of spin label moieties on particular amino acid residues.31 On the molecular biology end of the methodological repertoire, a classical but time-consuming approach is to assess the functional importance of all lysine residues in a protein by systematic, site-directed mutagenesis. In the example of the influenza A polymerase studied here, there are 48 lysine residues in the PB1 subunit. The advantage of the Q-POP assay when compared with the classical mutagenesis approach is immediately apparent from our data. Seven lysine residues in five regions of the protein were identified as changing on the interaction with the PA subunit, thus prioritizing these residues for further functional analysis. Thus, any functional effect observed in follow-up mutagenesis studies can be linked to the altered reactivity observed by the Q-POP assay.
In the particular case of PB1, our Q-POP assay revealed that lysine residues K265 and K471 of PB1 became about threefold less accessible to the biotinylation reagent in the PB1-PA heterodimer when compared with the PB1 monomer. Lysine residues K288, as well as K479, K480, and K481, displayed a similar, but less pronounced, though still significant change in accessibility. Remarkably, our functional validation of K265 and K481 by site-directed mutagenesis revealed that the overall interaction of PB1 with PA remained intact in our 2P and 3P assembly assay. However, we should point out that subtle effects on PB1-PA dimerization caused by mutations in PB1 might not be detected in this assay because the PB1 and PA subunits might interact through multiple contacts.22 Thus, a K11D mutation of PB1, in a region of PB1 known to interact through multiple contacts with PA, did not significantly impair PB1-PA interaction.22 Nevertheless, mutations in the K265 region compromised polymerase activity both as a heterotrimeric complex and when present as a ribonucleoprotein complex. Interestingly, the K481A mutation completely inhibited the polymerase activity in vivo, but not in vitro. This discrepancy could be explained by the different nature of the two assays.16 Thus, the K481 mutation might affect elongation, which would not be adequately tested in vitro because this assay only requires extension to give a very short nucleotide product (14 nt). Alternatively, the K481A mutation might result in some cell-specific effects on polymerase activity (e.g., host protein interactions with the viral polymerase).16
It should be noted that our mass spectrometry data (Table I and Supporting Information Table and Figure) are derived from one definitive experiment. Nevertheless, 9 of the 10 peptides with altered light/heavy ratios (see Table I) were either duplicate peptides, albeit with a different methionine or lysine oxidation states, or were in overlapping peptides. Only at lysine 471 was our result based on a single peptide. Moreover, in other repeat experiments (data not shown), we observed alterations in the light/heavy ratios for some of the biotinylated peptides reported in Table I. However, the full set of biotinylated peptides reported in Table I were not observed in these repeat experiments, probably because of technical limitations caused by low final yields of the biotinylated peptides, due either to lower starting amounts of protein, less efficient biotinylation, or lower recoveries of the biotinylated peptides.
Overall, these functional mutagenesis results, taken together with the Q-POP data, suggest that the two regions studied here in detail, both initially identified by the Q-POP assay, play critical roles in fulfilling polymerase functions through perturbation of PB1-PA conformation. However, it remains uncertain whether these two regions are directly or indirectly involved in the actual subunit–subunit interactions. The available evidence suggests that these residues are probably not directly involved. Future high-resolution structural analysis of the heterodimer or heterotrimeric polymerase complex is needed to resolve this.
The Q-POP assay also revealed a significantly increased accessibility of K121 of PB1 to the biotinylation reagent in the PB1-PA heterodimer compared with the PB1 monomer. This is best explained by a conformational change in PB1, induced by PA interaction, that results in the region of PB1 around K121 becoming more exposed in the heterodimer. Alternatively, PB1 could conceivable exist as a dimer or a higher order structure, by itself, in which case K121 may be shielded; in the presence of PA, PB1 would preferentially heterodimerize so that K121 of PB1 becomes more exposed. Other possibilities are that K121 becomes more accessible in the heterodimer because host proteins that copurify with PB1 monomer, for example, Hsp9032–34 or RanBP5,1 or other unknown host factors might be preferentially displaced by PA in the formation of the PB1-PA heterodimer. Thus, in theory, the Q-POP assay might give information not only of PB1-PA interactions but also of interactions sites in PB1 that may be involved in interaction with host proteins, such as Hsp90 or RanBP5. However, this scenario is unlikely because no obvious differences in copurified host proteins (e.g., Hsp90, RanBP5) were observed between partially purified PB1-PA and PB1 samples.2
All the regions identified in Table I, including PB1 K265, are distinct from regions of PB1 previously known to affect function.11,22,35–37 However, K265 is not far from a vRNA promoter-binding site between residues 233 and 24938 and might, we speculate, affect promoter binding, although direct experimental evidence would be needed to establish this. Residues K479–481, however, lie within the region 474–484 encompassing motif D common to many RNA-dependent RNA polymerases.36 This motif D is likely to be involved in some aspect of influenza RNA polymerase function. The fact that the reactivity of PB1 residues K479, K480, and K481 to biotinylation are all altered on binding PA suggests a new function for PA in modulating polymerase function. However, it is still possible that not all residues with altered reactivity will have functional importance; they may be spatially near to a functionally important conformational change occurring upon binding to PA but are not directly involved.
We should also point out that some lysine residues may be buried and therefore inaccessible to the chemical reagent. In addition, some functionally important sites may either not contain lysine or remain undetected. In the case of PB1 tested here, we could not detect the lysine residue K11, present in a N-terminal portion of PB1 that is known to be required for PA interaction and polymerase activity.11–13,22 This limitation is due to the inability of trypsin to cleave C-terminal of biotinylated lysine residues and the fact that this region of PB1 lacks lysine residues. After K11, there are no further lysine residues in the N-terminal region of PB1 until K121, and the first arginine residue is at R45. It may be possible to overcome this problem of uneven distribution of lysines by using a different protease for proteolytic cleavage. In our study, we tested endoproteinase GluC for this purpose, but this did not yield satisfactory amounts of peptides in our hands (data not shown).
In summary, the Q-POP assay described here should be generally applicable to study the effect of protein–protein interactions on protein structure, even with small amounts of less than 100% purity, because of its ability to detect and quantify subtle effects combined with its unbiased screening character. This method bridges the gap between the identification of partners of a given protein and the detailed analysis of the function of single amino acids in that protein. Moreover, the method will potentially facilitate the search for drug target sites in a pharmaceutically relevant protein, such as the polymerase PB1 of influenza A viruses. We speculate that some of the peptides identified in Table I might be potential antivirals. Introduction of those, or related, peptides intracellularly might inhibit influenza virus replication because they might compete with PB1 in the correct assembly of the PB1-PA heterodimer, or its subsequent further assembly into a functional trimeric complex.
Materials and Methods
Plasmids
The plasmids coding for PB1, PB2, PA, NP (nucleoprotein), and NA (neuraminidase) of influenza virus A/WSN/33 (pcDNA-PB1-TAP, pcDNA-PA, pcDNA-PB2, pcDNA-NP, pPOLI-NA-RT) have been described previously.2,39 PB1 is expressed in this system as a fusion protein with a TAP tag.40
Preparation of SILAC-labeled recombinant PB1 monomers and PB1-PA heterodimers
Human embryonic kidney 293T cells were passaged in DMEM (SILAC Kit, Invitrogen) containing either 12C6-lysine or 13C6-lysine for six generations and then transfected with plasmids expressing influenza virus polymerase subunits PB1-TAP or PB1-TAP in combination with PA to obtain partially purified PB1-TAP monomer (labeled with 12C6-lysine) and PB1-PA heterodimer (labeled with 13C6-lysine). The samples were partially purified by using the TAP method.1,2,40 Samples were analyzed by SDS-PAGE, followed by silver-staining with SilverXpress (Invitrogen). The purity of the PB1 and PB1-PA was estimated as about 20%. Major contaminants are tobacco etch virus protease, Hsp90, Hsp70, and RanBP5.1,32–34,41
Biotinylation and isolation of peptides
Biotinylation was performed with the N-hydroxysuccinimidobiotin (NHS-biotin) (Pierce) according to the manufacturer's instructions. In brief, freshly made up 10 mM NHS-biotin in anhydrous DMSO was added to (typically) 500 μL partially purified PB1 or PB1-PA heterodimer in 0.15 M NaCl, 1 mM DTT, 0.01 M Hepes, pH 8.0 to 0.2 mM final concentration for 30 min at room temperature. After quenching (2 mM lysine), protein was precipitated with 80% isopropanol and separated by 7% SDS-PAGE. After detection with Simply Blue Coomassie (Invitrogen), the PB1-TAP band (in low microgram quantities) was excised and in-gel digested with trypsin (Promega).1 The peptides were resuspended in 2× phosphate buffered saline for avidin affinity purification of biotinylated peptides with monomeric avidin cartridges (Applied Biosystems) according to the manufacturer's instructions.
LC MALDI MS/MS analysis
Peptides were resuspended in 5% acetonitrile/0.1% trifluoroacetic acid (TFA) and injected into a nanoflow liquid chromatography system (Agilent 1100). Peptides were separated on a Zorbax SB-300 C-18 nanoflow column (0.075 × 150 mm, Agilent) using an increasing concentration of solvent B (90% acetonitrile, 0.085% TFA) at a flow rate of 300 nL/min. The eluate was spotted with a spotting device onto an anchorchip target (600 μm anchor diameter). The spotting time was 30 s per position, and matrix (1 mg/mL α-cyano-4-hydroxycinnamic acid in 70% isopropanol, 10% acetone, 0.1% TFA) was cospotted. A peptide standard was spotted afterward on the dedicated positions for external calibration. Automated MS and MS/MS acquisition was controlled via the Software Warp-LC (Bruker). The spectra were acquired on an Ultraflex TOF/TOF mass spectrometer (Bruker Daltonics). MS spectra were acquired, annotated, and a compound list for MS/MS was generated. Potential SILAC pairs were quantified at this level. Peptides were then identified from the MS/MS spectra using Mascot (Matrix Science) searching against the NCBI nonredundant database, restricted to virus proteins. Subsequently, in silico digests were matched against the compound list, and any further potential peptides that had not been fragmented automatically were chosen for manual MS/MS analysis. Criteria for the acceptance of peptide hits besides the mass accuracy parameters (75 ppm, two missed cleavage sites permitted) were as follows: (1) the presence of the signatory biotinyl-lysine immonium ion, (2) no hit for a cellular protein, and (3) satisfactory coverage of the visible fragment ions by the proposed PB1 peptide sequence. Quantitation and statistical evaluation of the SILAC pairs assigned on this basis was performed with Warp-LC using its default statistical significance settings, with a confidence interval >95% as a cutoff. The mean abundance ratio of all biotinylated SILAC pairs was 1.25 (±0.4) in the experiment reported in Table I and the Supporting Information Table and Figure.
Primer extension assay
Transfection of human kidney 293T cells with pcDNA-PB1-TAP (wild type or mutant), pcDNA-PB2, pcDNA-PA, and pcDNA-NP (of influenza virus A/WSN/33) and a reporter plasmid, RNA isolation and primer extension were performed as described earlier, except that a pPOLI-NA reporter plasmid27,42 was used instead of pPOLI-CAT-RT. RNA was isolated 24 h post-transfection. For quantitation, values were standardized to 5S RNA.16
ApG-primed transcription
In vitro reactions were performed with recombinant wild-type or mutant polymerase and short 15 and 14 nt synthetic 5′ and 3′ ends of a model vRNA promoter, as previously described.43
Acknowledgments
The authors thank Julian Robinson for sequencing and Ervin Fodor and Ben Thomas for helpful discussion.
References
- 1.Deng T, Engelhardt OG, Thomas B, Akoulitchev AV, Brownlee GG, Fodor E. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J Virol. 2006;80:11911–11919. doi: 10.1128/JVI.01565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng T, Sharps J, Fodor E, Brownlee GG. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J Virol. 2005;79:8669–8674. doi: 10.1128/JVI.79.13.8669-8674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fodor E, Smith M. The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J Virol. 2004;78:9144–9153. doi: 10.1128/JVI.78.17.9144-9153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez S, Zurcher T, Ortin J. Identification of two separate domains in the influenza virus PB1 protein involved in the interaction with the PB2 and PA subunits: a model for the viral RNA polymerase structure. Nucleic Acids Res. 1996;24:4456–4463. doi: 10.1093/nar/24.22.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MT, Bishop K, Medcalf L, Elton D, Digard P, Tiley L. Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res. 2002;30:429–438. doi: 10.1093/nar/30.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohtsu Y, Honda Y, Sakata Y, Kato H, Toyoda T. Fine mapping of the subunit binding sites of influenza virus RNA polymerase. Microbiol Immunol. 2002;46:167–175. doi: 10.1111/j.1348-0421.2002.tb02682.x. [DOI] [PubMed] [Google Scholar]
- 7.Tsetlin V, Hucho F. Chemische modifikation von proteinen und proteinkomplexen. In: Lottspeich F, Zorbas H, editors. Bioanalytik. Heidelberg: Spektrum-Verlag; 1998. pp. 103–130. [Google Scholar]
- 8.Geoghegan KF. Current protocols in protein science. N.J., USA: Wiley Interscience, Wiley-Blackwell; 2001. Chapter 5. [Google Scholar]
- 9.Ong SE, Foster LJ, Mann M. Mass spectrometric-based approaches in quantitative proteomics. Methods. 2003;29:124–130. doi: 10.1016/s1046-2023(02)00303-1. [DOI] [PubMed] [Google Scholar]
- 10.Abdel-Ghafar AN, Chotpitayasunondh T, Gao Z, Hayden FG, Nguyen DH, de Jong MD, Naghdaliyev A, Peiris JS, Shindo N, Soeroso S, Uyeki TM. Update on avian influenza A (H5N1) virus infection in humans. N Engl J Med. 2008;358:261–273. doi: 10.1056/NEJMra0707279. [DOI] [PubMed] [Google Scholar]
- 11.Ghanem A, Mayer D, Chase G, Tegge W, Frank R, Kochs G, Garcia-Sastre A, Schwemmle M. Peptide-mediated interference with influenza A virus polymerase. J Virol. 2007;81:7801–7804. doi: 10.1128/JVI.00724-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, Ge R, Rao Z, Liu Y. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 13.Obayashi E, Yoshida H, Kawai F, Shibayama N, Kawaguchi A, Nagata K, Tame JRH, Park S-Y. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature. 2008;454:1127–1131. doi: 10.1038/nature07225. [DOI] [PubMed] [Google Scholar]
- 14.Palese P, Shaw M. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Field virology. 5th ed. Philadelphia: Lippincott, Williams, & Wilkins; 2006. pp. 1647–1689. [Google Scholar]
- 15.Hara K, Schmidt FI, Crow M, Brownlee GG. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J Virol. 2006;80:7789–7798. doi: 10.1128/JVI.00600-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maier HJ, Kashiwagi T, Hara K, Brownlee GG. Differential role of the influenza A virus polymerase PA subunit for vRNA and cRNA promoter binding. Virology. 2008;370:194–204. doi: 10.1016/j.virol.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 17.Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 18.Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Ly Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 19.Torreira E, Schoehn G, Fernandez Y, Jorba N, Ruigrok RW, Cusack S, Ortin J, Llorca O. Three-dimensional model for the isolated recombinant influenza virus polymerase heterotrimer. Nucleic Acids Res. 2007;35:3774–3783. doi: 10.1093/nar/gkm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Digard P, Blok VC, Inglis SC. Complex formation between influenza virus polymerase proteins expressed in Xenopus oocytes. Virology. 1989;171:162–169. doi: 10.1016/0042-6822(89)90523-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guu TS, Dong L, Wittung-Stafshede P, Tao YJ. Mapping the domain structure of the influenza A virus polymerase acidic protein (PA) and its interaction with the basic protein 1 (PB1) subunit. Virology. 2008;379:135–142. doi: 10.1016/j.virol.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 22.Perez DR, Donis RO. Functional analysis of PA binding by influenza a virus PB1: effects on polymerase activity and viral infectivity. J Virol. 2001;75:8127–8136. doi: 10.1128/JVI.75.17.8127-8136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuzuhara T, Kise D, Yoshida H, Horita T, Murazaki Y, Nishimura A, Echigo N, Utsunomiya H, Tsuge H. Structural basis of the influenza A virus RNA polymerase PB2 RNA-binding domain containing the pathogenicity-determinant lysine 627 residue. J Biol Chem. 2009;284:6855–6860. doi: 10.1074/jbc.C800224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarendeau F, Crepin T, Guilligay D, Ruigrok RW, Cusack S, Hart DJ. Host determinant residue lysine 627 lies on the surface of a discrete, folded domain of influenza virus polymerase PB2 subunit. PLoS Pathog. 2008;4:e1000136. doi: 10.1371/journal.ppat.1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, Lewis J, Ruigrok RW, Ortin J, Hart DJ, Cusask S. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol. 2008;15:500–506. doi: 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 26.Hung CW, Schlosser A, Wei J, Lehmann WD. Collision-induced reporter fragmentations for identification of covalently modified peptides. Anal Bioanal Chem. 2007;389:1003–1016. doi: 10.1007/s00216-007-1449-y. [DOI] [PubMed] [Google Scholar]
- 27.Fodor E, Mingay LJ, Crow M, Deng T, Brownlee GG. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase promotes the generation of defective interfering RNAs. J Virol. 2003;77:5017–5020. doi: 10.1128/JVI.77.8.5017-5020.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA:tRNA complex by mass spectrometry and a primary amine selective reagent. Proc Natl Acad Sci USA. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 30.Zhao Z, McKee CJ, Kessl JJ, Santos WL, Daigle JE, Engelman A, Verdine G, Kvaratskhelia M. Subunit-specific protein footprinting reveals significant structural rearrangements and a role for N-terminal Lys-14 of HIV-1 integrase during viral DNA binding. J Biol Chem. 2008;283:5632–5641. doi: 10.1074/jbc.M705241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Chase G, Deng T, Fodor E, Leung BW, Mayer D, Schwemmle M, Brownlee G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology. 2008;377:431–439. doi: 10.1016/j.virol.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 33.Mayer D, Molawi K, Martinez-Sobrido L, Ghanem A, Thomas S, Baginsky S, Grossmann J, Garcia-Sastre A, Schwemmle M. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J Proteome Res. 2007;6:672–682. doi: 10.1021/pr060432u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naito T, Momose F, Kawaguchi A, Nagata K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol. 2007;81:1339–1349. doi: 10.1128/JVI.01917-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li ML, Rao P, Krug RM. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J. 2001;20:2078–2086. doi: 10.1093/emboj/20.8.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poch O, Sauvaget I, Delarue M, Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989;8:3867–3874. doi: 10.1002/j.1460-2075.1989.tb08565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerry PS, Willsher N, Fodor E. A cluster of conserved basic amino acids near the C-terminus of the PB1 subunit of the influenza virus RNA polymerase is involved in the regulation of viral transcription. Virology. 2008;373:202–210. doi: 10.1016/j.virol.2007.11.030. [DOI] [PubMed] [Google Scholar]
- 38.Jung TE, Brownlee GG. A new promoter-binding site in the PB1 subunit of the influenza A virus polymerase. J Gen Virol. 2006;87:679–688. doi: 10.1099/vir.0.81453-0. [DOI] [PubMed] [Google Scholar]
- 39.Fodor E, Devenish L, Engelhardt OG, Palese P, Brownlee GG, Garcia-Sastre A. Rescue of influenza A virus from recombinant DNA. J Virol. 1999;73:9679–9682. doi: 10.1128/jvi.73.11.9679-9682.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 41.Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem. 2002;277:45306–45314. doi: 10.1074/jbc.M206822200. [DOI] [PubMed] [Google Scholar]
- 42.Vreede FT, Jung TE, Brownlee GG. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J Virol. 2004;78:9568–9572. doi: 10.1128/JVI.78.17.9568-9572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol. 2002;76:8989–9001. doi: 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]